

Abstract
Early blight of potato is caused by the fungal pathogen Alternaria solani and is an increasing problem worldwide. The primary strategy to control the disease is applying fungicides such as succinate dehydrogenase inhibitors (SDHI). SDHI‐resistant strains, showing reduced sensitivity to treatments, appeared in Germany in 2013, shortly after the introduction of SDHIs. Two primary mutations in the SDH complex (SdhB‐H278Y and SdhC‐H134R) have been frequently found throughout Europe. How these resistances arose and spread, and whether they are linked to other genomic features, remains unknown. For this project, we performed whole‐genome sequencing for 48 A. solani isolates from potato fields across Europe to better characterize the pathogen's genetic diversity in general and understand the development and spread of the genetic mutations that lead to SDHI resistance. The isolates can be grouped into seven genotypes. These genotypes do not show a geographical pattern but appear spread throughout Europe. We found clear evidence for recombination on the genome, and the observed admixtures might indicate a higher adaptive potential of the fungus than previously thought. Yet, we cannot link the observed recombination events to different Sdh mutations. The same Sdh mutations appear in different, non‐admixed genetic backgrounds; therefore, we conclude they arose independently. Our research gives insights into the genetic diversity of A. solani on a genome level. The mixed occurrence of different genotypes, apparent admixture in the populations, and evidence for recombination indicate higher genomic complexity than anticipated. The conclusion that SDHI tolerance arose multiple times independently has important implications for future fungicide resistance management strategies. These should not solely focus on preventing the spread of isolates between locations but also on limiting population size and the selective pressure posed by fungicides in a given field to avoid the rise of new mutations in other genetic backgrounds.
Keywords: agriculture, alternaria solani, fungicide resistance, plant pathology, population genetics – empirical, potato
1. INTRODUCTION
Early blight is one of the major diseases in potato‐growing areas. The causal agent of early blight is the fungal pathogen Alternaria solani. If the weather conditions are favourable and A. solani remains uncontrolled, yield losses can reach up to 40% (Kapsa, 2004; Leiminger & Hausladen, 2012, 2014). Typical symptoms of potato early blight are dark brown to black lesions with concentric rings. In the beginning, the necrotic area is often surrounded by a yellow, chlorotic halo (Rotem, 1994). Due to climatic changes Delgado‐Baquerizo et al. (2020) predicted a global increase of soil‐borne pathogens, for example, Alternaria spp., which would imply increasing disease pressure in the field. The potato crop is the fourth most important field crop in European agriculture based on yield (tons/year), after wheat, maize, and barley (Eurostat 2020). To guarantee the yield, farmers adhere to good agricultural practices, for example, crop rotation and sufficient nutrient supply, but in most cases the application of fungicides is needed.
For early blight control, three main fungicide groups are available: the Quinone outside Inhibitors (QoIs), the Succinate Dehydrogenase Inhibitors (SDHIs), and the Demethylation Inhibitors (DMIs), of which QoIs and SDHIs are most widely used and until recently considered most effective.
The development of resistance or reduced sensitivity to the compounds has been reported in A. solani and other fungi for all of these fungicide groups. DMI resistance appears to be related to changes in expression of the target site, Cyp51, possibly linked with a mutation (Zhang et al., 2020). Mutations in Cyp51 genes are common in other pathogens to which DMIs are applied (Blake et al., 2018; Pereira et al., 2017). For the other two fungicide groups, mutations in the pathogen target sites have already been confirmed in A. solani. The F129L mutation in the cytochrome b of the mitochondrial electron transport complex III (Bartlett et al., 2002) frequently occurs in the field (Leiminger and Hausladen, 2014; Odilbekov et al., 2016). Leiminger and Hausladen (2014) showed a reduced efficacy of azoxystrobin (a QoI) by almost 50% for the F129L‐mutant isolates in vivo in comparison to wild‐type isolates. Concerning the SDHIs, several different mutations have been identified in the subunits b, c, and d of the sdh‐gene in the mitochondrial electron transport complex II: SdhB‐H278Y, SdhB‐H278R, SdhC‐H134R, SdhC‐H134Q, SdhD‐D123E, SdhD‐H133R (Mallik et al., 2014; Metz et al., 2019). For these SDH mutations, a negative impact on fungicide efficiency has also been shown in several studies (Landschoot et al., 2017; Metz et al., 2019).
In a European study, 70% of A. solani isolates sampled between 2014 and 2015 contained one of the SDHI mutations and 40% also had the cytochrome b F129L mutation, leading to QoI resistance and thus possessing a dual fungicide resistance (Landschoot et al., 2017). A more recent, long‐term Swedish field study showed that nearly all Swedish isolates now carry the F129L mutation (Edin et al., 2019). In Germany also nearly all samples carry the F129L mutation and 43% show mutations in SDH subunits (Nottensteiner et al., 2019). In the United States, a study of over 1000 isolates revealed that between 2013 and 2015 the presence of the F129L mutation rose from 92 to 99% and mutations in any of the SDH subunits were also present in 99% of the isolates (Bauske et al., 2017).
The genetic diversity of A. solani in the field is relatively understudied, possibly because A. solani historically has been described as an asexual species and limited variation was expected. Yet, considerable genetic diversity was found in South African isolates using random amplified microsatellite (RAMS) primers (van der Waals et al., 2004). Random amplification of polymorphic DNA (RAPD) profiling confirmed this surprisingly high genetic diversity of field isolates in Germany (Leiminger et al., 2016) or Wisconsin (Weber & Halterman, 2012). Also on tomato in India, a marker‐based study revealed higher within and between state diversity in A. solani than anticipated by the authors (Upadhyay et al., 2019). Barcode sequencing revealed the presence of multiple A. solani haplotypes in a single field (Adhikari et al., 2020).
Odilbekov found that the genetic composition of the pathogen populations in the field changed after fungicide treatment. Their study revealed an increase of genotypes with reduced sensitivity over time (Odilbekov et al., 2019). However, the use of Amplified Fragment Length Polymorphisms (AFLP) limited the resolution of the genetic diversity analysis, and only two main genotypes could be described. Genome‐wide sequencing approaches, such as Genotyping By Sequencing (GBS) or full genome sequence analyses, are very powerful tools that are able to reveal genomic variation that previously remained hidden (Everhart et al., 2020). Indeed, a GBS study revealed that microevolutionary factors might play an important role in population structure of various Alternaria spp. (Adhikari et al., 2019).
Whereas fungicide resistance is on the rise in A. solani, it is not clear whether the causal mutations occur in one or few genetic backgrounds and spread or whether they arose multiple times independently in different genetic backgrounds. Understanding such microevolutionary forces at play in and between populations and their roles in fungicide resistance evolution is important. If mutations arise only once and then spread, this should have implications for management practices, for instance on transportation and movement of tubers and infected material.
Here we combine a genomic diversity study with analyses of fungicide resistance targets. We make use of the recently published A. solani reference genome (Wolters et al., 2018) and a Europe‐wide selection of A. solani samples to study the genetic diversity of the isolates in general and the occurrence of SDHI resistance in a genomic context.
2. METHODS
2.1. Fungal material
Forty‐eight different isolates were collected from five different localities across Europe (Figure 1). All samples were reported to possess the F129L mutation in cytochrome b, associated with QoI resistance. The status of SDHI resistance were partly mixed (reported wild‐type isolates as well as reported mutants) and partly unknown. All isolates were collected between 2014 and 2017 when SDHI resistance was reported to be on the rise, but not fully spread. This would allow screening of genetic backgrounds prior eventual fungicide‐related bottlenecks. Isolates DE_NM014 – DE_NM020 were collected from potato fields in Bavaria, Germany, DE_NM006 – DE_NM013 from fields in Lower Saxony in Germany. SE_EL001 – SE_EL012 isolates originate from Southern Sweden, BE_SL001 – BE_SL008 from fields in Belgium, and RS_ZI001 – RS_ZI008 were collected from two localities in Serbia. US_JW001 – US_JW004 and US_NM022 were collected in the United States. A detailed overview can be found in Table 1. All isolates were sampled in their respective fields from symptomatic potato leaves. Leaves were dried between tissue paper and surface sterilized before placement on Synthetic Nutrient Agar (SNA) (0.2 g/L glucose, 0.2 g/L sucrose, 0.5 g/L MgSO4‐7H2O, 0.5 g/L KCl, 1.0 g/L KH2PO4, 1.0 g/L KNO3, 22.0 g/L agar; 600 µl/L 1 M NaOH). A single spore was collected from each isolate and propagated under sterile condition for further usage.
FIGURE 1.
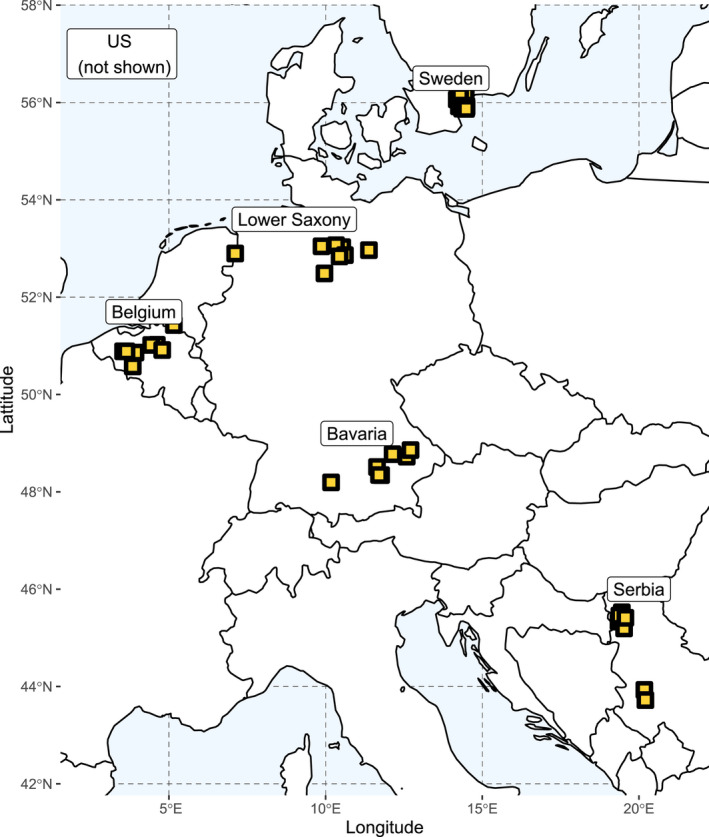
Overview of the sampling locations used in this study
TABLE 1.
Overview of the isolates used in this study
| CODE | Name | SRA Number | Original Name | Locality | Country | Year of Isolation | SDHI Reported | SDHI Detected | QoI | Haplotype Adhikari | Haplotype Multigene | GT group | Potato | Latitude | Longitude | Sequence depth |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BE | SL001 | SRS9529518 | 15.19s | Vollezele | Belgium | 2015 | Unknown | H134R | F129L | AsAlHs4 | MG_H1 | GT7 | Bintje | 50.760587 | 4.025269 | 54 |
| BE | SL002 | SRS9529519 | 14.23s | Leefdaal | Belgium | 2014 | Unknown | wt | F129L | unknown | MG_H1 | no GT | Bintje | 50.848069 | 4.604352 | 87 |
| BE | SL003 | SRS9529520 | 15.43s | Wannegem‐Lede | Belgium | 2015 | Unknown | H134R | F129L | AsAlHs4 | MG_H2 | GT7 | Bintje | 50.891398 | 3.55009 | 49 |
| BE | SL004 | SRS9529521 | 15.183s | Leefdaal | Belgium | 2015 | Unknown | wt | F129L | AsAlHs4 | MG_H1 | GT2 | Bintje | 50.848069 | 4.604352 | 79 |
| BE | SL005 | SRS9529522 | 15.186s | Kasterlee | Belgium | 2015 | Unknown | H278Y | F129L | AsAlHs4 | MG_H1 | GT4 | Fontane | 51.252769 | 4.959916 | 80 |
| BE | SL006 | SRS9529523 | 15.235s | Merksplas | Belgium | 2015 | Unknown | H278R | F129L | AsAlHs4 | MG_H2 | GT6 | Bintje | 51.371243 | 4.877228 | 42 |
| BE | SL007 | SRS9529524 | 15.1s | Leefdaal | Belgium | 2015 | wt | H278Y | F129L | AsAlHs4 | MG_H2 | GT6 | Bintje | 50.848069 | 4.604352 | 69 |
| BE | SL008 | SRS9529525 | 15.20s | Herne | Belgium | 2015 | wt | wt | F129L | AsAlHs4 | MG_H2 | GT2 | Bintje | 50.724535 | 4.040529 | 44 |
| DE | NM006 | SRS9529526 | 676_1 | Rupennest bei Lathen | Lower Saxony | 2015 | wt | wt | F129L | AsAlHs4 | MG_H2 | GT2 | Unknown | 52.87937461 | 7.308617612 | 54 |
| DE | NM007 | SRS9529527 | 687_1 | Immensen/Lehrte | Lower Saxony | 2015 | H134R | H134R | F129L | AsAlHs4 | MG_H1 | GT7 | Unknown | 52.38191091 | 10.01725194 | 55 |
| DE | NM008 | SRS9529529 | 691_1 | Hamerstorf | Lower Saxony | 2015 | H134R | H134R | F129L | AsAlHs4 | MG_H1 | GT7 | Unknown | 52.87658323 | 10.42499062 | 52 |
| DE | NM009 | SRS9529530 | 692_1 | Gusborn | Lower Saxony | 2015 | H134R | H134R | F129L | AsAlHs4 | MG_H1 | GT7 | Unknown | 53.07766577 | 11.19777163 | 43 |
| DE | NM010 | SRS9529531 | 711_2 | Hamerstorf | Lower Saxony | 2015 | wt | H278Y | F129L | AsAlHs4 | MG_H1 | GT3 | Unknown | 52.87658323 | 10.42499062 | 55 |
| DE | NM011 | SRS9529532 | 720_3 | Hamerstorf | Lower Saxony | 2015 | H278Y | H278Y | F129L | AsAlHs4 | MG_H1 | GT3 | Unknown | 52.87658323 | 10.42499062 | 52 |
| DE | NM012 | SRS9529533 | 754_2 | Solltau | Lower Saxony | 2015 | H278Y | H278Y | F129L | AsAlHs4 | MG_H1 | GT3 | Unknown | 52.99580915 | 9.856700578 | 52 |
| DE | NM013 | SRS9529534 | 774_1 | Hamerstorf | Lower Saxony | 2015 | H278Y | H278Y | F129L | AsAlHs4 | MG_H1 | GT3 | Unknown | 52.87658323 | 10.42499062 | 54 |
| DE | NM014 | SRS9529535 | 732_1 | Laberweinting | Bavaria | 2015 | H134R | H134R | F129L | AsAlHs4 | MG_H1 | GT7 | Unknown | 48.78143934 | 12.31529157 | 56 |
| DE | NM015 | SRS9529536 | 736_1 | Hallbergmoos | Bavaria | 2015 | wt | wt | F129L | AsAlHs4 | MG_H2 | GT6 | Unknown | 48.31047191 | 11.72974033 | 54 |
| DE | NM016 | SRS9529537 | 737_1 | Ismaning | Bavaria | 2015 | H278Y | H278Y | F129L | AsAlHs4 | MG_H1 | GT4 | Unknown | 48.24057911 | 11.70475989 | 52 |
| DE | NM017 | SRS9529538 | 739_3 | Feldkirchen | Bavaria | 2015 | wt | wt | F129L | AsAlHs4 | MG_H2 | GT6 | Unknown | 48.83017658 | 12.5302467 | 54 |
| DE | NM018 | SRS9529540 | 746_1 | Aiterhofen | Bavaria | 2015 | wt | wt | F129L | AsAlHs4 | MG_H1 | GT3 | Unknown | 48.8417768 | 12.62818583 | 56 |
| DE | NM019 | SRS9529496 | 749_3 | Freising | Bavaria | 2015 | wt | N75S | F129L | AsAlHs4 | MG_H2 | GT6 | Unknown | 48.39358972 | 11.70637674 | 46 |
| DE | NM020 | SRS9529495 | 615 | Kirchheim | Bavaria | 2014 | wt | wt | F129L | AsAlHs4 | MG_H1 | GT3 | Unknown | 48.32771814 | 10.30486316 | 56 |
| RS | ZI001 | SRS9529505 | 45–1 | Bački Maglić | Serbia | 2016 | wt | wt | F129L | AsAlHs4 | MG_H1 | GT1 | Brooke | 45.367993 | 19.513177 | 67 |
| RS | ZI002 | SRS9529506 | 45–12 | Bački Maglić | Serbia | 2016 | wt | wt | F129L | AsAlHs4 | MG_H1 | GT1 | VR 808 | 45.367993 | 19.513177 | 69 |
| RS | ZI003 | SRS9529517 | 54–1 | Guča | Serbia | 2016 | wt | wt | F129L | AsAlHs4 | MG_H1 | GT6 | Marabel | 43.76386 | 20.234928 | 61 |
| RS | ZI004 | SRS9529528 | 54–10 | Guča | Serbia | 2016 | wt | wt | F129L | AsAlHs4 | MG_H1 | GT6 | Marabel | 43.76386 | 20.234928 | 64 |
| RS | ZI005 | SRS9529539 | 45–4 | Bački Maglić | Serbia | 2016 | Unknown | wt | F129L | AsAlHs4 | MG_H1 | GT4 | Brooke | 45.367993 | 19.513177 | 83 |
| RS | ZI006 | SRS9529501 | 45–13 | Bački Maglić | Serbia | 2016 | Unknown | wt | F129L | AsAlHs4 | MG_H1 | GT4 | VR808 | 45.367993 | 19.513177 | 60 |
| RS | ZI007 | SRS9529500 | 47–10 | Bački Maglić | Serbia | 2016 | Unknown | wt | F129L | AsAlHs4 | MG_H1 | GT4 | Lady Claire | 45.367993 | 19.513177 | 80 |
| RS | ZI008 | SRS9529502 | 38–1 | Bački Maglić | Serbia | 2016 | Unknown | wt | F129L | AsAlHs4 | MG_H1 | GT4 | Opal | 45.367993 | 19.513177 | 85 |
| SE | EL001 | SRS9529503 | 2017_152.1 | Nymö | Sweden | 2017 | wt | wt | F129L | AsAlHs4 | MG_H1 | GT1 | Kuras | 56.017577 | 14.332252 | 69 |
| SE | EL002 | SRS9529504 | 2017_152.3 | Nymö | Sweden | 2017 | wt | wt | F129L | AsAlHs4 | MG_H1 | GT1 | Kuras | 56.017577 | 14.332252 | 84 |
| SE | EL003 | SRS9529507 | 2017_212.2 | Nymö | Sweden | 2017 | wt | wt | F129L | AsAlHs4 | MG_H1 | GT2 | Kuras | 56.017577 | 14.332252 | 66 |
| SE | EL004 | SRS9529509 | 2017_213.1 | Nymö | Sweden | 2017 | wt | wt | F129L | AsAlHs4 | MG_H1 | GT2 | Kuras | 56.017577 | 14.332252 | 71 |
| SE | EL005 | SRS9529508 | 2017_131.4 | Nymö | Sweden | 2017 | H134R | H134R | F129L | AsAlHs4 | MG_H1 | GT7 | Kuras | 56.017577 | 14.332252 | 66 |
| SE | EL006 | SRS9529510 | 2017_134.1 | Nymö | Sweden | 2017 | H134R | H134R | F129L | AsAlHs4 | MG_H1 | GT1 | Kuras | 56.017577 | 14.332252 | 69 |
| SE | EL007 | SRS9529511 | 2017_144.2 | Nymö | Sweden | 2017 | H134R | H134R | F129L | AsAlHs4 | MG_H1 | GT7 | Kuras | 56.017577 | 14.332252 | 79 |
| SE | EL008 | SRS9529512 | 2017_152.4 | Nymö | Sweden | 2017 | H134R | H134R | F129L | AsAlHs4 | MG_H1 | GT7 | Kuras | 56.017577 | 14.332252 | 65 |
| SE | EL009 | SRS9529513 | 2017_124.2 | Nymö | Sweden | 2017 | H278Y | H278Y | F129L | AsAlHs4 | MG_H1 | GT4 | Kuras | 56.017577 | 14.332252 | 67 |
| SE | EL010 | SRS9529514 | 2017_132.2 | Nymö | Sweden | 2017 | H278Y | H278Y | F129L | AsAlHs4 | MG_H1 | GT3 | Kuras | 56.017577 | 14.332252 | 79 |
| SE | EL011 | SRS9529515 | 2017_142.2 | Nymö | Sweden | 2017 | H278Y | H134R | F129L | AsAlHs4 | MG_H1 | GT7 | Kuras | 56.017577 | 14.332252 | 66 |
| SE | EL012 | SRS9529516 | 2017_151.3 | Nymö | Sweden | 2017 | H278Y | H278Y | F129L | AsAlHs4 | MG_H1 | GT2 | Kuras | 56.017577 | 14.332252 | 76 |
| US | JW001 | SRS9529493 | CONR4D | Unknown | Idaho | 2015 | Unknown | H134R | F129L | AsAlHs4 | MG_H1 | GT5 | Unknown | Unknown | Unknown | 53 |
| US | JW002 | SRS9529494 | CONRI4 | Unknown | Idaho | 2015 | Unknown | H134R | F129L | AsAlHs4 | MG_H1 | GT1 | Unknown | Unknown | Unknown | 54 |
| US | JW003 | SRS9529497 | OONR2J | Unknown | Idaho | 2015 | Unknown | I280V | F129L | AsAlHs4 | MG_H1 | GT5 | Unknown | Unknown | Unknown | 50 |
| US | JW004 | SRS9529498 | T2R3B | Unknown | Idaho | 2015 | Unknown | H134R | F129L | AsAlHs4 | MG_H1 | GT5 | Unknown | Unknown | Unknown | 49 |
| US | NM022 | SRS9529499 | 628 | Unknown | unknown | 2015 | H134R | H134R | F129L | AsAlHs4 | MG_H1 | GT7 | Unknown | Unknown | Unknown | 52 |
Observed and reported mutations refer to mutations in different SDHI subunits. H278Y and I280V occur in subunit B, H134R in Subunit C, and N75S in subunit D. Unknown indicates that a mutation was assumed in these isolates, but the exact nature was not established prior this study.
2.2. DNA extraction and sequencing
For high‐quality DNA extraction, 200 ml of potato dextrose broth was inoculated with small agar plugs from 10 to 14 days old A. solani cultures, grown on SNA. Liquid cultures were incubated for 48 hours at 28°C on a rotary shaker (110 rpm). Afterwards, mycelium was filtered through a cheese cloth and squeezed to remove most of the liquid. After freeze drying, mycelium was grinded with liquid nitrogen and a bit of clean sea sand to break the cells most efficiently. 50–100 mg of lyophilized powder was added to 1 ml of DNA extraction buffer (containing extraction buffer (0.35 M sorbitol, 0.1 M Tris‐HCl pH7.5, 5 mM EDTA), nucleic lysis buffer (2% CTAB, 2 M NaCl, 0.2 M Tris‐HCl pH7.5, 50 mM EDTA), and sarkosyl (10%) in a ratio of 2.5:2.5:1), 20µl proteinase K (25 mg/ml), and 20µl RNAse A (20 mg/ml) in a 2 ml tube. Everything was mixed well and incubated for 1hour at 60°C – inverting the tubes occasionally to gently mix the content. Afterwards samples were centrifuged for 10 minutes at maximum speed, and the supernatant was transferred into a new tube. An equal amount of PCI (Phenol:Chloroform:Isoamylalcohol (24:24:1)) was added to the tube and incubated overnight on a rotary shaker at 4°C (gentle rotation to avoid damaging DNA). On the following day, samples were spun down for 15 minutes at maximum speed and upper phase was transferred to a new tube. An equal amount of SEVAG (Chloroform:Isoamylalcohol (24:1)) was added and tubes were incubated for at least 1 hour on a rotary shaker at 4°C (gentle rotation). In a next step, samples were centrifuged for 10 minutes at maximum speed and supernatant was transferred to a new tube. For precipitation, 0.7 volume of isopropanol (room temperature) was added and samples were spun down for 15 minutes at 9,600 g. After washing the DNA pellet with 70% ethanol, remaining liquid was removed by pipetting. To entirely remove the ethanol, tubes were placed in a heating block at 37°C to let them dry completely. DNA pellet was dissolved in 100 µl distilled water and stored at 4°C. A Qubit was used to determine the quantity of the extracted DNA. Sequencing libraries were prepared by BGI Ltd (Hong Kong) using their proprietary protocol. The sequencing was done on the DNBSEQ platform, using paired end 150 bp reads, aiming for up to 3 Gb of data per sample.
2.3. Mapping and SNP processing
The quality of the raw sequence data was checked using FastQC version 0.11.5 Andrews, 2016). The Burrows‐Wheeler alignment tool (version 1) (Li & Durbin, 2009) was used to map all samples to our reference genome NL03003 (Wolters et al., 2018). Reads deduplication was performed using the picard tool Markduplicate (version 2.19.2). Subsequently, the single‐nucleotide polymorphisms (SNPs) were called using GATKs Haplotype‐Caller (version 4.1.7.0) in GVCF mode (McKenna et al., 2010) with default settings, except for having ploidy set to one. The SNPs were filtered using GATK with modified parameters compared to the recommended filtering criteria. SNPs were filtered out when they met the following parameters: low mapping quality rank sum test (MQRankSum < −12.5), low quality by depth (QD <2.0), low read pos rank sum test (ReadPosRankSum < −8.0), high Fisher strand difference (FS >60.0), low RMS mapping quality (MQ <40.0), high strand odds ratio (SOR >3.0), high haplotype score (HaplotypeScore >13.0). Additionally, the insertion or deletions (indels) were filtered out using GATK with the following parameters: low quality depth (QD <2.0), low read pos rank sum test (ReadPosRankSum < −20.0), and high Fisher strand difference (FS >200.0). Afterwards, SNP clusters and SNPs close to indels were removed using SnpSift (version 4.3 t) filter (Cingolani, Patel, et al., 2012). The summary of the variant analyses was generated by SnpEff (version 4.3 t) (Cingolani, Platts, et al., 2012).
2.4. Visual inspection
The vcfR package (version 1.12.0) was used to read and visualize the vcf file for quality control (Knaus & Grünwald, 2017). The read depth (DP), the mapping quality (MQ), the Phred‐scaled quality (QUAL), and the variants were visualized. This identified several low confidence areas with very high DP. The masker() function was used to filter out data outside the confidence range: DP (0–2500) and MQ (50–60).
2.5. Phylogeny of A. solani samples and extraction of SDHI gene data
In order to validate the nature of our samples, we extracted three commonly used barcode genes, gapdh, rbp2, and tef1, that can be used to distinguish Alternaria spp. (Woudenberg et al., 2013). References for gapdh (KC584139), rbp2 (KC584430), and tef1 (KC584688) were extracted form NCBI search against the reference genome using BLAST+ (Camacho et al., 2009). The chromosome number, start, and stop coordinates from the BLAST output were extracted and stored as an interval file. Then the GATK tool called FastaAlternateReferenceMaker was used to replace the reference bases with the alternative bases at variant sites available in the vcf files over the specified interval (McKenna et al., 2010). The intervals were as follows (chr: start stop): GAPDH; CP022029.1:862512–863090, RPB2; CP022028.1:224005–224869 TEF1; CP022026.1: 1668499–1668833. All sequences were loaded in ab12phylo (Kaindl et al., 2021), and a multi‐gene phylogeny was constructed using built‐in RaxML‐NG (Kozlov et al., 2019) including A. solani reference sequences and the sequences of two closely related sister species A. dauci and A. porri (GAPDH (KC584111, KC584132), RBP2 (KC584392, KC584421), TEF1 (KC584651, KC584679) for A. dauci and A. porri respectively). To analyse the genomic regions coding for the SDH subunits, we performed a BLAST search of sequences each of the SDH subunits against the A. solani reference and used the identified start and stop site to extract alternative reference fasta files using GATK (McKenna et al., 2010).
2.6. Genetic diversity summary statistics
Basic genomic statistics were calculated with the R package PopGenome (version 2.7.5) (Pfeifer et al., 2014). For whole‐genome analysis, vcf files were split into smaller chunks using vcftools (version 0.1.16) (Danecek et al., 2011) and imported using the readData function (include.unknown=F, format=”VCF”, SNP.DATA=T, big.data=T). Statistics were calculated via neutrality.stats() and diversity.stats() and for further use transformed into R‐dataframes. Populations were defined via the set‐populations() function. The Site Frequency Spectrum (SFS) was generated by extraction of AC‐values from the vcf file. This was done using the commands strsplit(), do.call(), and table() in base R. Histograms were plotted using ggplot2 (version 3.3.5) (Wickham, 2009).
Sliding window analysis of SNPs per site was performed using the sliding.window.transform() option of PopGenome (w=10000, j=10000, type=2, whole.data=T). To achieve that, vcf chunks of chromosomes were imported separately and analysed discretely. Therefore, a function was defined to allow the analysis of each chromosome and to re‐aggregate generated data, subsequently. This is necessary since the native function for data‐import (readVCF) does not apply for haploid datasets. Segregating sites per window were extracted and normalized per site. ggplot2 was used to visualize our findings.
2.7. Population phylogeny
We extracted the SNPs as the alternative reference genomes using the GATK FastaAlternateReferenceMaker tool to create an alignment of the whole isolates’ genomes and to construct a population phylogeny (DePristo et al., 2011). To create a maximum likelihood phylogeny RAxML version 8 (Stamatakis, 2014) was used with ‐p 1122590 ‐f a ‐x 1122590 ‐m GTRGAMMA ‐# 100 ‐s input ‐n output and 100 bootstraps. The phylogenetic tree was visualized using the R packages ggtree version 1.16.6 (Yu, 2020) and treeio version 1.8.2 (Wang et al., 2020).
2.8. Principal Component Analysis (PCA)
As a dimensionality‐reduction technique, a principal component analysis (PCA) was performed using the R packages gdsfmt (version 2.18.0) and SNPRelate (version 1.18.1) (Zheng et al., 2012). GGplot2 (version 3.3.5) was used for visualization. To avoid the overlapping labels on the PCA, the R package ggrepel (version 0.9.1) was used (Slowikowski, 2018).
2.9. LDHelmet
To test for signals of recombination on the genome, measured as the recombination rate in ρ per bp, we used LDHelmet (Chan et al., 2012). Like described previously (Stam et al., 2019), fasta sequences were retrieved for each isolate using FastaAlternateReferenceMaker from GATK (McKenna et al., 2010). Next, LDHelmet was executed looping over each of the chromosomes in windows of 50 SNPs, using recommended parameters for the recombination search space. We specified a burn in of 100,000 iterations and 1,000,000 true iterations and a block penalty of 50. The output was visualized using ggplot2.
2.10. LEA
Ancestry analysis was performed using the R package LEA (version 3.2.0) (Frichot & François, 2015). For that, vcf files were converted into a genotypic matrix using vcftools (‐‐plink) (Danecek et al., 2011) and the LEA‐function “ped2geno”. This file was then transformed manually (9=1, 2=0). In the LEA package, sparse nonnegative matrix factorization (sNMF) was performed and a cross‐entropy criterion was calculated to identify the best statistical model describing ancestral populations of the dataset. The minimal cross‐entropy for the dataset was determined with multiple repetitions (K1:15, ploidy=1, entropy=T, rep=10). Admixture analysis was performed for k=6, k=7, and k=8 [Q(obj, k, run=which.min(cross.enropy(obj, k)))] and ordered according to individually assigned genotypes or sample origin. Visualization and inference with mutations was done using the tidyverse package (version 1.3.1) (Wickham et al., 2019) and ggplot2.
2.11. poppR
A Minimum Spanning Network (MSN) analysis was performed using poppR (version 2) (Kamvar et al., 2014, 2015). Whole‐genome data was extracted from the vcf file using vcfR (version 1.12.0) (Knaus & Grünwald, 2017), omitting sites with >2 alleles called. Ploidy was set to 1, and distances between the samples were calculated using dist(). The MSN was calculated using the function poppr.msn() and plotted with plot_poppr_msn().
2.12. Splitstree
Phylogenetic trees were generated with splitstree (version 4.17.1) (Huson & Bryant, 2006). Aligned fasta sequences were retrieved for each isolate using FastaAlternateReferenceMaker from GATK (McKenna et al., 2010) and used for network construction using default settings in splitstree.
2.13. Distance matrix
An SNP distance matrix was constructed from the alternative reference sequences with the extract alternative reference fasta option from GATK (McKenna et al., 2010). Two versions were created. One including all sites, one removing all incomplete cases (e.g., removing sites that were called in one isolate, but not in another). The matrix was created using snp‐dists ‐a (https://github.com/tseemann/snp‐dists).
2.14. Data availability
Raw short read data are deposited to NCBI SRA (project number PRJNA746421). SNP call files are deposited to Zenodo.org under doi 10.5281/ (Stam et al., 2021).
3. RESULTS
3.1. A. solani is highly polymorphic across Europe
To assess the genome‐wide diversity of A. solani in Europe, we selected samples to represent a cross section of the continent. In Europe, 43 isolates were collected from potato fields – 8 from Belgium, 8 from Northern Germany (Lower Saxony), 7 from Southern Germany (Bavaria), 8 from Serbia, and 12 from Sweden (Figure 1, Table 1). We also included five isolates originating from the United States. All isolates were sequenced using DNBSeq and had an average read depth of 62x coverage, after mapping and filtering (Table 1). On average we found 47,098 SNPs in a single isolate, when comparing to the reference, corresponding to one SNP every ~700 bases. The number of SNPs in most isolates ranged from 32,079 SNPs (in DE_NM019) to 52,911 (in DE_NM017). BE_SL002 appeared to be an outlier with 157,798 SNPs compared to the reference. However, extraction of the sequences of typical barcode markers used in phylogenetic analyses of Alternaria species (RPB2, GAPDH, and EF1) confirmed that BE_SL002 is A. solani. With this combination of barcodes, we can define two haplotypes (seen as two major branches in Figure S1A). BE_SL002 fits firmly within one of the two haplotypes and does not cluster with one of the related sister species. Alignment with the reference sequences for GAPDH as provided by Adhikari et al. (2020) reveals that all of our isolates belong to what they identified as the AlAsHs4 haplotype, which they assigned to potato hosts (Figure S1B).
In total, we found 262,928 (136,180 without BE_SL002) SNPs between all isolates. Due to the comparably large number of unique SNPs in BE_SL002, further analyses were done without this sample, to allow better comparison.
We found a transition versus transversion (ts/tv) ratio of 3.05. Forty‐eight percent of the SNPs are singletons. The Site Frequency Spectrum (SFS) for the sample set shows a homogenous decrease first. However, the overall number of SNPs is not decreasing continuously, in fact, some minor peaks are occurring for higher SNP frequencies (Figure S2). This indicates that there are no large population expansions (as would be the case with increased number of singletons). The slightly uneven distribution might be an effect of partial clonality in the sample set. Next, we calculated several population genetics summary statistics for the sample set. Overall, Watterson θ per site equals 0.019 and π equals 0.016. The overall Tajimas’ D of the dataset is rather neutral with a value of 0.629.
On average we found one SNP every ~241 bp. SNP density varies per chromosome, ranging from one every 197 bp for chromosome 4 to one every 277 bp for chromosome 2. Using sliding window analysis with 10kb windows, confirmed that SNP‐density varies along the chromosomes and is highest and lowest in chromosome 6 and 1 respectively (Figure S3). The different genes coding for the different SDH subunits that are known to be subject to fungicide resistance mutations are located on chromosomes 7 (SdhB), 2 (SdhC), and 5 (SdhD). Thus, the fungicide target genes are not located specifically on very conserved or very diverse chromosomes.
3.2. A. solani shows differences in diversity between locations
Next, we wanted to know whether the genetic variation is similar for each of the five sample locations in Europe, and in our US samples. To this end, we summarized all statistics per location (Table 2). Even excluding BE_SL002, the Belgian samples had the highest number of SNPs (103,971 SNPs in total), followed by Bavaria (101,623). Lower Saxony and Serbia have 62,378 and 63,997 SNPs respectively. Interestingly, in Sweden, from where we analysed 12 samples, rather than 7 or 8, the number of segregating sites is only 71,232. This might in part be due to the fact that the Swedish samples were all collected in one locality, but it should be noted that with the exception for 1 sample this was also the case for Serbia. Moreover, a clear link between the number of segregating sites and the geographical spread between the samples of a certain locality cannot be made. To illustrate, the furthest distance between samples in Belgium is 107 km, the maximum distance between samples in Lower Saxony is 262 km, yet Belgian isolates remain more diverse.
TABLE 2.
Diversity statistics per locality
| Country | Segregating Sites | Theta Watterson per Site | Theta Pi per Site | Tajimas’ D | Clones (n) | Samples |
|---|---|---|---|---|---|---|
| Bavaria | 101623 | 0.0221 | 0.0227 | 0.1611 | 1 (2) | 7 |
| Belgium | 103971 | 0.0226 | 0.0234 | 0.213 | 0 (‐) | 7 |
| Lower Saxony | 62378 | 0.0128 | 0.0118 | −0.4428 | 2 (2, 3) | 8 |
| Serbia | 63997 | 0.01315 | 0.01312 | −0.00956 | 2 (2, 4) | 8 |
| Sweden | 71232 | 0.0126 | 0.0153 | 1.0242 | 3 (3, 2, 2) | 12 |
| USA | 55964 | 0.0143 | 0.0135 | −0.4143 | 1 (2) | 5 |
| Total | 136180 | 0.0164 | 0.0192 | 0.6295 | ‐ | 47 |
In line with the higher number of SNPs, we also found that the Belgian samples have the highest nucleotide diversity (Watterson's θ or π). Interestingly, we found some variation in Tajima's D. However, Tajima's D is negative or close to 0 in most localities. This indicates a relatively high number of singletons and is in line with the overall observations from the SFS. However, a Tajima's D of 1.024 in Sweden seems to violate the assumption of neutral theory suggesting less low‐frequency polymorphisms, probably due to higher clonality in these samples.
3.3. Signatures for sexual recombination
A. solani is considered to be an asexual pathogen, but some reports suggest parasexual reproduction is occurring in different populations and maybe even between related species (Alvarenga et al., 2016; Meng et al., 2015; Zhao et al., 2021). Therefore, we looked for signatures for recombination using LDHelmet (Chan et al., 2012). These analyses show clear evidence for recombination on the genome, indicated by high ρ/bp peaks, suggesting that parasexual recombination is likely, though signals on some of the chromosomes are very low (Figure S5). Next, we looked specifically at the chromosomes that harbour the genes coding for the two Sdh subunits with the most mutants detected. The gene for SdhC lies on chromosome 2, at position 3,213,044. Chromosome 2 contains only two major peaks at 2,883,609 and 3,851,760. Such limited number of possible sites of recombination and them being 500,000 bp away from the gene indicate that there is no specific recombination around these sites. The gene coding for SdhB lies on chromosome 7 at position 1,345,474. Chromosome 7 only has lower ρ/bp peaks, and we see no indication that fungicide resistance mutations affected the recombination around the SdhB locus as compared to other loci (Figure S5).
3.4. Classification of A. solani genotypes
We hypothesized that there is variation in genetic diversity and the genetic background of the isolates between each locality. Therefore, we wanted to define the overall population structure and see whether genotype distribution of A. solani matches with respective sample origin, for example, genotypes showing regional clustering. We first reduced the complexity of the data for PCA analyses. Combined the first three components explain 49% of the variation (Figure S4). The plot of the two first principal components (Figure 2A) shows three or four clusters, each of varying size and spread. All clusters that can be identified contain isolates from different geographical origins. When plotting PCA 1 versus 3, it can be seen that BE_SL002 is the only sample driving the difference on the third principal component and thus a clear outlier (Figure S4 (B)). To further corroborate these findings, we set to construct a phylogenetic tree based on all identified SNPs in the isolates (Figure 2B). As expected, isolates that grouped close together in the PCA also group in the phylogenetic tree. These findings are supported by high bootstrap values. In the phylogenetic tree, BE_SL002 groups together with the isolates that are closest to it in the PCA, yet it has a long branch length. Colour coding by locality in the PCA and on the branches of the tree shows that most localities indeed harbour several unrelated isolates.
FIGURE 2.
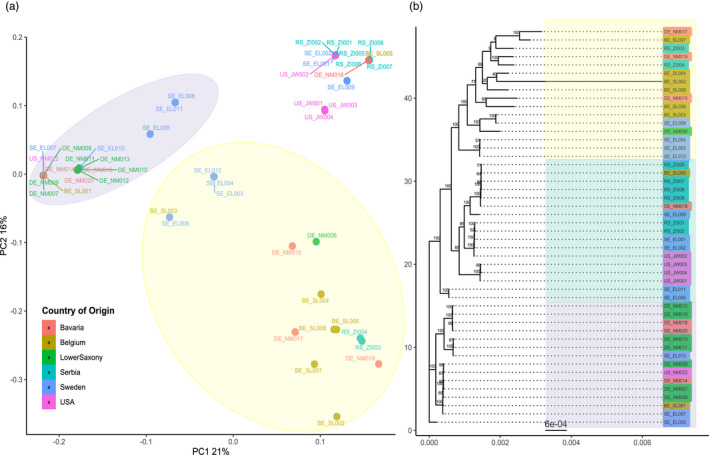
Principal component analysis (PCA) and phylogenetic tree constructed of the 48 Alternaria solani isolates. (a) Scatter plots of the first two principal components made using SNPRelate and ggplot2 packages in R. The x‐ and y‐axis represents the PC1 (with variance explained 21%) and PC2 (with variance explain 16%). The isolates are colour‐coded according to their region of origin. Highlighted clusters in yellow, green, and purple are based on the clustering on the phylogenetic tree. (b) Phylogeny of reconstructed whole‐genome sequences for each of the 48 isolates, made using RAxML (GTRGAMMA) with 100 bootstraps. The phylogenetic tree is constructed using treeio and ggtree packages in R. The colour coding of the isolates corresponds to the region of origin. The x‐axis and scale bar indicate the branch length
To further define the population structure of the isolates, we calculated admixture using the R package LEA (excluding BE_SL002). To estimate the most likely number of ancestral populations of the dataset, the cross‐entropy criterion was calculated. The lowest cross‐entropy was observed at K=7 (Figure S6), thus admixture analysis was conducted for K = 7 populations as well at K=6 and K=8. With K = 7, we can see groupings similar to those observed in the PCA. Several clusters show mainly uniform, un‐admixed ancestry. For example, the samples BE_SL005, RS_ZI008, RSZI006, and DE_NM016 (all yellow in Figure 3B) cluster closely together in the PCA and form a monophyletic clade in the phylogeny. All genotypes are present in distinguishable groups. When comparing K = 6 and K = 7, we observe one cluster (US_JW001, US_JW003, US_WJ004) dividing into clear not‐admixed genotypes. In some samples, increasing K can be associated with increasing admixture of various genotypes (e.g., in DE_NM006). For K=7, 13 isolates show a dominant ancestor only with minimal admixture. Eighteen isolates show admixture of 2 ancestors and 16 samples show admixture with 3–4 ancestors. Admixture from five ancestors can only be found in RS_ZI004. Note that the results of this whole‐genome genotyping do not correlate with the haplotyping results for the three barcode genes, suggesting the three barcodes alone cannot accurately capture the diversity of A. solani in a field (Figure S1, Table 1).
FIGURE 3.
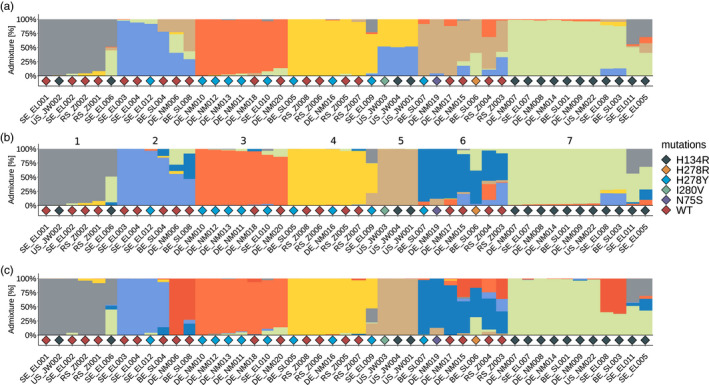
Ancestry analysis on A. solani samples. Ancestry analysis was performed on A. solani isolates (n=47, BE_SL002 excluded) using the R‐package LEA [snmf(K=1:15, rep=10, ploidy=1)] for 6 (a), 7 (b), and 8 (c) ancestral populations. Colours represent individual genotypes assigned by sparse nonnegative matrix factorization. Bar plots show the admixture of each genotype in every isolate. Diamonds represent mutations against SDHIs found in the data set. The numbers in panel B denominate the assigned genotype (GT) as used in Table 1 and other parts of this manuscript
Next, we assigned each isolate to the respectively most dominant of the seven genotype groups and calculated diversity statistics for each of them (Table 3). The nucleotide diversity differs between each genotype group. Genotype 6 (dark blue) has the highest numbers of SNPs, 96,239, probably because it is also the most admixed. Interestingly, the number of SNPs of the genotypes does not scale with sample size, for example, genotype 3 (n=7) has only 14,459 SNPs. Therefore, the nucleotide diversity per site diverges strongly between the genotypes, ranging from 0.002 (genotype 4) to 0.023 in genotype 6. Tajimas’ D is ranging from −1.709 in genotype 4 up to 2.212 in genotype 3. Genotypes 2, 6, and 7 show neutral Tajimas’ D; the calculation for Genotype 5 is not possible due to sample size.
TABLE 3.
Diversity statistics per genotype
| Spalte1 | Genotype | Segregating Sites | Theta Watterson per Site | Theta Pi per Site | Tajimas’ D | Clones (n) | Samples |
|---|---|---|---|---|---|---|---|
| 1 | Genotype 1 | 21461 | 0.00549 | 0.00459 | −1.24863 | 1 (5) | 6 |
| 2 | Genotype 2 | 72352 | 0.0169 | 0.0169 | 0.0182 | 1 (3) | 6 |
| 3 | Genotype 3 | 14459 | 0.00314 | 0.00432 | 2.21246 | 2 (2, 5) | 7 |
| 4 | Genotype 4 | 15640 | 0.0034 | 0.00242 | −1.709 | 1 (6) | 7 |
| 5 | Genotype 5 | 933 | 0.000331 | 0.000331 | NA | 1 (2) | 3 |
| 6 | Genotype 6 | 96239 | 0.0224 | 0.0229 | 0.142 | 0 (‐) | 7 |
| 7 | Genotype 7 | 44435 | 0.00808 | 0.007 | −0.65001 | 1 (7) | 11 |
Seeing the limited diversity in some genotype groups, we asked how many isolates could be considered clones of each other. To this extent we created a pairwise SNP‐distance table for isolates (Table S1). This reveals that on average there are 40,282 SNPs between the isolates (35,365 when taking only complete cases (e.g., sites that have been called in all isolates reliably). The lowest number of SNPs between two isolates is 822 (135 with complete cases). Pairwise comparisons with such low number of differences suggest the existence of true clonal isolates. Looking in the genotype groups constructed by LEA, we see that 4 genotype groups contain 3–4 samples that have less than 1500 SNPs between them, and one genotype group shows 2122 SNPS shared between 6 of its samples. Thus, our Europe‐wide sample set consists of a considerable number of likely true clones.
To better visualize the diversity per location, we also sorted the isolates per locality (Figure S7). It should be noted that each location contains a mix of different genotypes. The dominating genotypes of a locality vary slightly, but it is not possible to assign a single dominant genotype to a single region, thus, refuting our hypothesis that A. solani genotypes would show regional clustering. Variation in the diversity within geographical regions can also be observed. The samples from Belgium and Sweden show the most complex admixture structure, followed by Bavaria. The high complexity in Sweden is particularly interesting, because in absolute number of segregating sites, this population did not appear to be the most diverse. Interestingly, some of the clones described above can be found on different sides of the European continent (e.g., BE_SL005 clusters with some of the Serbian isolates) or even across continents (US_JW002 clusters with Serbian and Swedish isolates).
3.5. SDHI mutations arose independently in different genotypes
To understand the rise and spread of SHDI fungicide resistance, we overlaid the genotyping data with our data on SDHI‐target mutations. The H134R and H278Y mutations occur 15 and 10 times in our dataset, respectively. H134R is found in every locality, except Serbia; H278Y can be found in all locations but Serbia and the United States. Furthermore, we found three mutations (H278R, I280V & N75S) that occur only once in the dataset. In 20 samples, we found no mutation related to SDHI fungicides. Mutation‐free isolates were collected in all different localities except for the United States.
Overlaying the data shows that the mutations are not linked to the background genotypes that we defined previously. The different sdh SNPs were found in different genetic backgrounds (Figure 3B). For example, H134R occurs in the grey, brown, and green genotype. H278Y occurs in the blue, red, and yellow genotype. This strongly indicates that mutations in the SDH subunits arose independently in different genetic A. solani backgrounds.
To obtain additional lines of evidence for our findings, we constructed a minimum spanning network (MSN) based on all SNP data (Figure 4). These networks confirm that the LEA method reliably assigned genotypes to our isolates. Only two small differences can be observed in the MSN (Figure 4A). Isolates DE_NM006 and DE_NM015 appear to sit within the samples assigned to genotype 7 rather than genotype 2 and genotype 6 respectively. Looking at the results from LEA, it becomes evident that these two samples are in fact the two most admixed samples, and therefore the placement of these samples on the MSN should be taken with some caution. When looking at the occurrence of the SDHI mutations on the MSN (Figure 4B), the individual mutations can be seen on the different branches.
FIGURE 4.
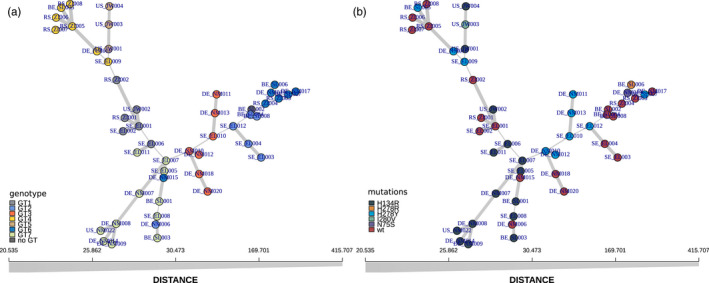
Minimum spanning network analyses (MSN) for our A. solani samples. MSN showing the relatedness of the samples as produced by the R package poppR. Both panels show the same network, with identical scaling (x axis). Each circle represents an individual isolate. Colours in (a) correspond to previously assigned genotypes, colours in (b) highlight the isolate's SDHI mutations
As a final line of evidence, we used splitstree (Huson & Bryant, 2006) to analyse the population structure of A. solani (Figure 5). Splitstree also shows a clear separation of the seven previously assigned genotype groups, with the most admixed isolates positioned between the groups. Moreover, it clearly shows evidence for recombination between some of the isolates, as illustrated by the connections between some of the branches. The phi test for recombination confirms that there is significant evidence for recombination. Lastly, the analyses also suggest that, as we observed with LDHelmet, the different Sdh mutations did not spread through recombination. Similar to the MSN analyses, Figure 5 clearly shows that the H134R and H278Y mutations appear multiple times at different branches. These branches are not connected through recombination events, and thus we conclude that they arose independently.
FIGURE 5.
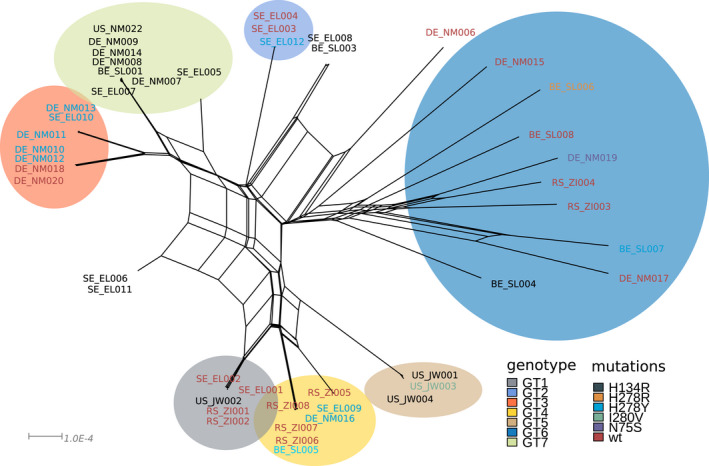
Splitstree analyses for the A solani isolates. Splitstree analysis reveals the same genetic clusters as our ancestry and MSN analysis. Several isolates show clear admixture or signals of recombination, as shown by the lines connecting the individual branches. The circles represent the dominant genotype as assigned with LEA, isolates that have no coloured background are admixed. The text colour for our each isolate indicates the respective SDHI mutation of the isolate
4. DISCUSSION
In this study we present the first Europe‐wide genetic diversity study of the early blight pathogen Alternaria solani based on whole‐genome data. We used 43 isolates collected in five different localities ranging from Sweden to Serbia. Seeing that A. solani is considered a clonal pathogen (synonymous: mitosporic fungus, Deuteromycete, fungi imperfecti), we expected to see certain geographical patterns related to the genetic diversity, for example, patterns of isolation by distance or predominance of a certain genotype in a locality. The strongest geographical patterns can be observed in invasive pathogens, like the rice and wheat blast fungus Magnaporthe oryzae, where multiple genetic lineages can be found in Southeast Asia, but single invasive lineages spread elsewhere and sometimes became epidemic (Gladieux et al., 2018; Islam et al., 2016). However, clear geographical patterns in population structure can also be observed in cosmopolitan pathogen species like the oomycete potato pathogen Phytophthora infestans, the causal agent of late blight, or in other ascomycete pathogens such as the sugar beet pathogen Cercospora beticola (Knight et al., 2019) and the well‐studied wheat pathogen Zymoseptoria tritici (Vagndorf et al., 2018).
We did not detect any geographical clustering of A. solani isolates in this study. This could be an artefact of the relatively small sample size per locality. However, some pathogens do already show clear separation even when taken such small samples (Mariette et al., 2016). High genotype diversity and lack of isolation by distance has previously been observed for A. solani in an SSR (simple‐sequence repeats)‐based study with populations from China. Assumed clones were detected in multiple populations separated by thousands of kilometres and random association among loci was found in half of the populations assayed (Meng et al., 2015). Our whole‐genome data now suggest that these results were not an artefact of the low‐resolution markers used.
Such lack of isolation by distance has been seen as well for several other supposedly clonal ascomycete pathogens. On a regional scale similar to our sample sites in Germany, this has been observed for the Brassica pathogen Leptosphaeria maculans in a collection from northern and southern France (Travadon et al., 2011) and on a global scale for the barley pathogen Ramularia collo‐cygni (Stam et al., 2019). Vice versa, isolates from distant sites cluster together on the phylogenetic tree, suggesting long distance transport, for example, by wind or seed tubers.
Adhakiri et al. compared field population diversity of three Alternaria species on potato and tomato in North Carolina and Wisconsin and found that A. solani had relatively lower diversity than A. alternata and A. linariae. Yet, four different haplotypes were found in one field and haplotype divergence was found in all three species (Adhikari et al., 2020). Our analyses revealed up to nine definable genotypes in A. solani in our sample set. Deeper analyses showed that some of the collected isolates from these genotypes are at least likely purely clonal. They contain very few SNPs between them. Whether such clones perform worse or better under specific climatic conditions or on specific cultivars remains to be investigated.
Based on the results of our study, we conclude that the mutations in the genes coding for the different SDH subunits that lead to resistance or higher tolerance to SDHIs can be found in multiple genetic backgrounds. This indicates that the mutations occurred multiple times at different locations, rather than that a single resistant isolate emerged and spread throughout Europe. Evolution of tolerance against QoI fungicides was initially suggested to have arisen once, due to its association with an arbitrarily assigned genotype (GT I), and then spread. However, recent work showed that the F129L mutation in the cytochrome b target gene responsible for this resistance could since 2016 also be observed in another genotype (Nottensteiner et al., 2019). Our study further confirms the genetic heterogeneity of QoI tolerant isolates, as all of the isolates in this study show the F129L mutation. As a consequence, all isolates with SDHI mutations possess dual fungicide resistance, which makes controlling the pathogen increasingly difficult.
A recent study in Z. tritici concluded that azole resistance is likely the result of so‐called “hotspot evolution” with convergent changes in small sets of loci and more population‐specific allele frequency changes (Hartmann et al., 2020). Fungicides can form a major bottleneck for fungal populations, but the ability to recombine allowed population‐specific adaptation in Z. tritici populations. Even though we found that fungicide resistance or fungicide tolerance mutations arose multiple times independently, future studies looking deeper into the effects of fungicide induced bottlenecks in A. solani are now required.
One additional outcome of our study is clear evidence for recombination. Previous studies have suggested (para‐)sexual recombination needs to happen in the field in order to explain the observed diversity patterns (Alvarenga et al., 2016; Meng et al., 2015; Zhao et al., 2021), but the use of markers with low resolution left this open for confirmation. We used whole‐genome data and three different methods to test possible recombination. All methods indicate that the observed genetic diversity arose in part through recombination. Recombination events likely make is easier for fungicide resistances to be spreading faster through different genetic backgrounds. This, combined with the fact that true clones can be found across continents, potentially amplifies the spread of more aggressive genotypes. For example, Bauske et al. (2017) already found that A. solani isolates possessing the SdhD‐D123E mutation were significantly more aggressive in vivo compared with wild‐type isolates.
Fungicide pressure and the aggressiveness of A. solani isolates under certain conditions or on a certain host cultivar alone might not be the only determinants for A. solani diversity in the field. Early blight lesions often appear to contain a mix of Alternaria species. A. solani often co‐occurs with A. alternata (e.g., Zheng & Wu, 2013). Tymon et al. also isolated several other small and large spored species from lesions on potato fields (Tymon et al., 2016). And also in tomato early blight symptoms can be associated with multiple species, the large spored A. solani, A. linariae, and A. grandis, and the small spored A. alternata (Bessadat et al., 2017). Ding et al. found, that in some years the disease severity in the field is more strongly correlated to the presence of early inoculum of A. alternata (Ding et al., 2020). Using barcode‐sequencing, they found that A. alternata possessed a greater diversity in the field (Ding et al., 2020). In all studies mentioned above, the disease phenotypes of A. solani isolates were more severe and A. solani is the more dominant partner in the interaction. Studies on Alternaria leaf spot on rape seed showed that also this disease is not caused just by A. brassicae, but that it can also be caused by A. japonica and that the ratio between the species and aggressiveness of the species was temperature dependent (Al‐lami et al., 2020). Findings like these indicate that genotypic diversity of A. solani can potentially also be shaped by the presence of diverse Alternaria species and that further research is needed to understand the effects of coinfection on early blight epidemiology. The presence of different co‐infecting Alternaria spp. might cause different evolutionary pressure on the isolates.
We presented a first genome‐wide diversity analysis for the early blight pathogen A. solani in Europe. We revealed surprising genetic diversity patterns throughout Europe and show that fungicide resistance evolved multiple times independently, rather than evolving once followed by spreading. These findings can help inform policy and fungicide management practices. The finding of multiple fungicide resistance mutation events highlights that there is “not one person to blame” for the rise of fungicide resistance in A. solani. This emphasizes the need to reduce the evolutionary pressure in each individual field and keeping the population size as small as possible at all times (McDonald & Linde, 2002). New strategies using evolutionary knowledge as well as the intensive application of already non‐fungicide available measures are required for good integrative disease management on a continental scale (Green et al., 2020). Only highly integrated approaches of pest management, including measures of all kinds, are capable of reducing the evolutionary potential of A. solani epidemics. To do this without increasing too much the evolutionary pressure in the field, fungicide application time frames and quantities need to be carefully adjusted as they play an important role in the development of resistances and diseases. Unfortunately, in our dataset, loss of fungicide sensitivity cannot be linked to the degree of fungicide‐use, since data is not fully available for all localities in this project.
Our analysis also shows that some true clonal isolates can be found thousands of kilometres apart. Understanding A. solani dispersal patterns over long distances will become increasingly more relevant. Recent studies have shown clear differences in aggressiveness in different A. solani isolates in general, for example, not associated with specific fungicide target mutations (Mphahlele et al., 2020). With the high‐resolution (whole genome) genotype data presented in this study, more meaningful comparisons can be made to study the link between pathogen genotype and aggressiveness. The data will also allow for studies tracing exact clones of A. solani over time, which will lead to a better understanding of pathogen dispersal patterns and should contribute to improving management strategies.
CONFLICT OF INTEREST
The authors have no conflict of interest.
Supporting information
Fig S1‐S7
Table S1
ACKNOWLEDGMENTS
We like to thank Regina Dittebrandt and Lena Forster for help with A. solani propagation.
Einspanier, S. , Susanto, T. , Metz, N. , Wolters, P. J. , Vleeshouwers, V. G. A. A. , Lankinen, Å. , Liljeroth, E. , Landschoot, S. , Ivanović, Ž. , Hückelhoven, R. , Hausladen, H. , & Stam, R. (2022). Whole‐genome sequencing elucidates the species‐wide diversity and evolution of fungicide resistance in the early blight pathogen Alternaria solani . Evolutionary Applications, 15, 1605–1620. 10.1111/eva.13350
Severin Einspanier and Tamara Susanto contributed equally to this study.
DATA AVAILABILITY STATEMENT
Raw short read data are deposited to NCBI SRA (project number PRJNA746421). SNP call files are deposited to Zenodo.org under doi 10.5281/ (Stam et al., 2021).
REFERENCES
- Adhikari, T. B. , Ingram, T. , Halterman, D. , & Louws, F. J. (2020). Gene genealogies reveal high nucleotide diversity and admixture haplotypes within three alternaria species associated with tomato and potato. Phytopathology®, 110, 1449–1464. [DOI] [PubMed] [Google Scholar]
- Adhikari, T. B. , Knaus, B. J. , Grünwald, N. J. , Halterman, D. , & Louws, F. J. (2019). Inference of population genetic structure and high linkage disequilibrium among alternaria spp. Collected from Tomato and Potato Using Genotyping by Sequencing. bioRxiv, 827, 790. [Google Scholar]
- Al‐lami, H. F. D. , You, M. P. , & Barbetti, M. J. (2020). Temperature drives contrasting alternaria leaf spot epidemic development in canola and mustard rape from alternaria japonica and A. brassicae. Plant Disease, 104, 1668–1674. [DOI] [PubMed] [Google Scholar]
- Alvarenga, T. V. M. , Ribeiro, S. R. R. D. P. , Souza, E. A. D. , Pereira, F. D. C. , & Pinto, C. A. B. P. (2016). Characterization of Alternaria isolates and reaction of potato genotypes to early blight. Ciência Rural, 46, 1783–1789. 10.1590/0103-8478cr20151447 [DOI] [Google Scholar]
- Andrews, S. (2016). FastQC: A quality control tool for high throughput sequence data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc
- Bartlett, D. W. , Clough, J. M. , Godwin, J. R. , Hall, A. A. , Hamer, M. , & Parr‐Dobrzanski, B. (2002). The strobilurin fungicides. Pest Management Science, 58, 649–662. 10.1002/ps.520 [DOI] [PubMed] [Google Scholar]
- Bauske, M. J. , Mallik, I. , Yellareddygari, S. K. R. , & Gudmestad, N. C. (2017). Spatial and temporal distribution of mutations Conferring QoI and SDHI resistance in alternaria solani across the United States. Plant Disease, 102, 349–358. [DOI] [PubMed] [Google Scholar]
- Bessadat, N. , Berruyer, R. , Hamon, B. , Bataille‐Simoneau, N. , Benichou, S. , Kihal, M. , Henni, D. E. , & Simoneau, P. (2017). Alternaria species associated with early blight epidemics on tomato and other Solanaceae crops in northwestern Algeria. European Journal of Plant Pathology, 148, 181–197. 10.1007/s10658-016-1081-9 [DOI] [Google Scholar]
- Blake, J. J. , Gosling, P. , Fraaije, B. A. , Burnett, F. J. , Knight, S. M. , Kildea, S. , & Paveley, N. D. (2018). Changes in field dose‐response curves for demethylation inhibitor (DMI) and quinone outside inhibitor (QoI) fungicides against Zymoseptoria tritici, related to laboratory sensitivity phenotyping and genotyping assays. Pest Management Science, 74, 302–313. [DOI] [PubMed] [Google Scholar]
- Camacho, C. , Coulouris, G. , Avagyan, V. , Ma, N. , Papadopoulos, J. , Bealer, K. , & Madden, T. L. (2009). BLAST+: architecture and applications. BMC Bioinformatics, 10(1). 10.1186/1471-2105-10-421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, A. H. , Jenkins, P. A. , & Song, Y. S. (2012). Genome‐wide fine‐scale recombination rate variation in Drosophila melanogaster. PLoS Genetics, 8, e1003090. 10.1371/journal.pgen.1003090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani, P. , Patel, V. M. , Coon, M. , Nguyen, T. , Land, S. J. , Ruden, D. M. , & Lu, X. (2012). Using Drosophila melanogaster as a Model for genotoxic chemical mutational studies with a new program, SnpSift. Frontiers in Genetics, 3, 35. 10.3389/fgene.2012.00035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani, P. , Platts, A. , Wang, L. L. , Coon, M. , Nguyen, T. , Wang, L. , Land, S. J. , Lu, X. , & Ruden, D. M. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso‐2; iso‐3. Fly, 6, 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek, P. , Auton, A. , Abecasis, G. , Albers, C. A. , Banks, E. , DePristo, M. A. , Handsaker, R. E. , Lunter, G. , Marth, G. T. , Sherry, S. T. , McVean, G. , & Durbin, R. (2011). The variant call format and VCFtools. Bioinformatics, 27, 2156–2158. 10.1093/bioinformatics/btr330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado‐Baquerizo, M. , Guerra, C. A. , Cano‐Díaz, C. , Egidi, E. , Wang, J.‐T. , Eisenhauer, N. , Singh, B. K. , & Maestre, F. T. (2020). The proportion of soil‐borne pathogens increases with warming at the global scale. Nature Climate Change, 10, 550–554. 10.1038/s41558-020-0759-3 [DOI] [Google Scholar]
- DePristo, M. A. , Banks, E. , Poplin, R. , Garimella, K. V. , Maguire, J. R. , Hartl, C. , Philippakis, A. A. , del Angel, G. , Rivas, M. A. , Hanna, M. , McKenna, A. , Fennell, T. J. , Kernytsky, A. M. , Sivachenko, A. Y. , Cibulskis, K. , Gabriel, S. B. , Altshuler, D. , & Daly, M. J. (2011). A framework for variation discovery and genotyping using next‐generation DNA sequencing data. Nature Genetics, 43, 491–498. 10.1038/ng.806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding, S. , Meinholz, K. , & Gevens, A. J. (2020). Spatiotemporal distribution of potato‐associated alternaria species in Wisconsin. Plant Disease, 105, 149–155. [DOI] [PubMed] [Google Scholar]
- Edin, E. , Liljeroth, E. , & Andersson, B. (2019). Long term field sampling in Sweden reveals a shift in occurrence of cytochrome b genotype and amino acid substitution F129L in Alternaria solani, together with a high incidence of the G143A substitution in Alternaria alternata. European Journal of Plant Pathology, 155, 627–641. [Google Scholar]
- Everhart, S. , Gambhir, N. , & Stam, R. (2020). Population genomics of filamentous plant pathogens—a brief overview of research questions, approaches, and pitfalls. Phytopathology®, 111, 12–22. [DOI] [PubMed] [Google Scholar]
- Frichot, E. , & François, O. (2015). LEA: an R package for landscape and ecological association studies. Methods in Ecology and Evolution, 6, 925–929. 10.1111/2041-210X.12382 [DOI] [Google Scholar]
- Gladieux, P. , Ravel, S. , Rieux, A. , Cros‐Arteil, S. , Adreit, H. , Milazzo, J. , Thierry, M. , Fournier, E. , Terauchi, R. , Tharreau, D. (2018). Coexistence of multiple endemic and pandemic lineages of the rice blast pathogen. MBio, 9, e01806–e1817. 10.1128/mBio.01806-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green, K. K. , Stenberg, J. A. , & Lankinen, Å. (2020). Making sense of Integrated Pest Management (IPM) in the light of evolution. Evolutionary Applications, 13, 1791–1805. 10.1111/eva.13067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann, F. E. , Vonlanthen, T. , Singh, N. K. , McDonald, M. C. , Milgate, A. , & Croll, D. (2020). The complex genomic basis of rapid convergent adaptation to pesticides across continents in a fungal plant pathogen. Molecular Ecology, 30, 5390. [DOI] [PubMed] [Google Scholar]
- Huson, D. H. , & Bryant, D. (2006). Application of phylogenetic networks in evolutionary studies. Molecular Biology and Evolution, 23, 254–267. 10.1093/molbev/msj030 [DOI] [PubMed] [Google Scholar]
- Islam, M. T. , Croll, D. , Gladieux, P. , Soanes, D. M. , Persoons, A. , Bhattacharjee, P. , Hossain, M. S. , Gupta, D. R. , Rahman, M. M. , Mahboob, M. G. , Cook, N. , Salam, M. U. , Surovy, M. Z. , Sancho, V. B. , Maciel, J. L. N. , NhaniJúnior, A. , Castroagudín, V. L. , Reges, J. T. D. A. , Ceresini, P. C. , … Kamoun, S. (2016). Emergence of wheat blast in Bangladesh was caused by a South American lineage of Magnaporthe oryzae. BMC Biology, 14, 84. 10.1186/s12915-016-0309-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaindl, L. , Small, C. & Stam, R. (2021). AB12PHYLO: an integrated pipeline for Maximum Likelihood phylogenetic inference from ABI trace data. bioRxiv: 2021.03.01.433007.
- Kamvar, Z. N. , Brooks, J. C. , & Grünwald, N. J. (2015). Novel R tools for analysis of genome‐wide population genetic data with emphasis on clonality. Frontiers in Genetics, 6, 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamvar, Z. N. , Tabima, J. F. , & Grünwald, N. J. (2014). Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ, 2, e281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapsa, J. (2004). Early blight (Alternaria spp.) in potato crops in Poland and results of chemical protection. Journal of Plant Protection Research, 44(3), 231–238. [Google Scholar]
- Knaus, B. J. , & Grünwald, N. J. (2017). vcfr: a package to manipulate and visualize variant call format data in R. Molecular Ecology Resources, 17, 44–53. [DOI] [PubMed] [Google Scholar]
- Knight, N. L. , Vaghefi, N. , Kikkert, J. R. , Bolton, M. D. , Secor, G. A. , Rivera, V. V. , Hanson, L. E. , Nelson, S. C. , & Pethybridge, S. J. (2019). Genetic diversity and structure in regional cercospora beticola populations from beta vulgaris subsp. vulgaris suggest two clusters of separate origin. Phytopathology®, 109, 1280–1292. [DOI] [PubMed] [Google Scholar]
- Kozlov, A. M. , Darriba, D. , Flouri, T. , Morel, B. , & Stamatakis, A. (2019). RAxML‐NG: a fast, scalable and user‐friendly tool for maximum likelihood phylogenetic inference. Bioinformatics, 35, 4453–4455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landschoot, S. , Carrette, J. , Vandecasteele, M. , De Baets, B. , Höfte, M. , Audenaert, K. , & Haesaert, G. (2017). Boscalid‐resistance in Alternaria alternata and Alternaria solani populations: An emerging problem in Europe. Crop Protection, 92, 49–59. 10.1016/j.cropro.2016.10.011 [DOI] [Google Scholar]
- Leiminger, J. H. , Auinger, H.‐J. , Wenig, M. , Bahnweg, G. , & Hausladen, H. (2016). Genetic variability among Alternaria solani isolates from potatoes in Southern Germany based on RAPD‐profiles. Journal of Plant Diseases and Protection, 120, 164–172. 10.1007/BF03356470 [DOI] [Google Scholar]
- Leiminger, J. H. , & Hausladen, H. (2012). Early blight control in potato using disease‐orientated threshold values. Plant Disease, 96, 124–130. 10.1094/PDIS-05-11-0431 [DOI] [PubMed] [Google Scholar]
- Leiminger, J. H. , & Hausladen, H. (2014). Untersuchungen zur Befallsentwicklung und Ertragswirkung der Dürrfleckenkrankheit (Alternaria spp.) in Kartoffelsorten unterschiedlicher Reifegruppe. Gesunde Pflanzen, 66, 29–36. 10.1007/s10343-014-0314-0 [DOI] [Google Scholar]
- Li, H. , & Durbin, R. (2009). Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics (Oxford, England), 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallik, I. , Arabiat, S. , Pasche, J. S. , Bolton, M. D. , Patel, J. S. , & Gudmestad, N. C. (2014). Molecular characterization and detection of mutations associated with resistance to succinate dehydrogenase‐inhibiting fungicides in Alternaria solani. Phytopathology, 104, 40–49. [DOI] [PubMed] [Google Scholar]
- Mariette, N. , Androdias, A. , Mabon, R. , Corbière, R. , Marquer, B. , Montarry, J. , & Andrivon, D. (2016). Local adaptation to temperature in populations and clonal lineages of the Irish potato famine pathogen Phytophthora infestans. Ecology and Evolution, 6, 6320–6331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald, B. A. , & Linde, C. (2002). Pathogen population genetics, evolutionary potential, and durable resistance. Annual Review of Phytopathology, 40, 349–379. [DOI] [PubMed] [Google Scholar]
- McKenna, A. , Hanna, M. , Banks, E. , Sivachenko, A. , Cibulskis, K. , Kernytsky, A. , Garimella, K. , Altshuler, D. , Gabriel, S. , Daly, M. , & DePristo, M. A. (2010). The Genome Analysis Toolkit: A MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Research, 20, 1297–1303. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng, J.‐W. , Zhu, W. , He, M.‐H. , Wu, E.‐J. , Duan, G.‐H. , Xie, Y.‐K. , Jin, Y.‐J. , Yang, L.‐N. , Shang, L.‐P. , & Zhan, J. (2015). Population genetic analysis reveals cryptic sex in the phytopathogenic fungus Alternaria alternata. Scientific Reports, 5, 18250. 10.1038/srep18250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metz, N. , Adolf, B. , Chaluppa, N. , Hückelhoven, R. , & Hausladen, H. (2019). Occurrence of sdh mutations in german alternaria solani isolates and potential impact on boscalid sensitivity in vitro, in the greenhouse, and in the field. Plant Disease, 103, 3065–3071. [DOI] [PubMed] [Google Scholar]
- Mphahlele, G. H. , Kena, M. A. , & Manyevere, A. (2020). Evaluation of aggressiveness of Alternaria solani isolates to commercial tomato cultivars. Archives of Phytopathology and Plant Protection, 53, 570–580. [Google Scholar]
- Nottensteiner, M. , Absmeier, C. , & Zellner, M. (2019). QoI fungicide resistance mutations in alternaria solani and alternaria alternata are fully established in potato growing areas in bavaria and dual resistance against SDHI fungicides is upcoming. Gesunde Pflanzen, 71, 155–164. 10.1007/s10343-019-00475-5 [DOI] [Google Scholar]
- Odilbekov, F. , Edin, E. , Garkava‐Gustavsson, L. , Hovmalm, H. P. , & Liljeroth, E. (2016). Genetic diversity and occurrence of the F129L substitutions among isolates of Alternaria solani in south‐eastern Sweden. Hereditas, 153(1), 10.1186/s41065-016-0014-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odilbekov, F. , Edin, E. , Mostafanezhad, H. , Coolman, H. , Grenville‐Briggs, L. J. , & Liljeroth, E. (2019). Within‐season changes in Alternaria solani populations in potato in response to fungicide application strategies. European Journal of Plant Pathology, 155, 953–965. 10.1007/s10658-019-01826-8 [DOI] [Google Scholar]
- Pereira, D. A. , McDonald, B. A. , & Brunner, P. C. (2017). Mutations in the CYP51 gene reduce DMI sensitivity in Parastagonospora nodorum populations in Europe and China. Pest Management Science, 73, 1503–1510. [DOI] [PubMed] [Google Scholar]
- Pfeifer, B. , Wittelsbürger, U. , Ramos‐Onsins, S. E. , & Lercher, M. J. (2014). PopGenome: an efficient Swiss army knife for population genomic analyses in R. Molecular Biology and Evolution, 31, 1929–1936. 10.1093/molbev/msu136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotem, J. (1994). The genus Alternaria: biology, epidemiology, and pathogenicity. American Phytopathological Society. [Google Scholar]
- Slowikowski, K. (2018). ggrepel Repel overlapping text labels away from each other. Available on ggrepel.slowkow.com
- Stam, R. , Einspanier, S. , & Susanto, T. (2021) Supplementary Data for: Whole genome sequencing elucidates the species‐wide diversity and evolution of fungicide resistance in the early blight pathogen Alternaria solani. [DOI] [PMC free article] [PubMed]
- Stam, R. , Sghyer, H. , Tellier, A. , Hess, M. , & Hückelhoven, R. (2019). The current epidemic of the barley pathogen ramularia collo‐cygni derives from a population expansion and shows global admixture. Phytopathology®, 109, 2161–2168. [DOI] [PubMed] [Google Scholar]
- Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics, 30, 1312–1313. 10.1093/bioinformatics/btu033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travadon, R. , Sache, I. , Dutech, C. , Stachowiak, A. , Marquer, B. , & Bousset, L. (2011). Absence of isolation by distance patterns at the regional scale in the fungal plant pathogen Leptosphaeria maculans. Fungal Biology, 115, 649–659. 10.1016/j.funbio.2011.03.009 [DOI] [PubMed] [Google Scholar]
- Tymon, L. S. , Peever, T. L. , & Johnson, D. A. (2016). Identification and Enumeration of Small‐Spored Alternaria Species Associated with Potato in the U.S. Northwest. Plant Disease, 100, 465–472. [DOI] [PubMed] [Google Scholar]
- Upadhyay, P. , Ganaie, S. H. , & Singh, N. (2019). Diversity assessment among alternaria solani isolates causing early blight of tomato in India. Proceedings of the National Academy of Sciences, India Section B: Biological Sciences. 89, 987–997. [Google Scholar]
- Vagndorf, N. , Heick, T. M. , Justesen, A. F. , Andersen, J. R. , Jahoor, A. , Jørgensen, L. N. , & Orabi, J. (2018). Population structure and frequency differences of CYP51 mutations in Zymoseptoria tritici populations in the Nordic and Baltic regions. European Journal of Plant Pathology, 152, 327–341. 10.1007/s10658-018-1478-8 [DOI] [Google Scholar]
- van der Waals, J. E. , Korsten, L. , & Slippers, B. (2004). Genetic diversity among alternaria solani isolates from potatoes in South Africa. Plant Disease, 88, 959–964. [DOI] [PubMed] [Google Scholar]
- Wang, L.‐G. , Lam, T.‐Y. , Xu, S. , Dai, Z. , Zhou, L. , Feng, T. , Guo, P. , Dunn, C. W. , Jones, B. R. , Bradley, T. , Zhu, H. , Guan, Y. I. , Jiang, Y. , & Yu, G. (2020). Treeio: An R package for phylogenetic tree input and output with richly annotated and associated data. Molecular Biology and Evolution, 37, 599–603. 10.1093/molbev/msz240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber, B. , & Halterman, D. A. (2012). Analysis of genetic and pathogenic variation of Alternaria solani from a potato production region. European Journal of Plant Pathology, 134, 847–858. 10.1007/s10658-012-0060-z [DOI] [Google Scholar]
- Wickham, H. (2009). ggplot2: Elegant Graphics for Data Analysis. Springer‐Verlag. [Google Scholar]
- Wickham, H. , Averick, M. , Bryan, J. , Chang, W. , McGowan, L. , François, R. , Grolemund, G. , Hayes, A. , Henry, L. , Hester, J. , Kuhn, M. , Pedersen, T. , Miller, E. , Bache, S. , Müller, K. , Ooms, J. , Robinson, D. , Seidel, D. , Spinu, V. , … Yutani, H. (2019). Welcome to the tidyverse. Journal of Open Source Software, 4, 1686. 10.21105/joss.01686 [DOI] [Google Scholar]
- Wolters, P. J. , Faino, L. , van den Bosch, T. B. M. , Evenhuis, B. , Visser, R. G. F. , Seidl, M. F. , & Vleeshouwers, V. G. A. A. (2018). Gapless genome assembly of the potato and tomato early blight pathogen Alternaria solani. Molecular Plant‐Microbe Interactions: MPMI, 31, 692–694. [DOI] [PubMed] [Google Scholar]
- Woudenberg, J. H. C. , Groenewald, J. Z. , Binder, M. , & Crous, P. W. (2013). Alternaria redefined. Studies in Mycology, 75, 171–212. 10.3114/sim0015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, G. (2020). Using ggtree to Visualize Data on Tree‐Like Structures. Current Protocols in Bioinformatics, 69, e96. 10.1002/cpbi.96 [DOI] [PubMed] [Google Scholar]
- Zhang, Y. , Zhou, Q. , Tian, P. , Li, Y. , Duan, G. , Li, D. , Zhan, J. , & Chen, F. (2020). Induced expression of CYP51 associated with difenoconazole resistance in the pathogenic Alternaria sect. on potato in China. Pest Management Science, 76, 1751–1760. [DOI] [PubMed] [Google Scholar]
- Zhao, D. , Fan, S. , Zhang, D. , Pan, Y. , Gu, Q. , Wang, J. , Yang, Z. , & Zhu, J. (2021). Parasexual reproduction in Alternaria solani: Simple sequence repeat molecular evidence for haploidization. Mycologia, 113, 949–955. [DOI] [PubMed] [Google Scholar]
- Zheng, H. H. , & Wu, X. H. (2013). First report of alternaria blight of potato caused by alternaria tenuissima in China. Plant Disease, 97, 1246. [DOI] [PubMed] [Google Scholar]
- Zheng, X. , Levine, D. , Shen, J. , Gogarten, S. M. , Laurie, C. , & Weir, B. S. (2012). A high‐performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics (Oxford, England), 28, 3326–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S7
Table S1
Data Availability Statement
Raw short read data are deposited to NCBI SRA (project number PRJNA746421). SNP call files are deposited to Zenodo.org under doi 10.5281/ (Stam et al., 2021).
Raw short read data are deposited to NCBI SRA (project number PRJNA746421). SNP call files are deposited to Zenodo.org under doi 10.5281/ (Stam et al., 2021).