

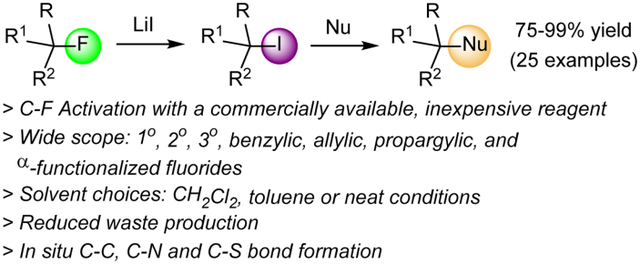
Abstract
A highly efficient method for C-F bond functionalization of a broad variety of activated and unactivated aliphatic substrates with inexpensive lithium iodide is presented. Primary, secondary, tertiary, benzylic, propargylic and α-functionalized alkyl fluorides react in chlorinated or aromatic solvents at room temperature or upon heating to the corresponding iodides which are isolated in 91–99% yield. The reaction is selective for aliphatic monofluorides and can be coupled with in situ nucleophilic iodide replacements to install carbon-carbon, carbon-nitrogen and carbon-sulfur bonds with high yields. Alkyl difluorides, trifluorides, even in activated benzylic positions, are inert under the same conditions and aryl fluoride bonds are also tolerated.
Keywords: C-F bond functionalization, alkyl fluorides, halide exchange, nucleophilic substitution, lithium iodide
Graphical Abstract
Fluorinated compounds have found numerous applications, in particular in the health and materials sciences.1 Their outstanding chemical properties and general usefulness have led to a steady demand for increasingly diverse organofluorines and consequently nurtured the development of synthetic methods that incorporate fluorine into a wide range of organic scaffolds. As a result, chemists now have access to a large pool of commercially available or easily prepared fluorinated building blocks. This relatively new scenario creates unprecedented synthetic opportunities in which the carbon-fluoride bond, considered thermodynamically stable and chemically inert for a long time, has become the prime target for selective functionalization. Although initial progress with synthetically useful carbon-fluoride bond cleavage was mostly confined to reactions with aryl and alkenyl substrates,2,3 intriguing methods that exploit allylic, propargylic or benzylic Csp3-F bonds have emerged.4–12 In addition, several laboratories including ours have been able to demonstrate carbon-fluoride bond functionalization with unactivated aliphatic substrates although the need for harsh conditions, low functional group tolerance and competing side reactions continue to limit the general scope in some cases.13–31
Having established a profound interest in organofluorine chemistry,32–40 including C-F bond activation methodology and synthetic or chiroptical sensing applications thereof,19,21,41–43 we decided to investigate the possibility of practical alkyl fluoride to iodide conversion which would also enable subsequent nucleophilic displacements or other reactions. Hilmersson and coworkers showed that this is possible with YbI3(THF)3 which has to be prepared from expensive ytterbium(II) iodide, Scheme 1.16,17 Inspired by their work and based on our recent finding that LiI facilitates Suzuki cross-couplings,21,44 we hypothesized that carbon-fluoride to carbon-iodide transformation might be generally possible with lithium iodide, which would exploit the formation of thermodynamically stable, nontoxic LiF as a driving force to energetically compensate for the cleavage of the strong C-F bond. In addition, LiI is significantly cheaper and generates less waste than ytterbium iodide due to its small molecular weight. We now report that LiI can be used to convert benzylic and propargylic as well as unactivated primary, secondary and tertiary alkyl fluorides to the corresponding iodides in high yields that typically exceed 90%. Moreover, we show that various functionalities including neighboring electron-withdrawing moieties such as ketone, ester and nitrile groups, for example in ethyl fluoroacetate and fluoroacetophenone, are tolerated. The Csp3-F bond activation and functionalization can be accomplished in chlorinated solvents, toluene or under solvent-free conditions, and coupled with in situ nucleophilic displacements to form carbon-carbon and carbon-heteroatom bonds.
Scheme 1.
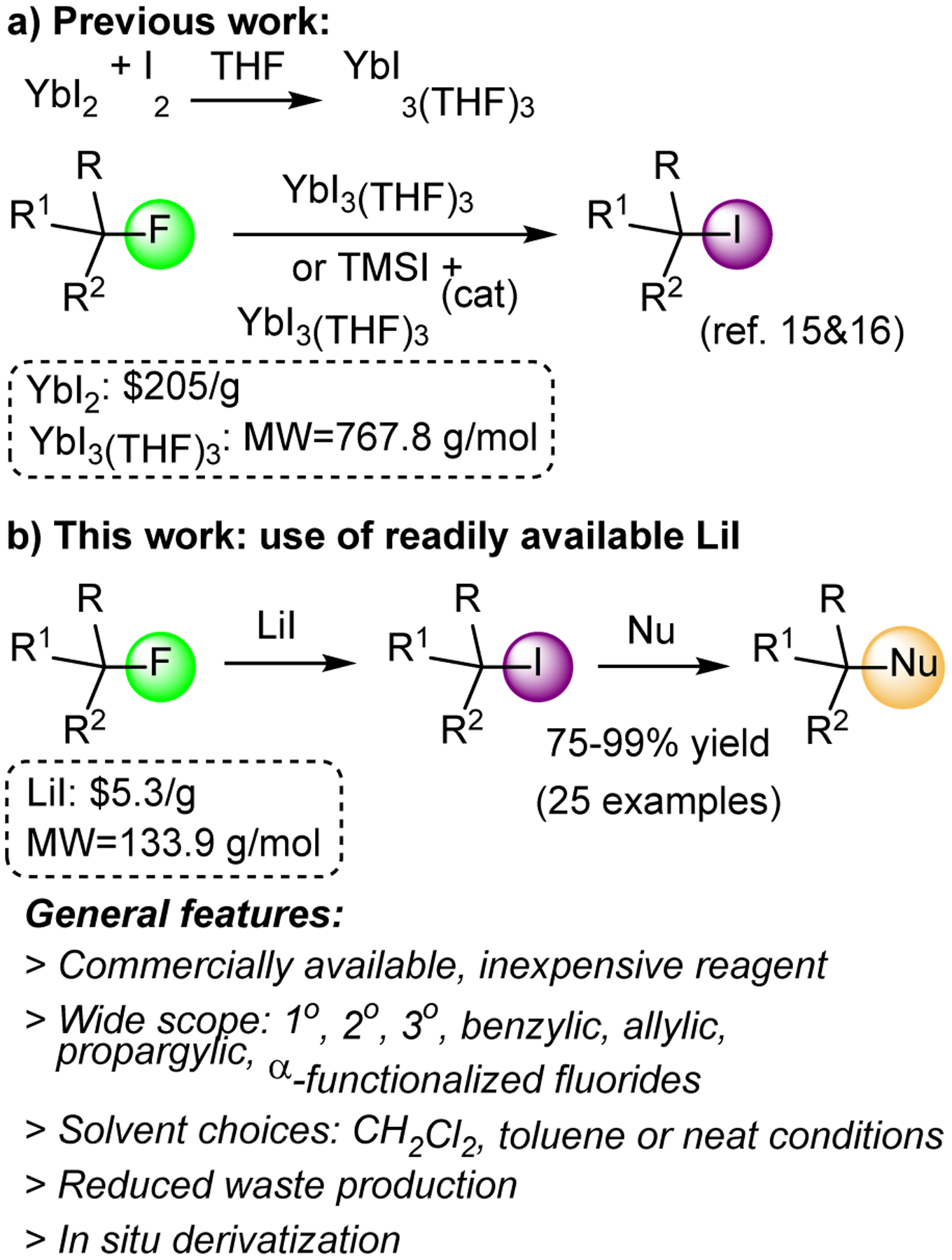
C-F Bond functionalization with metal iodides.49
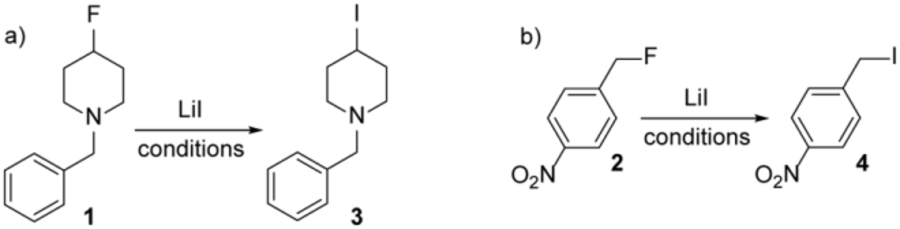
At the beginning of this study we chose to test the C-F bond functionalization of 1-benzyl-4-fluoropiperidine, 1, and 4-nitrobenzyl fluoride, 2, representing an unactivated and an activated alkyl fluoride substrate, respectively, with lithium iodide. To gather a comprehensive overview of the reactivities of these alkyl fluorides the formation of the corresponding iodides 3 and 4 was screened using fourteen different solvents as well as neat conditions at room and elevated temperatures, Table 1. Excellent results without noticeable by-product formation were obtained when 1 was treated with two equivalents of LiI in chlorinated and aromatic solvents (entries 1–4 and 14–16). The Csp3-F bond transformation is significantly faster in dichloromethane and the iodide 3 was produced in quantitative amounts within 6 hours at 25 °C. Heating of the reaction mixture in toluene to 60 °C, however, gave this secondary iodide in 99% yield in the same time period (entry 16). The reaction is somewhat slowed down when only one equivalent of LiI is added (entry 2). It is noteworthy that LiI is a common chemical in synthetic laboratories and fairly inexpensive (Scheme 1).45 We therefore concluded that the use of two equivalents can be easily justified, especially when this allows highly efficient functionalization of a wide scope of alkyl fluorides, vide infra. We also noticed that this reaction proceeds smoothly in the absence of solvent (entry 17). By contrast, no conversion was observed when coordinating solvents such as ethers, DMF, DMSO, acetonitrile, NMP, alcohols or pyridine were employed. This may be attributed to relatively strong solvation of the lithium ion by these solvents which would a) impede its Lewis acidity and ability to activate the Csp3-F bond and b) diminish the thermodynamic driving force expected from the exothermic conversion of LiI to the stable LiF salt. With a set of successful reaction conditions in hand, we continued to investigate the transformation of the benzylic substrate 2. Very efficient conversion to 4-nitrobenzyl iodide, 4, was observed in dichloromethane or toluene at room temperature or upon heating (entries 18–22). In analogy to the previously reported reaction with ytterbium iodide, we assume that the LiI mediated iodination follows an ionic mechanism that is likely to favor either an SN1 or SN2 pathway based on the substitution in the substrate structure. Accordingly, high-yielding iodination was still observed when one equivalent of the radical scavenger TEMPO was added into the reaction mixtures, indicating the absence of significant contributions from a radical mechanism (entries 19 and 21).
Table 1.
Optimization of the Csp3-F bond transformation with lithium iodidea
| |||||
|---|---|---|---|---|---|
| Entry | Substrate | Solvent | Time (h) | Product | Yield (%) |
| 1 | 1 | CH2Cl2 | 6 | 3 | 99 |
| 2 | 1 | CH2Cl2 | 6 | 3 | 80b |
| 3 | 1 | CHCl3 | 24 | 3 | 99 |
| 4 | 1 | ClCH2CH2Cl | 24 | 3 | 99 |
| 5 | 1 | DMF | 48 | 3 | 0 |
| 6 | 1 | DMSO | 48 | 3 | 0 |
| 7 | 1 | CH3CN | 48 | 3 | 0 |
| 8 | 1 | NMP | 48 | 3 | 0 |
| 9 | 1 | 1,4-dioxane | 48 | 3 | 0 |
| 10 | 1 | Et2O | 48 | 3 | 0 |
| 11 | 1 | MeOH | 48 | 3 | 0 |
| 12 | 1 | EtOH | 48 | 3 | 0 |
| 13 | 1 | pyridine | 48 | 3 | 0 |
| 14 | 1 | benzene | 48 | 3 | 98 |
| 15 | 1 | toluene | 48 | 3 | 65 |
| 16 | 1 | toluene | 6 | 3 | 99c |
| 17 | 1 | neat | 18 | 3 | 99d |
| 18 | 2 | CH2Cl2 | 48 | 4 | 99 |
| 19 | 2 | CH2Cl2 | 5 | 4 | 99e |
| 20 | 2 | toluene | 21 | 4 | 99 |
| 21 | 2 | toluene | 24 | 4 | 94e |
| 22 | 2 | toluene | 6 | 4 | 93c |
Conditions: 1 (0.2 mmol), LiI (0.4 mmol) in 0.4 mL solvent at 25 °C.
1.2 equiv. of LiI used,
The reaction was carried out at 60 °C,
The reaction was carried out in the absence of solvent with 3.0 equivalent of LiI,
1.0 equivalent of TEMPO was added.
To evaluate the substrate scope of the C-F bond functionalization, we decided to test a variety of primary, secondary, tertiary, benzylic, propargylic and α-functionalized alkyl fluorides (Scheme 2). We were pleased to find that n-alkyl fluorides and even unprotected 3-fluoropropanol react with LiI almost quantitatively. Under solvent-free conditions, the iodides 5-8 were isolated in 93–99% yield. Very similar results were obtained with three 1-(2-fluoroethoxy)benzenes in toluene at 100 °C and the corresponding alkyl iodides 9-11 were produced in 94–98% yield. The synthesis of the secondary iodide 3 obtained in our optimization study was repeated on the gram scale in dichloromethane at room temperature. This gave 1-benzyl-4-iodopiperidine in virtually the same yield, demonstrating that the reaction can be scaled without compromising results. Cyclohexyl iodide, 12, another secondary alkyl iodide, was isolated in 92% yield using the same protocol. Tertiary fluorides are also excellent substrates for mild C-F bond iodination and we found that the reaction with LiI affords (3-iodo-3-methylbutyl)benzene, 13, and iodoadamantane, 14, in near quantitative amounts at room temperature. Based on our success with the benzyl fluoride 2 during the optimization study, we expected that other activated compounds would be excellent candidates as well. In fact, the benzylic, allylic and propargylic iodides 15-17 were generated in 91–99% yield at room temperature using dichloromethane as solvent. We also applied commercially available fluoroacetophenone, fluoronitrile and ethyl fluoroacetate in our procedure. These α-functionalized fluorides reacted smoothly to the iodides 18-20 with 91–99% yield.
Scheme 2.
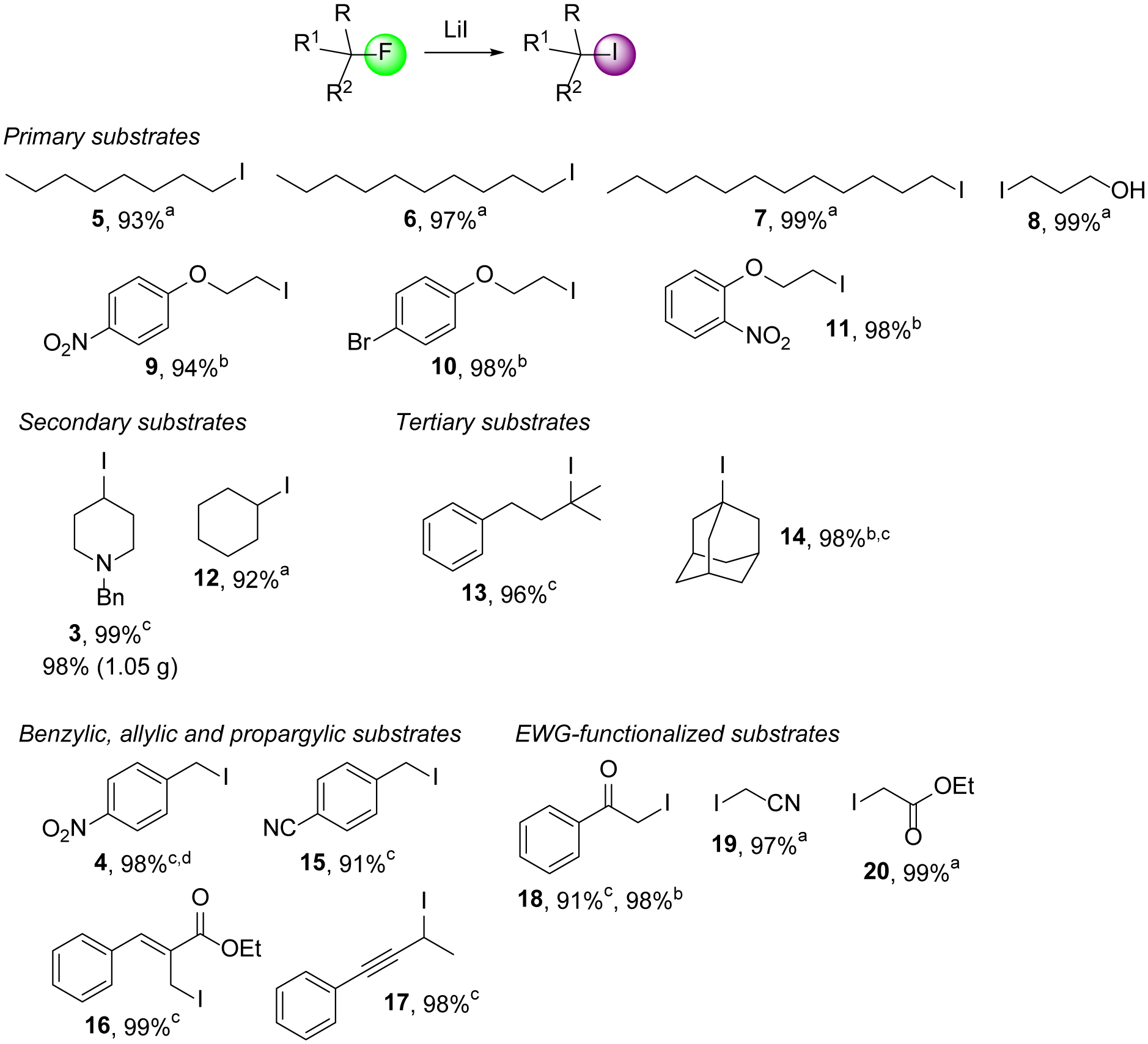
Iodination of primary, secondary, tertiary, benzylic, propargylic and α-functionalized alkyl fluorides. Conditions: aneat, 25 °C, 3.0 eq. LiI; btoluene, 100 °C, 2.0 eq. LiI; cCH2Cl2, 25 °C, 2.0 eq. LiI; dtoluene, 25 °C, 2.0 eq. LiI.
As mentioned above, the C-F bond functionalization with lithium iodide can be performed in dichloromethane or under neat conditions at room temperature. Alternatively, it can be conducted in toluene at higher temperatures which typically gives similar results but in shorter reaction times. For example, 14 was formed in 98% yield in dichloromethane at room temperature after 2 days while the same yield was obtained when lithium iodide and fluoroadamantane were heated in toluene to 100 °C for 1 hour. This was also the case with the synthesis of 2-iodo-1-phenylethan-1-one, 18, which was isolated in 91% yield after stirring with LiI in dichloromethane at 25 °C for 2 days and in 98% yield when the iodination was conducted in toluene at 100 °C for 4 hours, see SI for details.
Another noticeable feature of this reaction is the selectivity for alkyl monofluorides. While 1-benzyl-4-fluoropiperidine, 1, is converted almost quantitatively to its iodide derivative 3 the corresponding geminal difluoride 21 proved inert and only starting material was recovered under the same reaction conditions (Scheme 3). We made the same observations with difluoro- and trifluoroacetophenone, 24 and 25, which did not react with LiI upon heating to 100 °C in toluene. This is in stark contrast to the 98% yield obtained with the corresponding monofluoride. Similarly, trifluorotoluene 26 representing an activated benzylic substrate does not react. The high selectivity toward alkyl monofluorides is further demonstrated by the inertness of fluorobenzene, 27, which proves that Csp2-F bonds are perfectly tolerated by this method.
Scheme 3.
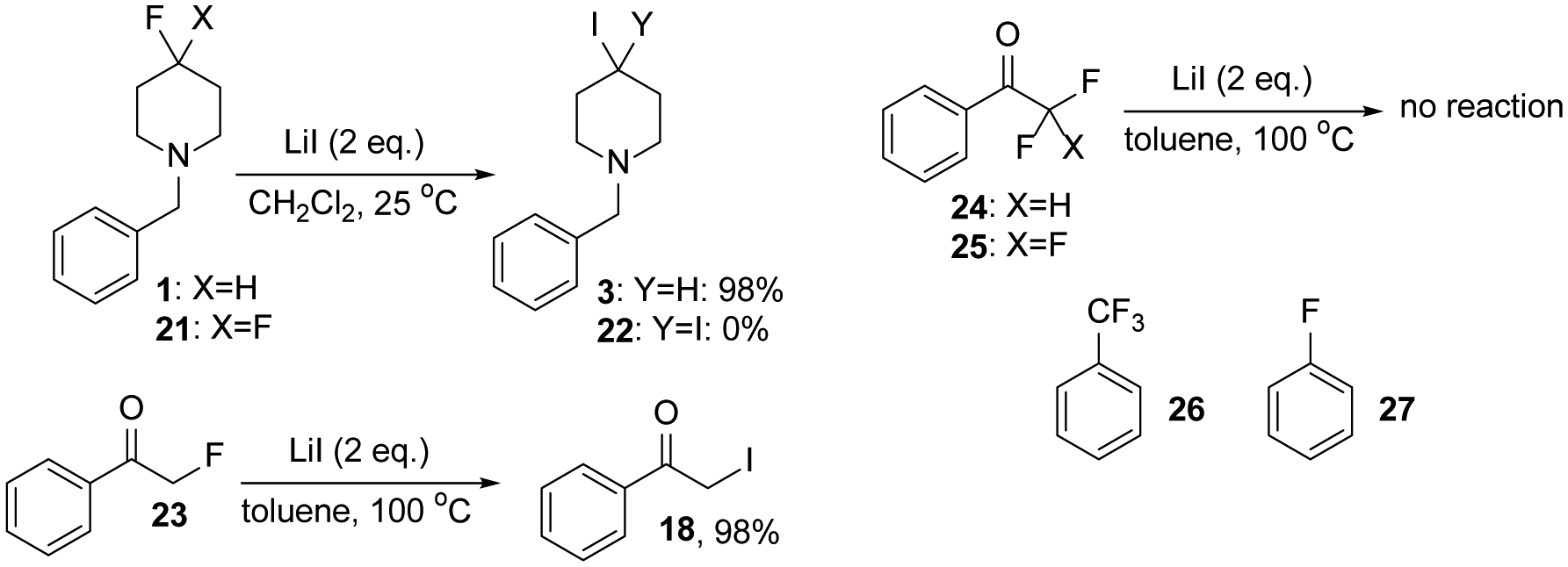
Selectivity of the Csp3-F bond iodination method toward alkyl monofluorides.
Finally, we investigated the possibility to couple the LiI mediated C-F bond activation with subsequent iodide replacement to achieve in situ carbon-carbon or carbon-heteroatom bond formation. Treatment of the fluorides 2 and 30 with LiI in the presence of benzo[d]oxazole-2(3H)-thione, 28, at 100 °C in toluene gave 29 and 31 in almost quantitative amounts (Scheme 4). The oxindole derivative 33 was obtained in 87% yield following the same protocol. We successfully employed the amines 34 and 37 under various conditions to produce 35, 36 and 38 in 85–97% yield. We also found that fluoroacetonitrile and 28 produce 40 in 75% yield. It is noteworthy that Paquin reported C-F bond functionalizations with benzylic substrates in aqueous isopropyl alcohol solution or in the presence of ethylene glycol.46,47 When we applied these protocols to the reaction of 28 and 39, formation of less than 10% of 40 was observed after 4 days. These results demonstrate that the fluoride/iodide exchange with LiI is broadly useful and furnishes synthetically useful alkyl iodides that can be derivatized without prior isolation to install carbon-carbon, carbon-nitrogen or carbon-sulfur bonds.
Scheme 4.
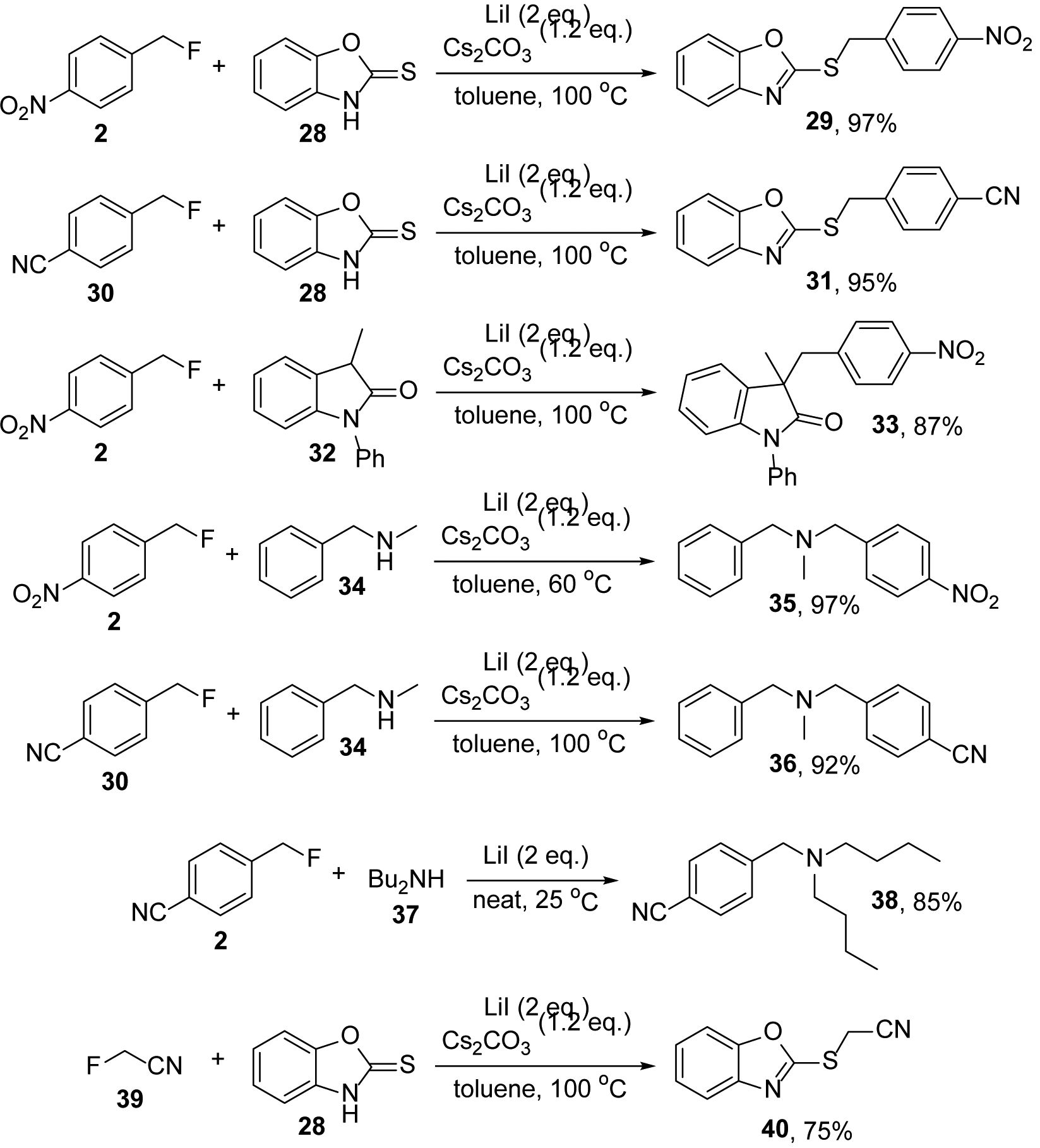
Examples of LiI mediated C-F bond activation coupled with subsequent carbon-carbon, carbon-nitrogen and carbon-sulfur bond formation.
In summary, we have developed a practical method that uses inexpensive lithium iodide for Csp3-F bond functionalization of a wide range of activated and unactivated aliphatic substrates. The substrate scope includes primary, secondary, tertiary, benzylic, allylic, propargylic and α-functionalized alkyl fluorides that were transformed in very high yields to synthetically useful iodides. This challenging task is achieved under relatively mild conditions eliminating the common need of strong Lewis acids or harsh conditions often used for C-F bond scission with unactivated substrates. The fluoride/iodide exchange is selective for aliphatic monofluorides whereas alkyl difluorides, trifluorides, even in activated benzylic positions, as well as aryl fluorides proved inert under the same conditions. The possibility of coupling this chemistry with nucleophilic iodide replacements was demonstrated with high-yielding carbon-carbon, carbon-nitrogen and carbon-sulfur bond formations. Altogether, we believe that this mild and broadly applicable Csp3-F bond functionalization protocol is likely to attract increasing attention to organofluorine chemistry and will stimulate the development of new C-F bond activation methodologies and late-stage functionalization strategies.
Commercially available organofluorines, lithium iodide, reagents and solvents were used as purchased without further purification. The solvents were stored over 4Å molecular sieves prior to use. NMR spectra were obtained at 400 MHz (1H NMR) and 100 MHz (13C NMR) in deuterated chloroform. All reaction products were purified by column chromatography on silica gel (particle size 40–63 μm) as described below.
Procedures
General procedures for the Csp3-F iodination
Method A: LiI (0.4 mmol) and the alkyl fluoride (0.2 mmol) were added into 0.4 mL of anhydrous CH2Cl2 under nitrogen atmosphere. The reaction mixture was stirred at 25 °C in the dark until completion. The solvent was removed and the crude products were purified by silica flash column chromatography as described below.
Method B: LiI (0.4 mmol) and the alkyl fluoride (0.2 mmol) were added into 0.4 mL of anhydrous toluene under nitrogen atmosphere. The reaction mixture was stirred at 100 °C in the dark until completion. The solvent was removed and the crude products were purified by silica flash column chromatography as described below.
Method C: A mixture of LiI (0.6 mmol) and the alkyl fluoride (0.2 mmol) was stirred at 25 °C without solvent in the dark until completion. The crude products were purified by silica flash column chromatography as described below.
1-Benzyl-4-iodopiperidine (3).
Compound 3 was obtained after column purification using hexanes/ethyl acetate (9:1) as mobile phase as a pale yellow oil in 99% yield (59 mg, 0.19 mmol) from 1-benzyl-4-fluoropiperidine (39 mg, 0.2 mmol) and LiI (54 mg, 0.4 mmol) in 0.4 mL of dry CH2Cl2 at 25 °C after 6 hours by following method A. Rf = 0.8 (hexanes/EtOAc, 4:1). The spectroscopic data of 1-benzyl-4-iodopiperidine (3) are in accordance with the literature.48
1H NMR (400 MHz, chloroform-d): δ = 7.32 − 7.25 (m, 4H), 7.23 (m, 1H), 4.27 (br, 1H), 3.46 (s, 2H), 2.68 − 2.51 (m, 2H), 2.33 − 2.04 (m, 6H).
13C NMR (100 MHz, chloroform-d): δ = 138.3, 129.0, 128.2, 127.0, 63.1, 53.7, 38.3, 28.3.
1-(Iodomethyl)-4-nitrobenzene (4).
Compound 4 was obtained after column purification using hexanes/ethyl acetate (19:1) as mobile phase as a pale yellow solid in 98% yield (51 mg, 0.19 mmol) from 1-(fluoromethyl)-4-nitrobenzene (31 mg, 0.2 mmol) and LiI (54 mg, 0.4 mmol) in 0.4 mL of dry CH2Cl2 at 25 °C after 2 days by following method A. Rf = 0.7 (hexanes/EtOAc, 4:1). The spectroscopic data of 1-(iodomethyl)-4-nitrobenzene (4) are in accordance with the literature.49
1H NMR (400 MHz, chloroform-d): δ = 8.15 (d, J = 8.4 Hz, 2H), 7.52 (d, J = 8.4 Hz, 2H), 4.48 (s, 2H).
13C NMR (100 MHz, chloroform-d): δ = 147.4, 146.9, 129.7, 124.2, 2.1.
1-Iodooctane (5).
Compound 5 was obtained after column purification using hexanes as mobile phase as a colorless oil in 93% yield (45 mg, 0.18 mmol) from 1-fluorooctane (26 mg, 0.2 mmol) and LiI (80 mg, 0.6 mmol) after 3 days by following method C. The spectroscopic data of 1-iodooctane (5) are in accordance with the literature.50
1H NMR (400 MHz, chloroform-d): δ = 3.19 (t, J = 7.0 Hz, 2H), 1.82 (m, 2H), 1.39 (m, 2H), 1.34 − 1.24 (m, 8H), 0.88 (t, J = 7.0 Hz, 3H).
13C NMR (100 MHz, chloroform-d): δ = 33.7, 31.9, 30.7, 29.2, 28.7, 22.8, 14.2, 7.5.
1-Iododecane (6).
Compound 6 was obtained after column purification using hexanes as mobile phase as a colorless oil in 97% yield (52 mg, 0.19 mmol) from 1-fluorodecane (32 mg, 0.2 mmol) and LiI (80 mg, 0.6 mmol) after 3 days by following method C. The spectroscopic data of 1-iododecane (6) are in accordance with the literature.51
1H NMR (400 MHz, chloroform-d): δ = 3.18 (t, J = 7.1 Hz, 2H), 1.82 (m, 2H), 1.39 (m, 2H), 1.34 − 1.24 (m, 12H), 0.88 (t, J = 7.1 Hz, 3H).
13C NMR (100 MHz, chloroform-d): δ = 33.7, 32.0, 30.7, 29.7, 29.6, 29.4, 28.7, 22.8, 14.2, 7.4.
1-Iodododecane (7).
Compound 7 was obtained after column purification using hexanes as mobile phase as a colorless oil in 99% yield (58 mg, 0.19 mmol) from 1-fluorododecane (38 mg, 0.2 mmol) and LiI (80 mg, 0.6 mmol after 3 days by following method C. The spectroscopic data of 1-iodododecane (7) are in accordance with the literature.52
1H NMR (400 MHz, chloroform-d): δ = 3.19 (t, J = 7.0 Hz, 2H), 1.82 (m, 2H), 1.39 (m, 2H), 1.33 − 1.25 (m, 16H), 0.88 (t, J = 7.1 Hz, 3H).
13C NMR (100 MHz, chloroform-d): δ = 33.7, 32.1, 30.7, 29.8, 29.8, 29.7, 29.6, 29.5, 28.7, 22.8, 14.3, 7.4.
3-Iodopropan-1-ol (8).
Compound 8 was obtained after extraction of the crude reaction mixture with chloroform as a colorless oil in 99% yield (37 mg, 0.2 mmol) from 3-fluoropropan-1-ol (16 mg, 0.2 mmol) and LiI (80 mg, 0.6 mmol) after 18 hours by following method C. The spectroscopic data of 3-iodopropan-1-ol (8) are in accordance with the literature.53
1H NMR (400 MHz, chloroform-d) δ = 3.91 (m, 2H), 3.52 (m, 1H), 3.34 (t, J = 6.6 Hz, 2H), 2.22 (m, 2H).
13C NMR (100 MHz, chloroform-d) δ = 63.8, 35.3, 3.3.
1-(2-Iodoethoxy)-4-nitrobenzene (9).
Compound 9 was obtained after column purification using hexanes/ethyl acetate (9:1) as mobile phase as a pale yellow solid in 94% yield (55 mg, 0.18 mmol) from 1-(2-fluoroethoxy)-4-nitrobenzene (37 mg, 0.2 mmol) and LiI (54 mg, 0.4 mmol) in 0.4 mL of dry toluene at 100 °C after 2 days by following method B. Rf = 0.7 (hexanes/EtOAc, 4:1). The spectroscopic data of 1-(2-iodoethoxy)-4-nitrobenzene (9) are in accordance with the literature.54
1H NMR (400 MHz, chloroform-d): δ = 8.20 (d, J = 8.8 Hz, 2H), 6.96 (d, J = 8.8 Hz, 2H), 4.34 (t, J = 6.8 Hz, 2H), 3.45 (t, J = 6.8 Hz, 2H).
13C NMR (100 MHz, chloroform-d): δ = 163.0, 142.1, 126.1, 114.8, 69.2, −0.3.
1-Bromo-4-(2-iodoethoxy)benzene (10).
Compound 10 was obtained after column purification using hexanes/ethyl acetate (49:1) as mobile phase as a colorless solid in 98% yield (64 mg, 0.19 mmol) from 1-bromo-4-(2-fluoroethoxy)benzene (44 mg, 0.2 mmol) and LiI (54 mg, 0.4 mmol) in 0.4 mL of dry toluene at 100 °C after 5 days by following method B. Rf = 0.9 (hexanes/EtOAc, 19:1). The spectroscopic data of 1-bromo-4-(2-iodoethoxy)benzene (10) are in accordance with the literature.55
1H NMR (400 MHz, chloroform-d): δ = 7.38 (d, J = 8.8 Hz, 2H), 6.78 (d, J = 8.8 Hz, 2H), 4.21 (t, J = 6.8 Hz, 2H), 3.40 (t, J = 6.8 Hz, 2H).
13C NMR (100 MHz, chloroform-d): δ = 157.2, 132.6, 116.8, 113.8, 69.0, 0.8.
1-(2-Iodoethoxy)-2-nitrobenzene (11).
Compound 11 was obtained after column purification using hexanes/ethyl acetate (9:1) as mobile phase as a pale yellow solid in 98% yield (57 mg, 0.19 mmol) from 1-(2-fluoroethoxy)-2-nitrobenzene (37 mg, 0.2 mmol) and LiI (54 mg, 0.4 mmol) in 0.4 mL of dry toluene at 100 °C after 21 hours by following method B. Rf = 0.7 (hexanes/EtOAc, 4:1).
1H NMR (400 MHz, chloroform-d): δ = 7.83 (dd, J = 8.4, 1.7 Hz, 1H), 7.53 (m, 1H), 7.09 − 7.05 (m, 2H), 4.38 (t, J = 7.2 Hz, 2H), 3.45 (t, J = 7.2 Hz, 2H).
13C NMR (100 MHz, chloroform-d): δ = 151.3, 140.5, 134.2, 125.8, 121.4, 115.4, 70.5, −0.4.
HRMS (ESI-TOF) m/z: [M+Na]+ calcd for C8H8INO3Na 315.9441, found 315.9440.
Iodocyclohexane (12).
Compound 12 was obtained after column purification using hexanes as mobile phase as a colorless oil in 92% yield (38 mg, 0.18 mmol) from fluorocyclohexane (20 mg, 0.2 mmol) and LiI (80 mg, 0.4 mmol) after 2 days by following method C. The spectroscopic data of iodocyclohexane (12) are in accordance with the literature.56
1H NMR (400 MHz, chloroform-d): δ = 4.36 (tt, J = 9.5, 3.8 Hz, 1H), 2.24 − 2.08 (m, 2H), 2.05 − 1.90 (m, 2H), 1.74 − 1.57 (m, 3H), 1.48 − 1.32 (m, 3H).
13C NMR (100 MHz, chloroform-d): δ = 39.8, 32.8, 27.4, 25.4.
(3-Iodo-3-methylbutyl)benzene (13).
Compound 13 was obtained after column purification using hexanes as mobile phase as a colorless oil in 96% yield (52 mg, 0.19 mmol) from (3-fluoro-3-methylbutyl)benzene (33 mg, 0.2 mmol) and LiI (54 mg, 0.4 mmol) in 0.4 mL of dry CH2Cl2 at 25 °C after 6 hours by following method A. Rf = 0.9 (hexanes). The spectroscopic data of (3-iodo-3-methylbutyl)benzene (13) are in accordance with the literature.57
1H NMR (400 MHz, chloroform-d): δ = 7.33 − 7.12 (m, 5H), 2.83 (t, J = 8.2 Hz, 2H), 1.98 (s, 6H), 1.90 (t, J = 8.2 Hz, 2H).
13C NMR (100 MHz, chloroform-d): δ = 141.5, 128.6, 128.6, 126.1, 52.5, 51.5, 38.2, 35.3.
1-Iodoadamantane (14).
Compound 14 was obtained after column purification using hexanes as mobile phase as a colorless solid in 98% yield (51 mg, 0.19 mmol) from 1-fluoroadamantane (31 mg, 0.2 mmol) and LiI (49 mg, 0.4 mmol) in 0.4 mL of dry toluene at 100 °C after 1 hour by following method B. Rf = 0.9 (hexanes). The spectroscopic data of 1-iodoadamantane (14) are in accordance with the literature.58
1H NMR (400 MHz, chloroform-d): δ = 2.64 (br, 6H), 1.95 (br, 3H), 1.81 (br, 6H).
13C NMR (100 MHz, chloroform-d): δ = 52.7, 51.2, 35.8, 33.3.
4-(Iodomethyl)benzonitrile (15).
Compound 15 was obtained after column purification using hexanes/ethyl acetate (19:1) as mobile phase as a colorless solid in 91% yield (44 mg, 0.18 mmol) from 4-(fluoromethyl)benzonitrile (27 mg, 0.2 mmol) and LiI (54 mg, 0.4 mmol) in 0.4 mL of dry CH2Cl2 at 25 °C after 24 hours by following method A. Rf = 0.6 (hexanes/EtOAc, 9:1). The spectroscopic data of 4-(iodomethyl)benzonitrile (15) are in accordance with the literature.59
1H NMR (400 MHz, chloroform-d): δ = 7.58 (d, J = 8.2 Hz, 2H), 7.46 (d, J = 8.2 Hz, 2H), 4.43 (s, 2H).
13C NMR (100 MHz, chloroform-d): δ = 144.8, 132.7, 129.6, 118.6, 111.8, 2.8.
Ethyl (Z)-2-(iodomethyl)-3-phenylacrylate (16).
Compound 16 was obtained after extraction of the crude reaction mixture with chloroform as a colorless oil in 99% yield (63 mg, 0.2 mmol) from ethyl (Z)-2-(fluoromethyl)-3-phenylacrylate (42 mg, 0.2 mmol) and LiI (80 mg, 0.6 mmol) after 18 hours by following method C. The spectroscopic data of ethyl (Z)-2-(iodomethyl)-3-phenylacrylate (16) are in accordance with the literature.60
1H NMR (400 MHz, chloroform-d) δ = 7.81 (s, 1H), 7.57 (dd, J = 7.7, 1.7 Hz, 2H), 7.50 − 7.43 (m, 2H), 7.41 (m, 1H), 4.41 (q, J = 7.1 Hz, 2H), 4.34 (s, 2H), 1.41 (t, J = 7.1 Hz, 3H).
13C NMR (100 MHz, chloroform-d) δ = 166.3, 140.6, 134.9, 130.4, 129.6, 129.3, 129.0, 61.6, 14.4, −0.1.
(3-Iodobut-1-yn-1-yl)benzene (17).
Compound 17 was obtained after column purification using hexanes as mobile phase as a colorless oil in 98% yield (50 mg, 0.19 mmol) from (3-fluorobut-1-yn-1-yl)benzene (30 mg, 0.2 mmol) and LiI (54 mg, 0.4 mmol) in 0.4 mL of dry CH2Cl2 at 25 °C after 24 hours by following method A. Rf = 0.9 (hexanes). The spectroscopic data of (3-iodobut-1-yn-1-yl)benzene (17) are in accordance with the literature.61
1H NMR (400 MHz, chloroform-d): δ = 7.47 − 7.37 (m, 2H), 7.40 − 7.22 (m, 3H), 4.89 (q, J = 7.0 Hz, 1H), 2.16 (d, J = 7.0 Hz, 3H).
13C NMR (100 MHz, chloroform-d): δ = 131.7, 128.7, 128.4, 122.7, 92.2, 85.5, 29.8, 3.4.
2-Iodo-1-phenylethan-1-one (18).
Compound 18 was obtained after column purification using hexanes/ethyl acetate (19:1) as mobile phase as a colorless oil in 98% yield (48 mg, 0.19 mmol) from 2-fluoro-1-phenylethan-1-one (28 mg, 0.2 mmol) and LiI (54 mg, 0.4 mmol) in 0.4 mL of dry toluene at 60 °C after 4 hours by following method B. Rf = 0.5 (hexanes/EtOAc, 9:1). The spectroscopic data of 2-iodo-1-phenylethan-1-one (18) are in accordance with the literature.62
1H NMR (400 MHz, chloroform-d): δ = 7.99 (dd, J = 7.6, 1.6 Hz, 2H), 7.59 (ddd, J = 7.8, 7.6, 1.6 Hz, 1H), 7.49 (ddd, J = 7.6, 7.6, 1.6 Hz, 2H), 4.37 (s, 2H).
13C NMR (100 MHz, chloroform-d): δ = 193.0, 134.0, 133.6, 129.2, 129.0, 1.8.
2-Iodoacetonitrile (19).
Compound 19 was obtained after column purification using hexanes as mobile phase as a colorless oil in 97% yield (32 mg, 0.19 mmol) from 2-fluoroacetonitrile (12 mg, 0.2 mmol) and LiI (80 mg, 0.6 mmol) after 18 hours by following method C. The spectroscopic data of 2-iodoacetonitrile (19) are in accordance with the literature.63
1H NMR (400 MHz, chloroform-d): δ = 3.5 (s, 2H).
13C NMR (100 MHz, chloroform-d): δ = 116.2, −31.6.
Ethyl 2-iodoacetate (20).
Compound 20 was obtained after column purification using hexanes as mobile phase as a colorless oil in 99% yield (42 mg, 0.19 mmol) from ethyl 2-fluoroacetate (21 mg, 0.2 mmol) and LiI (80 mg, 0.6 mmol) after 18 hours by following method C. Rf = 0.7 (hexanes). The spectroscopic data of ethyl 2-iodoacetate (20) are in accordance with the literature.64
1H NMR (400 MHz, chloroform-d): δ = 4.20 (q, J = 7.1 Hz, 2H), 3.68 (s, 2H), 1.28 (t, J = 7.1 Hz, 3H).
13C NMR (100 MHz, chloroform-d): δ = 168.9, 62.3, 14.0, −5.1.
General procedure for the synthesis of 29 and 31
To a mixture of benzylfluoride (0.3 mmol), 2-mercaptobenzoxazole (0.36 mmol) and LiI (0.6 mmol) in dry toluene (1.0 mL) was added Cs2CO3 (0.36 mmol) under nitrogen atmosphere. The mixture was stirred at 100 °C for 24–48 hours in the dark. Completion of the reaction was ascertained by TLC and 19F NMR analysis and the solvent was removed in vacuo. The crude product was purified by flash chromatography on silica gel using hexanes-ethyl acetate as mobile phase as described below.
2-((4-Nitrobenzyl)thio)benzo[d]oxazole (29).
Compound 29 was obtained after column purification using hexanes/ethyl acetate (4:1) as mobile phase as a colorless solid in 97% yield (83 mg, 0.29 mmol) from 1-(fluoromethyl)-4-nitrobenzene (47 mg, 0.3 mmol) and 2-mercaptobenzoxazole (54 mg, 0.36 mmol) in 1.0 mL of dry toluene at 100 °C after 48 hours following the general procedure described above. Rf = 0.5 (hexanes/EtOAc, 1:1). The spectroscopic data of 2-((4-nitrobenzyl)thio)benzo[d]oxazole (29) are in accordance with the literature.65
1H NMR (400 MHz, chloroform-d): δ = 8.16 (dd, J = 6.8, 2.0 Hz, 2H), 7.63 (dd, J = 6.8, 2.0 Hz, 2H), 7.59 (dd, J = 7.5, 1.6 Hz, 1H), 7.42 (dd, J = 7.5, 1.6 Hz, 1H), 7.32 − 7.19 (m, 2H), 4.58 (s, 2H).
13C NMR (100 MHz, chloroform-d): δ = 163.5, 152.2, 147.6, 144.1, 141.8, 130.1, 124.6, 124.4, 124.0, 118.8, 110.1, 35.6.
4-((Benzo[d]oxazol-2-ylthio)methyl)benzonitrile (31).
Compound 31 was obtained after column purification using hexanes/ethyl acetate (4:1) as mobile phase as a colorless solid in 95% yield (75 mg, 0.29 mmol) from 4-(fluoromethyl)benzonitrile (41 mg, 0.3 mmol) and 2-mercaptobenzoxazole (54 mg, 0.36 mmol) in 1.0 mL of dry toluene at 100 °C after 24 hours following the general procedure described above. Rf = 0.3 (hexanes/EtOAc, 4:1). The spectroscopic data of 4-((benzo[d]oxazol-2-ylthio)methyl)benzonitrile (31) are in accordance with the literature.65
1H NMR (400 MHz, chloroform-d): δ = 7.64 − 7.54 (m, 5H), 7.41 (dd, J = 7.5, 1.6 Hz, 1H), 7.31 − 7.20 (m, 2H), 4.54 (s, 2H).
13C NMR (100 MHz, chloroform-d): δ = 163.6, 152.1, 142.0, 141.7, 132.6, 129.9, 124.6, 124.3, 118.7, 118.6, 111.8, 110.1, 35.9.
Synthesis of 3-methyl-3-(4-nitrobenzyl)-1-phenylindolin-2-one (33):
A solution of 1-(fluoromethyl)-4-nitrobenzene (31 mg, 0.2 mmol), 3-methyl-1-phenylindolin-2-one (53 mg, 0.24 mmol), LiI (54 mg, 0.4 mmol) and Cs2CO3 (78 mg, 0.24 mmol) was stirred in 2 ml of dry toluene at 100 °C for 48 hours. Completion of the reaction was ascertained by TLC and 19F NMR analysis and the solvent was removed in vacuo. The crude product was purified by flash chromatography on silica gel using hexanes-ethyl acetate (9:1) as mobile phase. Compound 33 was obtained as a colorless solid in 87% yield (62 mg, 0.17 mmol). Rf = 0.3 (hexanes/EtOAc, 4:1).
1H NMR (400 MHz, Chloroform-d): δ = 7.91 (dd, J = 7.5, 1.6 Hz, 2H), 7.42 (dd, J = 7.6, 7.6 Hz, 2H), 7.36 − 7.32 (m, 2H), 7.15 − 7.10 (m, 2H), 7.04 (dd, J = 7.5, 1.6 Hz, 2H), 6.95 (dd, J = 7.5, 1.6 Hz, 2H), 6.55 (m, 1H), 3.40 (d, J = 12.7 Hz, 1H), 3.18 (d, J = 12.7 Hz, 1H), 1.66 (s, 3H).
13C NMR (100 MHz, Chloroform-d): δ = 178.5, 146.9, 144.0, 143.1, 134.1, 131.9, 130.8, 129.7, 128.4, 128.2, 126.3, 123.3, 123.1, 122.8, 109.6, 50.2, 44.8, 23.5.
HRMS (ESI-TOF): m/z: [M+H]+ calcd for C22H19N2O3 359.1390, found 359.1390.
General procedure for the synthesis of amines 35, 36 and 38
To a mixture of the benzylfluoride (1 eq), N-benzylmethylamine (2–3 eq) and LiI (2 eq) in dry toluene (1.0 mL) was added Cs2CO3 (1.2 eq) under nitrogen atmosphere. The mixture was stirred at 60–100 °C for 16–18 hours in the dark. Completion of the reaction was ascertained by TLC and 19F NMR analysis and the solvent was removed in vacuo. The crude product was purified by flash chromatography on silica gel using hexanes-ethyl acetate as mobile phase as described below.
N-Benzyl-N-methyl-1-(4-nitrophenyl)methanamine (35).
Compound 35 was obtained after column purification using hexanes/ethyl acetate (19:1) as mobile phase as a colorless oil in 97% yield (49 mg, 0.19 mmol) from 1-(fluoromethyl)-4-nitrobenzene (31 mg, 0.2 mmol) and N-benzylmethylamine (73 mg, 0.6 mmol) in 1.0 mL of dry toluene at 60 °C after 16 hours by following the general procedure described above. Rf = 0.3 (hexanes/EtOAc, 4:1). The spectroscopic data of N-benzyl-N-methyl-1-(4-nitrophenyl)methanamine (35) are in accordance with the literature.66
1H NMR (400 MHz, chloroform-d): δ = 8.17 (d, J = 8.2 Hz, 2H), 7.53 (d, J = 8.2 Hz, 2H), 7.39 − 7.21 (m, 5H), 3.58 (s, 2H), 3.55 (s, 2H), 2.19 (s, 3H).
13C NMR (100 MHz, chloroform-d): δ = 147.6, 147.2, 138.9, 129.4, 128.9, 128.5, 127.3, 123.6, 62.2, 61.0, 42.5.
4-((Benzyl(methyl)amino)methyl)benzonitrile (36).
Compound 36 was obtained after column purification using hexanes/ethyl acetate (19:1) as mobile phase as a colorless oil in 92% yield (43 mg, 0.18 mmol) from 4-(fluoromethyl)benzonitrile (27 mg, 0.2 mmol) and N-benzylmethylamine (48 mg, 0.4 mmol) in 1.0 mL of dry toluene at 100 °C after 18 hours by following the general procedure described above. Rf = 0.4 (hexanes/EtOAc, 4:1). The spectroscopic data of 4-((benzyl(methyl)amino)methyl)benzonitrile (36) are in accordance with the literature.67
1H NMR (400 MHz, chloroform-d): δ = 7.59 (d, J = 8.1 Hz, 2H), 7.47 (d, J = 8.1 Hz, 2H), 7.35 − 7.30 (m, 4H), 7.25 (m, 1H), 3.54 (s, 2H), 3.53 (s, 2H), 2.18 (s, 3H).
13C NMR (100 MHz, chloroform-d): δ = 145.4, 138.9, 132.2, 129.4, 128.9, 128.5, 127.3, 119.1, 110.9, 62.2, 61.3, 42.5.
N-Butyl-N-(4-cyanobenzyl)butan-1-amine (38).
To a mixture of LiI (54 mg, 0.4 mmol) and 4-(fluoromethyl)benzonitrile (27 mg, 0.2 mmol) was added dibutylamine (78 mg, 0.6 mmol) under nitrogen atmosphere. The mixture was stirred at 25 °C for 4 hours in the dark. Completion of the reaction was ascertained by TLC and 19F NMR analysis. The crude product was purified by flash chromatography on silica gel using hexanes-ethyl acetate (9:1) as mobile phase. Compound 38 was obtained as a colorless oil in 85% yield (41 mg, 0.17 mmol). Rf = 0.2 (hexanes/EtOAc, 4:1). The spectroscopic data of N-butyl-N-(4-cyanobenzyl)butan-1-amine (38) are in accordance with the literature.68
1H NMR (400 MHz, chloroform-d) δ = 7.58 (d, J = 8.2 Hz, 2H), 7.45 (d, J = 8.2 Hz, 2H), 3.57 (s, 2H), 2.39 (t, J = 7.3 Hz, 4H), 1.49 − 1.34 (m, 4H), 1.33 − 1.23 (m, 4H), 0.87 (t, J = 7.3 Hz, 6H).
13C NMR (100 MHz, chloroform-d) δ = 146.8, 132.1, 129.3, 119.3, 110.5, 58.6, 54.0, 29.4, 20.6, 14.1.
2-(Benzo[d]oxazol-2-ylthio)acetonitrile (40).
To a mixture of fluoroacetonitrile (21 mg, 0.36 mmol), 2-mercaptobenzoxazole (54 mg, 0.36 mmol) and LiI (95 mg, 0.72 mmol) in dry toluene (2.0 mL) was added Cs2CO3 (140 mg, 0.43 mmol) under nitrogen atmosphere. The mixture was stirred at 100 °C for 48 hours in the dark. Completion of the reaction was ascertained by TLC and 19F NMR analysis and the solvent was removed in vacuo. The crude product was purified by flash chromatography on silica gel using hexanes-ethyl acetate (9:1) as mobile phase. Compound 40 was obtained as a colorless solid in 75% yield (51 mg, 0.27 mmol). Rf = 0.4 (hexanes/EtOAc, 4:1). The spectroscopic data of 2-(benzo[d]oxazol-2-ylthio)acetonitrile (40) are in accordance with the literature.69
1H NMR (400 MHz, chloroform-d) δ = 7.66 (m, 1H), 7.49 (m, 1H), 7.38 − 7.27 (m, 2H), 4.11 (s, 2H).
13C NMR (100 MHz, chloroform-d) δ = 160.7, 152.6, 141.5, 125.0, 124.9, 119.2, 115.4, 110.4, 17.8.
Supplementary Material
Funding Information
US National Institutes of Health (GM106260).
Footnotes
Supporting Information
Experimental procedures and NMR spectra.
Conflict of Interest
The authors declare no conflict of interest.
References
- (1).Zhou Y; Wang J; Gu Z; Wang S; Zhu W; Acena JL; Soloshonok VA; Izawa K; Liu H Chem. Rev 2016, 116, 422. [DOI] [PubMed] [Google Scholar]
- (2).Amii H; Uneyama K Chem. Rev 2009, 109, 2119. [DOI] [PubMed] [Google Scholar]
- (3).Ahrens T; Kohlmann J; Ahrens M; Braun T Chem. Rev 2015, 115, 931. [DOI] [PubMed] [Google Scholar]
- (4).Champagne PA; Benhassine Y; Desroches J; Paquin J-F Angew. Chem. Int. Ed 2014, 53, 13835. [DOI] [PubMed] [Google Scholar]
- (5).Hamel JD; Paquin J-F Chem. Commun 2018, 54, 10224. [DOI] [PubMed] [Google Scholar]
- (6).Zi Y; Lange M; Schultz C; Vilotijevic I Angew. Chem. Int. Ed 2019, 58, 10727. [DOI] [PubMed] [Google Scholar]
- (7).Butcher TW; Yang JL; Amberg WM; Watkins NB; Wilkinson ND; Hartwig JF Nature 2020, 583, 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Vasilopoulos A; Golden DL; Buss JA; Stahl AS Org. Lett 2020, 22, 5753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Houle C; Savoie PR; Davies C; Jardel D; Champagne PA; Bibal B; Paquin J-F Chem. Eur. J 2020, 26, 10620. [DOI] [PubMed] [Google Scholar]
- (10).Gu W; Haneline MR; Douvris C; Ozerov OV J. Am. Chem. Soc 2009, 131, 11203. [DOI] [PubMed] [Google Scholar]
- (11).Nishimine T; Fukushi K; Shibata N; Taira H; Tokunaga E; Yamano A; Shiro M; Shibata N Angew. Chem. Int. Ed 2014, 53, 517. [DOI] [PubMed] [Google Scholar]
- (12).Mandal D; Gupta R; Jaiswal AK; Young RD J. Am. Chem. Soc 2020, 142, 2572. [DOI] [PubMed] [Google Scholar]
- (13).Traff AM; Janjetovic M; Hilmersson G Chem. Commun 2015, 51, 13260. [DOI] [PubMed] [Google Scholar]
- (14).Dryzhakov M; Richmond E; Li G; Moran JJ Fluorine Chem 2017, 193, 45. [Google Scholar]
- (15).Jaiswal AK; Prasad PK; Young RD Chem. Eur. J 2019, 25, 6290. [DOI] [PubMed] [Google Scholar]
- (16).Traff AM; Janjetovic M; Ta L; Hilmersson G Angew. Chem. Int. Ed 2013, 52, 12073. [DOI] [PubMed] [Google Scholar]
- (17).Janjetovic M; Ekebergh A; Traff AM; Hilmersson G Org. Lett 2016, 18, 2804. [DOI] [PubMed] [Google Scholar]
- (18).Dryzhakov M; Moran J ACS Catal 2016, 6, 3670. [Google Scholar]
- (19).Balaraman K; Wolf C Angew. Chem. Int. Ed 2017, 56, 1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Willcox DR; Nichol GS; Thomas SP ACS Catal 2021, 11, 3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Balaraman K; Wolf C Org. Lett 2021, 23, 8994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Terao J; Ikumi A; Kuniyasu H; Kambe NJ Am. Chem. Soc 2003, 125, 5646. [DOI] [PubMed] [Google Scholar]
- (23).Matsubara K; Ishibashi T; Koga Y Org. Lett 2009, 11, 1765. [DOI] [PubMed] [Google Scholar]
- (24).Blessley G; Holden P; Walker M; Brown JM; Gouverneur V Org. Lett 2012, 14, 2754. [DOI] [PubMed] [Google Scholar]
- (25).Erickson LW; Lucas EL; Tollefson EJ; Jarvo ER J. Am. Chem. Soc 2016, 138, 14006. [DOI] [PubMed] [Google Scholar]
- (26).Iwasaki T; Yamashita K; Kuniyasu H; Kambe N Org. Lett 2017, 19, 3691. [DOI] [PubMed] [Google Scholar]
- (27).Wolfe MMW; Shanahan JP; Kampf JW; Szymczak NK J. Am. Chem. Soc 2020, 142, 18698. [DOI] [PubMed] [Google Scholar]
- (28).Terao J; Watabe H; Kambe NJ Am. Chem. Soc 2005, 127, 3656. [DOI] [PubMed] [Google Scholar]
- (29).Iwasaki T; Shimizu R; Imanishi R; Kuniyasu H; Kambe N Angew. Chem. Int. Ed 2015, 54, 9347. [DOI] [PubMed] [Google Scholar]
- (30).Iwasaki T; Fukuoka A; Yokoyama W; Min X; Hisaki I; Yang T; Ehara M; Kuniyasu H; Kambe N Chem. Sci 2018, 9, 2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Iwasaki T; Min X; Fukuoka A; Kuniyasu H; Kambe N Angew. Chem. Int. Ed 2016, 55, 5550. [DOI] [PubMed] [Google Scholar]
- (32).Cook AM; Wolf C Angew. Chem. Int. Ed 2016, 55, 2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Ding R; Wolf C Chem. Commun 2016, 52, 3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Balaraman K; Moskowitz M; Liu Y; Wolf C Synthesis 2016, 48, 2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Balaraman K; Ding R; Wolf C Adv. Synth. Catal 2017, 359, 4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Ding R; Wolf C Org. Lett 2018, 20, 892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Moskowitz M; Balaraman K; Wolf CJ Org. Chem 2018, 83, 1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Balaraman K; Moskowitz M; Wolf C Adv. Synth. Catal 2018, 360, 4705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Ding R; De los Santos ZA; Wolf C ACS Catal 2019, 9, 2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Steber SE; Pham ANDL; Nelson E; Wolf C Chirality 2021, 33, 891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Ding R; Bakhshi PR; Wolf CJ Org. Chem 2016, 82, 1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Thanzeel FY; Sripada A; Wolf CJ Am. Chem. Soc 2019, 141, 16382. [DOI] [PubMed] [Google Scholar]
- (43).Hassan DS; Kariapper FS; Lynch CC; Wolf C Synthesis 2022, 54, DOI: 10.1055/a-1754-2271. [DOI] [Google Scholar]
- (44).During the preparation of this manuscript Davis reported similar results, see Zerban JJ; Bagnall B; Davis TA Tetrahedron Lett 2022, 91, 153639. [Google Scholar]
- (45).Prices for 1g amounts of LiI ($5.3/g; $0.7/mmol) and YbI2 ($205/g; $87.5/mmol) were obtained from Sigma-Aldrich, November 2021.
- (46).Champagne PA; Pomarole J; Therien M-E; Benhassine Y; Beaulieu S; Legault CY; Paquin J-F Org. Lett 2013, 15, 2210. [DOI] [PubMed] [Google Scholar]
- (47).Champagne PA; Drouin M; Legault CY; Audubert C; Paquin J-FJ Fluorine Chem 2015, 171, 113. [Google Scholar]
- (48).Gonnard L; Guérinot A; Cossy J Chem. - Eur. J 2015, 21, 12797. [DOI] [PubMed] [Google Scholar]
- (49).Strazzolini P; Runcio A Eur. J. Org. Chem 2003, 526. [Google Scholar]
- (50).Huang Z; Negishi E Org. Lett 2006, 8, 3675. [DOI] [PubMed] [Google Scholar]
- (51).Zhao Y; Snieckus V Org. Lett 2014, 16, 390. [DOI] [PubMed] [Google Scholar]
- (52).Depken C; Krätzschmar F; Rieger R; Rode K; Breder A Angew. Chem., Int. Ed 2018, 57, 2459. [DOI] [PubMed] [Google Scholar]
- (53).Arian D; Kovbasyuk L; Mokhir AJ Am. Chem. Soc 2011, 133, 3972. [DOI] [PubMed] [Google Scholar]
- (54).Autino JC; Bruzzone L; Romanelli GP; Jios JL; Ancinas HA Anales de Química, Intern. Ed 1998, 94, 292. [Google Scholar]
- (55).Bezencon O; Bur D; Weller T; Richard-Bildstein S; Remeň L; Sifferlen T; Corminboeuf O; Grisostomi C; Boss C; Prade L; Delahye S; Treiber A; Strickner P; Binkert C; Hess P; Steiner B; Fischli WJ Med. Chem 2009, 52, 3689. [DOI] [PubMed] [Google Scholar]
- (56).Ortega N; Feher-Voelger A; Brovetto M; Padrón JI; Martín VS; Martín T Adv. Synth. Catal 2011, 353, 963. [Google Scholar]
- (57).Chen J; Lin J-H; Xiao J-C Org. Lett 2018, 20, 3061. [DOI] [PubMed] [Google Scholar]
- (58).Mizukami Y; Song Z; Takahashi T Org. Lett 2015, 17, 5942. [DOI] [PubMed] [Google Scholar]
- (59).Combe SH; Hosseini A; Song L; Hausmann H; Schreiner PR Org. Lett 2017, 19, 6156. [DOI] [PubMed] [Google Scholar]
- (60).Ying T; Bao W; Wang Z; Zhang YJ Chem. Res 2005, 96. [Google Scholar]
- (61).Larock RC; Chow MS Organometallics 1986, 5, 603. [Google Scholar]
- (62).Zou H; He W; Dong Q; Wang R; Yi N; Jiang J; Pen D; He W Eur. J. Org. Chem, 2016, 116. [Google Scholar]
- (63).Reis AKCA; Rittner R Spectrochim. Acta, Part A 2007, 66, 681. [DOI] [PubMed] [Google Scholar]
- (64).Meyer D; Renaud P Angew. Chem., Int. Ed 2017, 56, 10858. [DOI] [PubMed] [Google Scholar]
- (65).Wang X; Wu C-Y; Li Y-S; Dong Z-B Eur. J. Org. Chem, 2020: 6770. [Google Scholar]
- (66).Thai T-T; Mérel DS; Poater A; Gaillard S; Renaud J-L Chem. Eur. J, 2015, 21, 7066. [DOI] [PubMed] [Google Scholar]
- (67).Ide T; Barham JP; Fujita M; Kawato Y; Egami H; Hamashima Y Chem, Sci 2018, 9, 8453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Chaudhary S; Milton MD; Garg P Chemistry Select 2017, 2, 650. [Google Scholar]
- (69).Yatam S; Jadav SS; Gundla R; Gundla KP; Reddy GM; Ahsan MJ; Chimakurthy J Chemistry Select 2018, 3, 10305. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.