

Abstract
Objectives:
Castleman disease (CD) is a heterogeneous group of disorders involving systemic inflammation and lymphoproliferation. Recently, clonal mutations have been identified in unicentric CD (UCD) and idiopathic multicentric CD (iMCD), suggesting a potential underlying neoplastic process.
Methods:
Patients with UCD or iMCD with next generation sequencing (NGS) data on tissue DNA and/or circulating tumor DNA (ctDNA) were included.
Results:
Five patients were included, 4 with iMCD and 1 with UCD. Four patients (80%) were women; median age was 40 years. Three of five patients (60%) had ≥1 clonal mutation detected on biopsy among the genes included in the panel. One patient with iMCD had a 14q32-1p35 rearrangement and a der(1)dup(1)(q42q21)del(1) (q42) (1q21 being IL-6R locus) on karyotype. This patient also had a NF1 K2459fs alteration on ctDNA (0.3%). Another patient with iMCD had a KDM5C Q836* mutation, and one patient with UCD had a TNS3-ALK fusion but no ALK expression by immunohistochemistry.
Conclusions:
We report 4 novel somatic alterations found in patients with UCD or iMCD. The 1q21 locus contains IL-6R, and duplication of this locus may increase IL-6 expression. These findings suggest that a clonal process may be responsible for the inflammatory phenotype in some patients with UCD and iMCD.
Keywords: Castleman Disease, next generation sequencing, genomics
Introduction
Castleman disease (CD) represents a heterogeneous group of lymphoplasmacytic proliferative disorders with a wide range of clinical presentations. Patients can exhibit asymptomatic single station lymphadenopathy [unicentric Castleman disease (UCD)] or a life-threatening inflammatory syndrome with multi-system organ failure [multicentric Castleman disease (MCD)]. CD is anatomically classified based on the number of lymph nodes regions involved into UCD and MCD. MCD is further subclassified by etiology into human herpesvirus-8 (HHV-8) associated MCD (HHV8-MCD), idiopathic MCD (iMCD), and polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, skin changes (POEMS) syndrome associated MCD (POEMS-MCD)1,2.
The etiologies of HHV-8 and POEMS syndrome associated MCD are well established. HHV8-MCD often presents in immunocompromised individuals, most commonly in patients with human immunodeficiency virus (HIV) infection, resulting in uncontrolled HHV-8 replication and production of virally encoded interleukin-6 (vIL-6). Overproduction of vIL-6 initiates a cascade of events resulting in severe systemic inflammation and ultimately organ failure3. Treatment of HHV8-MCD with rituximab-based regimens has resulted in improved outcomes4. In POEMS-MCD, an underlying clonal plasma cell population leads to excessive vascular endothelial growth factor (VEGF) and interleukin-12 (IL-12) production resulting in organomegaly, peripheral neuropathy, and skin disease5. Treatment is directed towards the underlying plasma cell clone.
In contrast to HHV8-MCD and POEMS-MCD, the etiology of UCD and the trigger for excess IL-6 and subsequent inflammatory syndrome in iMCD remains unknown. UCD was thought to be a benign reactive proliferation; however, recent data suggest a clonal process originating from the stromal cells within the lymph node6. Removal of the affected lymph node is typically curative, consistent with a localized etiology. Three processes have been proposed as potential disease drivers in iMCD: infection with a virus other than HHV-8, systemic inflammatory disease mechanisms via autoantibodies or inflammatory germline gene mutations, or a paraneoplastic process from a population of clonal cells7. An underlying virus other than HHV-8 seems unlikely based on the results of recent data8, and the role of autoantibodies is currently under investigation. Regardless of the trigger, inflammation, often mediated by IL-6, is directly implicated in all cases of iMCD9. The anti-IL-6 monoclonal antibody siltuximab has revolutionized the treatment of iMCD10–16. However, approximately 50% of patients will fail or lose their response to siltuximab. Therefore, it is important to elucidate the underlying pathogenesis of iMCD to help develop novel therapies.
Recently, somatic clonal mutations of UCD and iMCD have been reported17–30. Here, we describe two cases of iMCD and one case of UCD with novel chromosomal structural abnormalities and point mutations.
Materials and Methods
Patients
All patients who were diagnosed with UCD and iMCD at University of California San Diego (UCSD) were retrospectively reviewed. The pathology slides or pathology reports (if pathology slides were not available) were re-reviewed by a hematopathologist (HYW) and the diagnostic criteria of UCD and iMCD were re-reviewed (AMG and HYW) to confirm the diagnosis according to the Castleman Disease Collaborative Network (CDCN) criteria outlined by Fajgenbaum et al.31 (Supplemental Table 1). Patients who had comprehensive genomic studies were included in the final analysis. The study was carried out under the PREDICT study NCT02478931 approved by the institutional review board and any investigational studies administered for which the patients gave consent.
Immunohistochemistry
Immunohistochemistry (IHC) using formalin-fixed paraffin-embedded (FFPE) tissue blocks was carried out using Ventana BenchMark Special Satins platform (Roche).
Cytogenetics
Fresh tissue, if available at the time of excisional biopsy, was used for karyotyping using Giemsa banding method. Twenty metaphase cells were analyzed according to standard protocol.
Genomic sequencing
Genomic sequencing by next generation sequencing (NGS) was performed either by FoundationOneHeme panel of genes (FoundationMedicine)32 or UCSD comprehensive panel of genes. For FoundationOneHeme panel, briefly stated, deoxyribonucleic acid (DNA) was extracted from FFPE specimens and NGS was performed on selected exons of 406 cancer-related genes to evaluate for single nucleotide variants (SNVs), short insertions and deletions (indels), copy number alterations (CNAs), gene fusions, and rearrangements. For UCSD hematolymphoid panel of genes, solution-based hybrid capture was used on extracted DNA from FFPE specimens to select mutational hotspot regions for a selected panel of 397 genes. The DNA library was then sequenced using sequencing-by-synthesis technology. Gene fusions, rearrangements, SNV, indels, and CNAs were identified and if necessary, clinically significant variants were re-sequenced using Sanger technique.
Cell free DNA (cfDNA) analysis from peripheral blood was performed using Guardant360 assay (Guardant Health)33. Briefly, at least 5.0 ng of DNA was extracted from peripheral blood and oligonucleotide barcoding of each DNA strand was performed. Selected exons from 68 or 73 cancer-related genes were sequenced with HiSeq2500 (Illumina, USA), evaluating for SNVs, fusions, indels, and CNAs.
Results
Demographic and clinical data
A total of 8 patients with UCD and 6 patients with iMCD were identified. On further review, 1 patient considered to have iMCD did not meet the formal diagnostic criteria and was excluded. Among the 13 patients, 1 patient with UCD and 4 patients with iMCD had NGS performed on biopsy sample and were included for further analysis (Supplemental Figure 1).
Characteristics of the five patients are summarized in Table 1. Diagnostic studies performed are summarized in Supplemental Table 2. Four of five patients (80%) were women; median age was 40 years. The patient with UCD had resection of the lymph node and has no evidence of recurrent disease at 3.3 years of follow up. Three of the four patients with iMCD received siltuximab: one patient is maintained on siltuximab at 2 years of follow up with partial response, one patient discontinued siltuximab due to unknown reasons and is currently on sirolimus, and one patient required change of therapy due to progression of disease, currently in remission after rituximab based therapy. One patient with iMCD is on active surveillance with no therapy at all.
Table 1.
Patient characteristics
| Patient | Age (years)1/Sex | Diagnosis2 | Histology | Monotypia by flow cytometry / monoclonality of B-cells | Treatment | OS (years)3 | Tissue4 | Genomics5 | TMB (mut/mb) | cfDNA6 | Comment |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 29/F | iMCD | Hyaline vascular | Polytypic / ND | Siltuximab Sirolimus Anakinra |
2.7+ | LN | No alterations | 3 | No alterations | |
| 2 | 30/F | iMCD | Plasma cell | ND | Siltuximab Rituximab + chemotherapy Rituximab monotherapy |
4.5+ | LN | No alterations | N/A | No alterations | |
| 3 | 58/F | iMCD | Plasma cell | Polytypic / polyclonal | Siltuximab | 1.9+ | LN | NGS: 14q32–1p35 rearrangement Karyotype: der(1)dup(1)(q42q21)del(1)(q42) |
N/A | NF1 K2459fs (0.3%) | IL-6 R resides on 1q21 |
| 4 | 40/M | iMCD | Plasma cell | ND | Observation | 6.5+ | Skin | NGS: KDM5C Q836 | N/A | No alterations | |
| 5 | 40/F | UCD | Hyaline vascular | Polytypic / ND | Resection | 3.3+ | LN | NGS: TNS3-ALK fusion | 1 | ND | TNS3-ALK fusion would be predicted to inactivate ALK |
Abbreviations: ALK: Anaplastic lymphoma kinase; cfDNA: cell free DNA; F: female; fs: frame shift; iMCD: idiopathic multicentric Castleman disease; ND: not done; LN: lymph node; M: male; mb: megabase pair; mut: mutation; N/A: not available; ND: not done; NF1: neurofibromin 1; NGS: next generation sequencing; OS: overall survival; TNS3: Tensin3; TMB: tumor mutation burden; UCD: unicentric Castleman disease
Age at diagnosis
Cases of iMCD had to meet the diagnostic criteria outlined by D. Fajgenbaum et al.31
From diagnosis
Tissue used for next generation sequencing.
Patients 1,2,4, and 5 underwent sequencing using the Foundation One Heme panel. Patient 3 underwent sequencing using the University of California San Diego Comprehensive NGS mutation panel analysis.
Patients 1,2,3, and 4 underwent cfDNA analysis using the Guardant360 assay.
Genomic alterations
Three of the 5 patients (60%) demonstrated chromosomal and/or genomic clonal alterations at levels of either karyotype and/or point mutations by NGS (Table 1). Patient #3 had duplication of 1q at 1q42q21 and deletion of 1q42 locus on karyotype (Figure 1). By NGS, there was 14q32-1p35 reciprocal rearrangement. Of note, immunoglobulin heavy (IgH) chain gene resides on 14q32 locus; however, there is no monoclonal rearrangement of IgH by polymerase chain reaction (PCR) in this patient. Patient #3 also had neurofibromin 1 (NF1) K2459fs identified in 0.3% of cfDNA but not in lymph node tissue. Patient #4 had KDM5C Q836 mutation and patient #5 with UCD had Tensin3 (TNS3)-Anaplastic lymphoma kinase (ALK) fusion identified on NGS (Figure 1). There is no ALK expression by IHC with appropriate positive control, and there were no monoclonal T-cell populations by flow cytometry. In addition, there was no monoclonal rearrangement of T-cell receptor gamma gene (TRG) in this patient.
Figure 1.
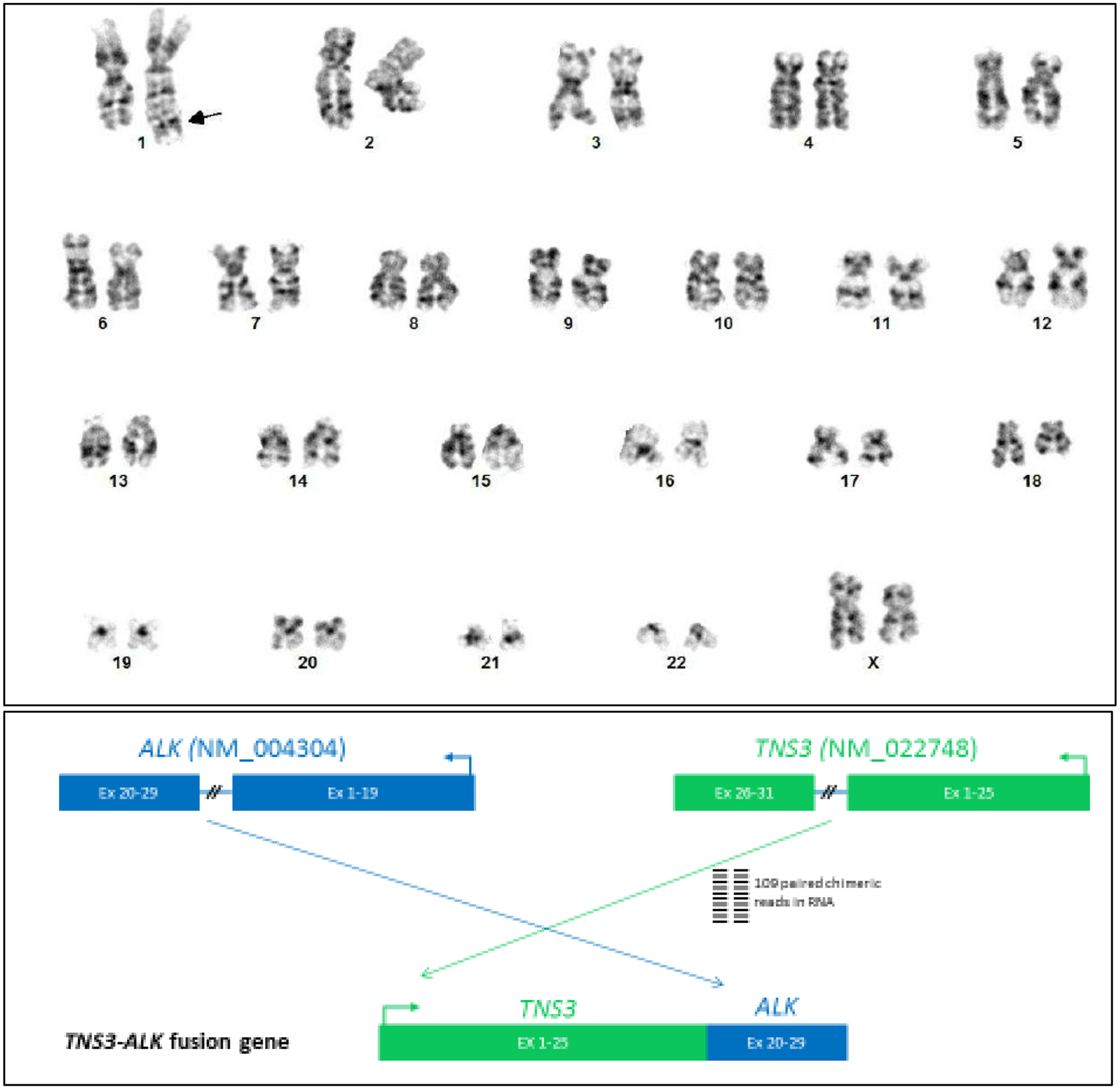
(Top panel) Chromosomal alterations from patient #3 by karyotype. Among 14 metaphase cells from unstimulated cell culture by G-banding, two cells showed a derivative chromosome 1 with inverted duplication of segment 1q21-q42 and suspected deletion 1q42 (arrow).
Case presentation
Patient #3 had particularly notable clinical and genomic findings. She is a 58-year-old female with history of bronchiectasis, violaceous skin lesions, polyclonal hypergammaglobulinemia, and thrombocytosis who was referred to our center for evaluation of iMCD. She did not have any personal or family history of blood disorders. She had chronic cough for 15 years and was diagnosed with bronchiectasis six years prior. She also reported painless, non-pruritic, hyperpigmented patches of the torso and groin that developed 13 years ago. On physical exam, she had diffuse, 1–2 cm violaceous patches over her torso and extremities (Figure 2). Laboratory evaluation demonstrated platelets of 578,000 /mm3, immunoglobulin G (IgG) of 2900mg/dL (reference range: 700–1600mg/dL), polyclonal hypergammaglobulinemia on serum protein electrophoresis (SPEP), and IL-6 level of 20pg/mL (reference range: ≤ 5pg/mL). Extensive infectious and rheumatologic evaluations previously performed were unrevealing. Positron emission tomography/computed tomography (PET/CT) scan showed bilateral axillary, mediastinal, retroperitoneal, and iliac lymphadenopathy. Biopsy of the cervical lymph node and skin lesion demonstrated polytypic interfollicular plasmacytosis with follicular hyperplasia, felt to be consistent with iMCD. IHC for HHV-8 was negative. Karyotype of lymph node tissue revealed duplication of 1q42q21, a locus which contains IL-6R, and deletion of 1q42 (Figure 1), and NGS revealed 14q32-1p35 rearrangement. NF1 K2459fs mutation was identified in 0.3% of the cfDNA but not in the lymph node tissue. She was diagnosed with iMCD and was started on siltuximab 11mg/kg every three weeks. Her cough improved, thrombocytosis resolved, and skin lesions decreased. Subsequent PET/CT demonstrated near resolution of her lymphadenopathy. She continues to receive siltuximab with excellent disease control at 2-year follow up.
Figure 2.
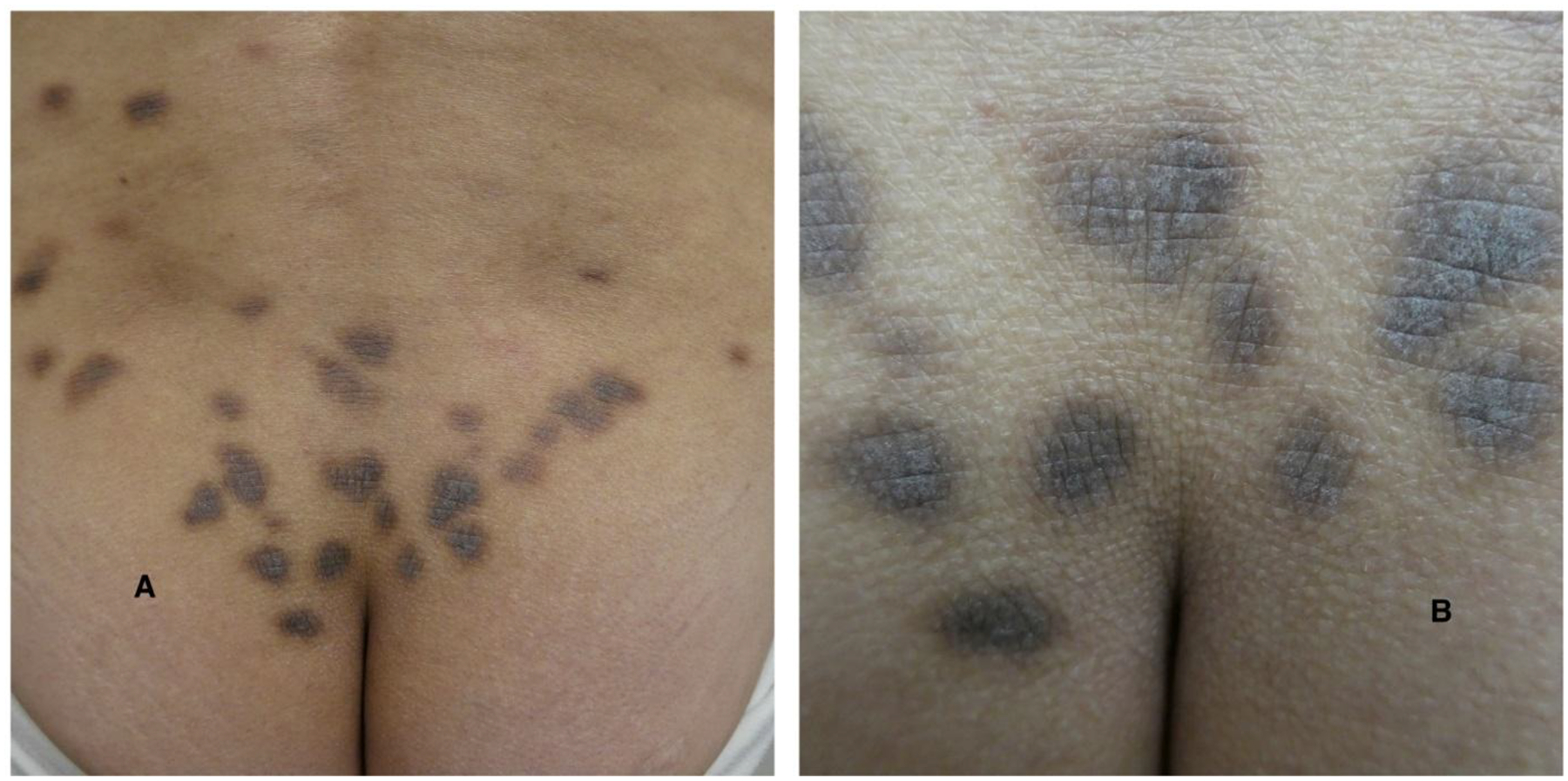
A distant (A) and close (B) view of the gluteal cutaneous lesions of Castleman disease presenting as infiltrative hyperpigmented dermal plaques.
Discussion
MCD includes a group of diseases with systemic inflammatory phenotypes, ranging from mild symptoms to life-threatening multi-organ failure. Some subtypes of MCD have defined etiologies such as HHV-8 infection in HHV8-MCD or a neoplastic plasma cell clone in POEMS-MCD. However, the driver of increased IL-6 production and subsequent cytokine storm in iMCD has not been defined. Similarly, the inciting event for UCD and its relationship to MCD is unknown. Recently, an association between a germline mutation of Mediterranean Fever (MEFV) gene, a gene implicated in familial Mediterranean fever , and iMCD was reported21. However, it is unclear if this case truly represents iMCD or familial Mediterranean fever due to the clinical overlap between these diseases. Similarly, a germline mutation in FAS, a gene implicated in autoimmune lymphoproliferative syndrome (ALPS), was recently reported in an iMCD patient and his father with UCD, but these patients may be more appropriately considered to have ALPS17. Thus far, a clear genomic alteration causing iMCD has not been found. In addition, the pathogen hypothesis has proven to be unlikely. A recent study analyzing fresh frozen lymph node samples from patients with UCD and iMCD by VirCapSeq, a high-throughput RNA sequencing approach, revealed that HHV-8 was not detected in any of the samples. In addition, no other acute viral infections were identified as likely causing UCD or iMCD8.
The paraneoplastic hypothesis is gaining attention as increasing numbers of clonal alterations have been reported in patients with UCD and iMCD, where the underlying clonal neoplastic process could potentially lead to lymph node findings characteristic of CD and increased IL-6 in iMCD (Supplemental Table 3). Notably, Li et al.6 reported platelet-derived growth factor receptor β (PDGFRB) Asn666Ser mutations in 17% (7/41) patients with UCD using whole exome sequencing (WES). These authors further demonstrated that the PDFGRB Asn666Ser mutations were localized to non-hematopoietic stromal cells. It is hypothesized that a clonal population of stromal cells may lead to increased cytokine production at least locally; however, this hypothesis has yet to be proven. In our UCD patient (#5) who underwent NGS, a PDGFRB alteration was not detected, but a novel ALK gene fusion was found.
In our cohort, two somatic alterations in iMCD were identified that have not been previously reported. Immunophenotyping studies using flow cytometry found that there were no monotypic B-cells or aberrant T-cell populations. Molecular studies using PCR have shown that there were no monoclonal rearrangements for IgH/IgK and TRG.
Patient #3 demonstrated very interesting findings. She was found to have dup(1q42q21), a locus which contains IL-6R. Copy number variations of this locus have been shown to increase expression of IL-6 and IL-6R34, which may explain the increased IL-6 seen in our patient and her excellent response to siltuximab. Similarly, a report by Nakamura et al.24 demonstrated a somatic mutation in the IL-6 locus in a patient with iMCD. In their report, the affected lymph node was found to have t(7;14)(p22;q22) by karyotype. The patient also had an elevated IL-6 level. The locus 7p21-22 contains IL6, thus the translocation could potentially lead to increased production of IL-6. Yoshimi et al.29 also reported a mutation in MEK2 P128L, which has been shown to be associated with hyperactivated MAP kinase signaling and increased proliferation. This data suggests that some cases of iMCD may be the result of a clonal process resulting in increased cytokine production including IL-6 or overexpression of IL-6R. However, this remains to be proven. Besides dup(1q42q21) and del(1q42), patient #3 also harbored 14q32-1p35 rearrangement. The 14q32 locus contains many genes besides IgH, and the genes from 1p35 is unknown. The significance of this rearrangement is unclear because there was no monoclonal rearrangement of IgH, thus the gene involved from 14q32 locus is probably not IgH. This patient was also found to have NF1 K2459fs mutation on cfDNA assay but not in tissue. NF1 is a tumor suppressor gene involved in the Rat sarcoma (RAS)/mitogen-activated protein kinase (MAPK) pathway.35 Somatic mutations of NF1 are found in various cancers such as melanoma, lung adenocarcinoma, and leukemia35. It is unclear how the frame shift mutation affects protein function. Since the NF1 mutation was not detected in NGS of the lymph node, its association with CD remains unclear. It may represent a technical artifact or early presentation of an undiagnosed malignancy.
Patient #4 was found to have a somatic mutation in KDM5C (Q836), agene encoding histone lysine demethylase that controls gene expression by histone modification36. Somatic mutations are linked to clear cell renal cell carcinoma and prostate adenocarcinoma37,38. The significance of this mutation in iMCD is unclear, as currently the KDM5C gene does not have any known association with inflammatory conditions.
Patient #5 harbored a novel chromosomal translocation t(2;7)(p23;p12) involving ALK from 2p23 and TNS3 from 7p12. TNS3 was reported as a thyroid-specific gene39. Fusions and less frequently mutations of ALK1 have long been known to be oncogenic and are seen in an increasing number of hematolymphoid and solid malignancies including lymphoma of T- and B-cell lineages, carcinoma, and neuroblastoma41. While the role of the ALK gene rearrangement in the pathogenesis of this UCD case remains to be elucidated, it is clear that the ALK in the TNS3-ALK fusion protein is not functional based on both the genomic structure (only exons 20–29 of ALK were fused with TNS3) and functional assay (protein expression) as revealed by IHC.
There are several other reports that have described clonal alterations in patients with UCD and iMCD. Patel et al.25 reported a mutation in JAK1 V310I in a patient with a condition demonstrating CD-like features in skin lesions only and no lymphadenopathy, which can sometimes be referred to as cutaneous CD. Treatment with siltuximab resulted in a complete response ongoing at seven years16. Interestingly, the patient’s pretreatment serum IL-6 level was normal. JAK1 is a crucial signaling component of the IL-6/IL-6R /gp130 machinery. JAK1 V310I may induce a conformation change with functional activation effect leading to enhanced sensitivity to the IL-6 ligand.
There are limitations in our study including small number of cases and lack of functional studies of genomic alternations. Furthermore, the panel only evaluated a select number of genes so genes not included in the panel may have mutations. Conversely, mutations present in genes included in the panel may not have been detected due to mutation abundance below the sensitivity of the assay.
Conclusion
In summary, we describe several novel gene mutations and chromosomal abnormalities from 2 iMCD and 1 UCD cases by karyotyping and NGS. Our new findings, in addition to the previously reported gene mutations, will advance the understanding of the pathogenesis of CD. As first-line treatment with siltuximab is only effective in approximately half of iMCD patients10,11, further genomic interrogation is warranted as a basis of identifying new therapeutic targets.
Supplementary Material
What is the NEW aspect of your work?
This manuscript describes novel somatic mutations found in Castleman disease, suggesting potential clonal and neoplastic etiology.
What is the CENTRAL finding of your work?
Three of the five patients with iMCD or UCD, who had next generation sequencing and cytogenetics demonstrated clonal mutations (14q32-1p35 rearrangement and a der(1)dup(1)(q42q21)del(1)(q42), KDM5C Q836* mutation, and TNS3-ALK fusion).
What is (or could be) the SPECIFIC clinical relevance of your work?
As the etiology and pathogenesis of unicentric and idiopathic multicentric Castleman disease remains unclear, these findings advance our understanding in this subject that potentially a clonal and neoplastic process triggers the development of Castleman disease.
ACKNOWLEDGEMENTS
Doctor Bob and The Shillman Foundation for their grant which made this work possible.
Funding:
This study was funded in part by the Joan and Irwin Jacobs Research Fund and by the National Institutes of Health grant P30 CA023100 (RK)
COI:
Dr. Goodman receives speaking and consulting fees from Seattle Genetics and consulting fees from EUSA Pharma. Dr. Sokol is an employee of Foundation Medicine. Dr. Kurzrock receives research funding from Genentech, Merck Serono, Pfizer, Boehringer Ingelheim, TopAlliance, Takeda, Incyte, Debiopharm, Medimmune, Sequenom, Foundation Medicine, Konica Minolta, Grifols, Omniseq, and Guardant, as well as consultant and/or speaker fees and/or advisory board for X-Biotech, Neomed, Pfizer, Actuate Therapeutics, and Roche, has an equity interest in IDbyDNA and CureMatch Inc, serves on the Board of CureMatch and CureMetrix, and is a co-founder of CureMatch. Dr. Fajgenbaum receives research funding from EUSA Pharma and has a provisional patent pending related to JAK1/2 inhibition in iMCD.
CONFLICT OF INTEREST
AMG receives speaking and consulting fees from Seattle Genetics and consulting fees from EUSA Pharma. ESS is an employee of Foundation Medicine and a shareholder of Roche. PRC is a con-sultant for ParaPRO. DCF receives research funding from EUSA Pharma and has a provisional patent pending related to JAK1/2 inhibition in iMCD. RK receives research funding from Genentech, Merck Serono, Pfizer, Boehringer Ingelheim, TopAlliance, Takeda, Incyte, Debiopharm, Medimmune, Sequenom, Foundation Medicine, Konica Minolta, Grifols, Omniseq, and Guardant, as well as con-sultant and/or speaker fees and/or advisory board for X-Biotech, Neomed, Pfizer, Actuate Therapeutics, and Roche, has an equity interest in IDbyDNA and CureMatch Inc, serves on the Board of CureMatch and CureMetrix, and is a co-founder of CureMatch.
DATA AVAILABILITY STATEMENT
The data that supports the findings of this study are available in the supplementary material of this article.
References
- 1.Fajgenbaum DC, Shilling D. Castleman Disease Pathogenesis. Hematol Oncol Clin North Am. 2018;32(1):11–21. [DOI] [PubMed] [Google Scholar]
- 2.El-Osta HE, Kurzrock R. Castleman’s disease: from basic mechanisms to molecular therapeutics. Oncologist. 2011;16(4):497–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oksenhendler E, Carcelain G, Aoki Y, et al. High levels of human herpesvirus 8 viral load, human interleukin-6, interleukin-10, and C reactive protein correlate with exacerbation of multicentric castleman disease in HIV-infected patients. Blood. 2000;96(6):2069–2073. [PubMed] [Google Scholar]
- 4.Pria AD, Pinato D, Roe J, Naresh K, Nelson M, Bower M. Relapse of HHV8-positive multicentric Castleman disease following rituximab-based therapy in HIV-positive patients. Blood. 2017;129(15):2143–2147. [DOI] [PubMed] [Google Scholar]
- 5.Kanai K, Sawai S, Sogawa K, et al. Markedly upregulated serum interleukin-12 as a novel biomarker in POEMS syndrome. Neurology. 2012;79(6):575–582. [DOI] [PubMed] [Google Scholar]
- 6.Li Z, Lan X, Li C, et al. Recurrent PDGFRB mutations in unicentric Castleman disease. Leukemia. 2019;33(4):1035–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fajgenbaum DC, van Rhee F, Nabel CS. HHV-8-negative, idiopathic multicentric Castleman disease: novel insights into biology, pathogenesis, and therapy. Blood. 2014;123(19):2924–2933. [DOI] [PubMed] [Google Scholar]
- 8.Nabel CS, Sameroff S, Shilling D, et al. Virome capture sequencing does not identify active viral infection in unicentric and idiopathic multicentric Castleman disease. PLoS One. 2019;14(6):e0218660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yoshizaki K, Matsuda T, Nishimoto N, et al. Pathogenic significance of interleukin-6 (IL-6/BSF-2) in Castleman’s disease. Blood. 1989;74(4):1360–1367. [PubMed] [Google Scholar]
- 10.Kurzrock R, Voorhees PM, Casper C, et al. A phase I, open-label study of siltuximab, an anti-IL-6 monoclonal antibody, in patients with B-cell non-Hodgkin lymphoma, multiple myeloma, or Castleman disease. Clin Cancer Res. 2013;19(13):3659–3670. [DOI] [PubMed] [Google Scholar]
- 11.van Rhee F, Wong RS, Munshi N, et al. Siltuximab for multicentric Castleman’s disease: a randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2014;15(9):966–974. [DOI] [PubMed] [Google Scholar]
- 12.van Rhee F, Casper C, Voorhees PM, et al. Long-term safety of siltuximab in patients with idiopathic multicentric Castleman disease: a prespecified, open-label, extension analysis of two trials. Lancet Haematol. 2020;7(3):e209–e217. [DOI] [PubMed] [Google Scholar]
- 13.Fajgenbaum DC, Kurzrock R. Siltuximab: a targeted therapy for idiopathic multicentric Castleman disease. Immunotherapy. 2016;8(1):17–26. [DOI] [PubMed] [Google Scholar]
- 14.van Rhee F, Casper C, Voorhees PM, et al. A phase 2, open-label, multicenter study of the long-term safety of siltuximab (an anti-interleukin-6 monoclonal antibody) in patients with multicentric Castleman disease. Oncotarget. 2015;6(30):30408–30419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Rhee F, Fayad L, Voorhees P, et al. Siltuximab, a novel anti-interleukin-6 monoclonal antibody, for Castleman’s disease. J Clin Oncol. 2010;28(23):3701–3708. [DOI] [PubMed] [Google Scholar]
- 16.Ahmed B, Tschen JA, Cohen PR, et al. Cutaneous castleman’s disease responds to anti interleukin-6 treatment. Mol Cancer Ther. 2007;6(9):2386–2390. [DOI] [PubMed] [Google Scholar]
- 17.Baker TS, Gambino KJ, Schriefer L, et al. A novel FAS mutation with variable expressivity in a family with unicentric and idiopathic multicentric Castleman disease. Blood Adv. 2018;2(21):2959–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang KC, Wang YC, Hung LY, et al. Monoclonality and cytogenetic abnormalities in hyaline vascular Castleman disease. Mod Pathol. 2014;27(6):823–831. [DOI] [PubMed] [Google Scholar]
- 19.Chen WC, Jones D, Ho CL, et al. Cytogenetic anomalies in hyaline vascular Castleman disease: report of two cases with reappraisal of histogenesis. Cancer Genet Cytogenet. 2006;164(2):110–117. [DOI] [PubMed] [Google Scholar]
- 20.Cokelaere K, Debiec-Rychter M, De Wolf-Peeters C, Hagemeijer A, Sciot R. Hyaline vascular Castleman’s disease with HMGIC rearrangement in follicular dendritic cells: molecular evidence of mesenchymal tumorigenesis. Am J Surg Pathol. 2002;26(5):662–669. [DOI] [PubMed] [Google Scholar]
- 21.Koné-Paut I, Hentgen V, Guillaume-Czitrom S, Compeyrot-Lacassagne S, Tran TA, Touitou I. The clinical spectrum of 94 patients carrying a single mutated MEFV allele. Rheumatology (Oxford). 2009;48(7):840–842. [DOI] [PubMed] [Google Scholar]
- 22.Legras A, Tallet A, Didelot A, et al. Clinical and molecular characteristics of unicentric mediastinal Castleman disease. J Thorac Dis. 2018;10(4):2079–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagy A, Bhaduri A, Shahmarvand N, et al. Next-generation sequencing of idiopathic multicentric and unicentric Castleman disease and follicular dendritic cell sarcomas. Blood Adv. 2018;2(5):481–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakamura H, Nakaseko C, Ishii A, et al. [Chromosomal abnormalities in Castleman’s disease with high levels of serum interleukin-6]. Rinsho Ketsueki. 1993;34(2):212–217. [PubMed] [Google Scholar]
- 25.Patel M, Ikeda S, Pilat SR, Kurzrock R. JAK1 Genomic Alteration Associated With Exceptional Response to Siltuximab in Cutaneous Castleman Disease. JAMA Dermatol. 2017;153(5):449–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pauwels P, Dal Cin P, Vlasveld LT, Aleva RM, van Erp WF, Jones D. A chromosomal abnormality in hyaline vascular Castleman’s disease: evidence for clonal proliferation of dysplastic stromal cells. Am J Surg Pathol. 2000;24(6):882–888. [DOI] [PubMed] [Google Scholar]
- 27.Reichard KK, Robinett S, Foucar MK. Clonal cytogenetic abnormalities in the plasma cell variant of Castleman disease. Cancer Genet. 2011;204(6):323–327. [DOI] [PubMed] [Google Scholar]
- 28.Stone K, Woods E, Szmania SM, et al. Interleukin-6 receptor polymorphism is prevalent in HIV-negative Castleman Disease and is associated with increased soluble interleukin-6 receptor levels. PLoS One. 2013;8(1):e54610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yoshimi A, Trippett TM, Zhang N, et al. Genetic basis for iMCD-TAFRO. Oncogene. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.You L, Lin Q, Zhao J, Shi F, Young KH, Qian W. Whole-exome sequencing identifies novel somatic alterations associated with outcomes in idiopathic multicentric Castleman disease. Br J Haematol. 2020;188(5):e64–e67. [DOI] [PubMed] [Google Scholar]
- 31.Fajgenbaum DC, Uldrick TS, Bagg A, et al. International, evidence-based consensus diagnostic criteria for HHV-8-negative/idiopathic multicentric Castleman disease. Blood. 2017;129(12):1646–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frampton GM, Fichtenholtz A, Otto GA, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31(11):1023–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lanman RB, Mortimer SA, Zill OA, et al. Analytical and Clinical Validation of a Digital Sequencing Panel for Quantitative, Highly Accurate Evaluation of Cell-Free Circulating Tumor DNA. PLoS One. 2015;10(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rafiq S, Frayling TM, Murray A, et al. A common variant of the interleukin 6 receptor (IL-6r) gene increases IL-6r and IL-6 levels, without other inflammatory effects. Genes Immun. 2007;8(7):552–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Philpott C, Tovell H, Frayling IM, Cooper DN, Upadhyaya M. The NF1 somatic mutational landscape in sporadic human cancers. Hum Genomics. 2017;11(1):13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tumber A, Nuzzi A, Hookway ES, et al. Potent and Selective KDM5 Inhibitor Stops Cellular Demethylation of H3K4me3 at Transcription Start Sites and Proliferation of MM1S Myeloma Cells. Cell Chem Biol. 2017;24(3):371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Cubas AA, Rathmell WK. Epigenetic modifiers: activities in renal cell carcinoma. Nat Rev Urol. 2018;15(10):599–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goncalves TF, Goncalves AP, Fintelman Rodrigues N, dos Santos JM, Pimentel MM, Santos-Reboucas CB. KDM5C mutational screening among males with intellectual disability suggestive of X-Linked inheritance and review of the literature. Eur J Med Genet. 2014;57(4):138–144. [DOI] [PubMed] [Google Scholar]
- 39.Maeda I, Takano T, Yoshida H, Matsuzuka F, Amino N, Miyauchi A. Tensin3 is a novel thyroid-specific gene. J Mol Endocrinol. 2006;36(1):R1–8. [DOI] [PubMed] [Google Scholar]
- 40.Borsani G, Piovani G, Zoppi N, et al. Cytogenetic and molecular characterization of a de-novo t(2p;7p) translocation involving TNS3 and EXOC6B genes in a boy with a complex syndromic phenotype. Eur J Med Genet. 2008;51(4):292–302. [DOI] [PubMed] [Google Scholar]
- 41.Minoo P, Wang HY. ALK-immunoreactive neoplasms. Int J Clin Exp Pathol. 2012;5(5):397–410. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that supports the findings of this study are available in the supplementary material of this article.