

Abstract
Purpose
Complement alternative pathway (AP) dysregulation has been implicated in geographic atrophy, an advanced form of age-related macular degeneration. Danicopan is an investigational, first-in-class inhibitor of factor D, an essential AP activation enzyme. We assessed danicopan distribution to the posterior segment of the eye after oral dosing.
Methods
Tissue distribution of drug-derived radioactivity was evaluated using whole-body autoradiography following oral administration of [14C]-danicopan to pigmented and albino rats. Pharmacokinetics and ocular tissue distribution were studied in pigmented and albino rabbits following single and multiple oral dosing of danicopan. The melanin binding property was characterized in vitro.
Results
Radioactivity was distributed widely in rats and became nonquantifiable in most tissues 24 hours postdose except in the pigmented rat uvea (quantifiable 672 hours postdose). Danicopan binding to melanin was established in vitro. After single dosing, the maximum concentration (Cmax) and area under the curve (AUC) in neural retina and plasma were similar in both rabbit types. After multiple dosing, AUC in neural retina was 3.4-fold higher versus plasma in pigmented rabbits. Drug levels in choroid/Bruch's membrane (BrM)/retinal pigment epithelium (RPE) were similar to plasma in albino rabbits but higher in pigmented rabbits: Cmax and AUC were 2.9- and 23.8-fold higher versus plasma after single dosing and 5.8- and 62.7-fold higher after multiple dosing. In pigmented rabbits, ocular tissue exposures slowly declined over time but remained quantifiable 240 hours postdose.
Conclusions
The results demonstrate that danicopan crosses the blood–retina barrier and binds melanin reversibly, leading to a higher and more sustained exposure in melanin-containing ocular tissues (choroid/BrM/RPE) and in the neural retina as compared to in plasma after repeated oral dosing in pigmented animals.
Translational Relevance
These findings suggest that oral danicopan possesses potential for treating geographic atrophy because AP dysregulation in the posterior segment of the eye is reported to be involved in the disease pathogenesis.
Keywords: ACH-4471, ACH-0144471, danicopan, ocular pharmacokinetics, age-related macular degeneration, alternative pathway inhibition, ALXN2040, complement, drug depot, geographic atrophy, melanin binding, retina
Introduction
Geographic atrophy (GA) is an advanced form of age-related macular degeneration (AMD), a chronic disorder of the central retina characterized by drusen formation and changes in the choroid/Bruch's membrane (BrM)/retinal pigment epithelium (RPE) layers in the macula.1 These alterations ultimately lead to photoreceptor loss and, as the disease advances, involve the fovea, affecting central vision.2 Epidemiologic estimates suggest that up to 8 million people worldwide have GA, and bilateral eye involvement is common.1,2 AMD is a leading cause of vision loss, and as many as 20% of cases of legal blindness in AMD are attributed to GA specifically.1 The pathogenesis of GA is not completely understood, but it is thought to result from genetic and environmental factors, chronic inflammation, and oxidative stress.1 For example, components of drusen and lipofuscin, as well as other products of oxidative stress, are believed to cause inflammation mediated by the complement cascade.2 Genetic studies have found variants associated with AMD, such as those in complement factor H, as well as local and systemic alterations in components of the complement pathway, in patients with GA.1–3 In addition, results of nonclinical in vitro and in vivo studies have suggested that complement dysregulation, and the complement alternative pathway (AP) in particular, play a role in the pathogenesis and progression of AMD.1,2
There are currently no approved treatments for GA.3 The Age-Related Eye Disease Study formulations are supportive options for GA and can possibly reduce risk of progressing to advanced AMD for certain patient groups (AMD categories 3 and 4).4,5 Potential therapeutic agents for GA are in development, including anti-inflammatories, antioxidation/RPE protectors, lipofuscin/visual cycle inhibitors, choroidal blood flow restoration agents, and stem-cell therapies. Additionally, anti-complement activation agents have been or are being evaluated in clinical studies.1–3 Promising efficacy data have been reported for pegcetacoplan (APL-2; cyclic peptide against C3)6 and avacincaptad pegol (pegylated RNA aptamer against C5)7 via intravitreal administration.
Drug delivery to the posterior segment of the eye remains a challenge due to the blood–retina barrier (BRB), which strictly regulates the permeation of drug agents from the blood to the retina and can result in inadequate distribution to the desired tissues.8,9 Intravitreal drug administration relieves the need for drug agents to cross the BRB as compared to the systemic route.8 However, intravitreal injection is highly invasive and may require frequent administration, which may not be acceptable to many patients and may increase the risk for procedure-related adverse events while potentially reducing compliance and consequently treatment efficacy.9–11 In individuals with neovascular AMD receiving intravitreal treatment with ranibizumab, a monoclonal antibody Fab fragment that inhibits vascular endothelial growth factor-A and therefore angiogenesis, there was a consistent decline in the number of injections per patient after the first year that may have been associated with a decline in visual acuity.12 Successful delivery of a complement inhibitor via oral administration to the posterior segment of the eye may represent a better therapeutic option because of effective inhibition in the target tissues, as well as reduction in the treatment burden for patients, and may lead to better patient compliance and outcomes. Additionally, as stated above, bilateral eye involvement is common in GA; thus, oral administration will offer the benefit of targeting both eyes simultaneously.
Factor D is an essential enzyme that mediates activation of the complement AP.2 Danicopan (ALXN2040, ACH-0144471, ACH-4471) is an orally administered, first-in-class small-molecule factor D inhibitor currently under clinical investigation for the treatment of diseases driven by complement dysregulation, including paroxysmal nocturnal hemoglobinuria.13 Danicopan binds to factor D with high affinity (KD = 0.54 nM), inhibiting cleavage of factor B into Ba and Bb, thereby blocking AP activation, amplification, and finally terminal pathway activation.14,15 Consequently, factor D inhibition prevents all three effector functions of the complement pathway, including opsonization, inflammatory response and cell lysis (Fig. 1).
Figure 1.

Mechanism of action of danicopan. The alternative pathway (AP) is constitutively activated through slow spontaneous hydrolysis of C3 that forms C3(H2O). C3(H2O) binds to factor B (FB) and is cleaved by factor D (FD) to generate the initial AP C3 convertase C3(H2O)Bb. C3 cleavage produces C3b, which binds to FB and is cleaved by FD to form the membrane-bound C3 convertase C3bBb. C3bBb cleaves more C3 molecules, leading to rapid generation of C3b, resulting in an amplification loop. FD inhibitor danicopan binds to FD, thereby inhibiting its proteolytic activity and cleavage of FB at two points in the cascade. The cross-section of tissues in the posterior segment of the eye is drawn to depict the membrane-bound events.
The purpose of these studies was to determine the pharmacokinetics and ocular tissue distribution of orally administered danicopan in preclinical animal models to evaluate the potential for development of danicopan as a systemically administered treatment option for GA. We examined the permeability of danicopan across the BRB in vivo, as well as its binding to melanin in vitro and in vivo. Melanins consist of several negatively charged macromolecule polymers16 and are present in high concentrations in ocular pigmented tissues, including posteriorly located choroid and RPE.17,18 Melanin binding of a drug causes drug accumulation and increases drug retention in pigmented ocular tissues, thereby affecting drug response and toxicity.19 Additionally, melanin-bound drugs in pigmented ocular tissues can act as a large reservoir, leading to the continuous release of the free drug.20 We showed here that danicopan readily crosses the BRB and binds to melanin reversibly, leading to a higher and more sustained exposure not only in the ocular melanin-containing tissues of choroid and RPE but also in the neutral retina as compared to in plasma after multiple oral dosing to pigmented animals.
Methods
Animal Care
Rabbit ocular pharmacokinetics and tissue distribution experiments were conducted by Covance Laboratories, Inc. (Madison, WI), and [14C]-labeled danicopan rat experiments were conducted at QPS, LLC (Newark, DE). Animals were individually housed and cared for in accordance with the Care and Use of Laboratory Animals guidelines and the U.S. Department of Agriculture Animal Welfare Act. Experimental procedures were reviewed and approved by an Institutional Animal Care and Use Committee before initiation of the study. All protocols adhered to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. A total of 13 male Long–Evans pigmented rats and three male Wistar Han albino rats were supplied by Hilltop Lab Animals (Scottdale, PA). A total of 44 male pigmented Dutch Belted (DB) rabbits and 15 male New Zealand White (NZW) albino rabbits were supplied by Envigo RMS (Indianapolis, IN). For all rabbits, observations were recorded during the acclimation period, at predose, and at least daily postdose.
Danicopan and [14C]-Danicopan Synthesis
Danicopan was synthesized using standard methods.15 [14C]-labeled danicopan was prepared in an analogous manner, with the label located in the tertiary amide carbonyl core of the molecule. The radiochemical purity of [14C]-danicopan was 99.8%, and the specific activity was 57.9 mCi/mmol (99.44 µCi/mg). Single-dose formulations for oral dosing were prepared on the day of dosing for all animals. Compounds were suspended using 60:36.3:3.7 (v:v:v) polyethylene glycol 400:saline:dimethyl sulfoxide.
Tissue Distribution of Danicopan/[14C]-Danicopan in Rats by Quantitative Whole-Body Autoradiography
Tissue distribution of danicopan was measured by quantitative whole-body autoradiography following a single oral dose of 20 mg/kg [14C]-danicopan to pigmented Long–Evans and albino Wistar Han rats. This dose was chosen based on the results from an earlier plasma pharmacokinetic study with unlabeled danicopan; thus, it was expected to yield a good exposure signal (i.e., radioactivity) to evaluate danicopan tissue distribution. Rats were euthanized at 1, 2, 4, 8, 24, 72, 168, 336, 504, and 672 hours postdose, and carcasses were frozen in hexane/dry ice and stored at or below −20°C before processing and sectioning. Frozen samples were embedded in 1% carboxymethylcellulose matrix, mounted on a microtome stage (Leica CM3600 Cryomacrocut or Vibratome 9800; Leica Biosystems, Nussloch, Germany) maintained at −20°C, and sectioned in approximately 40-µm–thick sagittal sections. Sections were labeled with calibration standards of [14C]-glucose mixed with blood at various concentrations, visualized using a [14C]-sensitive phosphor-imaging plate (Fuji Biomedical, Stamford, CT), and scanned using a GE Amersham Molecular Dynamics Typhoon 9410 image acquisition system (Molecular Dynamics, Sunnyvale, CA), with a scanning resolution of 50 µm.
Tissue Distribution of Danicopan in Rabbits
Ocular tissue and plasma distributions of danicopan were evaluated in pigmented DB and albino NZW rabbits. Based on efficacious regimens in human studies and pharmacokinetic/pharmacodynamic modeling, danicopan was administered orally at 7.5, 15, or 50 mg/kg with single or multiple dosing to cover the projected range of therapeutic exposures in the posterior segment of the eye; for multiple dosing, rabbits received danicopan approximately every 12 hours for 14 days and a single dose on the morning of day 15. Rabbits were euthanized, and plasma and ocular tissues were collected at various time points either after a single dose or after the morning dose on day 15 (i.e., the last dose of the multiple-dosing regimen) with one exception: Samples were only collected at the approximate time to maximum concentration (tmax; hour 1) from NZW rabbits in the multiple-dosing study at 50 mg/kg. Danicopan concentrations were determined by liquid chromatography/mass spectrometry.
Liquid Scintillation Counting and Statistical Analysis
Radioactivity was expressed as the microgram equivalents of danicopan per gram sample (µg equivalent/g). The upper limit of quantification (ULOQ) and lower limit of quantification (LLOQ) were applied to the data based on previously validated calibration standards.21,22 The LLOQ and ULOQ were 0.105 and 4405 µg equivalent/g, respectively. Pigmented rat tissue concentration data (after oral dosing; µg equivalent/g) were used to calculate pharmacokinetic and radiation dosimetry parameters using Phoenix WinNonlin 6.3 (Pharsight Corporation, Mountain View, CA). For each individual tissue, terminal half-life (t1/2) was determined by noncompartmental analysis of the data using Phoenix NCA model 200.
In Vitro Melanin-Binding Studies
Materials were obtained from Sigma-Aldrich (St. Louis, MO). Solutions of two sources of melanin (natural Sepia officinalis and synthetic melanin), danicopan, and chloroquine (as a positive control) were used in the assay. Danicopan and chloroquine were incubated with or without melanin in 96-well plates. Melanin and bound compound were collected by centrifugation at 4000 rpm, diluted with acetonitrile containing the internal assay standard, and passed through a Waters XSelect HSS T3 2.5-µm gradient column (50 × 2.1 mm for danicopan and 30 × 2.1 mm for chloroquine; Waters Corporation, Milford, MA). Samples and internal standards were measured using a SCIEX API-5500 triple quadrupole mass spectrometer (Applied Biosystems, Waltham, MA) using multiple reaction-monitoring scan modes (danicopan, 580.2/360.2; chloroquine, 320.1/247.3), and data were captured using SCIEX Analyst 1.6.2. Data curves of mean free concentration versus bound concentration were fit with a one-sided hyperbolic model using Prism 5.0 (GraphPad, San Diego, CA) to obtain the KD as a measure for affinity and the maximum bound concentration binding (Bmax) as a measure of binding capacity.
Results
Following oral administration of a single dose of [14C]-danicopan at 20 mg/kg, a dose that was expected to yield a good exposure to evaluate danicopan tissue distribution based on earlier studies with unlabeled danicopan, drug-derived radioactivity was rapidly absorbed and widely distributed to tissues in both pigmented (n = 10) and albino (n = 3) rats. Radioactivity was evident 1 to 8 hours postdose and became nonquantifiable at 24 hours postdose in most tissues, as demonstrated by whole-body autoradiography (Fig. 2). In pigmented rats at 24 hours postdose, radioactivity localized primarily to the uvea tract (i.e., melanin-containing ocular tissues), pigmented skin, and liver (Fig. 2A); in albino rats, radioactivity was primarily localized to the liver (Fig. 2B). Quantification of radioactivity showed that [14C]-danicopan remained quantifiable in the uveal tract of pigmented rats at 672 hours (28 days) postdose (t1/2 = 576 hours) (Fig. 2C), whereas it became undetectable in plasma and whole blood after 8 hours postdose (Fig. 2).
Figure 2.
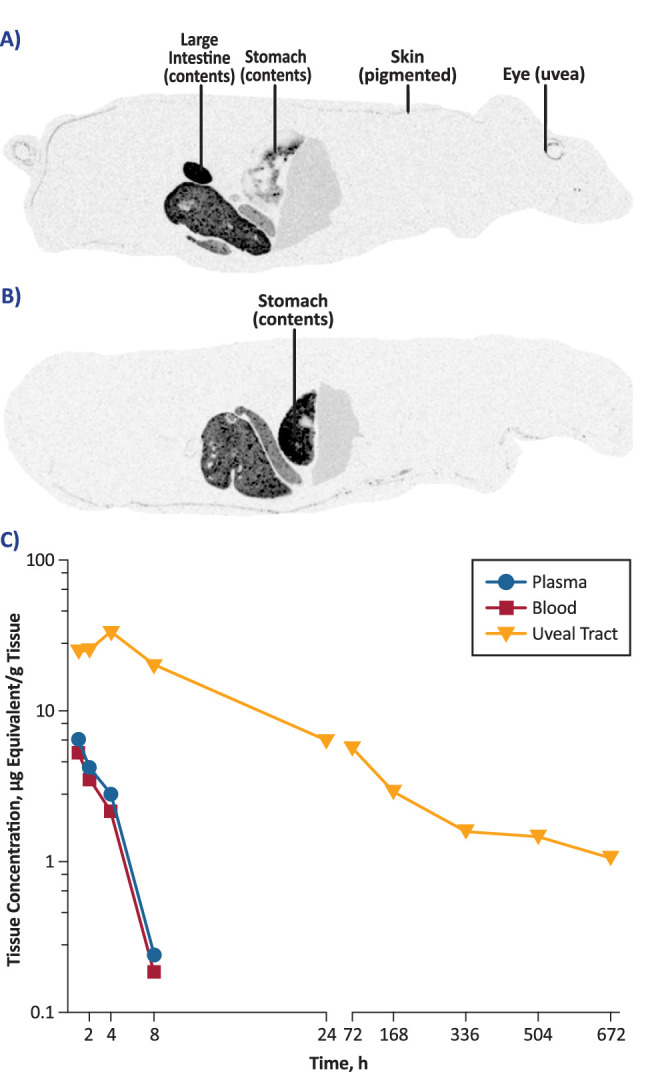
(A, B) Whole-body autoradiography showing tissue distribution of [14C]-danicopan 24 hours after oral administration of a single 20-mg/kg dose in pigmented rats (A) and albino male rats (B). Frozen sections approximately 40 µm in thickness were taken in the sagittal plane. (C) Quantitative whole-body autoradiography of [14C]-danicopan–derived radioactivity in tissues of pigmented rats following oral administration. Concentrations of radioactivity were determined by assessing image densitometry and were expressed as microgram equivalents of danicopan per gram of sample.
To evaluate whether danicopan binding to melanin could account for the observed persistence of [14C]-danicopan in the rat uveal tract, a pigmented tissue, danicopan binding was evaluated in vitro using natural (S. officinalis) and synthetic melanin; chloroquine was used as a positive control. Danicopan bound to natural and synthetic melanin, but with a 6.1- to 12.4-fold lower affinity, respectively, than chloroquine (Fig. 3). The binding capacity of danicopan to natural and synthetic melanin was 2.0-fold and 2.4-fold lower, respectively, than that of chloroquine.
Figure 3.

In vitro evaluation of danicopan and chloroquine binding to natural (Sepia officinalis) and synthetic melanin. Following gradient column purification, samples were quantified by mass spectrometry. Bmax, maximal bound ligand concentration.
To further investigate the distribution of danicopan in ocular tissues, danicopan was initially dosed orally to pigmented DB (n = 14) and albino NZW (n = 3) rabbits at 50 mg/kg twice daily for 15 days. Plasma and ocular tissue concentrations at tmax from both rabbit strains are compared in Figure 4. Similar to the observations from the tissue distribution study in rats with [14C]-danicopan, the highest concentrations of danicopan were evident in melanin-containing tissues (iris/ciliary body and choroid/BrM/RPE) in DB rabbits. In both DB and NZW rabbits, the concentrations in other ocular tissues, including the neural retina, were similar to plasma concentrations, indicating that danicopan crosses the BRB and distributes to the neural retina.
Figure 4.
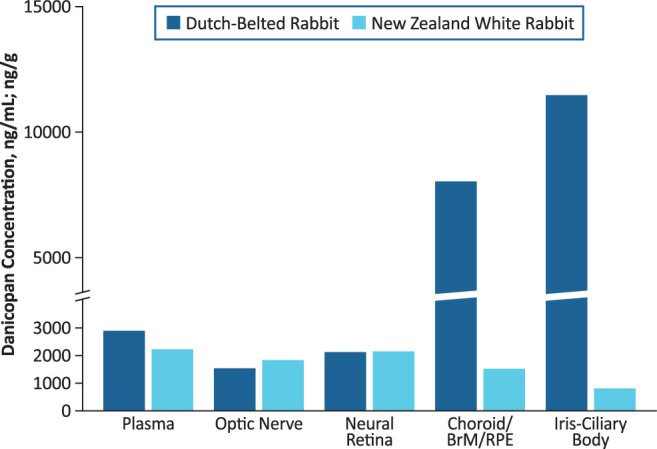
Plasma and ocular tissue concentrations of danicopan at time of maximum concentration following 15-day multiple dosing (50 mg/kg/dose) in pigmented and albino rabbits. Danicopan was administered twice daily at approximately 12-hour intervals for 14 days with a final single dose administered on day 15. Units are nanograms per milliliter (ng/mL) for plasma and nanograms per gram (ng/g) for ocular tissues.
To confirm these observations, danicopan was administered orally to both DB rabbits (single dose, n = 14; multiple doses, n = 14) and NZW rabbits (single dose, n = 14) at lower doses (15 mg/kg; single dose or multiple doses for 15 days). Plasma and ocular tissue concentrations of danicopan over time are shown in Figure 5, and the corresponding pharmacokinetic parameters are summarized in the Table. In NZW rabbits, maximum concentration (Cmax) values in neural retina and choroid/BrM/RPE were similar to those in plasma regardless of dosing duration, with the neural retina-to-plasma ratio ranging from 0.4 to 0.6. Cmax values in neural retina were also similar to those in plasma in DB rabbits after single and multiple doses (neural retina-to-plasma ratios, 0.5 and 0.7, respectively). In contrast, the Cmax values in DB rabbits in choroid/BrM/RPE were higher than in plasma after a single dose (choroid/BrM/RPE-to-plasma ratio, 2.9) and were even higher after multiple doses (ratio, 5.8). Overall, the data from experiments in rabbits are consistent with the tissue distribution experiments conducted in rats and confirm that danicopan crosses the BRB and achieves higher concentrations in melanin-containing choroid/BrM/RPE compared with plasma in pigmented animals.
Figure 5.

Plasma and ocular tissue concentrations of danicopan following single or 15-day multiple dosing (15 mg/kg/dose) in Dutch Belted (pigmented) rabbits (A, B) and New Zealand White (albino) rabbits (C, D). Danicopan was administered twice daily for 14 days at approximately 12-hour intervals with a final single dose administered on day 15. Units are nanograms per milliliter (ng/mL) for plasma and nanograms per gram (ng/g) for ocular tissues.
Table.
Pharmacokinetic Parameters of Oral Danicopan in DB and NZW Rabbits After a Single Dose or Multiple Doses
| Single Dose (15 mg/kg) | Multiple Doses (15 mg/kg BID)a | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DB Rabbit | NZW Rabbit | DB Rabbit | NZW Rabbit | |||||||||
| Pharmacokinetic Parametersb | Plasma | Neural Retina | Choroid/BrM/RPE | Plasma | Neural Retina | Choroid/BrM/RPE | Plasma | Neural Retina | Choroid/BrM/RPE | Plasma | Neural Retina | Choroid/BrM/RPE |
| Cmaxc | 318 | 145 | 917 | 310 | 200 | 172 | 845 | 576 | 4870 | 558 | 248 | 296 |
| tmax (h) | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| AUClastd | 1570 | 998 | 37,400 | 1430 | 665 | 1010 | 3990 | 13,400 | 250,000 | 3490 | 1830 | 1830 |
| MRTlast (h) | 5.58 | 7.83 | 49.7 | 4.01 | 1.49 | 5.87 | 6.51 | 71.0 | 68.5 | NC | NC | NC |
| Ratio over plasma | ||||||||||||
| Cmaxc | 1.0 | 0.5 | 2.9 | 1.0 | 0.6 | 0.6 | 1.0 | 0.7 | 5.8 | 1.0 | 0.4 | 0.5 |
| AUClastd | 1.0 | 0.6 | 23.8 | 1.0 | 0.5 | 0.7 | 1.0 | 3.4 | 62.7 | 1.0 | 0.5 | 0.5 |
AUClast, area under the curve from hour 0 to last measurable concentration; BID, twice daily; MRTlast, mean residence time from 0 to the time of the last measurable concentration; NC, not calculable due to insufficient data.
Dosing regimen was BID for 14 days, approximately 12 hours apart, with a single dose on day 15.
Pharmacokinetic data are presented as mean values.
Cmax units are ng/mL for plasma and ng/g for neural retina and choroid/BrM/RPE.
AUClast units are h·ng/mL for plasma and h·ng/g for neural retina and choroid/BrM/RPE.
In addition to Cmax values, area under the curve (AUC) values from both DB and NZW rabbits were determined (Table). In NZW rabbits, AUC values were comparable between ocular tissues (choroid/BrM/RPE or neural retina) and plasma after single or multiple doses, with tissue-to-plasma ratios ranging from 0.5 to 0.7. In contrast, AUC values in DB rabbits were different between ocular tissues and plasma. AUC values were higher in choroid/BrM/RPE compared with plasma in DB rabbits, with choroid/BrM/RPE-to-plasma ratios of 23.8 after a single dose and 62.7 after multiple doses. AUC values were also higher in neural retina compared with plasma after multiple doses (ratio, 3.4) in DB rabbits, although AUC was similar between neural retina and plasma after a single dose (neural retina-to-plasma ratio, 0.6). The higher exposure in neural retina of DB rabbits after multiple doses is likely due to prolonged mean residence time, which increased from 7.8 hours after a single dose to 71 hours after multiple doses, indicating that melanin-bound danicopan in choroid/BrM/RPE may serve as a depot to continuously replenish drug in neural retina.
The dose–exposure relationship in ocular tissues was further evaluated in DB rabbits by oral administration of danicopan twice daily at 7.5 (n = 14), 15 (n = 14), and 50 (n = 14) mg/kg/dose for 15 days (Fig. 6). At all three dosages and after the last dose, danicopan exposure was detected through 24 hours in plasma, but was sustained through 240 hours in choroid/BrM/RPE and neural retina. Further, plasma and neural retina exposure exhibited approximate dose linearity among the three doses evaluated, whereas choroid/BrM/RPE exposure showed less-than-dose proportionality between 15 and 50 mg/kg/dose (data not shown).
Figure 6.
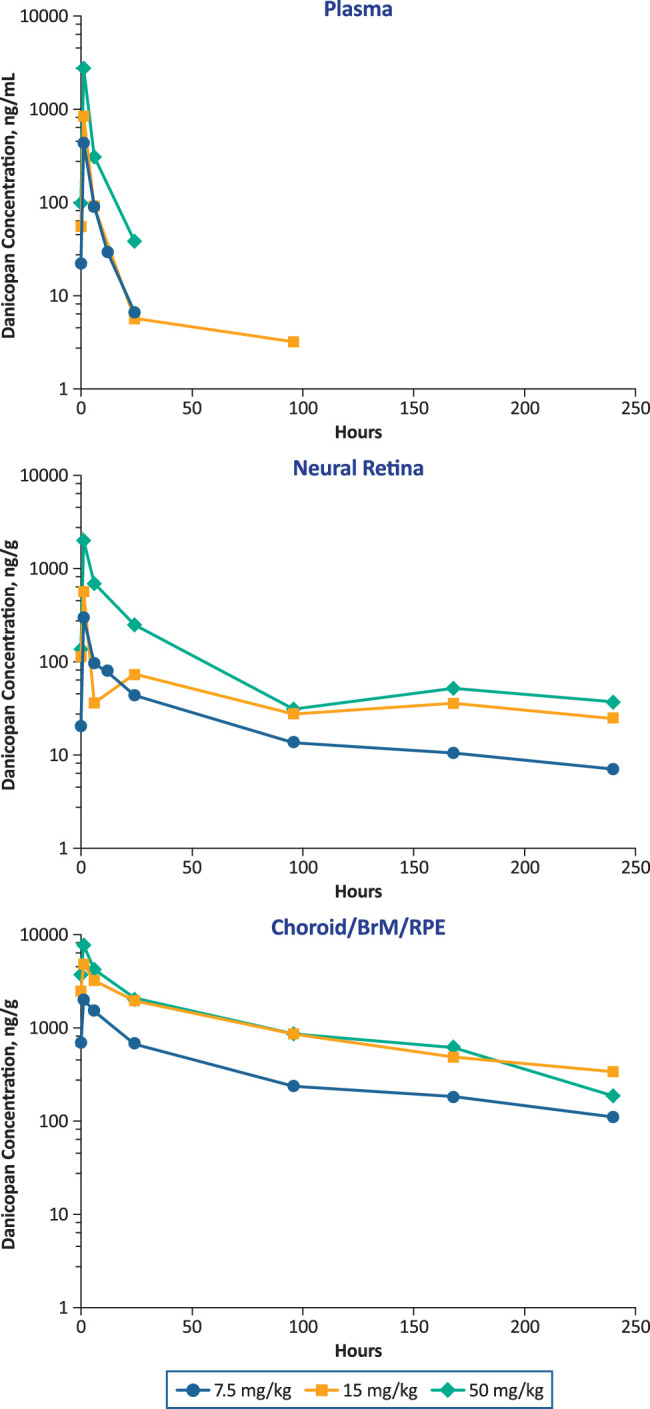
Danicopan dose dependence in plasma and ocular tissues in pigmented rabbits at day 15 at steady state. Rabbits received danicopan at the indicated doses twice daily for 14 days at approximately 12-hour intervals, with a final single dose administered on day 15.
In our studies across multiple animal species and strains, no safety issues pertaining to ophthalmic accumulation of danicopan were observed. In rabbits dosed with danicopan at 50 mg/kg twice daily for 15 days (n = 17), there was no evidence of ophthalmic abnormalities after examination with slit-lamp biomicroscopy, indirect opththalmoscopy, TONOVET rebound tonometry, or ultrasonic corneal pachymetry. In toxicology studies performed according to Good Laboratory Practice guidelines, there were no clinical or histopathological ophthalmic findings at several time points following oral administration in rats (1 month, 3 months, 6 months) and dogs (14 days, 3 months, 9 months). Moreover, no abnormalities were detected in dogs when examined with electroretinography (ERG) during week 38 when compared to pretreatment ERG measurements.
Discussion
The results of the studies presented here showed that, in preclinical animal models, danicopan readily crosses the BRB after oral dosing, leading to distribution in the neural retina. Additionally, danicopan was shown to bind melanin, leading to high and sustained exposure in melanin-containing ocular tissues such as choroid/BrM/RPE after systemic oral dosing. Moreover, danicopan exposure increased in the neural retina after repeat dosing in pigmented rabbits.
The BRB is composed of both the inner and outer BRB. The outer BRB is composed of tight junctions of RPE cells, whereas the inner BRB comprises retinal capillary endothelial cells. Similar to the blood–brain barrier, lack of fenestrations in the RPE and retinal endothelial cells prevents large and most small molecules from passing the barrier to enter the inner retinal tissues.23 Danicopan was evaluated in vivo and was found to be present at a similar level in the neural retinal and the plasma after a single oral dose in both pigmented and albino rabbits (Figs. 5A, 5C), demonstrating that danicopan is able to readily cross the BRB.
Several ocular tissues have high concentrations of melanin and are presented as pigmented tissues, including the choroid and RPE in the posterior and the iris and ciliary body in the anterior.17,18 Melanins consist of several negatively charged macromolecule polymers, and drugs commonly bind to melanin, as all basic and lipophilic drugs are expected to bind to melanin to some extent.16,24,25 Indeed, the accumulation of drugs in pigmented tissues has been known for a long time,26,27 and various clinical drugs have been shown to bind to melanin.28 Drug retention caused by melanin binding in pigmented ocular tissues has been explored to assist ocular drug delivery, especially for delivery to the posterior segment of the eye, a pathological site of GA.29–32 Preclinical studies have demonstrated that melanin binding can indeed extend the pharmacokinetic half-life of ophthalmic drugs in the posterior segment of the eye after systemic administration.20,33 Pazopanib, a small-molecule inhibitor of tyrosine kinases including the vascular endothelial growth factor receptor, was detected in melanin-containing tissues of pigmented rats, including the uveal tract, up to 35 days following the administration of a single oral dose (10 mg/kg).20 In mice, plasma levels of pazopanib were undetectable 3 days after a single loading dose (10 mg/kg); however, choroidal neovascularization lesion size was significantly reduced up to 2 weeks after choroidal neovascularization induction. The lesion size reductions were comparable to reductions achieved with twice-daily dosing of pazopanib (10 mg/kg), suggesting that uveal retention of pazopanib served as a sustained-release depot that allowed therapeutic levels of the drug to be sustained following a single oral dose.
In this study, after oral dosing to pigmented rats and rabbits, danicopan was retained in the pigmented tissues of the eyes due to its ability to bind to melanin. The binding of danicopan to melanin is reversible; in a tissue distribution study with [14C]-danicopan in pigmented rats, the radioactivity in the uveal tract diminished over time (Fig. 2C). Moreover, the danicopan concentration in the choroid/BrM/RPE decreased over time in pigmented rabbits (Figs. 5A, 5B). The danicopan reversibly bound to melanin was expected to provide a drug depot to prolong danicopan exposure in the neural retina. This was indeed the case; after repeat dosing, danicopan exposure in the neural retina exceeded the systemic (plasma) exposure in pigmented rabbits (Fig. 5B), whereas similar exposure was observed between the neutral retina and the plasma in nonpigmented rabbits (Fig. 5D). Of note, although melanin binding enhances the pharmacokinetics of danicopan, the elevated concentration of danicopan in ocular tissues did not elicit safety concerns in multiple animal studies across several species and strains.
There are limitations to this study. First, although rabbit is widely used as a preclinical model for ocular pharmacokinetics, characteristics of the BRB and melanin-binding properties may differ between rabbits and humans. Second, due to the lack of an inhibitory effect of danicopan on rodent factor D, the effectiveness of danicopan via oral dosing in inhibition of AP in the eye has not been tested in preclinical models established in rodents. Therefore, we have relied instead on the rabbit ocular pharmacokinetics model and its translation to a human pharmacokinetics/pharmacodynamics model to predict the effective regimen in humans. Our prediction of the regimen is yet to be confirmed in the ongoing clinical study.
In summary, we showed that danicopan, a complement factor D inhibitor, crosses the BRB and binds melanin reversibly, leading to a higher and more sustained exposure not only in melanin-containing ocular tissues such as choroid/BrM/RPE but also in the neural retina as compared to plasma after repeated oral dosing in pigmented animals. Additionally, no ocular safety signals were observed with oral dosing of danicopan in the multiple animal studies across several species and strains. Danicopan is currently being investigated in a phase 2 clinical study in patients with GA (NCT05019521).
Acknowledgments
The authors thank Jose Rivera, MS, for his contributions to the study. Writing support was provided by Miranda Tradewell, PhD, for The Curry Rockefeller Group, LLC (Tarrytown, NY), and was funded by Alexion, AstraZeneca Rare Disease (New Haven, CT).
Funded by Alexion, AstraZeneca Rare Disease, which was involved in the study design; in the collection, analysis, and interpretation of data; in the writing of the report; and in the decision to submit the article for publication.
Disclosure: D.D. Boyer, Alexion, AstraZeneca Rare Disease (E); Y.-P. Ko, Alexion, AstraZeneca Rare Disease (E); S.D. Podos, Alexion, AstraZeneca Rare Disease (E); M.E. Cartwright, Alexion, AstraZeneca Rare Disease (E); X. Gao, Alexion, AstraZeneca Rare Disease (E); J.A. Wiles, Alexion, AstraZeneca Rare Disease (E); M. Huang, Alexion, AstraZeneca Rare Disease (E)
References
- 1. Hanus J, Zhao F, Wang S.. Current therapeutic developments in atrophic age-related macular degeneration. Br J Ophthalmol. 2016; 100: 122–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Boyer DS, Schmidt-Erfurth U, van Lookeren Campagne M, Henry EC, Brittain C.. The pathophysiology of geographic atrophy secondary to age-related macular degeneration and the complement pathway as a therapeutic target. Retina. 2017; 37: 819–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fleckenstein M, Mitchell P, Freund KB, et al.. The progression of geographic atrophy secondary to age-related macular degeneration. Ophthalmology. 2018; 125: 369–390. [DOI] [PubMed] [Google Scholar]
- 4. Age-Related Eye Disease Study 2 Research Group. Lutein + zeaxanthin and omega-3 fatty acids for age-related macular degeneration: the Age-Related Eye Disease Study 2 (AREDS2) randomized clinical trial. JAMA. 2013; 309: 2005–2015. [DOI] [PubMed] [Google Scholar]
- 5. Age-Related Eye Disease Study Research Group. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch Ophthalmol. 2001; 119: 1417–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liao DS, Grossi FV, El Mehdi D, et al.. Complement C3 inhibitor pegcetacoplan for geographic atrophy secondary to age-related macular degeneration: a randomized phase 2 trial. Ophthalmology. 2020; 127: 186–195. [DOI] [PubMed] [Google Scholar]
- 7. Jaffe GJ, Westby K, Csaky KG, et al.. C5 inhibitor avacincaptad pegol for geographic atrophy due to age-related macular degeneration: a randomized pivotal phase 2/3 trial. Ophthalmology. 2021; 128: 576–586. [DOI] [PubMed] [Google Scholar]
- 8. Gaudana R, Ananthula HK, Parenky A, Mitra AK.. Ocular drug delivery. AAPS J. 2010; 12: 348–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mandal A, Pal D, Agrahari V, Trinh HM, Joseph M, Mitra AK.. Ocular delivery of proteins and peptides: challenges and novel formulation approaches. Adv Drug Deliv Rev. 2018; 126: 67–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shen J, Lu GW, Hughes P.. Targeted ocular drug delivery with pharmacokinetic/pharmacodynamic considerations. Pharm Res. 2018; 35: 217. [DOI] [PubMed] [Google Scholar]
- 11. Spooner KL, Guinan G, Koller S, Hong T, Chang AA.. Burden of treatment among patients undergoing intravitreal injections for diabetic macular oedema in Australia. Diabetes Metab Syndr Obes. 2019; 12: 1913–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schwartz R, Warwick A, Olvera-Barrios A, et al.. Evolving treatment patterns and outcomes of neovascular age-related macular degeneration over a decade. Ophthalmol Retina. 2021; 5: e11–e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Risitano AM, Kulasekararaj AG, Lee JW, et al.. Danicopan: an oral complement factor D inhibitor for paroxysmal nocturnal hemoglobinuria. Haematologica. 2021; 106: 3188–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yuan X, Gavriilaki E, Thanassi JA, et al.. Small-molecule factor D inhibitors selectively block the alternative pathway of complement in paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome. Haematologica. 2017; 102: 466–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wiles JA, Galvan MD, Podos SD, Geffner M, Huang M.. Discovery and development of the oral complement factor D inhibitor danicopan (ACH-4471). Curr Med Chem. 2020; 27: 4165–4180. [DOI] [PubMed] [Google Scholar]
- 16. Ito S, Wakamatsu K, d'ischia M, Napolitano A, Pezzella A.. Structure of melanins. In: Borovanský J, Riley PA, eds. Melanins and Melanosomes: Biosynthesis, Biogenesis, Physiological, and Pathological Functions. New York: Wiley; 2011: 167–185. [Google Scholar]
- 17. Schraermeyer U, Heimann K.. Current understanding on the role of retinal pigment epithelium and its pigmentation. Pigment Cell Res. 1999; 12: 219–236. [DOI] [PubMed] [Google Scholar]
- 18. Liu Y, Hong L, Wakamatsu K, et al.. Comparisons of the structural and chemical properties of melanosomes isolated from retinal pigment epithelium, iris and choroid of newborn and mature bovine eyes. Photochem Photobiol. 2005; 81: 510–516. [DOI] [PubMed] [Google Scholar]
- 19. Rimpela AK, Reinisalo M, Hellinen L, et al.. Implications of melanin binding in ocular drug delivery. Adv Drug Deliv Rev. 2018; 126: 23–43. [DOI] [PubMed] [Google Scholar]
- 20. Robbie SJ, Lundh von Leithner P, Ju M, et al.. Assessing a novel depot delivery strategy for noninvasive administration of VEGF/PDGF RTK inhibitors for ocular neovascular disease. Invest Ophthalmol Vis Sci. 2013; 54: 1490–1500. [DOI] [PubMed] [Google Scholar]
- 21. Kolbe H, Dietzel G.. Technical validation of radioluminography systems. Regul Toxicol Pharmacol. 2000; 31: S5–S14. [DOI] [PubMed] [Google Scholar]
- 22. Solon EG, Lee F.. Methods determining phosphor imaging limits of quantitation in whole-body autoradiography rodent tissue distribution studies affect predictions of 14C human dosimetry. J Pharmacol Toxicol Methods. 2001; 46: 83–91. [DOI] [PubMed] [Google Scholar]
- 23. Mitra AK. Ocular Transporters and Receptors: Their Role in Drug Delivery. Oxford: Woodhead Publishing; 2013:xxiii, 251. [Google Scholar]
- 24. Ings RM. The melanin binding of drugs and its implications. Drug Metab Rev. 1984; 15: 1183–1212. [DOI] [PubMed] [Google Scholar]
- 25. Zane PA, Brindle SD, Gause DO, O'Buck AJ, Raghavan PR, Tripp SL. Physicochemical factors associated with binding and retention of compounds in ocular melanin of rats: correlations using data from whole-body autoradiography and molecular modeling for multiple linear regression analyses. Pharm Res. 1990; 7: 935–941. [DOI] [PubMed] [Google Scholar]
- 26. Ullberg S, Lindquist NG, Sjostrand SE.. Accumulation of chorio-retinotoxic drugs in the foetal eye. Nature. 1970; 227: 1257–1258. [DOI] [PubMed] [Google Scholar]
- 27. Potts AM. The reaction of uveal pigment in vitro with polycyclic compounds. Invest Ophthalmol. 1964; 3: 405–416. [PubMed] [Google Scholar]
- 28. Pelkonen L, Tengvall-Unadike U, Ruponen M, et al.. Melanin binding study of clinical drugs with cassette dosing and rapid equilibrium dialysis inserts. Eur J Pharm Sci. 2017; 109: 162–168. [DOI] [PubMed] [Google Scholar]
- 29. Rimpela AK, Hagstrom M, Kidron H, Urtti A.. Melanin targeting for intracellular drug delivery: quantification of bound and free drug in retinal pigment epithelial cells. J Control Release. 2018; 283: 261–268. [DOI] [PubMed] [Google Scholar]
- 30. Bahrpeyma S, Rimpela AK, Hagstrom M, Urtti A.. Ocular melanin binding of drugs: in vitro binding studies. Acta Ophthalmol (Copenh). 2019; 97: 1–2. [Google Scholar]
- 31. Del Amo EM, Rimpela AK, Heikkinen E, et al.. Pharmacokinetic aspects of retinal drug delivery. Prog Retin Eye Res. 2017; 57: 134–185. [DOI] [PubMed] [Google Scholar]
- 32. Jakubiak P, Reutlinger M, Mattei P, Schuler F, Urtti A, Alvarez-Sanchez R.. Understanding molecular drivers of melanin binding to support rational design of small molecule ophthalmic drugs. J Med Chem. 2018; 61: 10106–10115. [DOI] [PubMed] [Google Scholar]
- 33. Jakubiak P, Cantrill C, Urtti A, Alvarez-Sanchez R.. Establishment of an in vitro–in vivo correlation for melanin binding and the extension of the ocular half-life of small-molecule drugs. Mol Pharm. 2019; 16: 4890–4901. [DOI] [PubMed] [Google Scholar]
- 34. Guymer RH, Mitchell P, Wong J, et al.. Efficacy and safety of intravitreal pegcetacoplan in geographic atrophy: results from the phase 3 DERBY and OAKS trials. Clin Exp Ophthalmol. 2022; 49: 833. [Google Scholar]
- 35. Yaspan BL, Williams DF, Holz FG, et al.. Targeting factor D of the alternative complement pathway reduces geographic atrophy progression secondary to age-related macular degeneration. Sci Transl Med. 2017; 9: eaaf1443. [DOI] [PubMed] [Google Scholar]
- 36. Holz FG, Sadda SR, Busbee B, et al.. Efficacy and safety of lampalizumab for geographic atrophy due to age-related macular degeneration: Chroma and Spectri phase 3 randomized clinical trials. JAMA Ophthalmol. 2018; 136: 666–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Desai D, Dugel PU.. Complement cascade inhibition in geographic atrophy: a review. Eye (Lond). 2022; 36: 294–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yehoshua Z, de Amorim Garcia Filho CA, Nunes RP, et al.. Systemic complement inhibition with eculizumab for geographic atrophy in age-related macular degeneration: the COMPLETE study. Ophthalmology. 2014; 121: 693–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. ClinicalTrials.gov. Intravitreal LFG316 in patients with age-related macular degeneration (AMD). Available at: https://clinicaltrials.gov/ct2/show/NCT01527500. Accessed June 14, 2022.
- 40. ClinicalTrials.gov. CLG561 proof-of-concept study as a monotherapy and in combination with LFG316 in subjects with geographic atrophy (GA). Available at: https://clinicaltrials.gov/ct2/show/NCT02515942. Accessed June 14, 2022.
- 41. ClinicalTrials.gov. A phase 3 safety and efficacy study of intravitreal administration of Zimura (complement C5 inhibitor). Available at: https://clinicaltrials.gov/ct2/show/NCT04435366. Accessed April 13, 2022.
- 42. ClinicalTrials.gov. A study to compare the efficacy and safety of intravitreal APL-2 therapy with sham injections in patients with geographic atrophy (GA) secondary to age-related macular degeneration. Available at: https://clinicaltrials.gov/ct2/show/NCT03525613. Accessed June 14, 2022.
- 43. ClinicalTrials.gov. Study to compare the efficacy and safety of intravitreal APL-2 therapy with sham injections in patients with geographic atrophy (GA) secondary to age-related macular degeneration. Available at: https://clinicaltrials.gov/ct2/show/NCT03525600. Accessed June 14, 2022.
- 44. Heiduschka P, Fietz H, Hofmeister S, et al.. Penetration of bevacizumab through the retina after intravitreal injection in the monkey. Invest Ophthalmol Vis Sci. 2007; 48: 2814–2823. [DOI] [PubMed] [Google Scholar]
- 45. Julien S, Biesemeier A, Taubitz T, Schraermeyer U.. Different effects of intravitreally injected ranibizumab and aflibercept on retinal and choroidal tissues of monkey eyes. Br J Ophthalmol. 2014; 98: 813–825. [DOI] [PubMed] [Google Scholar]
- 46. Demirs JT, Yang J, Crowley MA, et al.. Differential and altered spatial distribution of complement expression in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2021; 62: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]