

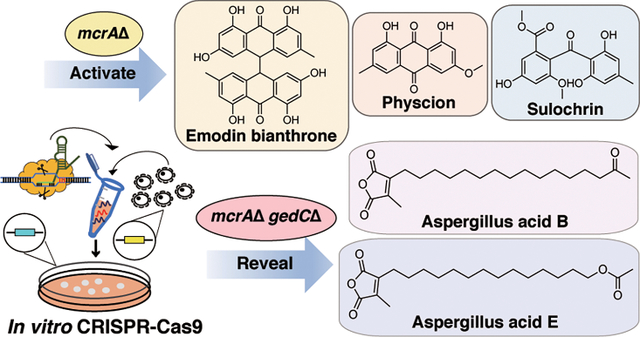
Abstract
Genome sequencing of filamentous fungi has demonstrated that most secondary metabolite biosynthetic gene clusters (BGCs) are silent under standard laboratory conditions. In this work, we have established an in vitro CRISPR-Cas9 system in Aspergillus wentii. To activate otherwise silent BGCs, we deleted the negative transcriptional regulator mcrA. Deletion of mcrA (mcrAΔ) resulted in differential production of 17 SMs in total when the strain was cultivated on potato dextrose media (PDA). Nine out of fifteen of these SMs were fully characterized, including emodin (1), physcion (2), sulochrin (3), physcion bianthrone (4), 14-O-demethylsulochrin (5), (trans/cis)-emodin bianthrone (6 and 7), and (trans/cis)-emodin physcion bianthrone (8 and 9). These compounds were all found to be produced by the same polyketide synthase (PKS) BGC. We then performed a secondary knockout targeting this PKS cluster in the mcrAΔ background. The metabolite profile of the dual-knockout strain revealed new metabolites that were not previously detected in the mcrAΔ parent strain. Two additional SMs were purified from the dual-knockout strain and were characterized as aspergillus acid B (16) and a structurally related but previously unidentified compound (17). For the first time, this work presents a facile genetic system capable of targeted gene editing in A. wentii. This work also illustrates the utility of performing a dual knockout to eliminate major metabolic products, enabling additional SM discovery.
Graphical Abstract
INTRODUCTION
Secondary metabolites (SMs) produced by filamentous fungi often exhibit interesting and potent bioactivities widely applicable to the pharmaceutical and food industries. Fungal SMs, for example, exhibit antibiotic, anticancer, and immunosuppressive properties.1,2 Initial efforts toward the identification of novel fungal SMs relied on methods such as the one strain-many compounds (OSMAC) approach.3,4 However, the majority of SM biosynthetic gene clusters (BGCs) are silent under normal laboratory growth conditions, which makes OSMAC only partially successful in identifying new SMs. New techniques are needed to activate otherwise silent BGCs and express and characterize their corresponding SMs. Several genomic approaches to activate SM BGCs have been developed, including heterologous expression, promotor substitution, and overexpression of transcription factors. Despite these advances, the activation of many SM BGCs and identification of downstream products remain elusive, particularly for species without well-developed molecular genetic systems.5,6
The number of sequenced fungal genomes continues to increase, enabling genomic and comparative genomics projects. The growth of these genomic resources is supported by efforts such as the Fungal Genome Initiative,7 1000 Fungal Genomes Project,8 and the Y1000 project along with hundreds of projects from individual labs and consortia. The genus Aspergillus has been heavily studied and encompasses a diverse collection of globally distributed species that occupy broad ecological niches.9,10 Many genus-sequencing efforts have been devoted to studying different Aspergillus species and the SMs they produce. Aspergilli were found to produce extracellular enzymes (section Nigri), pharmaceuticals (Aspergillus fumigatus and Aspergillus terreus), and toxic fungal compounds such as aflatoxins (Aspergillus flavus and Aspergillus parasiticus).11–13 However, genome sequencing efforts have revealed that the number of predicted BGCs greatly exceeds the number of known SMs, especially in some less well-studied species, such as Aspergillus wentii and Aspergillus steynii. 14,15
Recently, molecular genetic tools have rapidly progressed to enable rapid and highly programmable genome editing.16,17 CRISPR (clustered regularly interspaced short palindromic repeat)-Cas9 is one technique that has been successfully utilized in bacteria, fungi, and mammalian cells to edit DNA sequences.18 Cas9 is a DNA nuclease that cleaves target DNA sequences by combining with a guide RNA (gRNA) to form a ribonucleoprotein (RNP) complex. The gRNA consists of two parts: the CRISPR RNA (crRNA) and trans-activating CRISPR RNA (tracrRNA). While tracrRNA remains unchanged, the crRNA contains a 20-base pair protospacer that can be modified to complement a target gene. Prior to transformation, commercially available Cas9, crRNA, and tracrRNA were rapidly assembled in vitro, which makes the transformation process more efficient. The speed and efficiency of this CRISPR-RNP system have also been used to engineer several fungal species such as Penicillium chrysogenum and Aspergillus niger.19–21,27 To ensure DNA cleavage, the protospacer is followed by a PAM (protospacer-adjacent motif) sequence, which is typically NGG, where N is any nucleotide.22,23 Following DNA cleavage, the genome can be repaired through either the non-homologous end-joining (NHEJ) or homology-directed repair (HDR) DNA-repair pathways. While NHEJ usually introduces short deletions or insertions of genes to the target site, HDR integrates a repair template that is largely homologous to the target gene. It has recently been demonstrated that the genome can also be repaired by providing a microhomology template flanking the double-stranded break (DSB) site through microhomology-mediated end joining (MMEJ).17,24
In recent years, many regulators of secondary metabolism have been characterized in Aspergillus species. Among them, global regulators such as laeA and mcrA have been noted for their abilities to activate multiple SM BGCs simultaneously. McrA was found to encode a zinc-finger transcription factor that was able to regulate multiple BGCs at once in previous studies.25,26 Deletion of mcrA in Aspergillus nidulans was found to upregulate over 1000 genes, including those contained within 10 BGCs. Given the highly conserved nature of mcrA and its ability to regulate multiple BGCs simultaneously, the manipulation of the mcrA gene represents an attractive method to activate BGCs in other filamentous fungi.26
Although the genomes of thousands of fungal species have been sequenced, only a small fraction has genetic tools developed to fully exploit their secondary metabolism potential. Some wild-type fungal species have high resistance to indels (insertion or deletions of nucleotide bases) introduced by NHEJ, and some strains require species-specific promoters for expressing Cas9.27 Previous studies have illustrated that higher gene targeting efficiency was observed in Ku-knockout strains, where Ku is a dimeric protein complex required for NHEJ.28–30 Unfortunately, deletion of Ku genes in a wild-type strain with low transformation efficiency sometimes can be very difficult to achieve and requires laborious work for selecting correct transformants. As genetic manipulation of wild-type fungal strains remains difficult, new methods are needed to make programmable editing feasible and easy. As recent studies indicate that one can target specific genes directly in wild-type fungal strains using in vitro assembled Cas9-guide RNA ribonucleoproteins coupled with microhomology repair templates,17,22 we decided to apply this new technique to A. wentii, which previously exhibited low transformation efficiency in our laboratory using plasmidfacilitated CRISPR-Cas9.
In this work, we have sequenced and annotated an A. wentii strain, established an in vitro CRISPR-Cas9 system, deleted mcrA, and observed increased production of several metabolites produced by this mutant strain. Nine of the fifteen upregulated metabolites produced in sufficient quantities for purification and characterization by NMR were found to be part of the physcion/emodin pathway.31,32 To isolate the other BGC products activated in the mcrA deletion strain, we needed to divert the starter unit pool away from the physcion/emodin pathways by eliminating physcion/emodin genes. We performed a second knockout targeting the PKS gene involved in emodin biosynthesis in the mcrAΔ background using the same in vitro CRISPR technique. By creating a dual-knockout A. wentii strain (mcrAΔ gedCΔ), six LCMS-detectable metabolites found to have increased in the mcrAΔ strain disappeared, suggesting that these are also involved in the physcion/emodin pathway. Satisfyingly, two metabolites (Aspergillus acid B (16)33,34 and a structurally related new metabolite, designated Aspergillus acid E) could be isolated and characterized from the dual-knockout strain. Our data show that compounds that were upregulated by deletion of mcrA, but were previously hidden due to overlap with the major SMs upregulated, could be detected in the dual-knockout strain.
Our results indicate that this in vitro CRISPR-Cas9 system can be employed to generate dual knockout mutants and enable direct hypothesis testing of genes involved in production of specific SMs. This technique facilitated the upregulation, purification, and characterization of new metabolites that have previously eluded identification. Finally, we show that wild-type fungal species can be genetically accessed using the in vitro CRISPR-Cas9 system described in this study.
RESULTS AND DISCUSSION
Establishment of an In Vitro CRISPR-Cas9 System in A. wentii by Targeting the pksP Melanin Gene.
To assess the transformation efficiency for A. wentii using our in vitro CRISPR-Cas9 system, we targeted the homologue of a pigment-associated gene, pksP, that was first discovered in A. fumigatus (Afu2g17600). Since the gene is involved in dihydroxy naphthalene (DHN)-melanin biosynthesis, targeting pksP allows us to evaluate CRISPR-Cas9 efficiency by selecting transformants that are antibiotic-resistant and determining the fraction of transformants that are white. The melanin metabolite imparts a gray-greenish color to A. fumigatus conidia, while it may also account for a yellow-brownish color in other species.35,36 The homologue of pksP was identified in the genome by searching the annotated proteins with the Afu2g17600 protein sequence using BLASTp (RRID:SCR_001010). Each domain within this gene was carefully analyzed to confirm its identity.
Different from plasmid-facilitated CRISPR-Cas9, which usually results in deletions or insertion of a few nucleotides through NHEJ, the in vitro CRISPR-Cas9 system we utilized is predicted to delete the entire coding region of the gene through MMEJ. We designed two crRNAs targeting the 5′ untranslated region (UTR) and 3′ UTR of pksP separately. Both targeting sites were followed by functional PAM (NGG) sequences to permit Cas9 cleavage. To minimize off-target effects, we performed a BLAST analysis for the protospacer sequences within the A. wentii genome. The chosen protospacers adjacent to the PAMs were close (within the first three nucleotides) to the start and stop codons, with no obvious off-targets being identified.
Wild-type fungal strains with intact Ku genes favor NHEJ repair over homologous recombination (HR) following DNA double-strand breaks.29,30 However, the efficiency of gene replacement in wild-type strains can be improved by simultaneous two-site CRISPR cuts (at the 5′ and 3′ UTRs) with a repair template containing a proper selection marker.17,37 Phleomycin and hygromycin B were chosen as selection markers for A. wentii based on the sensitivity of the strain to these chemicals. Antibiotic resistance tests were performed in order to determine the effective concentrations of each antibiotic. Wild-type A. wentii protoplasts (1.0 × 106) were inoculated onto agar plates containing various phleomycin or hygromycin concentrations (0, 0.01, 0.05, 0.1, 0.2, and 0.3 mg/mL). The minimum effective concentration that inhibits growth was found to be 0.1 mg/mL for phleomycin and 0.3 mg/mL for hygromycin B (Supplemental Figure 1 and 2). As the wild-type A. wentii is more resistant to hygromycin B, we decided to use a phleomycin cassette as a selection marker for the first transformation targeting pksP. The repair template containing a phleomycin resistance gene (blue) and 50 bp homologous flanking sequences were amplified via PCR (Figure 1B).
Figure 1.
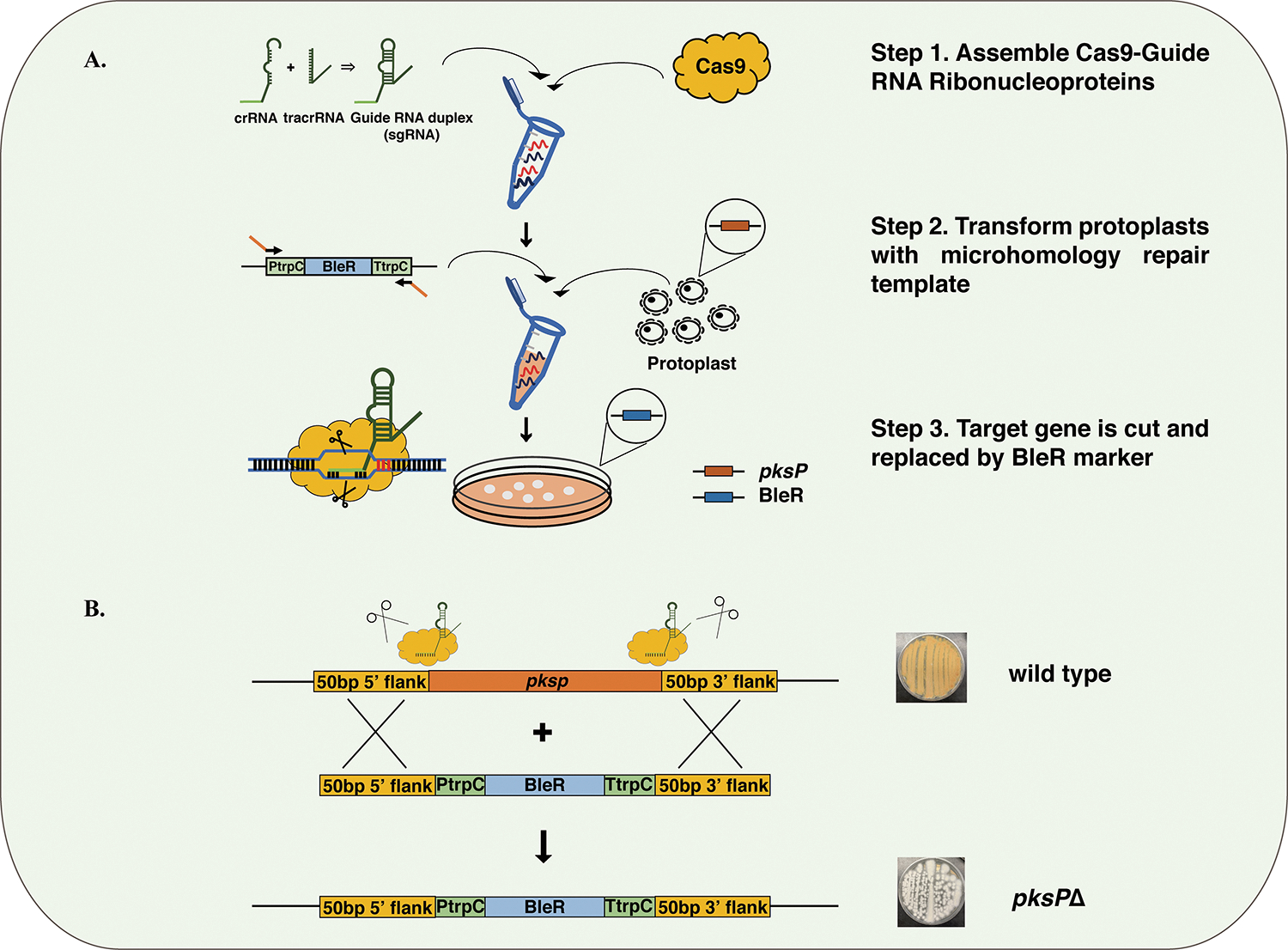
Overview of gene replacement coupled with in vitro assembled RNP cleavage. (A) Replacement of pksP with an antibiotic resistance gene by in vitro CRISPR coupled with dual Cas9 RNP cleavage. Dual crRNAs targeting the beginning and the end of pksP were assembled separately with tracrRNAs to become sgRNAs. Two sgRNAs were then assembled with Cas9 to form the RNP. The repair template, consisting of a phleomycin resistance gene (1477 bp), was amplified from plasmid pFC333. Wild-type A. wentii protoplasts were transformed with the RNPs together with the repair template. (B) The pksP gene in A. wentii was replaced by the phleomycin selection marker through DNA cleavages induced by the Cas9 RNP. The resulting white transformants, with the phleomycin resistance marker, were able to grow on media containing phleomycin.
The transformation was performed according to a slightly modified protocol for A. fumigatus.17 All transformants that appeared after 4 days on agar plates containing phleomycin were white, and these were re-streaked on the day after. All the restreaked transformants produced white colonies after a 5 day incubation. Nested primers were designed to amplify the pksP sequence in both the wild type and mutants. PksP amplicons were 6.8 and 1.6 kb for the wild-type and mutant strains, respectively. We determined that 1.6 kb is the combined length of bleR and the homologous flanking regions. The gene replacement efficiency was 100% among the six chosen colonies (Supplemental Figure 3). These results indicate that CRISPR-Cas9 with in vitro-assembled RNPs can be applied to wild-type A. wentii, resulting in exceptionally high gene targeting efficiency.
McrA Deletion Results in Upregulation of Secondary Metabolite Production.
To activate silent BGCs in A. wentii, we deleted the entire coding region of the master negative regulator mcrA. We used the A. nidulans mcrA (AN8694) protein sequence to locate the gene within the A. wentii genome via BLASTp. Because the in vitro CRISPR-Cas9 system had been verified as described above, we performed the same process for designing crRNAs targeting mcrA and the homologous repair template containing bleR with modification in the flanking tails.
Following A. wentii inoculation and transformation, colonies that appeared after 6 days were restreaked to new agar plates and allowed to grow for 5 more days. A new set of nested primers was designed for verifying the deletion of mcrA. Because the total lengths of mcrA and bleR amplicons are similar (2.3 and 2.4 kb, respectively), we designed the forward primer to bind to a sequence within the mcrA coding region and the reverse primer to bind within the repair template. Therefore, only the mutant strains with correct gene replacement would generate amplicons using the designed primers.
Following PCR amplification, no band was observed in wild-type A. wentii, as expected. Three out of eight restreaked transformants also did not generate any amplicons. The remaining transformants generated amplicons of the expected size (Supplemental Figure 4). Therefore, the gene replacement efficiency targeting mcrA is approximately 62.5%. The discrepancy in the knockout efficiency between targeting pksP and mcrA may have been caused by the retarded growth of the mcrAΔ strain. The mcrA mutant colonies appeared on phleomycin-containing plates 1 day later than the pksP mutants, which allowed an extra day for wild-type colonies to grow. However, the gene targeting efficiency remains high, especially when considering that the strain is not NHEJ-deficient.
To examine the effect that an mcrA deletion has on the secondary metabolite profile of A. wentii, we cultivated the wild-type and mcrAΔ strains separately in potato dextrose agar (PDA). We extracted SMs of both the wild-type and mcrAΔ strains from solid PDA plates and analyzed the extracts through HPLC-DAD-MS. Comparative metabolic analysis revealed an increase in the production of several metabolites (Figure 2). To identify these upregulated metabolites, the mcrAΔ strain was scaled up in 100 plates with 20 mL of solid PDA per plate. Subsequent solid–liquid extraction, column chromatography, and HPLC permitted isolation of each upregulated compound. Nuclear magnetic resonance (NMR) analysis led to the identification of nine metabolites, all related to the physcion/emodin biosynthetic pathway,31 including emodin (1), physcion (2), sulochrin (3), physcion bianthrone (4), 14-O-demethylsulochrin (5), (trans/cis)-emodin bianthrone (6 and 7), and (trans/cis)-emodin physcion bianthrone (8 and 9) (Figure 3 and Supplemental Figure 6–14). The results confirm that mcrA is capable of modulating SM production in A. wentii. Manipulation of mcrA opens opportunities for more fungal SM discovery.
Figure 2.
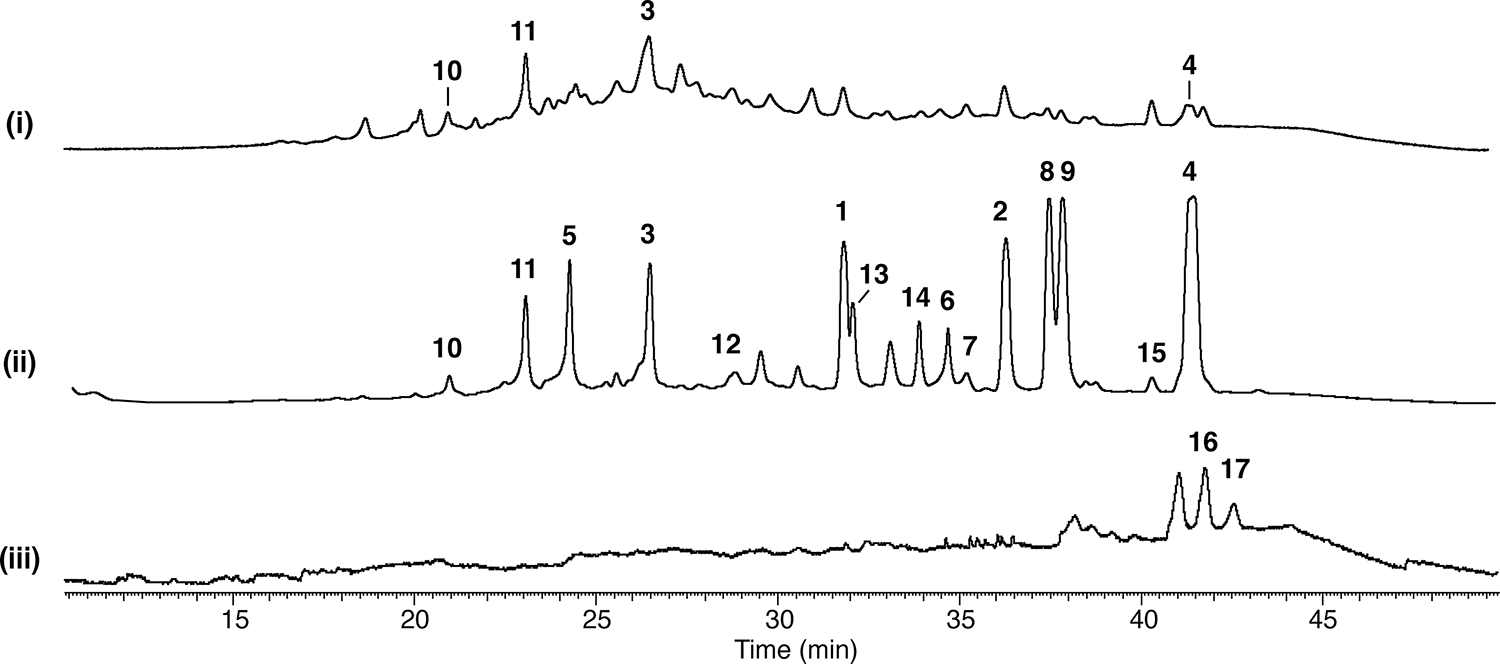
Paired HPLC profiles of A. wentii (wild type, mcrAΔ, and mcrAΔ gedCΔ dual knockout strains) extracts when grown on a solid PDA medium. (i) wild type, (ii) mcrAΔ, and (iii) mcrAΔ gedCΔ dual knockout strains. Compounds 1–15 are gedC-related, while compounds 16 and 17 are additional metabolites revealed in the dual knockout strain.
Figure 3.
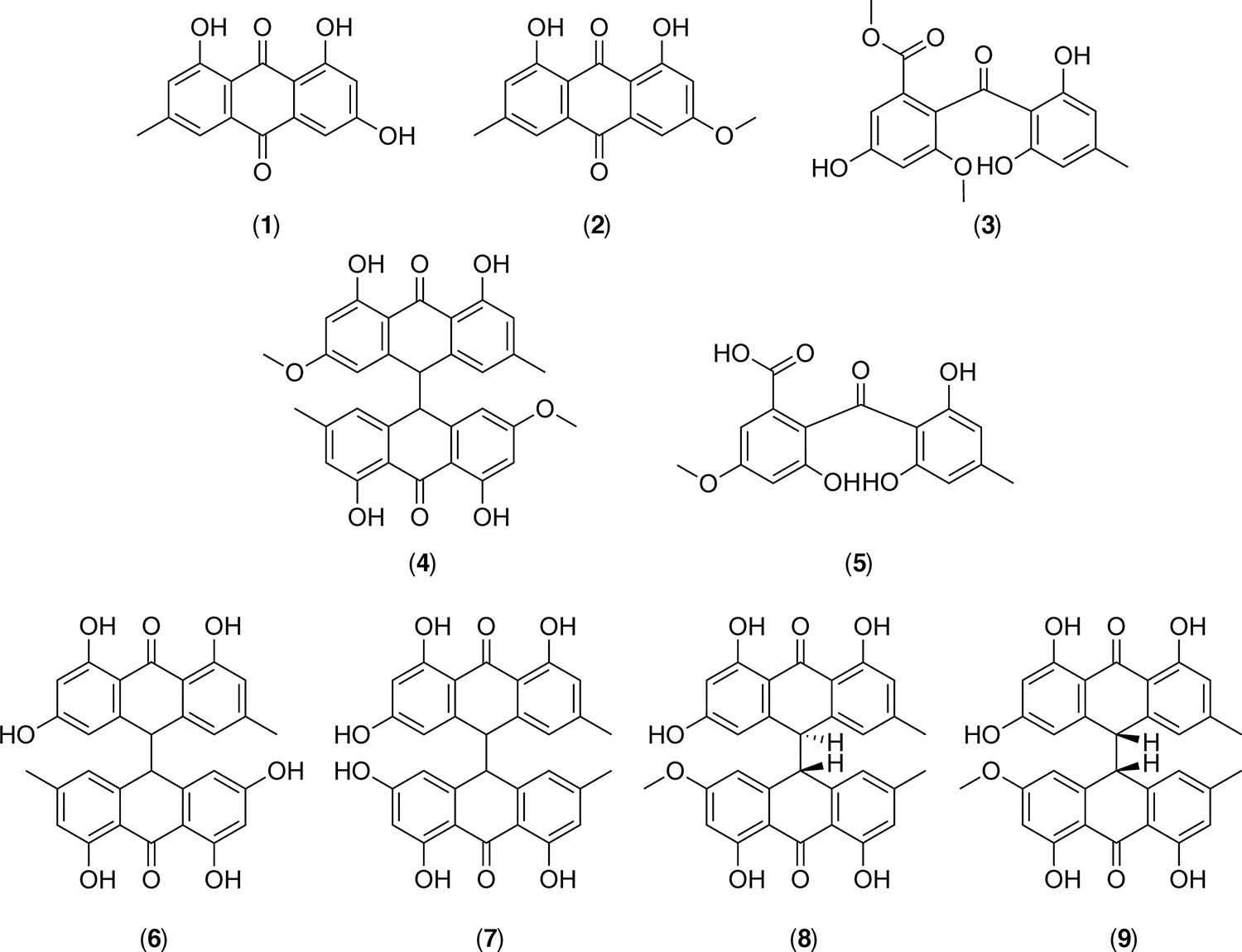
Chemical structures of the upregulated compounds in the A. wentii mcrAΔ strain. Emodin (1), physcion (2), sulochrin (3), physcion bianthrone (4), 14-O-demethylsulochrin (5), (trans)-emodin bianthrone (6), (cis)-emodin bianthrone (7), (trans)-emodin physcion bianthrone (8), (cis)-emodin physcion bianthrone (9).
McrA/gedC Homologue Double-Knockout Reveals Additional Upregulated SMs.
Several compounds that appeared to be unrelated to the physcion/emodin pathway were also detected by HPLC-DAD-MS analysis of the mcrAΔ strain. Because the amount of these metabolites was relatively low compared to physcion/emodin-related compounds, we decided to perform a secondary knockout, targeting the physcion/emodin gene cluster in the mcrAΔ strain parent. We used gedC (ATEG_08451), a gene encoding the polyketide synthase (PKS) that synthesizes a backbone precursor of geodin in A. terreus as a probe, to locate the physcion/emodin gene cluster in A. wentii.32 The proposed biosynthetic pathway of geodin involves intermediates such as emodin and sulochrin that were previously detected in the mcrAΔ strain. Geodin itself was not detected among the upregulated SMs, and the biosynthetic genes that directly form geodin such as gedL and gedJ were also missing in the A. wentii genome, suggesting that different final compounds might be produced (Supplemental Table 3). We designed crRNAs targeting the gedC homologue and amplified the repair template for hygromycin B (hygR) as a second selection marker. Transformed colonies appeared after 6 days, which were restreaked on plates containing 0.3 mg/mL hygromycin B. Successful gene replacement in transformants was confirmed by PCR amplification using nested primers (Supplemental Figure 5). Seven out of nine restreaked colonies generated amplicons of the expected size (2.9 kb) (a 78% gene replacement efficiency). The dual-knockout transformants carrying deletions of both mcrA and gedC were able to grow on plates containing both phleomycin and hygromycin B.
We then cultured the wild type, mcrAΔ, and mcrAΔ gedCΔ double-knockout strains on solid PDA plates and extracted SM from the agar plates. As anticipated, the gedC-related compounds were not detected, and several additional metabolites were revealed (Figure 2). We then performed a scale-up of the mcrAΔ gedCΔ strain. Two major compounds that were upregulated were purified and identified as aspergillus acid B (16)33,34 (Figure 4) and a structurally related new compound that we designate aspergillus acid E (17) (Figure 4 and Supplemental Figures 15–18). These two compounds were originally produced in low quantities and coeluted with physcion bianthrone (4), which allowed them to evade detection in the mcrAΔ mutant. Aspergillus acids B and E are found to be structurally related to the monomers that produce scytalidin and zopfiellin, which belong to an important class of natural products containing maleic anhydride moieties (Supplemental Table 4).38,39
Figure 4.
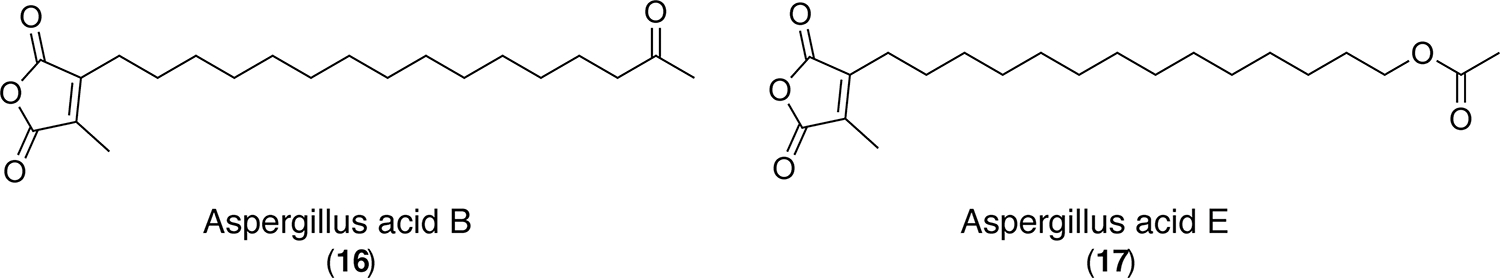
Chemical structures of compound 16 (Aspergillus acid B) and compound 17 (Aspergillus acid E) as determined by NMR.
With an additional selection marker, one can perform a second knockout targeting a different gene on a previously manipulated strain without sacrificing the transformation efficiency rate. As deletion of mcrA usually upregulates different BGCs, a dual-knockout CRISPR opens opportunities to characterize more SMs that have previously eluded identification due to overlapping with major compounds. Previous work has revealed that different compounds are upregulated on different carbon sources in an A. nidulans mcrA-deletion strain.26 Experiments focusing on purifying and identifying additional SMs produced by A. wentii mutant strains under different growth media conditions are ongoing and will be reported in due course.
CONCLUSIONS
We successfully applied an in vitro CRISPR-Cas9 technique to genetically engineer a wild-type A. wentii strain that previously exhibited low transformation efficiency via plasmid-facilitated CRISPR-Cas9. We deleted mcrA through MMEJ and examined its effect on secondary metabolism through metabolic profiling. Nine of fifteen upregulated SMs were characterized through NMR, including emodin (1), physcion (2), sulochrin (3), physcion bianthrone (4), 14-O-demethylsulochrin (5), (trans/cis)-emodin bianthrone (6 and 7), and (trans/cis)-emodin physcion bianthrone (8 and 9). The six additional metabolites were also revealed to be related to the physcion/emodin pathway by performing a second knockout targeting the physcion/emodin gene cluster. As production of the 15 upregulated SMs was abated in the dual-knockout strain, additional metabolites that were previously undetectable were revealed. By analyzing the dual-knockout strain, two more SMs were purified and identified as aspergillus acid B (16) and a new, structurally related compound designated aspergillus acid E (17). Overall, our results indicate that in vitro CRISPR-Cas9 systems can be highly successful in wild-type Aspergilli. This method allows the wild-type fungal species to be genetically accessed, bypassing the disruption of Ku genes, which makes gene edits easier and faster to achieve. Once the in vitro CRISPR-Cas9 system is established in a wild-type species, the strain might be adapted to multi-gene knockouts, and therefore, synergetic effects of two or multiple genes can be examined straightforwardly. It might be critical to generate transformation protocols using recycled markers so that the in vitro CRISPR system established would not be limited by the number of selection markers that can be applied to the target strains. Based on our results, generating a dual-knockout strain and eliminating the major compounds produced are beneficial for the detection and isolation of new compounds in filamentous fungi.
METHODS
Genome Assembly and Annotation.
For 5 days, wild-type A. wentii (IMI 49129) was cultured on potato dextrose agar (PDA). The spore suspension was harvested using ST buffer (8.5 g L−1 NaCl and 1 mL L−1 Tween 80). DNA extraction of fresh spores was performed using Qiagen DNeasy PowerLyzer Microbial Kit through the standard protocol. The quality of the extracted DNA and the final concentration were tested using Nanodrop (Thermo Scientific NanoDrop 2000c spectrophometer). Approximately 50 μL of DNA sample (50.9 ng/μL) was sent to Novogene for Illumina NovaSeq sequencing.
After retrieving the raw sequencing results from Novogene, downstream genome analysis was performed at the High-Performance Computing Center (HPCC) at UC Riverside (https://hpcc.ucr.edu). The fungal genome was assembled through AAFTF (v0.2.0),40 which relies on Trimmomatic v0.36 to trim reads and Bowtie v2.3.4.1 to filter against a databases of contaminants, assemble reads with SPAdes (v3.12.0),41 remove vector sequences by BLASTN against vector sequence databases, filter bacteria contamination with Sourmash v3.5.0,42 and polish the assembly with the short reads using Pilon. The final assembly was masked with RepeatMasker (v. open-1.0.11)43 and annotated with the Funannotate pipeline (v1.5.0).44 This approach uses HISAT2 (v2.2.1),45 Trinity (v2.11.0),46 and PASA (v2.4.1)47 to predict genes and identify homology for functional annotation. The annotated genome was analyzed with antiSMASH (v4.1.0)48 to search for SM gene clusters. The sequence reads, assembly, and annotation of the strain were deposited at NCBI GenBank under Bioproject PRJNA826222. The genome characteristics and predicted clusters were summarized in Supplemental Tables 1 and 2.
Molecular Genetic Procedures.
Phleomycin and hygromycin B resistance cassettes were used as selection markers in this work. A 1477 bp BleR microhomology repair template spanning 320 bp of the trpC promoter, 375 bp of the phleomycin resistance cassette, and 782 bp of the trpC terminator was amplified from pFc333 through PCR using primers containing 50 bp flanking regions (Supplemental Table 5). A 1375 bp HygR microhomology repair template, which spans 320 bp of the trpC promoter and 1020 bp of the hygromycin B resistance cassette, was amplified from pFc332. The PCR products were purified by gel extraction and eluted with Elution Buffer (Qiagen, cat. no. 19086).
The Cas9-gRNA ribonucleoprotein complexes were assembled with Alt-R-CRISPR-Cas9 components from Integrated DNA Technologies (IDT). The designed crRNA and universal tracrRNA were prepared as 100 μM stock solutions and stored at −20 °C until use. The Cas9 nuclease was diluted to 1 μg/μL using nuclease-free Cas9 working buffer (20 mM HEPES, 150 mM KCl, pH 7.5) and stored at −20 °C. The guide RNA duplex was assembled by mixing crRNA and tracrRNA in equal molar concentrations to reach a final concentration of 33 μM (Figure 1). The mixtures were heated at 95 °C for 5 min and cooled to room temperature. The final duplex was stored at −20 °C until use. The Cas9 RNP complexes were generated by combining 1.5 μL of each gRNA duplex separately with 0.75 μL of Cas9 and 11 μL of nuclease-free Cas9 working buffer. The mixtures were incubated at room temperature for 5 min to form the RNP complexes. Two RNP complexes were combined to form a final volume of 26.5 μL before they were added to A. wentii protoplasts during transformation.
Transformation of A. wentii.
Fresh spores of wild-type A. wentii were harvested from PDA plates. Spores of A. wentii (1 × 108) were inoculated into 50 mL of potato dextrose broth (PDB) in a 250 mL flask and incubated overnight at 30 °C with shaking at 135 rpm. Mycelia were harvested through filtration and resuspended in protoplasting buffer. The protoplasting buffer was prepared by adding 1.2 g of VinoTaste Pro (VTP) in 20 mL of 1.1 M KCl and 0.39 M citric acid monohydrate buffer (pH 5.8, adjusted with 1.1 M KOH). After vortexing for 15 min, the protoplasting buffer was centrifuged for 15 min at 1800g. The supernatant was filter-sterilized into a 50 mL flask together with the filtered mycelia. The flask was incubated at 30 °C with shaking at 100 rpm for 4 h. Five milliliters of the protoplast suspension was transferred into a 15 mL tube and gently overlaid with 5 mL of 0.4 M ST (0.4 M d-sorbitol and 100 mM Tris–HCl (pH 8)). Protoplasts were separated from the mycelial debris by centrifuging for 15 min at 4 °C and 800g. The protoplast layer at the interface was collected and transferred into a new tube. After gently adding 15 mL of ST (1.0 M d-sorbitol and 50 mM Tris–HCl (pH 80), the solution was centrifuged at room temperature at 800g for 10 min. The pellet was washed with 10 mL of ST and centrifuged at room temperature at 800g for 10 min. The protoplast pellet was resuspended in STC buffer (1.0 M d-sorbitol, 50 mM CaCl2, and 50 mM Tris–HCl (pH 8)), and 100 μL (approximately 1.0 × 106) of protoplasts were transferred to the tube containing Cas9 RNP mixture (26.5 μL). Three micrograms of the purified repair template and 25 μL of polyethylene glycol (PEG)-CaCl2 buffer (40% [wt/vol] PEG 3350, 50 mM CaCl2, and 50 mM Tris–HCl (pH 8)) were added immediately after transferring protoplasts. The mixture was incubated on ice for 50 min. After adding 1.25 mL of PEG-CaCl2, the mixture was then incubated at room temperature for 20 min. By adding STC buffer, the mixture was brought to 2 mL, and 500 μL of suspension was spread on SMM agar plates (GMM supplemented with 1.2 M sorbitol and 1.5% [wt/vol] agar). The agar plates were incubated at room temperature overnight, and the second layer of SMM top agar (GMM supplemented with 1.2 M sorbitol and 0.7% [wt/vol] agar) containing the selected antibiotic was overlaid. The plates were incubated at 30 °C for 4 days for spore generation.
Culturing and HPLC-DAD-MS Analysis.
A. wentii strains were incubated at 30 °C on PDA (Difco Potato Dextrose Agar) plates. Spores (1.0 × 107) were inoculated on each plate, and five plugs (7 mm diameter) were cut out for compound extraction after 5 days of culturing. The agar plugs were extracted with 5 mL of methanol with 1 h of sonication. The extract was collected, and the agar plugs were extracted with 5 mL of dichloromethane:methanol (1:1) followed by another 1 h of sonication. The extract was collected into the same clean vial, and the solvent was evaporated through a TurboVap LV (Caliper LifeSciences). The solid residues were dissolved in equal amounts (10 mL) of EtOAc and water. The EtOAc layer was collected and evaporated with a TurboVap LV. The final extract was redissolved in 400 μL of DMSO:MeOH (1:4), and 10 μL was injected into LC-DAD-MS for analysis. To obtain the LC/MS spectra, we used a ThermoFinnigan LCQ Advantage ion trap mass spectrometer with a reverse-phase C18 column (Alltech Prevail C18; column, 2.1 mm × 100 mm; particle size, 3 μm; flow rate 125 μL min−1). Solvent A was 5% MeCN–H2O, and solvent B was 95% MeCN–H2O. Both solvents contained 0.05% formic acid. The solvent gradient was as follows: 100% solvent A from 0 to 5 min, 0 to 25% solvent B from 5 to 6 min, 25 to 100% solvent B from 6 to 35 min, 100% solvent B from 35 to 40 min, 100 to 0% solvent B from 40 to 45 min, and re-equilibration with 100% solvent A from 45 to 50 min. The MS included a 5.0 kV capillary voltage, 60 arbitrary unit flow rate of the sheath gas, 10 arbitrary units of the auxiliary gas, and 350 °C of the ion transfer capillary temperature.
Compound Purification and Characterization.
A. wentii strains were cultured in 40 mm × 150 mm diameter Petri dishes with a total volume of 2 L of PDA medium at 30 °C for 5 days. The agar was chopped into pieces and extracted with methanol and dichloromethane:methanol (1:1) followed by 1 h of sonication as described above. The residue was extracted three times with EtOAc, and the combined EtOAc layers were evaporated in vacuo to yield a crude extract. Silica gel column chromatography was performed using dichloromethane and methanol as eluent (starting with 100% dichloromethane). Each fraction was further separated through preparative HPLC [Phenomenex Luna 5 μm C18 (2), 250 × 21.2; flow rate of 5.0 mL min−1; UV detector at 280 nm]. Nuclear magnetic resonance (NMR) spectral analysis was performed using a Varian Mercury Plus 400 spectrometer. High-resolution electrospray ionization mass spectra (HRESIMS) were obtained on a Thermo Scientific Q Exactive Hybrid Quadrupole-Orbitrap mass spectrometer at a flow rate of 5 μL min−1. MS conditions included a spray voltage of 5 kV, sheath gas flow rate at 15 au, auxiliary gas flow rate at 5 au, capillary temperature at 320 °C, s-lens RF level 60, scan range of 100.0–500.0 m/z, resolution of 140,000, AGC target of 5 × 105, and maximum injection time of 50 ms.
2-Tetradec-(17-acetoxy)yl-3-methylmaleic Anhydride (Aspergillus Acid E) (17).
White powder; UV λmaxMeOH nm: 239, 250; 1H NMR (CDCl3): Δ 1.25 (br s, 20H), 1.53–1.63 (m, 4H), 2.04 (s, 3H), 2.07 (s, 3H), 2.45 (t, J = 6 Hz, 2H), 4.05 (t, J = 6 Hz, 2H); 13C NMR (CDCl3): δ 9.5, 21.0, 24.4, 25.8, 27.5, 28.6, 29.2–29.5 (9 × CH2), 64.6, 140.4, 144.7165.8, 166.2, 171.2. See Supplemental Figure 19 for UV and ESIMS spectrum; HRESIMS obtained m/z [M + H]+ = 367.2487 (calcd 367.2485 for C21H35O5).
Supplementary Material
ACKNOWLEDGMENTS
This research was supported in part by the National Institute of Allergy and Infectious Diseases (grant R21AI156320) to C.C.C.W. and B.R.O., and by the Irving S. Johnson fund of the Kansas University Endowment Association to B.R.O.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.2c00456.
NMR spectroscopic data, genome assembly characteristics and secondary metabolite cluster-predicted tables, antibiotic sensitivity tests, diagnostic PCR amplifications, and UV–vis and ESIMS spectra of new and unknown compounds (PDF)
Contributor Information
Bo Yuan, Department of Pharmacology and Pharmaceutical Sciences, School of Pharmacy, University of Southern California, Los Angeles, California 90089, United States.
Nancy P. Keller, Department of Medical Microbiology and Immunology and Bacteriology, University of Wisconsin–Madison, Madison, Wisconsin 53706, United States.
Berl R. Oakley, Department of Molecular Biosciences, University of Kansas, Lawrence, Kansas 66045, United States.
Jason E. Stajich, Department of Microbiology and Plant Pathology, University of California Riverside, Riverside, California 92521, United States.
Clay C. C. Wang, Department of Pharmacology and Pharmaceutical Sciences, School of Pharmacy and Department of Chemistry, Dornsife College of Letters, Arts, and Sciences, University of Southern California, Los Angeles, California 90089, United States.
REFERENCES
- (1).Chavali AK; Rhee SY Bioinformatics tools for the identification of gene clusters that biosynthesize specialized metabolites. Briefings Bioinf. 2018, 19, 1022–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Keller NP; Turner G; Bennett JW Fungal secondary metabolism - from biochemistry to genomics. Nat. Rev. Microbiol. 2005, 3, 937–947. [DOI] [PubMed] [Google Scholar]
- (3).Bode HB; Bethe B; Höfs R; Zeeck A Big effects from small changes: possible ways to explore nature’s chemical diversity. ChemBioChem 2002, 3, 619–627. [DOI] [PubMed] [Google Scholar]
- (4).Hu Z; Ye Y; Zhang Y Large-scale culture as a complementary and practical method for discovering natural products with novel skeletons. Nat. Prod. Rep. 2021, 38, 1775–1793. [DOI] [PubMed] [Google Scholar]
- (5).Keller NP Fungal secondary metabolism: regulation, function and drug discovery. Nat. Rev. Microbiol. 2019, 17, 167–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Chiang Y-M; Wang CCC; Oakley BR (2014) Analyzing fungal secondary metabolite genes and gene clusters. In Natural Products, pp. 171–193, John Wiley & Sons, Ltd. [Google Scholar]
- (7).Galagan JE; Henn MR; Ma LJ; Cuomo CA; Birren B Genomics of the fungal kingdom: insights into eukaryotic biology. Genome Res. 2005, 15, 1620–1631. [DOI] [PubMed] [Google Scholar]
- (8).Grigoriev IV; Nordberg H; Shabalov I; Aerts A; Cantor M; Goodstein D; Kuo A; Minovitsky S; Nikitin R; Ohm RA; Otillar R; Poliakov A; Ratnere I; Riley R; Smirnova T; Rokhsar D; Dubchak I The Genome Portal of the Department of Energy Joint Genome Institute. Nucleic Acids Res. 2012, 40, D26–D32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).de Vries RP; Riley R; Wiebenga A; Aguilar-Osorio G; Amillis S; Uchima CA; Anderluh G; Asadollahi M; Askin M; Barry K; et al. Comparative genomics reveals high biological diversity and specific adaptations in the industrially and medically important fungal genus Aspergillus. Genome Biol. 2017, 18, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Kjærbølling I; Vesth TC; Frisvad JC; Nybo JL; Theobald S; Kuo A; Bowyer P; Matsuda Y; Mondo S; Lyhne EK; Kogle ME; Clum A; Lipzen A; Salamov A; Ngan CY; Daum C; Chiniquy J; Barry K; LaButti K; Haridas S; Simmons BA; Magnuson JK; Mortensen UH; Larsen TO; Grigoriev IV; Baker SE; Andersen MR Linking secondary metabolites to gene clusters through genome sequencing of six diverse Aspergillus species. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E753–E761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kjærbølling I; Vesth T; Frisvad JC; Nybo JL; Theobald S; Kildgaard S; Petersen TI; Kuo A; Sato A; Lyhne EK; et al. A comparative genomics study of 23 Aspergillus species from section Flavi. Nat. Commun. 2020, 11, 1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Vesth TC; Nybo JL; Theobald S; Frisvad JC; Larsen TO; Nielsen KF; Hoof JB; Brandl J; Salamov A; Riley R; et al. Investigation of inter- and intraspecies variation through genome sequencing of Aspergillus section Nigri. Nat. Genet. 2018, 50, 1688–1695. [DOI] [PubMed] [Google Scholar]
- (13).Kjærbølling I; Vesth T; Andersen MR Resistance Gene-Directed Genome Mining of 50 Aspergillus Species. mSystems 2019, 4, No. e00085–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Selva A; Traldi P; Camarda L; Nasini G New secondary metabolites of Aspergillus wentii Wehmer. The positive and negative ion mass spectra produced by electron impact. Biomed. Mass Spectrom. 1980, 7, 148–152. [DOI] [PubMed] [Google Scholar]
- (15).Miao FP; Liang XR; Liu XH; Ji NY Aspewentins A-C, norditerpenes from a cryptic pathway in an algicolous strain of Aspergillus wentii. J. Nat. Prod. 2014, 77, 429–432. [DOI] [PubMed] [Google Scholar]
- (16).Nødvig CS; Nielsen JB; Kogle ME; Mortensen UH A CRISPR-Cas9 System for Genetic Engineering of Filamentous Fungi. PLoS One 2015, 10, No. e0133085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Al Abdallah Q; Ge W; Fortwendel JR A Simple and Universal System for Gene Manipulation in Aspergillus fumigatus: In Vitro-Assembled Cas9-Guide RNA Ribonucleoproteins Coupled with Microhomology Repair Templates. mSphere 2017, 2, No. e00446–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Ramakrishna S; Kwaku Dad AB; Beloor J; Gopalappa R; Lee SK; Kim H Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Res. 2014, 24, 1020–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Pohl C; Kiel JAKW; Driessen AJM; Bovenberg RAL; Nygård Y CRISPR/Cas9 Based Genome Editing of Penicillium chrysogenum. ACS Synth. Biol. 2016, 5, 754–764. [DOI] [PubMed] [Google Scholar]
- (20).Kuivanen J; Korja V; Holmström S; Richard P Development of microtiter plate scale CRISPR/Cas9 transformation method for Aspergillus niger based on in vitro assembled ribonucleoprotein complexes. Fungal Biol. Biotechnol 2019, 6, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Nagy G; Szebenyi C; Csernetics Á; Vaz AG; Tóth EJ; Vágvölgyi C; Papp T Development of a plasmid free CRISPR-Cas9 system for the genetic modification of Mucor circinelloides. Sci. Rep. 2017, 7, 16800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Jinek M; Chylinski K; Fonfara I; Hauer M; Doudna JA; Charpentier E A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Ran FA; Hsu PD; Wright J; Agarwala V; Scott DA; Zhang F Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Sakuma T; Nakade S; Sakane Y; Suzuki K-IT; Yamamoto T MMEJ-assisted gene knock-in using TALENs and CRISPR-Cas9 with the PITCh systems. Nat. Protoc. 2016, 11, 118–133. [DOI] [PubMed] [Google Scholar]
- (25).Bok JW; Keller NP LaeA, a regulator of secondary metabolism in Aspergillus spp. Eukaryotic Cell 2004, 3, 527–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Oakley CE; Ahuja M; Sun WW; Entwistle R; Akashi T; Yaegashi J; Guo CJ; Cerqueira GC; Russo Wortman J; Wang CC; Chiang YM; Oakley BR Discovery of McrA, a master regulator of Aspergillus secondary metabolism. Mol. Microbiol. 2017, 103, 347–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Grahl N; Demers EG; Crocker AW; Hogan DA Use of RNA-Protein Complexes for Genome Editing in Non-albicans Candida Species. mSphere, 2 (3), e00218–17. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Shrivastav M; De Haro LP; Nickoloff JA Regulation of DNA double-strand break repair pathway choice. Cell research 2008, 18, 134–147. [DOI] [PubMed] [Google Scholar]
- (29).Choquer M; Robin G; Le Pêcheur P; Giraud C; Levis C; Viaud M Ku70 or Ku80 deficiencies in the fungus Botrytis cinerea facilitate targeting of genes that are hard to knock out in a wild-type context. FEMS Microbiol. Lett. 2008, 289, 225–232. [DOI] [PubMed] [Google Scholar]
- (30).Abbasi S; Parmar G; Kelly RD; Balasuriya N; Schild-Poulter C The Ku complex: recent advances and emerging roles outside of non-homologous end-joining. Cellular and molecular life sciences: CMLS 2021, 78, 4589–4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Du L; Zhu T; Liu H; Fang Y; Zhu W; Gu Q Cytotoxic polyketides from a marine-derived fungus Aspergillus glaucus. J. Nat. Prod. 2008, 71, 1837–1842. [DOI] [PubMed] [Google Scholar]
- (32).Nielsen MT; Nielsen JB; Anyaogu DC; Holm DK; Nielsen KF; Larsen TO; Mortensen UH Heterologous reconstitution of the intact geodin gene cluster in Aspergillus nidulans through a simple and versatile PCR based approach. PLoS One 2013, 8, No. e72871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Secondary metabolites, 3D structure: Trivial name – Citraconic anhydride metabolite 1. The Aspergillus Website. http://www.aspergillus.org.uk. March 2022 accessed. [Google Scholar]
- (34).Argade NP; Easwar S A facile synthesis and enzymatic resolution of naturally occurring remotely functionalized alkylmethylmaleic anhydrides from Aspergillus wentii: Aspergillus acids A-D. Synthesis 2006, 2006, 831–838. [Google Scholar]
- (35).Tsai HF; Wheeler MH; Chang YC; Kwon-Chung KJ A developmentally regulated gene cluster involved in conidial pigment biosynthesis in Aspergillus fumigatus. J. Bacteriol. 1999, 181, 6469–6477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Sugareva V; Hürtl A; Brock M; Hübner K; Rohde M; Heinekamp T; Brakhage AA Characterisation of the laccase-encoding gene abr2 of the dihydroxynaphthalene-like melanin gene cluster of Aspergillus fumigatus. Arch. Microbiol. 2006, 186, 345–355. [DOI] [PubMed] [Google Scholar]
- (37).Krappmann S; Sasse C; Braus GH Gene targeting in Aspergillus fumigatus by homologous recombination is facilitated in a nonhomologous end-joining-deficient genetic background. Eukaryotic Cell 2006, 5, 212–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Shiina T; Ozaki T; Matsu Y; Nagamine S; Liu C; Hashimoto M; Minami A; Oikawa H Oxidative Ring Contraction by a Multifunctional Dioxygenase Generates the Core Cycloocatadiene in the Biosynthesis of Fungal Dimeric Anhydride Zopfiellin. Org. Lett. 2020, 22, 1997–2001. [DOI] [PubMed] [Google Scholar]
- (39).de Mattos-Shipley K; Spencer CE; Greco C; Heard DM; O’Flynn DE; Dao TT; Song Z; Mulholland NP; Vincent JL; Simpson TJ; Cox RJ; Bailey AM; Willis CL Uncovering biosynthetic relationships between antifungal nonadrides and octadrides. Chem. Sci. 2020, 11, 11570–11578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Stajich J; Palmer J AAFTF-v0.2, 2018. https://zenodo.org/record/1620527 (accessed April 2022).
- (41).Prjibelski A; Antipov D; Meleshko D; Lapidus A; Korobeynikov A Using SPAdes De Novo Assembler. Curr. Protoc. Bioinform. 2020, 70 (1), e102. [DOI] [PubMed] [Google Scholar]
- (42).Brown CT; Irber L Sourmash: a library for MinHash sketching of DNA. JOSS 2016, 1 (5), 27. [Google Scholar]
- (43).Smit AFA; Hubley R RepeatModeler-v1.0, (2008–2015). http://www.repeatmasker.org (accessed April 2022).
- (44).Love J; Palmer J; Stajich J; Esser T; Kastman E; Winter D Funannotate-v1.5.0, 2018. https://zenodo.org/record/1342272 (accessed April 2022).
- (45).Kim D; Paggi JM; Park C; Bennett C; Salzberg SL Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37 (8), 907–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Grabherr MG; Haas BJ; Yassour M; Levin JZ; Thompson DA; Amit I; Adiconis X; Fan L; Raychowdhury R; Zeng Q; Chen Z; Mauceli E; Hacohen N; Gnirke A; Rhind N; di Palma F; Birren BW; Nusbaum C; Lindblad-Toh K; Friedman N; Regev A Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29 (7), 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Haas BJ; Salzberg SL; Zhu W; Pertea M; Allen JE; Orvis J; White O; Buell CR; Wortman JR Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9 (1), R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Blin K; Wolf T; Chevrette MG; Lu X; Schwalen CJ; Kautsar SA; Suarez Duran HG; de los Santos ELC; Kim HU; Nave M; Dickschat JS; Mitchell DA; Shelest E; Breitling R; Takano E; Lee SY; Weber T; Medema MH antiSMASH 4.0-improvements in chemistry prediction and gene cluster boundary identification. Nucleic Acids Res. 2017, 45 (W1), W36–W41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.