

Abstract
FGF19/FGF15 is an endocrine regulator of hepatic bile salt and lipid metabolism, which has shown promising effects in the treatment of NASH in clinical trials. FGF19/15 is transcribed and released from enterocytes of the small intestine into enterohepatic circulation in response to bile-induced FXR activation. Previously, the TSS of FGF19 was identified to bind Wnt-regulated TCF7L2/encoded transcription factor TCF4 in colorectal cancer cells. Impaired Wnt signaling and specifical loss of function of its coreceptor LRP6 have been associated with NASH. We, therefore, examined if TCF7L2/TCF4 upregulates Fgf19 in the small intestine and restrains NASH through gut-liver crosstalk. We examined the mice globally overexpressing, haploinsufficient, and conditional knockout models of TCF7L2 in the intestinal epithelium. The TCF7L2+/− mice exhibited increased plasma bile salts and lipids and developed diet-induced fatty liver disease while mice globally overexpressing TCF7L2 were protected against these traits. Comprehensive in vivo analysis revealed that TCF7L2 transcriptionally upregulates FGF15 in the gut, leading to reduced bile synthesis and diminished intestinal lipid uptake. Accordingly, VilinCreert2; Tcf7L2fl/fl mice showed reduced Fgf19 in the ileum, and increased plasma bile. The global overexpression of TCF7L2 in mice with metabolic syndrome-linked LRP6R611C substitution rescued the fatty liver and fibrosis in the latter. Strikingly, the hepatic levels of TCF4 were reduced and CYP7a1 was increased in human NASH, indicating the relevance of TCF4-dependent regulation of bile synthesis to human disease. These studies identify the critical role of TCF4 as an upstream regulator of the FGF15-mediated gut-liver crosstalk that maintains bile and liver triglyceride homeostasis.
Keywords: Fgf19, lipid absorption, NASH, tcf7l2
1 |. INTRODUCTION
NAFLD is the most common form of chronic liver disease, affecting about one-quarter of the world population.1 It is a spectrum of liver diseases ranging from steatosis, steatohepatitis (NASH) to cirrhosis and hepatocellular carcinoma. The pathogenesis of NAFLD is not well understood. Recent studies indicate the possible role of fibroblast growth factor 19 (FGF19) in the regulation of hepatic bile2 and lipid metabolism and NAFLD development in both humans and mice.3 Fibroblast growth factor (FGF) 19 is an atypical member of the fibroblast growth factor family that exhibits reduced heparin-binding, which allows it to diffuse beyond its site of origin in the ileum, enter portal circulation and act as endocrine hormones in the liver as a key regulator of bile acids biosynthesis and transport4 and lipid synthesis. FGF19 expression is induced by the nuclear receptor FXR upon binding of bile acid chenodeoxycholic acid. The induced FGF19 then plays a key role in the feedback suppression of the gene encoding cholesterol 7 alpha-hydroxylase (CYP7A1), the first and rate-limiting step in the biosynthesis of bile acids in the liver through a c-Jun N-terminal kinase (JNK)-dependent pathway.5 Mouse FGF15 is the orthologue of human FGF19 and resides on the syntenic region of the mouse genome. While mouse and human FGF 15/19 have only about 50% sequence similarity, there is conservation in their sequence and position of the FXR binding element and their function.6 Altered Wnt signaling has been associated with NAFLD and NASH and may be targeted for the treatment of liver fibrosis7 and cirrhosis.8 We have shown that mutations in Wnt coreceptor LRP6 are associated with NAFLD and NASH in humans and mice.9 Common genetic variants in the gene encoding the Wnt-regulated transcription factor TCF7L2 gene are strongly linked to type2 diabetes risk.10 Genome-wide TCF7L2 chromatin occupancy in colorectal cancer cells has identified 3′-TSS of FGF19 as one of the TCF4 binding sites,11 but this was not further confirmed by ChIP-qPCR nor the proximal promoter was examined in detail for potential TCF4 binding. In addition, whether it suppresses or upregulates TCF7L2 expression was not determined. Notably, it has been shown that individuals with familial combined hyperlipidemia have reduced transcript levels of TCF7L2.12 Therefore, the investigation of the TCF4 regulation of plasma lipids is of great relevance for understanding diabetes-dyslipidemia. Thus, we decided to examine the effect of the TCF7L2 encoded protein TCF4 on FGF15 expression, and plasma and liver fat in mice with global haploinsufficiency and overexpression of TCF7L2. We made an intriguing observation that TCF4 transcriptionally activates FGF19 and regulates the cross talk between gut and liver to maintain bile acid and lipid homeostasis.
2 |. MATERIALS AND METHODS
2.1 |. Animals
Mice overexpressing TCF7L2 under mouse TCF7L2 promoter (TCF7L2-bac mice) and TCF7L2 heterozygous knockout (TCF7L2+/−) mice were gifts from M. Nobrega’s laboratory at The University of Chicago, Chicago, IL.17 These mice were then backcrossed to C57Bl6 mice (n > 10). Wild-type (WT) littermates were used as controls. Generation of homozygous LRP6R611C mice was previously described.18 Generation of LRP6R611C; TCF7L2-bac was done by crossing LRP6R611C/R611C to C57Bl6 backcrossed TCF7L2-bac mice. The VilinCreert2 and Tcf7L2fl/fl were obtained from Jackson labs and the lab of Hans Clevers at the Hubrecht Institute in the Netherlands, respectively. All mice were housed at constant ambient temperature in a 12-h light, 12-h dark cycle. Both TCF7L2-bac male and female mice were studied. Six-week-old mice were fed a high-cholesterol diet (40% fat, 1.25% cholesterol, and 0.5% cholic acid) ad libitum (Research Diets) for 4 or 6 weeks. All studies in animals were conducted in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals.
2.2 |. mRNA extraction
The RNA was extracted by Qiagen RNAeasy Mini Kit (Catalog#74104), treated with RNase-free DNAase (Promega, M6106) for 30′ at 37°C, and purified again with the RNAeasy Mini Kit to remove DNAase (Catalog#74104). Approximately 1 μg of RNA was used for cDNA synthesis (iScript cDNA synthesis, Biorad, 1708890). A no-RT and no-RNA controls were included for each sample to ensure DNA digestion and no residual contamination. The cDNA was diluted to 5 ngm of cDNA/reaction well using qPCR. The following primer sequences were used:
Fgf19
F: CCCAGTCTGTGTCAGATGAAG
R: GAGGAAGCAGTTGGAGACATAG
Nr1H4/Fxr
F: GGGCAGAATCTGGATTTGGA
R: GTAGAAGCCCAGGTTGGAATAG
Srebp1
F: TGACCCGGCTATTCCGTGA
R: CTGGGCTGAGCAATACAGTT
Srebp2
F: TTCCAACTCTCCTCCTGTGGCT
R: CCAGCACAAATAAGCAGGTTTGTA
Fasn
F: GGAGTTCTCAGGCCGGGATA
R: GGGTACATCCCAGAGGAAGTCA
Acc1
F: TGGGGATCTCTGGCTTACAGG
R: AGCCAGACATGCTGGATCTCAT
Lima1
F: CAACAAGCTCAGTCTAGGAACA
R: CCAAAGCCCTCGTCATAGTT
Scarb1
F: CCCAGACATGCTTCCCATAAA
R: CCAGATGGATCCTGCTGAAAT
Tcf7L2
F: ATCGTCACACCGACAGTCAA
R: TTGGAGTCCTGATGCTTTGAG
Npc1L1
F: TAC CTC CTA CTG GGC CTG GAC AT
R: GGT AGG CTT GGT GCT TAC AGC AA
Cyp7a1
F: GCAGCCTCTGAAGAAGTGAATG
R: AGAGCCTCCTTGATGATGCTAT
Cyp7b1
F: CTGCCTGGAAAGCACTATTCTTGAG
R: CCTTTGGAGCATCGAAGATTTCCG
Cyp8b1
F: TGACTCATCCTGGAGGGCAGACCA
R: ATTTGGAGTAAGGGCATCGAGTAAG
Cyp27a1
F: AGACTGTGTCCCGCATCGTCCTGGT
R: GTGTCTATAGGAAGGGCACAGAG
Cd36
F: TCATATTGTGCTTGCAAATCCAA
R: GCTTTACCAAAGATGTAGCCAGTGT
Fabp2
F: AGGAATAAGTCAACTTCTCAGAGC
R: ACGAATGAGCCTGGCATTAG
Fatp2
F: GCTGACATCGTGGGACTGGT
R: TTCGACCCTCATGACCTGGC
Fatp5
F: ACCACTGGACTCCCAAAGCC
R: AGGACAGCACGTTGCTCACT
Fgf21
F: AATGTGTACCAGTCTGAAGCC
R: AGGAATCCTGCTTGGTCTTG
2.2.1 |. P-407 administration
The mice were fasted overnight, then injected with 1000 mg/kg body weight of P-407 (Spectrum, P1166). The blood was collected at 0′, 1, 2, and 5 h post injection. The triglycerides in the plasma were quantified by Fujifilm Wako Diagnostics (Catalog# 994–02891, 990–02991) kit.
2.2.2 |. Corn oil gavage with BODIPY-FA
Two hundred microliters of corn oil was administered using an oral-gavage needle into the alimentary canal of the mice. BODIPY-FA was diluted to 2 μg/gm body weight in corn oil. The BODIPY-FA was fed into mice fasted for 6 h. The proximal part of the jejunum was fixed in 4% formaldehyde to measure FA-absorption and the distal part of jejunum and ileum was subjected to microvilli isolation by standard procedures.13
2.2.3 |. TG, TC, bile salt estimation in plasma and feces
The animals were fasted for 6 h starting 9:00 a.m., mildly anesthetized in 30% isoflurane, and approx. Hundred microliters of blood was collected by retro-orbital bleeding using heparinized capillaries (Fisherbrand, Catalog#22–362-566) into heparin-coated tubes (Minicollect, Catalog# 450477). The blood was spun down at maximum speed for 15′ at 4°C at 12 000rpm and the plasma was collected into Eppendorf tubes and frozen. The TAG was measured by commercially available reagents from Fujifilm Wako Diagnostics (Catalog# 994–02891, 990–02991), total cholesterol by Cell Biolabs (Catalog# STA-384), and bile salts by Cell Biolabs (STA-631). The protocol was performed according to the manufacturer’s guidelines. To estimate bile acids in the feces, the fecal pellets from individually housed animals were collected for 2–3 days and flash-frozen in liquid nitrogen. The fecal pellets were pulverized on dry ice in a pestle-mortar and added to 500 μl of precooled chloroform: methanol (2:1). The solution was rotated at 60rpm at RT for 4–5 h, dried in dry nitrogen gas, and TAG was estimated by Fujifilm Wako Diagnostics (Catalog# 994–02891, 990–02991) kit.
2.2.4 |. Hepatic TG quantification
Approximately, 100 mg of snap-frozen tissue from the largest liver lobe was added to the 10% Triton solution, heated to 95°C for 5 min, and homogenized by the pestle and vortexed thoroughly. An aliquot of the homogenized solution was kept for protein quantification. The remaining solution was centrifuged at 13 000 rpm for 5 min and the supernatant was used for Triglyceride (TAG) quantification. TAG was quantified was Fujifilm Wako Diagnostics (Catalog# 994–02891, 990–02991). The hepatic TAG was normalized to total protein levels in the sample. Protein was estimated by Bicinchoninic Acid assay.
2.2.5 |. Cryosectioning and immunohistochemistry
Immediately after sacrifice, the mouse liver was perfused with 1Xphosphate-buffer saline (PBS) and added to a 10% formalin solution stored at 4°C. After overnight incubation, the fixed liver tissue was moved to a 30% sucrose solution overnight at 4°C, followed by a Tissue-Tek O.C.T. compound in plastic molds, allowed to freeze on dry ice and stored at −80°C. The O.C.T. submerged tissue was sectioned into 8 μm slices in a cryostat and dried on Superfrost Plus slides (Catalog# 22–037-246) for 12 to 16 h at room temperature (RT). The Oil red O staining was performed according to the manufacturer’s guidelines (Abcam, Catalog# AB150678). F4/80 (Abcam, Ab6640, Lot# GR3189625–1), Tcf7L2 (Abcam, Ab134275), Cyp7a1 (MAB9120-SP, Novus Biologicals). Immunostaining was done by standard procedures except for Tcf7L2 and Cyp7a1, when antigen retrieval was performed in 10 mM sodium- citrate buffer, pH = 6.
2.2.6 |. Western blot
Western blot was performed by standard procedures. The following antibodies were used: Acc1 (CST, 3662S, Lot# 4), Fasn (CST, 3189S, Lot# 2), b-actin (CST, 5125S, Lot# 6).
2.2.7 |. Metabolic cage studies
The mice were individually housed in metabolic cages in a relatively quiet room in the vivarium. Every day, the cages were cleaned, and the total food and water consumed were measured. The urine and fecal pellets were collected for further studies.
2.2.8 |. Chromatin immunoprecipitation assay
The ChIP assay was done as described in Ref. [14]. Briefly, the microvilli from distal jejunum and ileum were isolated from fasted (n = 2) and fast-refeed mice (n = 2 mice) (6 h fast +45 min post corn oil gavage) by standard procedures (O’Rourke, K. P., Bio-protocol 6(4), 2016). The microvilli were snap-frozen at −80°C. All the buffers and reagents were obtained from Active Motif, catalog#103930, unless specified otherwise. On the day of experimentation, the microvillus tissue was fixed in 1%formalin in 1X PBS for 15′, followed by the addition of the stop solution (Active Motif, catalog#103930). The tissue was homogenized with stainless-steel beads in the Tissue-Ruptor, followed by 2XPBS washes, and incubated in cell lysis buffer for 10′ at 4°C with protease inhibitors. The nuclei were released manually by tight-fitting pestle A, and spun down at 1250× g at 4°C for 5′. The nuclear pellet was resuspended in ChIP buffer with protease inhibitors. The chromatin in the nuclear extract was sonicated to achieve DNA fragmentation from 800–200 bp. Following this, the sonicated chromatin was subjected to immunoprecipitation with the following antibodies (IgG: ImmunoReagents Catalog#GtxRb-003-E3, lot# 47–180-042817; Catalog#Mu-003-B, Tcf7L2: ThermoFisher catalog# MA5–14975, Lot#WH3349044, FXR:Cell Signaling, catalog#72105 lot#2). The chromatin was diluted fivefold in ChIP buffer and 10 μg of antibody was used for 25 μg of chromatin. The immunoprecipitated DNA and the input DNA from all samples were reverse-cross linked by incubation with proteinase K at 55°C for 2 h, followed by incubation with 0.25M NaCl at 65°C overnight. After 16 h, the DNA was extracted from the aqueous phase after two rounds of phenol:choloform:isoamyl alcohol extractions. The DNA was precipitated with 0.037M glycogen, 0.1M NaCl, and 2X volumes of 95% ethanol. The purified DNA was subjected to qPCR, containing 2X Sybr green, 0.25 μM F+R primer, and diluted ChIP DNA, using the following primers:
Promoter1:
F: TACAGGAGACATCGCTCTCTAA
R: TGGGAGAATGTCATCAGGAAAG
Promoter2:
F: GGACTCTTCAGTAAAGCCTCTG
R: CAGCGCGGGCTATTAATCT
Intergenic site1:
F1: TTCGCCTCCTGCTCAGGTCCA
R1: TGA GGG AGG GGG AGG AAA TCG
Intergenic site2:
F2: GGCTCCTTGTCATCCCAGCTCTT
R2: ATT GTT GGC TCA TCC CAG AAG TAT CCA
Negative Control:
F: TCAGAGGTCAGAAGAGAACCT
R: GAGGGTTGGCAAGATGACTTA
2.2.9 |. In vivo FA uptake
The uptake of labeled tri-olein FA by different tissues was performed as reported in Ref. [15].
2.2.10 |. Tamoxifen treatment
The VilinCreert2; Tcf7L2fl/fl mice were administered tamoxifen as reported in Ref. [16].
2.2.11 |. NASH samples
The de-identified human NASH samples were obtained from Yale repository and diagnosed by pathologists at Yale New Haven Hospital. The Medical record number was accessed by designated personnel.
2.3 |. Statistical analyses
All statistical comparisons between two groups were done by unpaired t-test, two-tailed after testing for normality by Kolmogorov-Smirnov test. To correct for multiple comparisons, Holm-Sidak posthoc test was performed. p < .05 was considered significant.
3 |. RESULTS
3.1 |. TCF7L2 inhibits gut absorption of fat
Given the essential role of Wnt signaling in maintaining lipid homeostasis,17,18 we first examined fasting plasma triglyceride (TG) and total cholesterol (TC) in transgenic mice with multiple integrations of the endogenous Tcf7L2 gene.19 These mice are hereafter called Tcf7L2-bac. TCF7L2-bac mice showed lower fasting plasma TG and lower fasting TC compared to the wild-type littermates after 3 months on a high-fat diet (Figure 1A,B). To examine whether reduced plasma TG is caused by lower hepatic TG secretion, we measured plasma TG after injection of fasted mice with the lipolysis inhibitor poloxamer P-407. There was no significant difference in TG secretion between TCF7L2-bac mice and the wild-type littermates (Figure 1C). We next examined if the difference in TG can be explained by reduced TG absorption from the gut. We first verified that Tcf7L2 expression was elevated in the jejunum of Tcf7L2-bac mice by IHC (Figure 1D) and RT-qPCR (Figure 1E). To measure the absorption of triglycerides from the small intestine, we administered corn oil into mice and measured plasma TG at different time points. The mice were co-injected with poloxamer P-407 to block the lipolysis of the administered TG in corn oil. There was significantly lower plasma TG at 4 h post gavage in TCF7L2-bac mice (Figure 1F) suggesting reduced TG absorption compared to wild-type littermates. To further confirm impaired fat absorption in TCF7L2-bac mice, we administered BODIPY-FA (2 μg/gm body weight in corn oil) by oral gavage and collected jejunum 45 min later while lipid absorption is actively occurring in the gut. The intestinal tissue was fixed, and BODIPY-FA was visualized by confocal microscopy. A greatly reduced fluorescence was observed in the jejunum in TCF7L2-bac mice versus wild-type littermates (Figure 1G,H). Taken together, these findings indicate that a global increase in TCF7L2 causes reduced plasma TG and cholesterol by diminishing uptake of fatty acids in the gut.
FIGURE 1.
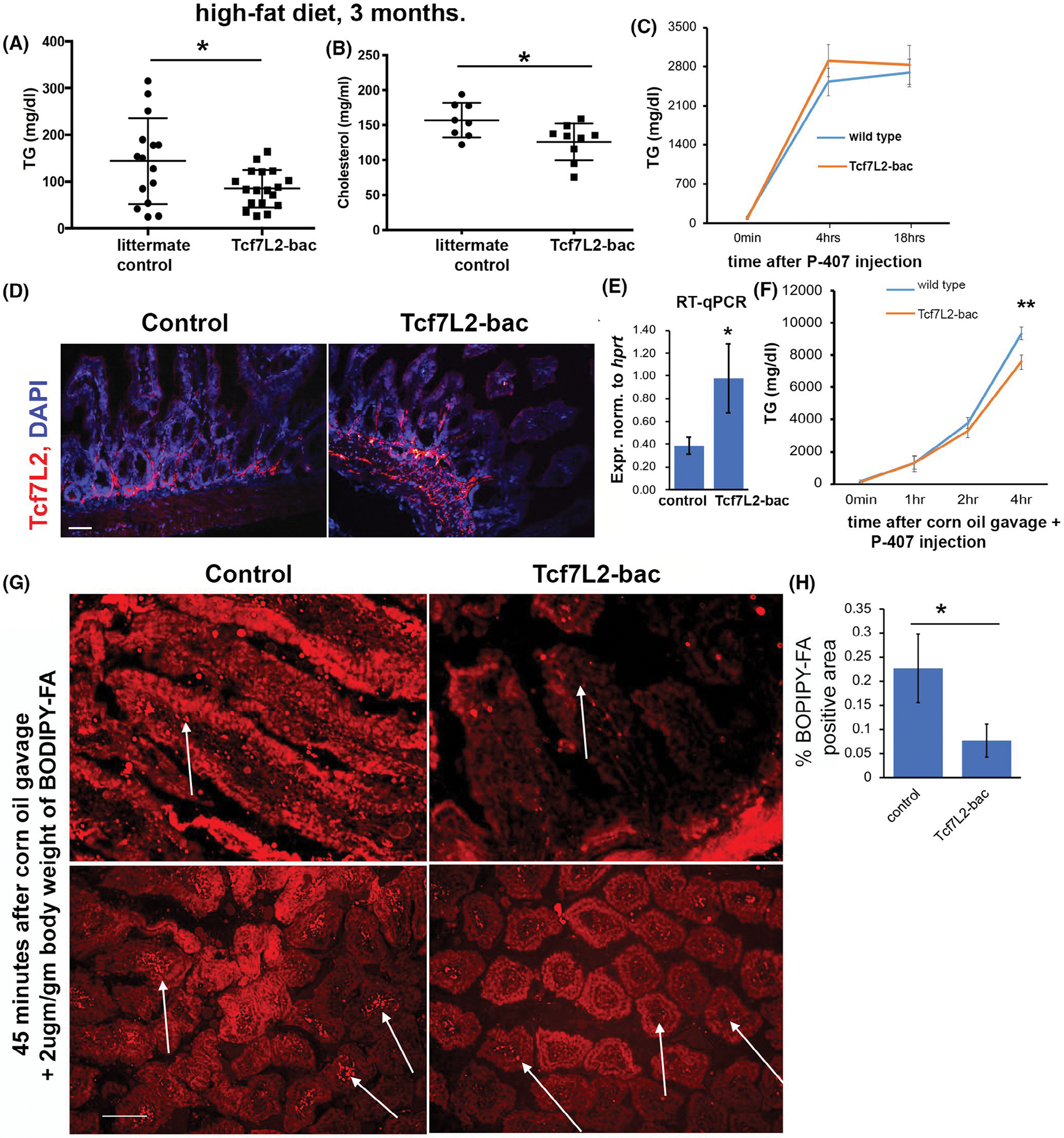
Tcf7L2 limits gut absorption of fatty acids. (A, B) Fasting plasma TG (n > 15 mice each genotype) and TC (n > 7 mice each genotype) in the littermate controls and Tcf7l2-bac mice. Each dot in the dot plot is a biological replicate. Unpaired t-test, two-tailed, normality-tested. (C) Hepatic TG secretion assay showing plasma TG at indicated time points after injection of mice, fasted for 6 h, with polyoxamer P-407. Mice were fed a high fat diet for 3 months. n > 6 mice each genotype. (D, E) Tcf7L2 expression in the jejunum of indicated mice by IHC (D) and RT-qPCR (E) (n > 6 mice each genotype). scale bar = 70 μm. Unpaired t-test, two-tailed. (F) Plasma TG at indicated time points after feeding mice with corn oil and co-injecting with polyoxamer P-407. The mice were fasted for 6 h prior to the start of experiment. n > 7 mice each genotype, holm-sidak posthoc test was performed. (G) BODIPY-FA uptake visualized by confocal microscopy in the jejunum of indicated mice 45 min after administration of corn oil + BODIPY-FA by oral gavage. Top panel: longitudinal section, bottom panel: cross-section. White arrows point to fluorescent BODIPY-FA in the controls and much-reduced fluorescence, indicating uptake of FA in Tcf7L2-bac. n > 5 mice each genotype, scale bar = 150 μm. (H) Quantification of percent area of BODIPY-FA in the indicated genotypes. Unpaired t-test, two-tailed, normality-tested
3.2 |. TCF7L2 reduces bile salts by increasing intestinal FGF15
The reduced FA uptake in the jejunum of Tcf7L2-bac mice prompted an investigation into the potential transcriptional targets of Tcf7L2 in the small intestine. Among the candidate genes, previously implicated in the regulation of FA uptake, such as Nr1H4, Scarb1, Lima1, Npc1L1, Fabp2, Fatp5, CD36, and Fatp2, only Fgf15 was upregulated by Tcf7l2 in both fast and re-feed conditions (Figure 2A). Fgf15 is a 24 kDa protein syntenic with FGF19 in humans and has similar tissue distribution and physiological roles.20 FGF15/19 are low heparin sulfate binding members of the FGF family of proteins that act as a circulating endocrine molecule that feedbacks to inhibit hepatic bile acid synthesis and hepatic lipogenesis in the liver through gut-liver crosstalk.2,21 In accordance with increased Fgf19 in the gut, the total bile salt content was significantly lower in the plasma (Figure 2B). We then measured bile acid content in the feces to exclude any influence from reduced reabsorption of bile acids from the gut. We reasoned that reduced bile reabsorption in the gut would result in increased bile acid content in the feces. The bile acid content was significantly reduced in the feces of TCF7L2-bac mice versus wild-type littermates (Figure 2C) suggesting reduced bile synthesis, as opposed to reabsorption causes, reduced plasma bile acids. Consistent with reduced bile synthesis, the expression of CYP7A1, the first and rate-limiting enzyme in the major bile acid synthesis pathway in the liver,2 was reduced in TCF7L2-bac mice versus wild-type littermates (Figure 2D). CYP7A1 was previously shown to be repressed by Fgf19 in the liver.22 The expression of other genes in the bile synthesis pathway such as Cyp8a1, Cyp27a1, Cyp7b1, Nr1H4, and cholesterol biosynthesis pathways such as Srebp2 and HMGCR were unchanged (Figure 2D). FGF19 also protects mice against diet-induced fatty liver disease23 by suppressing the expression of lipogenic enzymes24 and suppressing fatty acid synthesis.25 We, therefore, assessed the development of NAFLD by oil red O staining. TCF7L2-bac mice were protected against diet-induced NAFLD when compared to wild-type littermates (Figure 2E). Accordingly, Srebp1 and Fasn transcript levels were lower in TCF7L2-bac mice liver compared to wild-type littermates (Figure 2D). Taken together, our findings suggest that TCF7L2 upregulates Fgf19 and protects mice against hyperlipidemia and NAFLD by reducing bile acid synthesis and lipogenesis. Further, these findings identify a previously unknown function of FGF19 to reduce fatty-acid uptake in the gut which then confers protection against diet-induced hyperlipidemia and NAFLD in addition to previously described effects of Fgf19 on hepatic lipogenesis.
FIGURE 2.
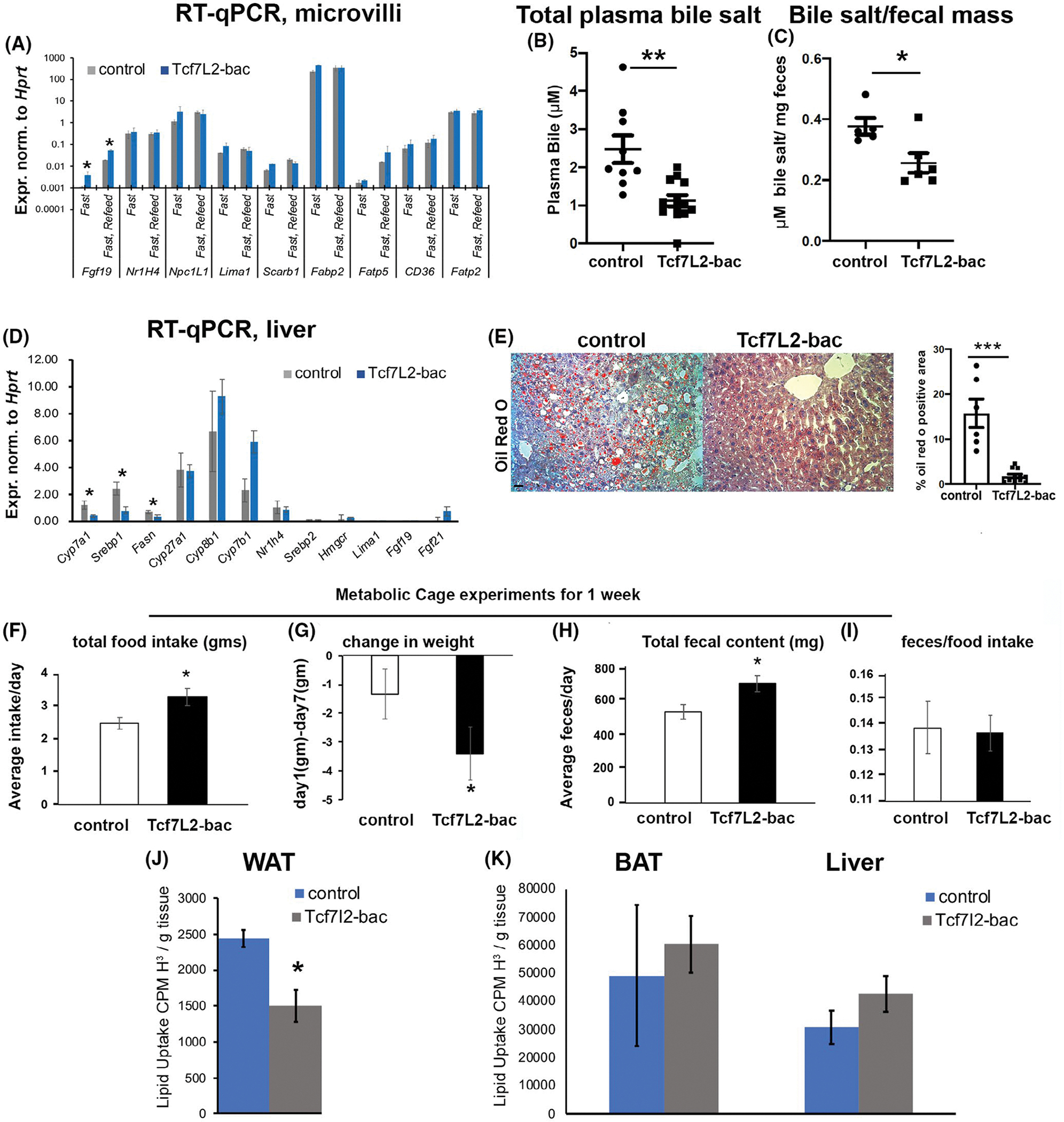
Tcf7L2 increases Fgf15 in the ileum to suppress triglyceride synthesis in the liver. (A) RT-qPCR showing mRNA levels of indicated genes from the isolated microvilli from distal jejunum and ileum of control and Tcf7l2-bac mice in either fast or fast/refeed conditions (n > 4 mice each condition and each genotype), unpaired t test, 2-tailed. (B, C) Total bile acid content in the plasma of fasted mice 45′ post prandially, n > 7 mice each condition (B) and fecal pellets, n > 5 mice each genotype (C) of littermate controls and Tcf7L2-bac mice, unpaired t test, 2-tailed. (D) mRNA expression of the designated genes in the liver of Tcf7L2-bac versus littermate mice after 6 h fast, n > 10 mice each genotype, unpaired t test, 2-tailed. (E) Oil Red O staining and quantification depicting neutral lipids in the liver of the designated mice, n > 5 mice each genotype, unpaired t test, 2-tailed, scale bar = 150 μm. (F–I) Measurement of indicated parameters measured when littermate control and Tcf7l2-bac mice were subjected to metabolic cages, n = 3 mice control and n = 6 mice for Tcf7l2-bac, unpaired t test, two-tailed. (J, K) Fatty acid uptake measured in epididymal WAT, BAT, and liver in the controls and Tcf7L2-bac mice. n = 3 mice each. unpaired t test, two-tailed
Previously, bile acids were shown to increase the expression of Glp-1 peptide by binding the TRG5 receptor in the L-cells of the distal small intestine, and are considered as a therapeutic option to reduce obesity by decreasing food intake.26 In accordance with reduced plasma bile-salt in Tcf7L2-bac mice, we observed increased food intake in Tcf7L2-bac mice when they were subjected to metabolic cage studies (Figure 2F). Strikingly, plasma Fgf21 levels were reduced in Tcf7L2-bac mice versus wild-type littermates (Figure S1A,B). We speculate this unexpected reduction of FGF21 to be a compensatory mechanism to normalize plasma bile acid concentration since FGF21 has been shown to repress bile synthesis.27 Despite increased food intake and reduced FGF21 levels, the Tcf7L2-bac mice showed higher weight loss during the course of metabolic cage studies, which is attributable to reduced fat absorption (Figure 2G). Accordingly, Tcf7L2-bac mice had higher total fecal content, while the ratio of fecal content to food intake remained unchanged compared to wild-type littermates (Figure 2H,I). Next, we determined if Tcf7L2 lowers adipose tissue mass as reported before,28 accounting for the reduced hepatic TAG in Tcf7L2-bac mice. To this end, we gavaged mice with labeled triglycerides15 and measured radioactivity in the peripheral tissues. Notably, the FA uptake was significantly reduced in epididymal white adipose tissue of Tcf7L2-bac mice (Figure 2J). No changes in FA uptake were detected in brown adipose tissue and liver (Figure 2K), indicating that reduced FA uptake in the adipose tissue does not result in lipotoxicity as predicted. Altogether, these data suggest that the reduced plasma bile acid in Tcf7L2-bac mice is due to impaired bile acid synthesis in the liver and not due to its excretion into the feces and that the reduced DNL and adiposity could contribute to lowered hepatic TAG in Tcf7L2-bac mice.
3.3 |. TCF4 binds promoter of Fgf15
We next examined whether TCF4 is directly bound to Fgf15 to affect its transcription. The binding of TCF4 to the Fgf15 locus was assayed by chromatin immunoprecipitation (ChIP) assay in the isolated microvilli from distal jejunum and the ileum of TCF4-bac mice subjected to fast or fast/refeed (6 h-fast; 45′ refeed post corn oil gavage). We probed regions in the proximal promoter upstream of putative Fgf15 transcription start site at −1.2 and −1.3 kb (Figure 3A). Previously, FXR-responsiveness was detected up to 8.8 kb downstream of the Fgf15 transcription start site.29 Both regions showed significant enrichment for TCF4 in the fast-refeed condition but not in the fasting condition (Figure 3B). The binding of TCF4 to Fgf19 promoter is associated with transcriptional activation of Fgf19 during refeeding (Figure 2A, compare Fgf19 expression during fast and re-feed) as indicated previously.2 Another region further upstream at −3.4 kb (negative control), showed no significant binding (Figure 3B). Intergenic sites at +765 bp and +2.6 kb, which were predicted to bind TCF4, based on sequence conservation for canonical TCF4, did not show significant binding perhaps due to weak interaction with TCF4 (Figure 3B). As expected, FXR bound the promoter elements29 very efficiently upon fast-refeed condition (compare % input between TCF4 and FXR) (Figure S2). Altogether, these results reveal TCF4 directly binds the Fgf19 promoter and is associated with transcriptional activation of Fgf19.
FIGURE 3.
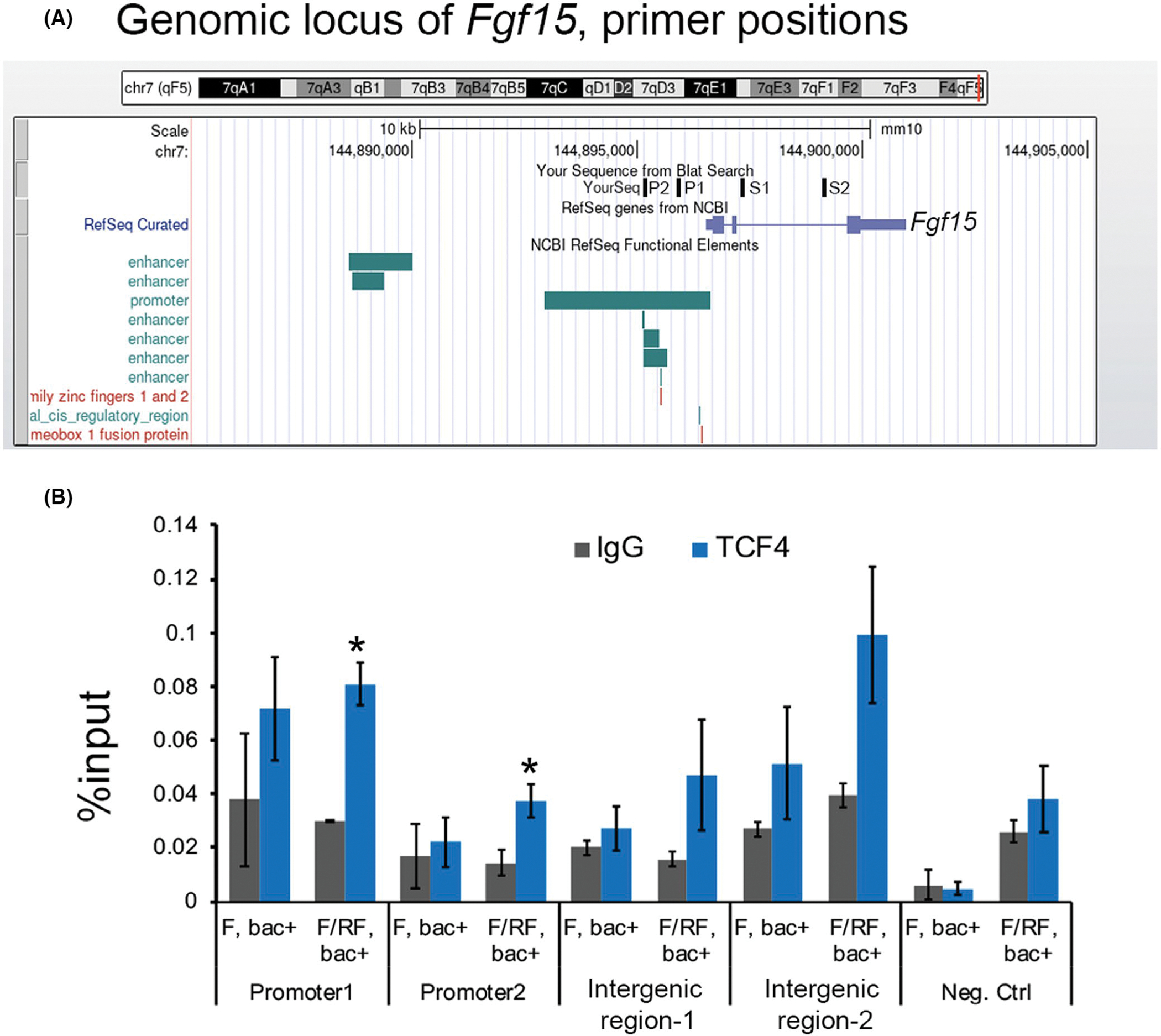
Tcf4 directly binds Fgf15 promoter. (A) snapshot of the UCSC genome browser showing the Fgf15 locus in the mouse genome; primers used to probe Tcf4 binding are indicated. Two primers in the promoter (P1, P2) and two in the intergenic regions of Fgf15 (S1, S2) were tested. (B) Chromatin Immunoprecipitation (ChIP) assay showing % input enrichment with IgG or TCF4 in fast (F) or Fast/Refeed (F/RF) conditions at the indicated regions at Fgf15 locus in the microvilli of distal jejunum and ileum. Unpaired t test, two-tailed, normality-tested, *p < .05 Unpaired t test, two-tailed, normality-tested, *p < .05
3.4 |. TCF7L2 deficient mice exhibit increased hepatic lipogenesis, triglyceride secretion and develop fatty liver disease
Homozygous TCF7L2 mice die shortly after birth. To establish the causal role of TCF7L2 in FGF19-dependent regulation of hepatic lipogenesis, we embarked on the characterization of heterozygous TCF7L2 KO mice (TCF7L2+/−). TCF7L2+/− mice appeared healthy, had normal body weight and appearance (data previously published by our lab30). 44 weeks old TCF7L2+/− mice had higher plasma TG, and higher total cholesterol compared to wild-type littermates on chow diet (Figure 4A,B). Consistent with regulation of bile acid synthesis in Tcf7l2-bac mice (see Figure 2), the plasma bile salts were increased (Figure 4C) and Fgf15 expression in the ileum was decreased (Figure 4D) in Tcf7l2+/− mice. Accordingly, the expression of genes in the bile synthesis pathway, Cyp8b, and Cyp27a1 were increased in the liver of fasted Tcf7l2+/− mice (Figure 4E,F). Since Fgf15 represses DNL in the liver,31 we observed increased de-novo lipogenesis, as measured by synthesis of palmitate from labeled acetate, in the isolated hepatocytes from Tcf7l2+/− mice (Figure 4G). The neutral lipid staining by Oil Red O in 12 weeks old mice revealed increased triglyceride content in the liver (Figure 4H,I) and increased hepatic TG secretion in Tcf7l2+/− mice (Figure 4J). Lipid profiling of FPLC-separated plasma lipoprotein fractions showed that, as expected, the increased TG and TC in TCF7L2+/− mice were predominantly associated with VLDL particles (Figure 4K). Taken together, these findings suggest that haploinsufficiency of TCF7L2 results in increased DNL due to the loss of gut TCF7L2 and the interruption of gut-liver feedback mechanism due to reduced FGF15/19. Taken together, these findings further confirm that TCF7L2 transcriptional regulation of intestinal FGF15/FGF19 plays a key role in modulating liver bile and lipid synthesis.
FIGURE 4.
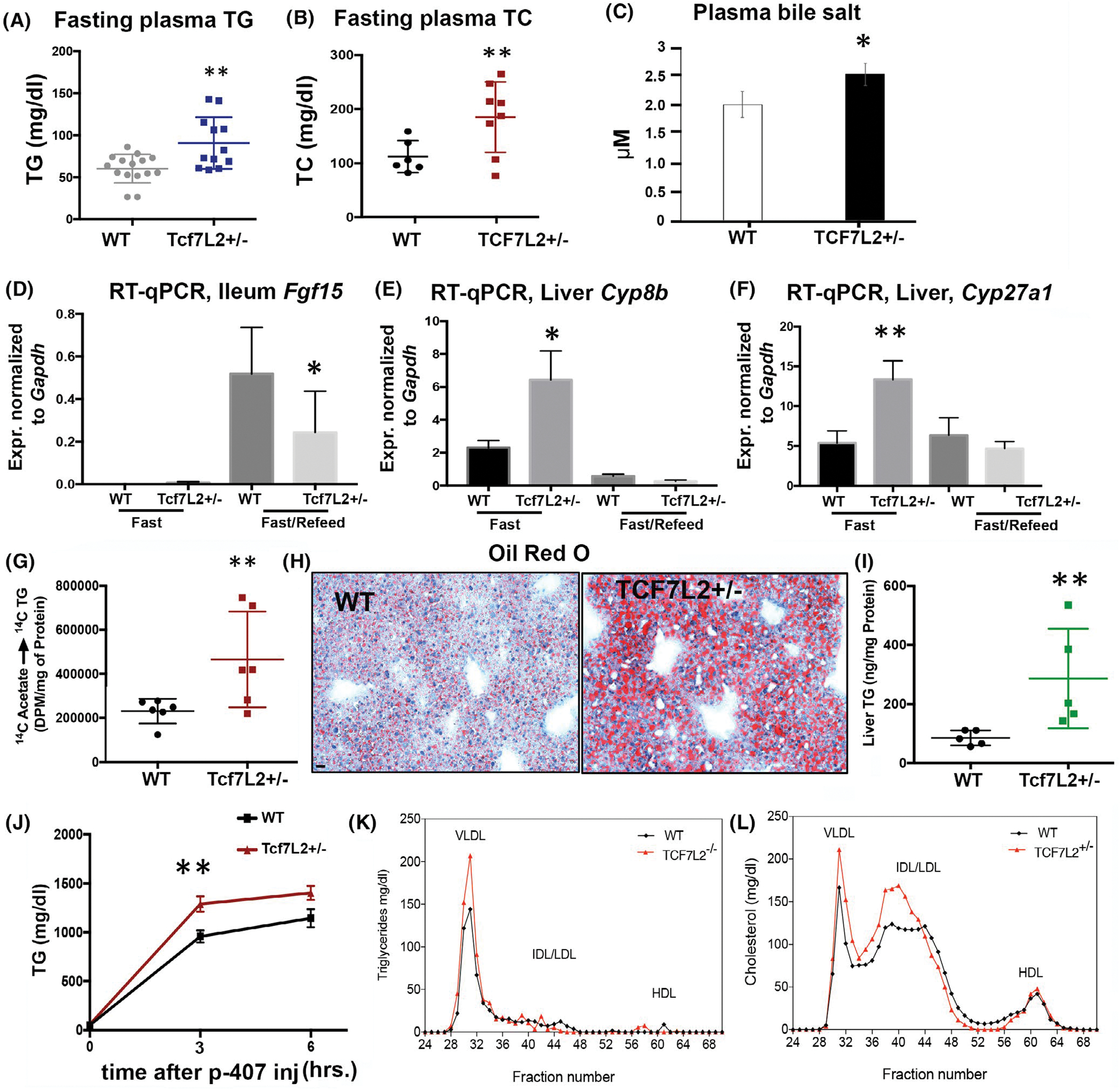
Tcf7L2 is necessary for the suppression of hepatic BA production by Fgf15 in the ileum. (A, B) Fasting plasma TG, and total cholesterol in TCF7L2+/− versus wild-type mice on chow diet. n > 12 mice each genotype, each dot is data from individual mice, unpaired t test, 2-tailed. (C) Bile salt concentration in TCF7L2+/− versus wild-type mice, unpaired t test, 2-tailed, n > 8 mice each genotype. (D) Ileal Fgf15 mRNA by RT-qPCR in the designated mice, unpaired t test, two-tailed, n = 8 mice each. (E, F) Liver Cyp27a1, and Cyp8b in fasted and fed states in TCF7L2+/− versus wild-type mice. unpaired t-test, two-tailed, n = 8 mice each. (G) TG synthesis assayed by 14C Acetate incorporation in palmitate in TCF7L2+/− versus wild-type mice on a high-fat diet. unpaired t test, two-tailed, n = 6 mice each genotype. (H, I) Oil red O staining (H) and TAG quantification (I) of the liver in TCF7L2+/− versus wild-type mice on high fat diet, scale bar = 150 μm, unpaired t test, two-tailed, n = 5 mice each genotype. (J) Plasma TG secretion after P-407 injection in TCF7L2+/− versus wild-type mice on chow diet. unpaired t test, two-tailed. (K, L) FPLC of plasma TG and cholesterol in TCF7L2+/− versus wild-type mice on high fat diet, n = 3 mice each. unpaired t test, two-tailed
3.5 |. The conditional knockout of Tcf7L2 in the gut epithelium reduces ileal Fgf19 expression and raises plasma bile levels
Next, we determined if Tcf7L2 in the intestinal epithelium is directly regulating Fgf19 expression and consequently hepatic bile synthesis. To this end, we crossed Tcf7L2fl/fl mice to VilinCreert2 to create tamoxifen (TAM)-inducible loss of Tcf7L2 in the intestinal epithelium as previously reported in Ref. [32]. (Figure 5A). To minimize the effects from the reduced proliferation of intestinal stem cells and consequently defective morphogenesis/functionality in intestinal villi due to loss of TCF4,32 we euthanized mice within 72 h post last TAM injection, 4 days before intestinal epithelia completely regenerates. 72 h post last TAM injection, the ileal mucosa showed reduced expression of Tcf7L2 and Fgf19 in VilinCreert2; Tcf7L2fl/fl compared to the Tcf7L2fl/fl mice (Figure 5B) despite defective morphogenesis of the villi as previously reported.32 The reduced Fgf19 correlated with increased post prandial plasma bile concentration, which also indicated a relatively intact enterohepatic circulation (Figure 5C). Altogether, these results verify Fgf19 to be a target of Tcf7L2 in the intestinal epithelium which suppresses the production of bile salts through entero-hepatic circulation.
FIGURE 5.
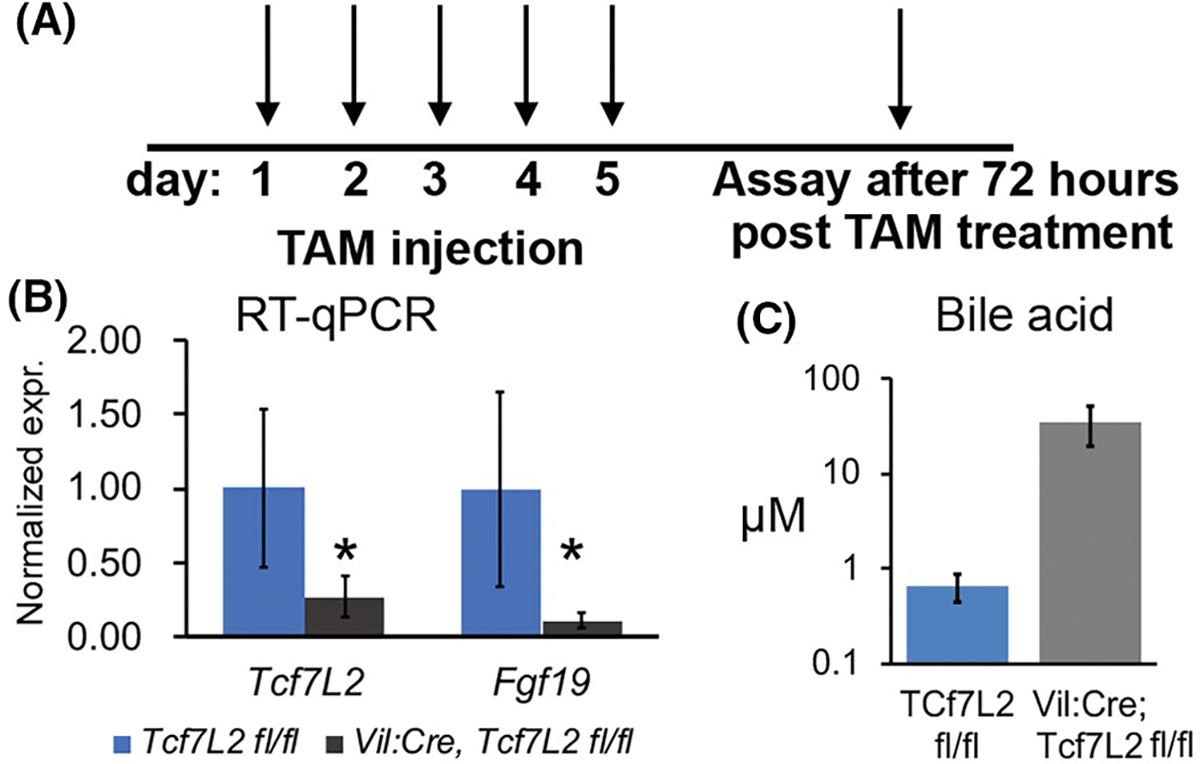
Loss of Tcf7L2 in the intestinal epithelium reduces Fgf15 expression and FA absorption. (A) Schematic displaying the strategy to knock out Tcf7L2 in the intestinal epithelium. (B) RT-qPCR showing Tcf7L2 and Fgf19 expression in the ileum of mice with the designated genotypes, n = 7 mice each genotype, unpaired t test, 2-tailed. (C) Plasma bile acids in the designated genotypes 45′ post corn oil gavage, n = 7 mice each genotype. Unpaired t test, two-tailed, normality-tested, *p < .05
3.6 |. TCF7L7L2 overexpression normalizesplasma triglyceride, liver fat, and inflammation of LRP6 RC/RC mice
We had previously shown that mice with the human mutation in LRP6 (R611C), the gene encoding the Wnt coreceptor LRP6, develop NAFLD and NASH and this can be rescued by Wnt3a injection in mice.18 The activation of canonical Wnt signaling results in translocation of beta catenin into the nucleus, binding of TCF7L2, and activation of the target genes. We investigated if TCF7L2 overexpression can rescue NASH and NAFLD in mice homozygous for p.R611C substitution in LRP6, the coreceptor for Wnt proteins (here referred to as LRP6 RC/RC mice). We crossbred TCF7L2-bac mice into LRP6 RC/RC mice and fed them a high-fat diet for 3 months. There was significantly lower liver fat assayed by Oil red O staining (Figure 6A), and TG content (Figure 6B) in LRP6 RC/RC; TCF7L2-bac compared to LRP6 RC/RC mice alone. The plasma TG was also significantly lower in LRP6 RC/RC; TCF7L2-bac versus in LRP6 RC/RC mice (Figure 6C). Consistent with these findings, TG secretion was significantly lower in LRP6 RC/RC; TCF7L2-bac versus LRP6 RC/RC mice (Figure 6D). Accordingly, the DNL enzyme ACC1 was downregulated and FASN showed a strong trend toward reduction in LRP6 RC/RC; TCF7L2-bac versus LRP6 RC/RC mice (Figure 6E). Conversely, the protein expression levels of ACC1, FASN, and DGAT1 were upregulated in the liver of Tcf7L2+/− mice versus wild-type littermates (Figure 6F). The levels of INSIG1, the suppressor of Srebp1 activity, were downregulated in the liver of Tcf7L2+/− mice (Figure 6F), which is consistent with the similarly reduced INSIG1 levels in LRP6 RC/RC mouse liver.18,33 In accordance with its effect on lowering DNL, the liver inflammation assayed by F4/80 was also rescued in LRP6 RC/RC mice by TCF7L2 (Figure 6G). Altogether, our findings suggest that Wnt-regulated transcription factor TCF7L2 can rescue NASH caused by impaired Wnt signaling in LRP6 RC/RC mice. These results confirm our earlier findings that impaired Wnt signaling induces NASH/NAFLD and that the Wnt pathway is a potential target for the treatment of NASH.
FIGURE 6.
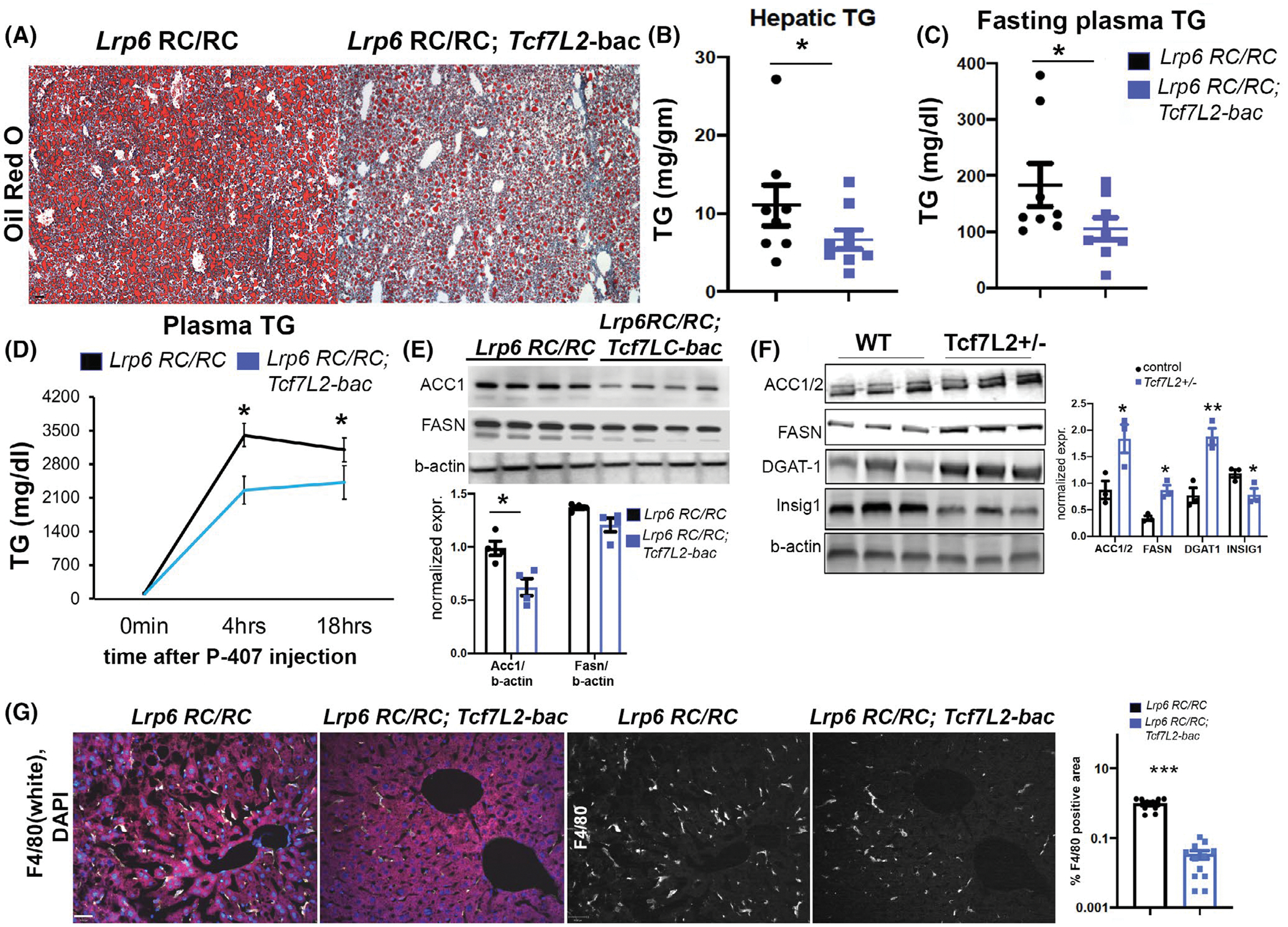
Tcf7L2-bac rescues steatohepatitis in Lrp6 RC/RC mice. (A, B) Oil red O (A) staining and hepatic TG (B) in the indicated mice. Each dot in the dotplot indicates a biological replicate, n = 7 for Lrp6 RC/RC and n = 9 for Lrp6 RC/RC; Tcf7L2-bac. Mice were fed a high-calorie diet (HCD) with 40% calories from fat, and 40% from carbohydrates, scale bar = 150 μm, unpaired t test, two-tailed. (C) Plasma TG after 6 h fast in the indicated mice fed with HCD, n > 7 mice each genotype, unpaired t test, two-tailed. (D) Time course of plasma TG after intraperitoneal injection of weight-normalized P-407 in the indicated mice, fed with HCD, after 6 h-fast, holm-sidak post hoc test was performed. (E) Expression and quantification of DNL enzymes Acc1 and Fasn in the indicated mice by WB. The mice were fasted overnight, followed by 6 h refeeding before sacrifice. n = 4 mice each genotype, unpaired t test, two-tailed. (F) Expression and quantification of enzymes in the DNL pathway and Insig1 expression in Tcf7L2+/− mice. n = 3 mice each genotype, unpaired t test, two-tailed. (G) Expression of pro-inflammatory macrophage marker F4/80 in the indicated mice by IF. The single-channel (F4/80) images are depicted in white in the last two panels, scale bar = 70 μm, unpaired t test, two-tailed, normality-tested, *p < .05
3.7 |. TCF4-Fgf19-Cyp7a1 axis is conserved in human NASH
The increased induction of the DNL pathway in the Tcf7L2 haploinsufficient mice and protection from diet-induced fatty liver disease in Tcf7L2-bac mice led us to investigate Tcf7L2 levels in human NASH samples versus control liver samples from individuals with no NASH or diabetes (Table S1). As expected, TCF4 levels were significantly reduced in the biopsy-proven human NASH samples (Figure 7A,B). Low levels of liver Fgf1934 and failure of residual Fgf19 to suppress Cyp7a1 have been previously reported in human NASH.3 Accordingly, CYP7A1 expression levels were higher in biopsy-proven human NASH samples compared to control liver (Figure 7C,D). In summary, these findings indicate that hepatic TCF4 is reduced and expression of CYP7A1, a direct target of Fgf19,2 fails to be suppressed in human NASH.
FIGURE 7.
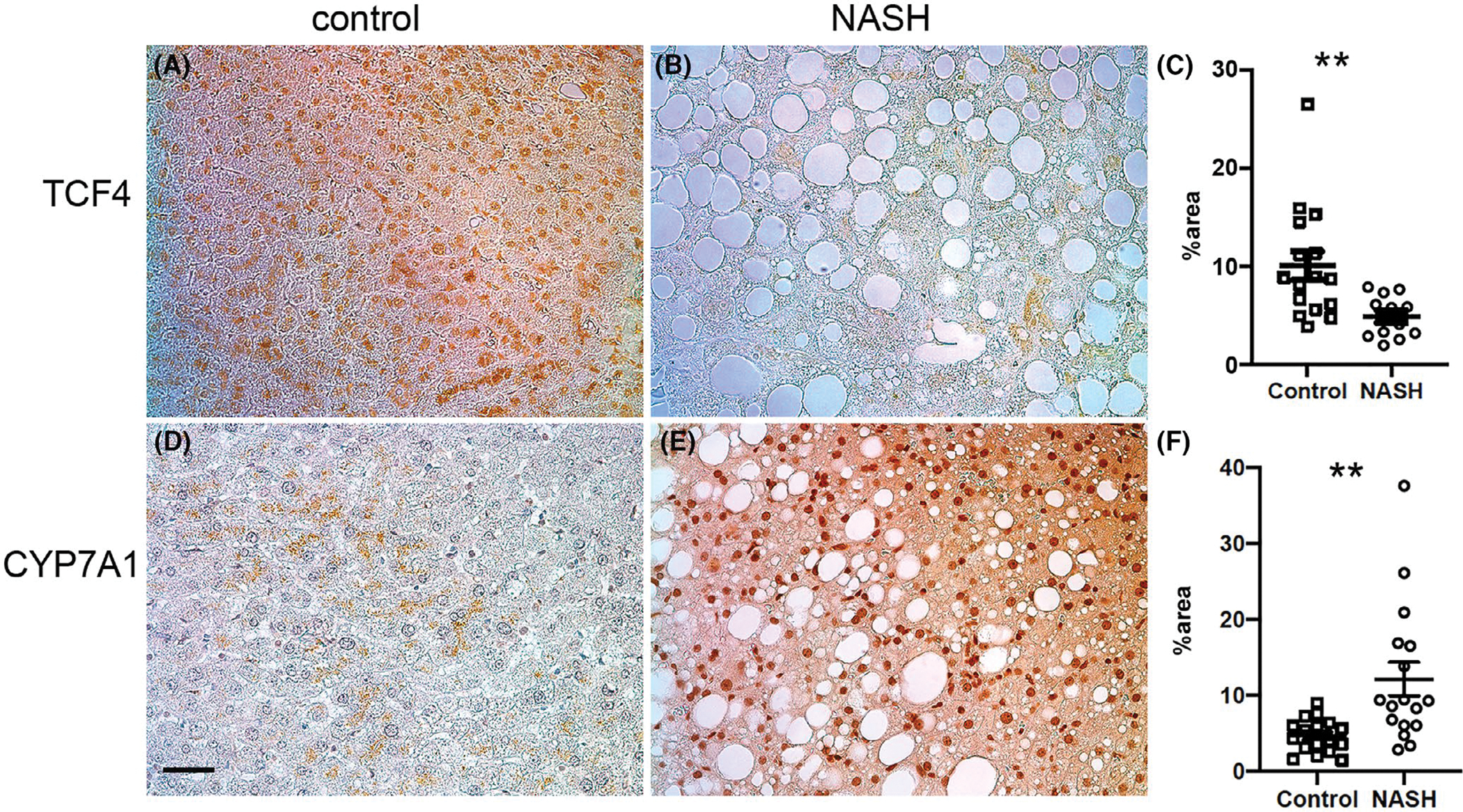
Expression of TCF4 and CYP7A1 in the non-NASH and NASH human samples. (A–C) Expression and quantification of TCF4. n = 6 controls; n = 19 samples. (D–F) Expression and quantification of CYP7A1, n = 6 controls, n = 19 NASH samples. Unpaired t test, two-tailed, *p < .05. scale bar = 70 μm
4 |. DISCUSSION
In this study, we show that TCF7L2-encoded protein TCF4 regulates a crosstalk between intestine and liver to maintain bile acid, liver, and plasma homeostasis. Our findings indicate that TCF7L2 is a transcriptional coactivator of FGF19/15, a circulating hormone that enters the portal circulation and suppresses bile synthesis in the liver by inhibiting the rate-limiting enzyme of bile biosynthesis Cyp7a1. Bile acids are synthesized from cholesterol in the liver and act as biological detergents to facilitate biliary cholesterol secretion and solubilize and absorb dietary cholesterol35 and triglycerides.36 Accordingly, reduced bile acid in TCF7L2-bac mice leads to reduced uptake of FA in the gut, reduced plasma cholesterol and TG, and is protective against diet-induced NAFLD (Figure 8). In contrast, loss of gut FGF15/19 in Tcf7l2+/− mice disrupts the gut-liver crosstalk in suppressing bile, TG, and cholesterol biosynthesis (Figure 8).
FIGURE 8.
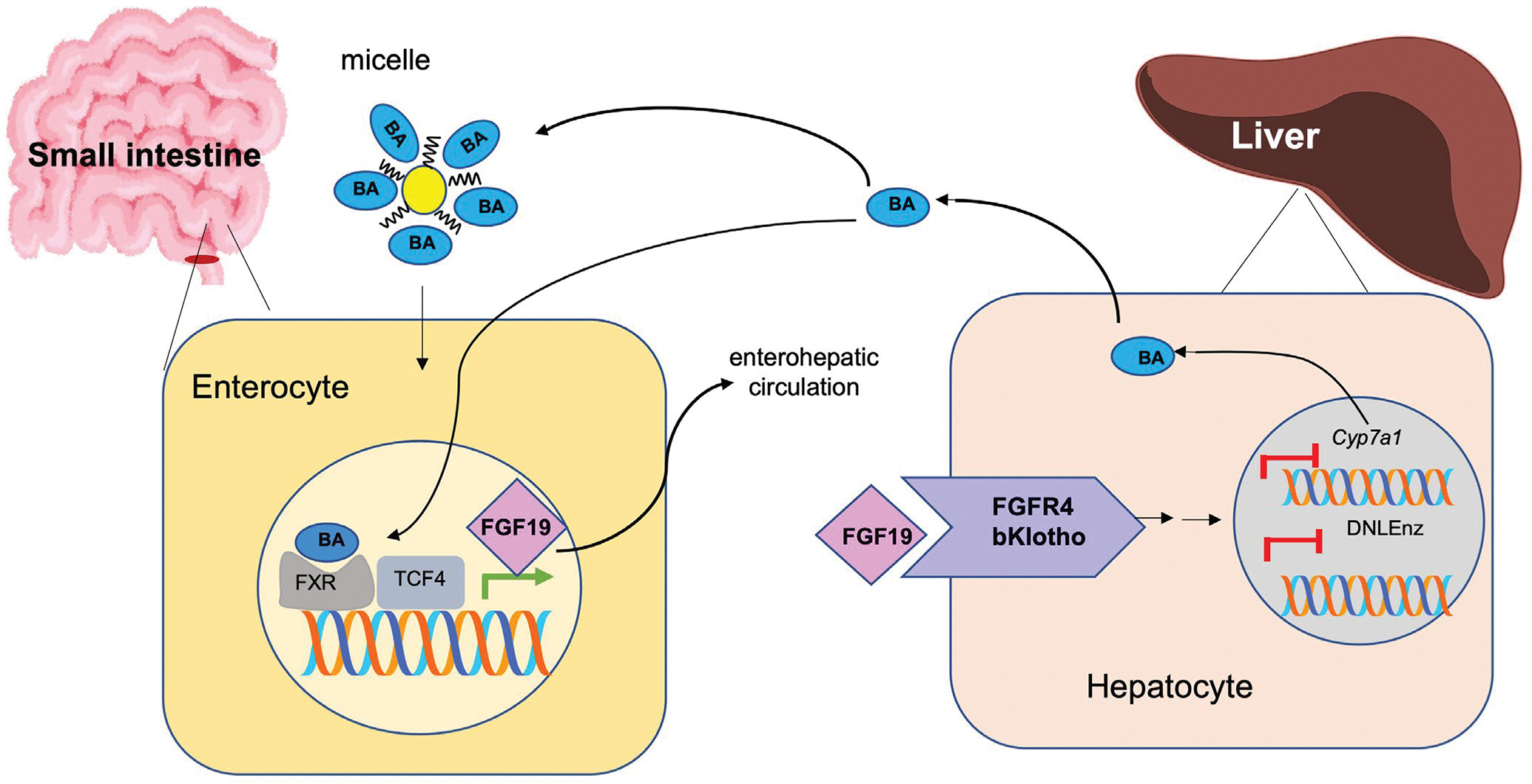
Model. Graphical representation of TCF4 mediated regulation of enterohepatic regulation of bile and lipid synthesis. TCF4 transcriptionally activates Fgf19 which then suppresses bile synthesis and lipid synthesis in the liver
The synthesis of FGF15/FGF19 in ileal enterocytes and their release into the enterohepatic circulation Is regulated by FXR.37 FXR is activated upon binding of bile acids and recruits transcriptional coactivators, which are mostly unknown.38 Here, we identify TCF4 as a transcription factor that is necessary for FGF19/15 transcription in the enterocytes of the ileum. TCF4 binds the promoter of Fgf19 in the ileal enterocytes during fast-refeed, a condition associated with transcriptional activation of Fgf19. Several pharmacologic studies in hyperglycemic, obese animal models have shown that FGF19 can improve metabolic rate, lower serum glucose levels by inhibiting hepatic glucose production,39 and reduce hepatic triglyceride and cholesterol by suppressing TG and cholesterol synthesis and increasing fatty acid oxidation.40 In contrast, reduced fasting FGF19 levels have been associated with the development of non-alcoholic fatty liver disease in obese adolescents.34 Others have shown that hepatic FGF19 resistance and increased hepatic BA production are associated with NAFLD.41 Interestingly, engineered FGF19 has been shown to resolve steatohepatitis and fibrosis in mice by eliminating bile acid toxicity and lipotoxicity.31 The positive metabolic effects of bariatric surgery are also in part attributed to increasing FGF19 levels.42 Accordingly, increased Fgf19 in our Tcf7L2-bac mice protected against NAFLD while haploinsufficiency of TCF7L2 or its disruption in the gut results in reduced FGF15/19 leading to increased hepatic bile acid, TG, and cholesterol synthesis. Reduced TCF4 in the liver of patients with NASH may suggest a universal disease pathway that can be targeted by activation of Wnt signaling. Interestingly, we noted reduced FGF21 levels in Tcf7L2-bac mice compared to their littermates. Since FGF21 has been shown to be a negative regulator of bile acid synthesis this finding can be attributed to a compensatory mechanism that we did not further explore. It is unlikely that TCF7L2 transcriptionally regulates FGF21 as genome-wide TCF7L2 chromatin occupancy in colorectal cancer cells has shown binding of FGF19 but not FGF21 promoter.11 Nevertheless, this finding may raise the question as to whether combined FGF19 and FGF21 may have a superior effect for the treatment of NASH compared to each of them alone.
Consistent with earlier findings43 increased intestinal expression of FGF19/15 was associated with decreased expression levels of the rate-limiting enzyme cholesterol 7 alpha-hydroxylase (Cyp7a1) and lower plasma bile acid levels in TCF7L2-bac mice versus wild-type littermates (Figure 8). Unexpectedly, the TCF7L2 haploinsufficiency was found to be associated with an upregulation of Cyp27a1, Cyp8b1, two enzymes that also increase levels of bile salts and intestinal cholesterol absorption but are not known to be regulated by FGF15/19. The exact mechanism of this complex regulation of bile acid synthesis enzymes by TCF7L2 remains unknown. In the human NASH, Cyp7a1 seems resistant to the suppressive actions of Fgf19, leading to increased bile acid production in human NASH samples.3,41 Consistent with this observation, CYP7a1 levels were elevated in human NASH indicating dysregulated entero-hepatic TCF7L2/Fgf19/CYP7a1 signaling cascade.
Elevated plasma TG levels have been identified as a risk for type2 diabetes and atherosclerosis. Genetic variants near the TCF7L2 gene are the strongest GWAS variants for type 2 diabetes44 and are associated with increased risk for other metabolic traits of the metabolic syndrome, including increased body mass and hypertension. The role of TCF7L2 risk variants in TCF7L2 transcript levels in patients with diabetes has been controversial, likely due to the small effect sizes of the risk alleles. The knockdown of Tcf7L2 in rodent islets and β cell lines by RNAi causes reduced insulin secretion.45 Similarly, the pancreas-specific Tcf7l2 knockout mice exhibit impaired β cell growth, reduced insulin secretion and diminished incretin response.46 In contrast, inducible liver-specific knockout of Tcf7L2 has been shown to be associated with hypoglycemia and reduced hepatic glucose production.47 These controversial findings suggest that TCF7L2 genetic variants may have independent effects involving multiple tissues. Nevertheless, the current finding is consistent with our previous report that impaired Wnt signaling in mice with a human LRP6 mutation (RC/RC mice) causes fatty liver disease and hypertriglyceridemia, which can be normalized by Wnt3a administration.18 Both RC/RC and TCF7L2+/− mice exhibit reduced Insig1 levels and enhanced DNL. These findings are in contrast with a previous report in which mice deficient for Insig1 were protected against NASH progression.48 One potential explanation for these differences is the direct protective effect of the Wnt signaling pathway on hepatic inflammation as previously demonstrated by our group.9,33 Alternatively, Insig1 may have DNL-independent effects on liver inflammation. Of note, the association between increased DNL and increased inflammation has been reported in other mouse models of fatty liver disease.49–51
Considering that TCF7L2 risk alleles are germline variants and globally expressed, we reasoned that the study of mice that either globally overexpress or are haploinsufficient for TCF7L2 is more relevant to human disease. Indeed, this approach allowed us to discover the TCF7L2 regulation of crosstalk between gut and liver in bile and lipid homeostasis, an effect that would have remained undiscovered using tissue-specific knockout models. In conclusion, our study identifies TCF4 as an important regulator of bile acid and an attractive target for the control of gut absorption of dietary lipids.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by 5R35HL135767 (NHLBI Outstanding Investigator Award) to A.M. and 5R35HL 135833 to H.N.G. We would like to thank Dr. Marcello A. Nobrega at the University of Chicago for providing us with TCF7L2-bac and heterozygous TCF7L2 knockout mice and Hans Clevers at the Hubrecht Institute in the Netherlands for providing us with TCF7L2 flox mice.
Funding information
NHLBI, Grant/Award Number: 5R35HL135767 and 5R35HL135833
Footnotes
DISCLOSURES
The authors declare no conflict of interest.
SUPPORTING INFORMATION
Additional supporting information may be found in the online version of the article at the publisher’s website.
DATA AVAILABILITY STATEMENT
No new data: Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
REFERENCES
- 1.Chalasani N, Younossi Z, Lavine JE, et al. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology. 2018;67:328–357. [DOI] [PubMed] [Google Scholar]
- 2.Inagaki T, Choi M, Moschetta A, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2:217–225. [DOI] [PubMed] [Google Scholar]
- 3.Schreuder TC, Marsman HA, Lenicek M, et al. The hepatic response to FGF19 is impaired in patients with nonalcoholic fatty liver disease and insulin resistance. Am J Physiol Gastrointest Liver Physiol. 2010;298:G440–G445. [DOI] [PubMed] [Google Scholar]
- 4.Jones S Mini-review: endocrine actions of fibroblast growth factor 19. Mol Pharm. 2008;5:42–48. [DOI] [PubMed] [Google Scholar]
- 5.Holt JA, Luo G, Billin AN, et al. Definition of a novel growth factor-dependent signal cascade for the suppression of bile acid biosynthesis. Genes Dev. 2003;17:1581–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wright TJ, Ladher R, McWhirter J, Murre C, Schoenwolf GC, Mansour SL. Mouse FGF15 is the ortholog of human and chick FGF19, but is not uniquely required for otic induction. Dev Biol. 2004;269:264–275. [DOI] [PubMed] [Google Scholar]
- 7.Wang JN, Li L, Li LY, Yan Q, Li J, Xu T. Emerging role and therapeutic implication of Wnt signaling pathways in liver fibrosis. Gene. 2018;674:57–69. [DOI] [PubMed] [Google Scholar]
- 8.Nishikawa K, Osawa Y, Kimura K. Wnt/beta-catenin signaling as a potential target for the treatment of liver cirrhosis using antifibrotic drugs. Int J Mol Sci. 2018;19(10):3103. doi: 10.3390/ijms19103103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang S, Song K, Srivastava R, et al. Nonalcoholic fatty liver disease induced by noncanonical Wnt and its rescue by Wnt3a. FASEB J. 2015;29(8):3436–3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grant SF, Thorleifsson G, Reynisdottir I, et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet. 2006;38:320–323. [DOI] [PubMed] [Google Scholar]
- 11.Hatzis P, van der Flier LG, van Driel MA, et al. Genome-wide pattern of TCF7L2/TCF4 chromatin occupancy in colorectal cancer cells. Mol Cell Biol. 2008;28:2732–2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huertas-Vazquez A, Plaisier C, Weissglas-Volkov D, et al. TCF7L2 is associated with high serum triacylglycerol and differentially expressed in adipose tissue in families with familial combined hyperlipidaemia. Diabetologia. 2008;51:62–69. [DOI] [PubMed] [Google Scholar]
- 13.O’Rourke KP, Ackerman S, Dow LE, Lowe SW. Isolation, culture, and maintenance of mouse intestinal stem cells. Bio Protoc. 2016;6(4):e1733. doi: 10.21769/bioprotoc.1733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhat N, Park J, Zoghbi HY, Arthur JS, Zaret KS. The chromatin modifier MSK1/2 suppresses endocrine cell fates during mouse pancreatic development. PLoS One. 2016;11:e0166703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aryal B, Singh AK, Zhang X, et al. Absence of ANGPTL4 in adipose tissue improves glucose tolerance and attenuates atherogenesis. JCI Insight. 2018;3(6):e97918. doi: 10.1172/jci.insight.97918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.el Marjou F, Janssen KP, Chang BH, et al. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis. 2004;39:186–193. [DOI] [PubMed] [Google Scholar]
- 17.Ye ZJ, Go GW, Singh R, Liu W, Keramati AR, Mani A. LRP6 protein regulates low density lipoprotein (LDL) receptor-mediated LDL uptake. J Biol Chem. 2012;287:1335–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang S, Song K, Srivastava R, et al. Nonalcoholic fatty liver disease induced by noncanonical Wnt and its rescue by Wnt3a. FASEB J. 2015;29:3436–3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Savic D, Ye H, Aneas I, Park SY, Bell GI, Nobrega MA. Alterations in TCF7L2 expression define its role as a key regulator of glucose metabolism. Genome Res. 2011;21:1417–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Potthoff MJ, Kliewer SA, Mangelsdorf DJ. Endocrine fibroblast growth factors 15/19 and 21: from feast to famine. Genes Dev. 2012;26:312–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kir S, Beddow SA, Samuel VT, et al. FGF19 as a postprandial, insulin-independent activator of hepatic protein and glycogen synthesis. Science. 2011;331:1621–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shin DJ, Osborne TF. FGF15/FGFR4 integrates growth factor signaling with hepatic bile acid metabolism and insulin action. J Biol Chem. 2009;284:11110–11120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alvarez-Sola G, Uriarte I, Latasa MU, et al. Fibroblast growth factor 15/19 (FGF15/19) protects from diet-induced hepatic steatosis: development of an FGF19-based chimeric molecule to promote fatty liver regeneration. Gut. 2017;66:1818–1828. [DOI] [PubMed] [Google Scholar]
- 24.Kim YC, Seok S, Zhang Y, et al. Intestinal FGF15/19 physiologically repress hepatic lipogenesis in the late fed-state by activating SHP and DNMT3A. Nat Commun. 2020;11:5969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bhatnagar S, Damron HA, Hillgartner FB. Fibroblast growth factor-19, a novel factor that inhibits hepatic fatty acid synthesis. J Biol Chem. 2009;284:10023–10033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brighton CA, Rievaj J, Kuhre RE, et al. Bile acids trigger GLP-1 release predominantly by accessing basolaterally located g protein-coupled bile acid receptors. Endocrinology. 2015;156:3961–3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen MM, Hale C, Stanislaus S, Xu J, Veniant MM. FGF21 acts as a negative regulator of bile acid synthesis. J Endocrinol. 2018;237:139–152. [DOI] [PubMed] [Google Scholar]
- 28.Geoghegan G, Simcox J, Seldin MM, et al. Targeted deletion of Tcf7l2 in adipocytes promotes adipocyte hypertrophy and impaired glucose metabolism. Mol Metab. 2019;24:44–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miyata M, Hata T, Yamakawa H, Kagawa T, Yoshinari K, Yamazoe Y. Involvement of multiple elements in FXR-mediated transcriptional activation of FGF19. J Steroid Biochem Mol Biol. 2012;132:41–47. [DOI] [PubMed] [Google Scholar]
- 30.Srivastava R, Rolyan H, Xie Y, et al. TCF7L2 (transcription factor 7-like 2) regulation of GATA6 (GATA-binding protein 6)-dependent and -independent vascular smooth muscle cell plasticity and intimal hyperplasia. Arterioscler Thromb Vasc Biol. 2019;39:250–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou M, Learned RM, Rossi SJ, DePaoli AM, Tian H, Ling L. Engineered FGF19 eliminates bile acid toxicity and lipotoxicity leading to resolution of steatohepatitis and fibrosis in mice. Hepatol Commun. 2017;1:1024–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Es JH, Haegebarth A, Kujala P, et al. A critical role for the Wnt effector Tcf4 in adult intestinal homeostatic self-renewal. Mol Cell Biol. 2012;32:1918–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Go GW, Srivastava R, Hernandez-Ono A, et al. The combined hyperlipidemia caused by impaired Wnt-LRP6 signaling is reversed by Wnt3a rescue. Cell Metab. 2014;19:209–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wojcik M, Janus D, Dolezal-Oltarzewska K, et al. A decrease in fasting FGF19 levels is associated with the development of non-alcoholic fatty liver disease in obese adolescents. J Pediatr Endocrinol Metab. 2012;25:1089–1093. [DOI] [PubMed] [Google Scholar]
- 35.Woollett LA, Wang Y, Buckley DD, et al. Micellar solubilisation of cholesterol is essential for absorption in humans. Gut. 2006;55:197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morgan RG, Borgstrom B. The mechanism of fat absorption in the bile fistula rat. Q J Exp Physiol Cogn Med Sci. 1969;54:228–243. [DOI] [PubMed] [Google Scholar]
- 37.Fon Tacer K, Bookout AL, Ding X, et al. Research resource: comprehensive expression atlas of the fibroblast growth factor system in adult mouse. Mol Endocrinol. 2010;24:2050–2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jung D, Inagaki T, Gerard RD, et al. FXR agonists and FGF15 reduce fecal bile acid excretion in a mouse model of bile acid malabsorption. J Lipid Res. 2007;48:2693–2700. [DOI] [PubMed] [Google Scholar]
- 39.Tomlinson E, Fu L, John L, et al. Transgenic mice expressing human fibroblast growth factor-19 display increased metabolic rate and decreased adiposity. Endocrinology. 2002;143:1741–1747. [DOI] [PubMed] [Google Scholar]
- 40.Fu L, John LM, Adams SH, et al. Fibroblast growth factor 19 increases metabolic rate and reverses dietary and leptin-deficient diabetes. Endocrinology. 2004;145:2594–2603. [DOI] [PubMed] [Google Scholar]
- 41.Appleby RN, Moghul I, Khan S, et al. Non-alcoholic fatty liver disease is associated with dysregulated bile acid synthesis and diarrhea: a prospective observational study. PLoS One. 2019;14:e0211348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bozadjieva N, Heppner KM, Seeley RJ. Targeting FXR and FGF19 to treat metabolic diseases-lessons learned from bariatric surgery. Diabetes. 2018;67:1720–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Song KH, Li T, Owsley E, Strom S, Chiang JY. Bile acids activate fibroblast growth factor 19 signaling in human hepatocytes to inhibit cholesterol 7alpha-hydroxylase gene expression. Hepatology. 2009;49:297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McCarthy MI, Rorsman P, Gloyn AL. TCF7L2 and diabetes: a tale of two tissues, and of two species. Cell Metab. 2013;17:157–159. [DOI] [PubMed] [Google Scholar]
- 45.da Silva Xavier G, Loder MK, McDonald A, et al. TCF7L2 regulates late events in insulin secretion from pancreatic islet beta-cells. Diabetes. 2009;58:894–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.da Silva Xavier G, Mondragon A, Sun G, et al. Abnormal glucose tolerance and insulin secretion in pancreas-specific Tcf7l2-null mice. Diabetologia. 2012;55:2667–2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boj SF, van Es JH, Huch M, et al. Diabetes risk gene and Wnt effector Tcf7l2/TCF4 controls hepatic response to perinatal and adult metabolic demand. Cell. 2012;151:1595–1607. [DOI] [PubMed] [Google Scholar]
- 48.Azzu V, Vacca M, Kamzolas I, et al. Suppression of insulin-induced gene 1 (INSIG1) function promotes hepatic lipid remodelling and restrains NASH progression. Mol Metab. 2021;48:101210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bhat N, Narayanan A, Fathzadeh M, et al. Dyrk1b promotes hepatic lipogenesis by bypassing canonical insulin signaling and directly activating mTORC2 in mice. J Clin Invest. 2021. e153724. [Epub ahead of print]. doi: 10.1172/JCI153724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ishimoto T, Lanaspa MA, Rivard CJ, et al. High-fat and high-sucrose (western) diet induces steatohepatitis that is dependent on fructokinase. Hepatology (Baltimore, MD). 2013;58:1632–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang ZH, Miyahara H, Takeo J, Katayama M. Diet high in fat and sucrose induces rapid onset of obesity-related metabolic syndrome partly through rapid response of genes involved in lipogenesis, insulin signalling and inflammation in mice. Diabetol Metab Syndr. 2012;4:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No new data: Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.