

SUMMARY
Genetic studies have highlighted microglia as pivotal in orchestrating Alzheimer’s disease (AD). Microglia that adhere to Aβ plaques acquire a transcriptional signature, “disease-associated microglia” (DAM), which largely emanates from the TREM2-DAP12 receptor complex that transmits intracellular signals through the protein tyrosine kinase SYK. The human TREM2R47H variant associated with high AD risk fails to activate microglia via SYK. We found that SYK-deficient microglia cannot encase Aβ plaques, accelerating brain pathology and behavioral deficits. SYK deficiency impaired the PI3K-AKT-GSK3β-mTOR pathway, incapacitating anabolic support required for attaining the DAM profile. However, SYK-deficient microglia proliferated and advanced to an Apoe-expressing prodromal stage of DAM; this pathway relied on the adaptor DAP10, which also binds TREM2. Thus, microglial responses to Aβ involve non-redundant SYK- and DAP10-pathways. Systemic administration of an antibody against CLEC7A, a receptor that directly activates SYK, rescued microglia activation in mice expressing the TREM2R47H allele, unveiling new options for AD immunotherapy.
Graphical Abstract
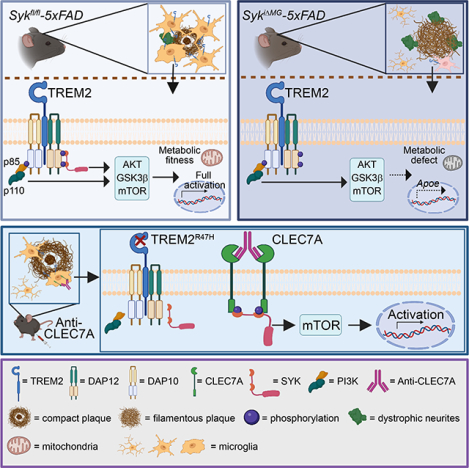
Microglia respond to Aβ through the activation of TREM2 mediated Syk and DAP10 signaling mechanisms. Antibody-mediated activation of Syk in mic expressing the Alzheimer’s disease associated human TREM2R47H allele rescues microglial activation.
INTRODUCTION
Alzheimer’s disease (AD) is the leading cause of late-onset dementia worldwide (2021). AD pathology features extracellular amyloid beta (Aβ) deposition and hyperphosphorylation of intraneuronal Tau, causing neuronal and synaptic loss, ultimately leading to cognitive impairment (Long and Holtzman, 2019). Microglia are professional phagocytes of the central nervous system (CNS) that contribute to CNS development, homeostasis and defense (Prinz et al., 2021; Thion et al., 2018). In response to Aβ pathology, microglia proliferate, associate with Aβ plaques, and acquire an activated transcriptional profile, commonly referred to as disease-associated microglia (DAM) (Keren-Shaul et al., 2017; Friedman et al., 2018; Krasemann et al., 2017; Marsh et al., 2022), Single-cell RNA sequencing (scRNA-seq) has further resolved DAM complexity, identifying four signatures of microglia in amyloid pathology, namely DAM proper (enriched for ApoE, Spp1, Clec7a, Cd11c, Cst7, Lpl), MHC class II (MHCII) microglia (enriched for MHC-II molecules and Cd74), type I interferon responsive (IFN-R) microglia (enriched for Ifitm3, Bst2, Stat1), and proliferating microglia (enriched for Mki67 and Top2a) (Mathys et al., 2017; Sala Frigerio et al., 2019; Ellwanger et al., 2021; Wang et al., 2020). Plaque-associated microglia may help contain Aβ plaques, and delay disease progression (Condello et al., 2015; Wang et al., 2016; Yuan et al., 2016). However, excessive microglia activation may aggravate neuronal damage at later stages of amyloid pathology (Krasemann et al., 2017; Spangenberg et al., 2019; Wes et al., 2016). To comprehend beneficial and detrimental functions of plaque-associated microglia, it is necessary to identify the underlying mechanisms of DAM generation.
The microglial receptor TREM2 binds phospholipids, apoptotic cells, Aβ, and lipoproteins such as ApoE, LDL and HDL (Atagi et al., 2015; Wang et al., 2015; Yeh et al., 2016) (Zhao et al., 2018). TREM2 associates with the transmembrane adaptor DAP12 (Ulland and Colonna, 2018), which recruits spleen tyrosine kinase (SYK) through a cytoplasmic ITAM motif (Chu et al., 1998; Turner et al., 2000; Yao et al., 2019). Blunted microglial responses to Aβ along with impaired acquisition of DAM signature are evident in mice deficient for Trem2 or expressing the hypofunctional TREM2R47H human variant (Ellwanger et al., 2021; Jay et al., 2017; Lee et al., 2021; Song et al., 2018; Wang et al., 2015). Other than TREM2, there are several immune receptors upregulated in DAM signaling via SYK, including CLEC7A, CD300LB, CD200R4, and the β2 integrin CD11c (Keren-Shaul et al., 2017; Mócsai et al., 2006; Rogers et al., 2005; Wright et al., 2003; Yamanishi et al., 2008). Based on these observations we hypothesized that SYK signaling may play a central role in microglial responses to Aβ.
To test this hypothesis, we crossed the 5×FAD mouse model of amyloid pathology (Oakley et al., 2006) with Cx3cr1CreERT2 × Sykfl/fl mice lacking Syk expression in microglia. Using complementary techniques, we demonstrated that SYK-deficient microglia develop an intermediate activation signature in response to Aβ, but fail to attain a complete DAM phenotype, indicating that microglia require cooperation of both SYK-dependent and - independent pathways to elicit an effective response to Aβ. Furthermore, we demonstrated that immunotherapies boosting SYK through CLEC7A improve microglia activation during Aβ pathology, providing a possible therapeutic avenue for AD.
RESULTS
Conditional Syk deletion blunts microglial responses to Aβ pathology and exacerbates plaque deposition
We first tested whether Aβ accumulation activates SYK in microglia. Immunoblot analysis revealed increased SYK phosphorylation in microglia from 5×FAD mice, whereas the total amount of SYK protein was unchanged (Figure 1A). Next, we crossed Sykfl/fl mice (Saijo et al., 2003) with Cx3cr1CreERT2 mice (Yona et al., 2013), which enable inducible deletion of Syk in Cx3cr1 positive cells by administration of tamoxifen (TAM). We refer to these mice as SykiΔMG whereas Sykfl/fl serve as controls. Microglia originate from yolk sac hematopoiesis (Ginhoux et al., 2010; Schulz et al., 2012) and are maintained through self-renewal (Askew et al., 2017; Tay et al., 2017), with no input from circulating monocytes (Goldmann et al., 2013; Gomez Perdiguero et al., 2015). Thus, TAM induction of Cx3cr1CreERT2 allows for permanent gene deletion in microglia, while bone marrow-derived monocytes that undergo Cre-mediated recombination are replaced within 4 weeks due to their constant turnover. We further crossed SykiΔMG mice to 5×FAD mice, which recapitulate features of Aβ pathology characteristic of AD. One-month-old SykiΔMG-5×FAD mice and Sykfl/fl-5×FAD control mice were fed with TAM-containing chow until two months of age and returned to regular chow until 6 or 9 months of age to allow accumulation of Aβ plaques (Figure 1B). Using flow cytometry, we showed that SYK protein was greatly reduced in microglia from SykiΔMG mice, whereas monocytes, neutrophils and B cells remained unaffected (Figure S1A, B). Border-associated macrophages (BAMs) exhibited a moderate SYK reduction, consistent with their embryonic origin and slow turnover (Goldmann et al., 2016; Masuda et al., 2022; Utz et al., 2020). However, given the extra-parenchymal localization of BAMs, partial Syk deletion in these cells is unlikely to significantly affect parenchymal Aβ pathology. Nine-month-old SykiΔMG-5×FAD mice had markedly less microgliosis and more fibrillar Aβ deposition (Figure S1C–F). In both the cortex and hippocampus, microglia density within a 15-μm radius from plaque borders was significantly reduced in SykiΔMG-5×FAD mice (Figure 1C–E). Likewise, Iba1+ methoxy-X04+ colocalized staining was diminished in SykiΔMG-5×FAD mice, indicating fewer plaque-associated microglia. This finding was further confirmed by staining for the transcription factor PU.1, which is expressed solely in microglia in the brain parenchyma. A reduced percentage of plaque-associated PU.1+ nuclei was apparent in SykiΔMG-5×FAD mice, whereas the density of PU.1+ nuclei away from the plaques was unchanged (Figure S1G–I). Of note, no difference in microglia numbers or Iba1 staining was found when comparing SykiΔMG and Sykfl/fl mice without amyloid pathology (Figure S1J–L). Thus, SYK appears redundant for microglial development and self-renewal in the steady state. Conversely, SYK signaling is required to induce microglia responses to Aβ plaques.
Figure 1. Conditional deletion of Syk in microglia abolishes their capacity to respond to Aβ plaques and uptake Aβ.
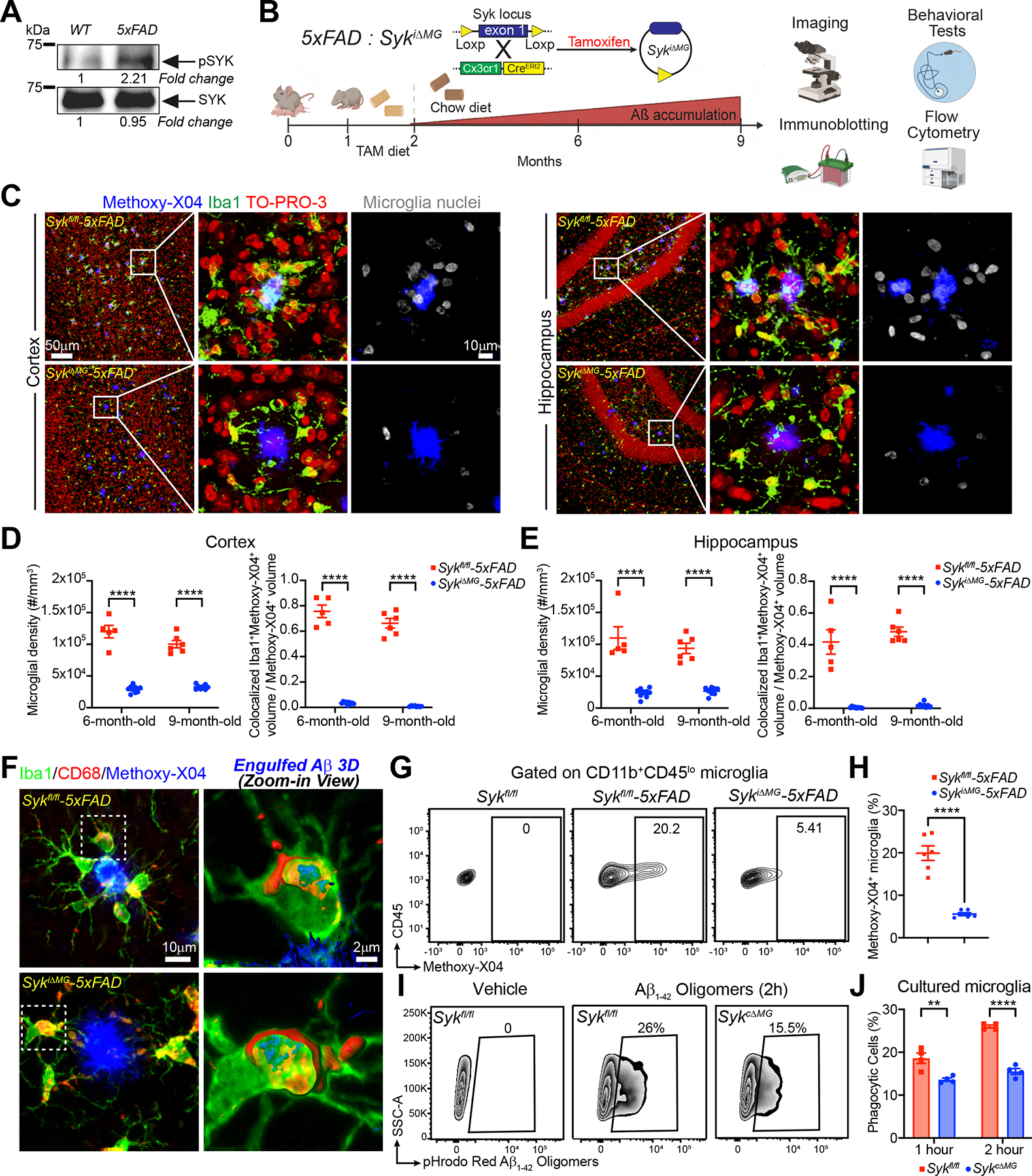
(A) Immunoblot of SYK and pSYK protein on sorted microglia from 8-month-old WT and 5×FAD mice. Representative of two independent experiments.
(B) Schematic of the experimental design.
(C) Representative confocal images of cortex and dentate gyrus from 9-month-old Sykfl/fl- and SykiΔMG-5×FAD mice.
(D and E) Quantification of microglia density within a 15-μm radius from the plaque surfaces and Iba1+methoxy-X04+ colocalization volume normalized by total methoxy-X04+ volume, in either cortex or hippocampus of 6- and 9- month-old mice of each genotype. Data points represent the average of two technical repeats (one experiment; n = 5–9 mice/genotype).
(F) 3D reconstruction of microglia from 6-month-old Sykfl/fl- and SykiΔMG-5×FAD mice, showing engulfed Aβ inside CD68+ phagosomes.
(G) Representative FACS plots of methoxy-X04+ microglia from 6-month-old Sykfl/fl, Sykfl/fl-5×FAD and SykiΔMG-5×FAD mice.
(H) Percentage of methoxy-X04+ microglia from each genotype (two independent experiments; n = 6 mice/genotype).
(I) Representative FACS plots of Sykfl/fl and SykcΔMG primary microglia cultured with pHrodo-Red labeled Aβ1–42 for 2 hours.
(J) Quantification of Aβ1–42 uptake by primary microglia from each genotype. Representative of three independent experiments (n = 4 technical repeats/group).
**, P < 0.01; ****, P < 0.0001- by two-way ANOVA with Sidak’s multiple comparisons test (D, E and J), or two-tailed unpaired Student’s t test (H). Data are presented as mean ± SEM.
See also Figure S1 in the supplemental information.
We asked whether SYK deficiency affected microglial phagocytosis. 3D reconstructions of Iba1, CD68, and methoxy-X04 staining revealed Aβ in microglial CD68+ phagosomes in both genotypes (Figure 1F). To quantify Aβ uptake, we injected mice intraperitoneally (ip.) with methoxy-X04 and analyzed microglia by flow cytometry. SykiΔMG-5×FAD mice had fewer methoxy-X04+ microglia, indicating less Aβ engulfment (Figure 1G, H). To validate this, we assessed pHrodo-labeled Aβ1–42 uptake in primary microglia cultures by flow cytometry. Since neonatal mice cannot feed on TAM-containing chow, we cultured microglia from P4 mice with constitutive deletion of Syk (Sykfl/fl × Cx3cr1Cre, referred to as SykcΔMG) (Yona et al., 2013). SYK-deficient microglia phagocytosed less Aβ1–42 than did control microglia (Figure 1I, J). Thus, SYK deficiency affects microglia clustering around Aβ plaques and Aβ phagocytosis.
Lack of SYK in microglia correlates with increased Aβ pathology, neuronal damage and behavioral deficits
We compared SykiΔMG-5×FAD and Sykfl/fl-5×FAD mice for neuronal damage and behavioral alterations. First, we examined brain accumulation of Aβ1–42 by immunofluorescence and found that SykiΔMG-5×FAD mice had a significant increase of Aβ1–42 deposition at both 6 and 9 months (Figure 2A, B). Aβ1–42 is prone to aggregation and adhesion to cell membranes (Masters and Selkoe, 2012; Mucke and Selkoe, 2012), which makes it highly neurotoxic. Thus, we examined the damage of axons and dendrites (collectively neurites) by staining for LAMP1, which labels vesicles accumulating in dystrophic neurites (Condello et al., 2015). LAMP1 staining volume was significantly amplified in SykiΔMG-5×FAD mice (Figure 2C, D), indicating increased neuronal dystrophy. Similar results were observed by staining for the N terminus of APP, which accumulates in dystrophic neurites (Figure S2A, B). SYK deficiency in microglia also impacted the shape and compactness of Aβ deposits (inert, dynamic, and filamentous plaques) (Figure S2C). SykiΔMG-5×FAD mice had fewer inert plaques at both 6 and 9 months, and more dynamic plaques at 9 months of age (Figure S2D).
Figure 2. SYK deficiency in microglia exacerbates Aβ pathology and leads to behavior and memory defects.
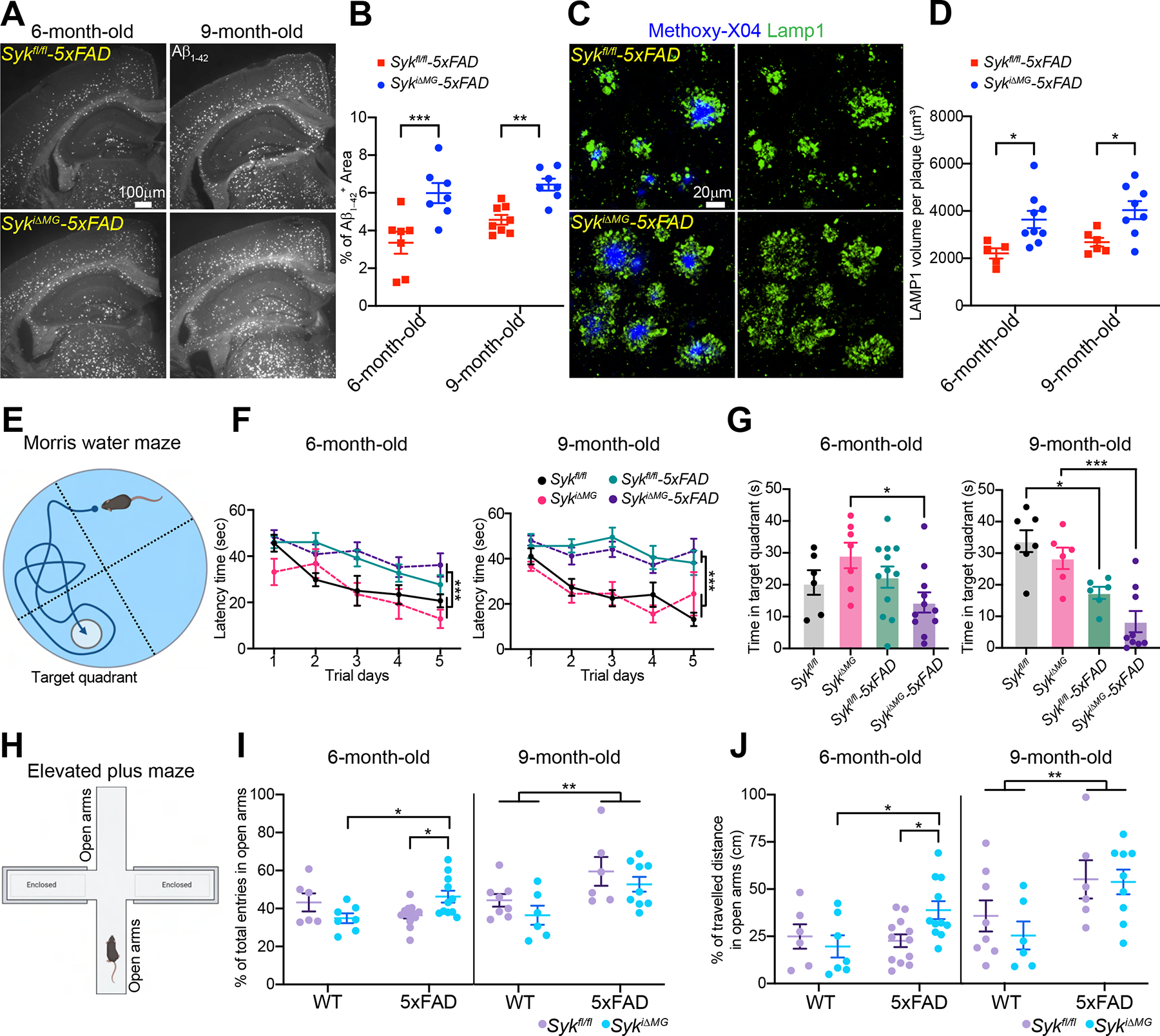
(A) Representative images of Aβ1–42 staining in 6- and 9-month-old Sykfl/fl-5×FAD and SykiΔMG-5×FAD mice.
(B) Percentage of Aβ1–42+ area in the cortex of 6- and 9-month-old mice of each genotype. Data points represent the average of three technical repeats (one experiment; n = 7–8 mice/genotype).
(C) Representative confocal images of methoxy-X04 and Lamp1+ dystrophic neurites in the cortex of 9-month-old Sykfl/fl-5×FAD and SykiΔMG-5×FAD mice.
(D) Volumes of dystrophic neurites per plaque in 6- and 9-month-old mice of each genotype. Data points represent the average of two technical repeats (one experiment; n = 5–9 mice/genotype).
(E-G) Morris water maze tests on 6- and 9-month-old Sykfl/fl and SykiΔMG mice with or without 5×FAD transgenes. F) Latency time to submerged platform; (G) time in target quadrant without submerged platform.
(H-J) Elevated plus maze test on 6- and 9-month-old Sykfl/fl and SykiΔMG mice with or without 5×FAD transgenes. (I) Percentage of entries into open arms; (J) percentage of travelled distance in open arms.
Sample size of behavioral tests: 6-month-old Sykfl/fl (n=6), SykiΔMG (n=7), Sykfl/fl-5×FAD (n=12) and SykiΔMG-5×FAD (n=11); 9-month-old Sykfl/fl (n=7), SykiΔMG (n=6), Sykfl/fl-5×FAD (n=6) and SykiΔMG-5×FAD (n=9). *, P < 0.05; **, P < 0.01; ***, P < 0.001 by two-way ANOVA with Sidak’s multiple comparisons test (B, D, F, I and J), or one-way ANOVA with Tukey’s multiple comparisons test (G). Data are presented as mean ± SEM.
See also Figure S2 in the supplemental information.
Lastly, we performed a battery of behavioral tests. 5×FAD mice had impaired learning ability in comparison to non-5×FAD mice at both 6 and 9 months of age by the end of five days trials in the Morris water maze (Figure 2E, F); no differences were noted between the Sykfl/fl and SykiΔMG groups at either age (Figure 2F). However, at the memory recall session without a submerged platform, 6-month-old SykiΔMG-5×FAD mice spent significantly less time in the target quadrant than did SykiΔMG mice, whereas no difference was found between Sykfl/fl-5×FAD and Sykfl/fl mice (Figure 2G). At 9-months, both SykiΔMG-5×FAD and Sykfl/fl-5×FAD groups spent less time in the target quadrant than did either SykiΔMG or Sykfl/fl mice, with SykiΔMG-5×FAD mice exhibiting the most significant defect (Figure 2G). Latency time to reach the visible platform did not reveal any impairment of visual or swimming ability (Figure S2E). Thus, SYK-deficiency in microglia accelerates memory impairment in 5×FAD mice.
Mice were also tested at the elevated plus maze, which assesses anxiety levels (Figure 2H). At 6 months, SykiΔMG-5×FAD mice made significantly more entries and traveled longer distances in the open arms than did Sykfl/fl-5×FAD and all non-5×FAD mice (Figure 2I, J). At 9 months, however, Sykfl/fl-5×FAD and SykiΔMG-5×FAD behaved similarly: both made significantly more entries and traveled longer distances in the open arms than did non-5×FAD mice (Figure 2I, J). All groups travelled comparable total distances, indicating no locomotor impairment (Figure S2F). We conclude that SYK deficiency in microglia exacerbates the Aβ1–42 load and neuronal damage, worsening behavioral deficits.
SYK deficiency in microglia dampens mTOR signaling and enhances autophagy
TREM2-DAP12 elicits the PI3K-AKT-mTOR signaling pathway, enhancing anabolic and energy production pathways required for microglia activation (Ulland et al., 2017). Thus, we asked whether SYK acts through a similar mechanism. First, we sorted microglia from SykiΔMG-5×FAD and Sykfl/fl-5×FAD mice (Figure S3A) and performed biochemical analyses. SYK deficiency was associated with reduced phosphorylation of AKT and its targets N-myc downstream regulated 1 (NDRG1) and GSK-3β, which are implicated in mTOR activation, protein synthesis and cell cycle progression (Figure 3A, B). Conversely, ERK was not activated in either genotype (Figure 3B). Microglia from SykiΔMG-5×FAD mice also had significantly reduced mitochondrial mass (Figure 3C, D). Further biochemical analysis was conducted in primary microglia cultures from neonatal mice with a constitutive deletion of Syk. Microglia were cultured with or without CSF1-containing medium (LCCM) to evaluate sensitivity to metabolic stress. SYK-deficient microglia had reduced phospho-AKT and the mTOR target phospho-S6 Kinase (S6K) regardless of the presence of CSF1, indicating a metabolic defect (Figure S3B). Thus, SYK-deficient microglia suffer impaired AKT-mTOR signaling. Concurrently, microglia activated autophagy. The LC3II /LC3I ratio was higher in SykiΔMG:5×FAD microglia, indicating increased autophagy (Figure 3E). Consistently, immunofluorescence analysis revealed accumulation of LC3+ autophagosomes in SykiΔMG:5×FAD microglia (Figure 3F, G and S3C). Transmission electron microscopy (TEM) corroborated that microglia from SykiΔMG-5×FAD mice contained abundant multivesicular/multilamellar structures indicative of autophagosomes, which were largely absent in the other genotypes (Figure 3H, I). TEM also showed that SykiΔMG:5×FAD microglia accumulated lipid-rich vesicles (Figure S3D, E). SYK deficiency may impair cholesterol efflux and lipid catabolism, a defect recently observed in TREM2-deficient microglia (Claes et al., 2021; Nugent et al., 2020; Safaiyan et al., 2021). We conclude that SYK-deficient microglia exhibit metabolic defects during Aβ pathology, such as impaired AKT-mTOR signaling, along with an increase in autophagy.
Figure 3. SYK deficiency impairs the PI3K-AKT-GSK3-β-mTOR axis, and augments autophagy in microglia of 5×FAD mice.
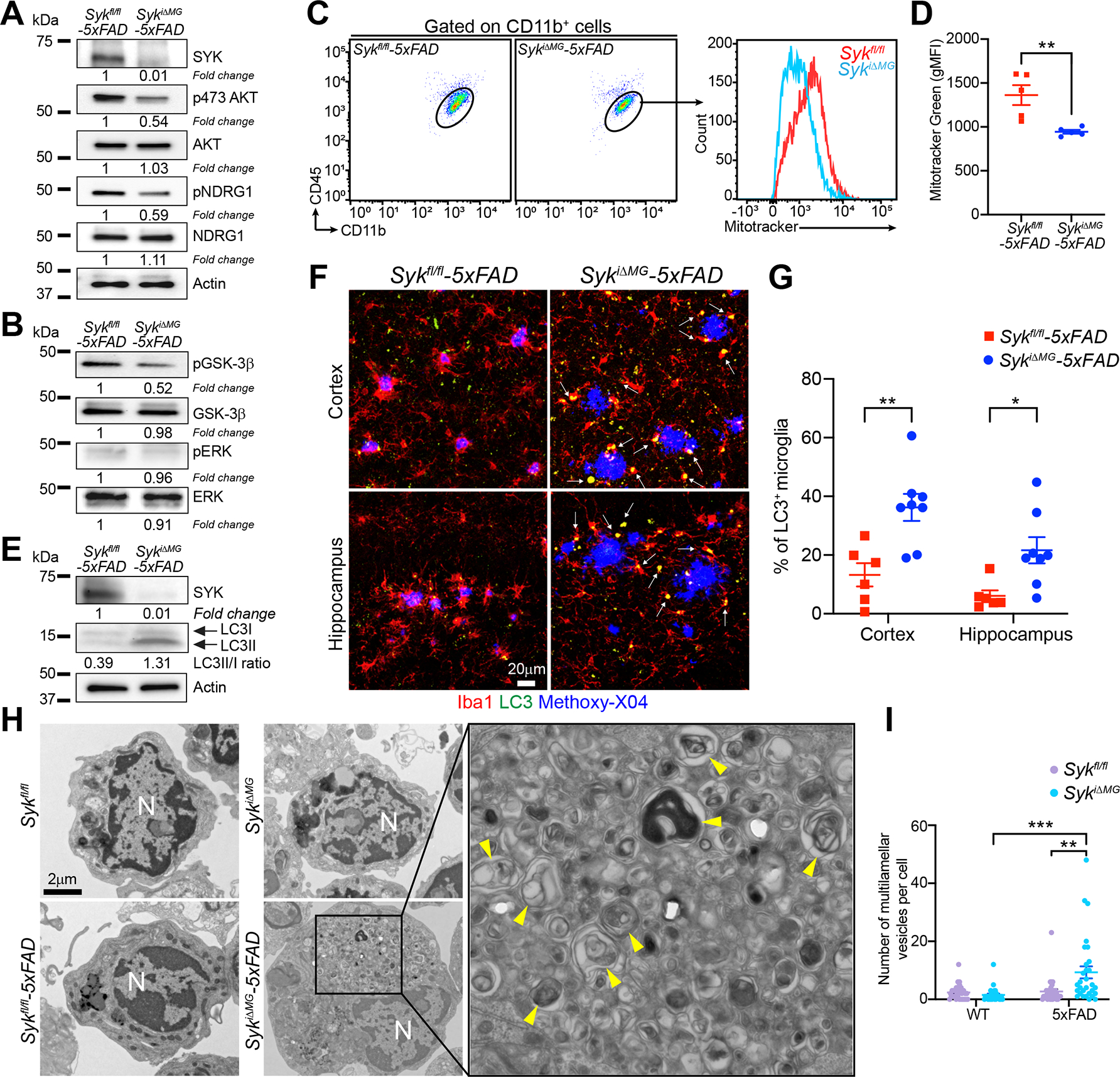
(A and B) Immunoblots of the indicated molecules on sorted microglia from 9-month-old Sykfl/fl-5×FAD and SykiΔMG-5×FAD mice. Representative of two independent experiments.
(C and D) Quantification of mitochondrial mass in microglia from 9-month-old Sykfl/fl-5×FAD and SykiΔMG-5×FAD mice assessed by MitoTracker Green incorporation (two independent experiments; n = 5 mice/genotype).
(E) Immunoblot of the LC3II/LC3I ratio on sorted microglia from 9-month-old Sykfl/fl- 5×FAD and SykiΔMG-5×FAD mice. Representative of two independent experiments.
(F) Representative confocal images of methoxy-X04, Iba1, and LC3 staining in the cortex and hippocampus of 9-month-old Sykfl/fl-5×FAD and SykiΔMG-5×FAD mice (arrows indicate microglial LC3+ vesicles).
(G) Percentage of LC3+ microglia in 9-month-old mice of each genotype. Data points represent the average of two technical repeats (one experiment; n = 6–8 mice/genotype).
(H) Representative TEM images of microglia sorted from 9-month-old Sykfl/fl and SykiΔMG mice, with or without 5×FAD transgenes (N = nucleus; yellow arrowheads = multivesicular structures).
(I) Number of multivesicular structures per microglia (n = 30 cells/genotype).
*, P < 0.05; **, P < 0.01; ***, P < 0.001 by two-tailed unpaired Student’s t test (D), or two-way ANOVA with Sidak’s multiple comparisons test (G and I). Data are presented as mean ± SEM.
See also Figure S3 in the supplemental information.
SYK is required for maintenance of microglial clustering around Aβ plaques
We asked whether SYK deficiency affects microglia fitness when induced during disease progression rather than before its onset. We fed SykiΔMG-5×FAD and Sykfl/fl-5×FAD control mice with TAM-containing chow from 4 to 5 months of age (Figure S4A), when mice have already developed Aβ deposits and microgliosis. One cohort of mice was analyzed at the end of TAM treatment (5 months of age), whereas a second cohort was returned to normal chow and analyzed at 9 months of age (Figure S4A). Quantification of plaque-associated microglia in cortex and hippocampus exposed subtle differences between genotypes at 5 months, whereas a significant reduction was observed in SykiΔMG-5×FAD mice at 9 months of age (Figure S4B–C). Furthermore, analysis of the colocalized Iba1 and methoxy-X04 staining revealed a reduction at both 5 and 9 months of age in cortex and hippocampus (Figure S4B–D). Lastly, we determined whether the total number of microglia was also affected by late Syk deletion. Quantification of PU.1+ nuclei revealed that microglia numbers were slightly but significantly reduced in SykiΔMG-5×FAD mice as early as 5 months of age. At 9 months, microglia numbers were elevated in both groups; however, SykiΔMG-5×FAD mice had significantly fewer microglia than did Sykfl/fl-5×FAD mice (Figure S4E, F). These results demonstrate that SYK is required for the maintenance of microglial clustering around Aβ plaques after the onset of disease.
SYK deficiency impairs generation of DAM
We examined the impact of SYK deficiency on microglial transcriptome during amyloid pathology by scRNA-seq. Single/live CD45+ cells were sorted from Sykfl/fl-5×FAD, Syk iΔMG-5×FAD, Sykfl/fl, and SykiΔMG cortices, and single-cell transcriptomes were generated using the 10x Genomics platform (Figure 4A). After quality control, a total of 89,615 single cells were plotted on uniform manifold approximation projection (UMAP) dimensions for visualization. Unsupervised clustering revealed a total of 15 distinct clusters across all mice (Figure S5A), which were annotated based on differential expression of known cell type-specific marker genes. The preponderance of cells captured by our scRNA-seq dataset were microglia, with significantly fewer BAMs, monocytes, T cells, B cells, and neutrophils (Figure S5B). None of the genotypes had a significant impact on non-microglial cell types.
Figure 4. SYK deficiency in microglia hampers microglial activation in 5×FAD mice.
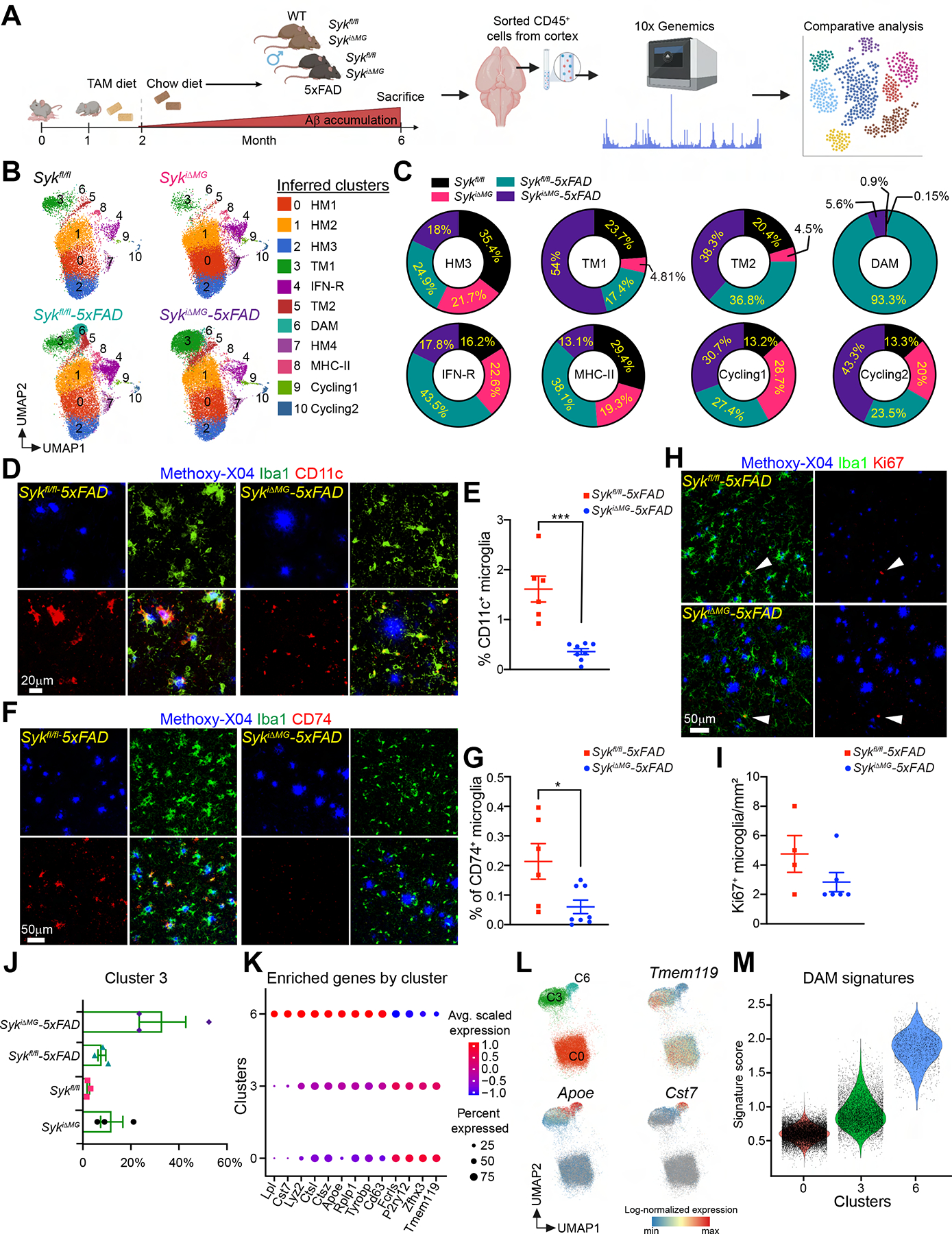
(A) Schematic of the experimental design.
(B) UMAP plots of microglia from each genotype (n = 3 mice/genotype). Cluster annotation based on expression of signature genes shown in Figure S5.
(C) Proportional contribution of each genotype to designated clusters.
(D and F) Representative confocal images of methoxy-X04, Iba-1 and CD11c or CD74 staining in the cortex of 6-month-old Sykfl/fl-5×FAD and SykiΔMG-5×FAD mice.
(E and G) Percentage of CD11c+ or CD74+ microglia in the cortex of each genotype. Data points represent the average of two technical repeats (one experiment; n = 6–8 mice/genotype).
(H) Representative confocal images of methoxy-X04, Iba-1, and Ki67 in the cortex of 6-month-old Sykfl/fl-5×FAD and SykiΔMG-5×FAD mice.
(I) Quantification of Ki67+ microglia from indicated mice. Data points represent the average of two technical repeats (representative of two independent experiments; white arrows indicate Ki67+ microglia; n = 4–6 mice/genotype).
(J) Relative abundance of cluster 3 (TM1) in all genotypes.
(K) Average scaled expression levels of selected signature genes for clusters 0 (HM1), 3 (TM1) and 6 (DAM).
(L) UMAP plots showing enrichment of selected signature in clusters 0 (HM1), 3 (TM1) and 6 (DAM).
(M) Violin plots of the DAM signature scores in clusters 0 (HM1), 3 (TM1) and 6 (DAM).
*, P < 0.05; ***, P < 0.001 by two-tailed unpaired Student’s t test. Data are presented as mean ± SEM.
See also Figure S5 and S6 in the supplemental information.
We next re-clustered microglia only, resulting in 11 distinct clusters comprising 57,830 cells (Figure 4B and S5C). Based on expression of specific marker genes (Chen and Colonna, 2021), we identified homeostatic (clusters 0, 1, 2, and 7), DAM (cluster 6), MHCII (cluster 8), IFN-R (cluster 4), and proliferating (cluster 9, 10) microglia (Figure 4B and S5C–H). We also detected two clusters of microglia with intermediate signatures between homeostatic and DAM, defined as transitioning microglia (TM1 and TM2, clusters 3 and 5), which may represent prodromal stages of DAM. We next examined the representation of DAM, TM1, TM2, MHCII, IFN-R, and proliferating microglia clusters in each genotype (Figure 4C). Notably, DAM appeared almost entirely represented in Sykfl/fl-5×FAD mice (93.3%), with a minimal contribution from SykiΔMG-5×FAD mice (5.6%). MHCII and IFN-R microglia were abundant in Sykfl/fl-5×FAD mice (38.1% and 43.5% respectively), but less represented in the other groups. All genotypes contributed similarly to cycling microglia, though Sykfl/fl contributed the least and SykiΔMG-5×FAD the most. Breakdown of TM1 among different genotypes showed that 54% of these cells were present in SykiΔMG:5×FAD microglia, while relatively small percentages were present in the other genotypes (Figure 4C). Thus, SYK deficiency in the 5×FAD background mainly affected the generation of DAM. However, TM1 microglia (bona fide prodromal to DAM) were expanded, and microglia cycling was unaffected, in fact elevated. This suggests that a basal microglia response to Aβ pathology is maintained in SykiΔMG-5×FAD mice, although microglia are not able to fully develop a DAM phenotype.
To validate these findings, we performed immunofluorescence staining for TMEM119 (homeostatic marker), TREM2, CLEC7A, CD11c (DAM markers), and CD74 (MHCII marker). Consistent with transcriptomic data, we found significantly fewer TREM2+ (Figure S6A–C), CLEC7A+ (Figure S6D, E), CD11c+ (Figure 4D, E), and CD74+ microglia (Figure 4F, G) in SykiΔMG-5×FAD mice, along with increased TMEM119+ microglia (Figure S6D, F). These changes were particularly evident in microglia adjacent to Aβ plaques (Figure S6D), whereas SYK deficiency had little impact on microglia distal to Aβ plaques. The morphology of non-plaque associated microglia was also similar in SykiΔMG-5×FAD and Sykfl/fl-5×FAD mice (Figure S6G). Immunofluorescence staining for the proliferation marker Ki67 exposed similar numbers of Ki67+ microglia in SykiΔMG-5×FAD and Sykfl/fl-5×FAD mice (Figure 4H, I), corroborating scRNA-seq data. We also delved into the characterization of TM1 cells of cluster 3, which were enriched in SykiΔMG-5×FAD mice (Figure 4J). Analysis of genes differentially expressed among cluster 3, homeostatic cluster 0, and DAM cluster 6, revealed that cluster 3 expressed higher levels of microglia activation genes (Apoe, Ctsz, Ctsl, and Lyz2) than did homeostatic microglia. However, these genes were expressed at lower levels in cluster 3 than in DAM. Conversely, the homeostatic genes Tmem119, P2ry12, and Fcrls were more highly expressed in cluster 3 than in DAM but at lower levels than in homeostatic microglia. DAM also expressed high levels of Cst7 and Lpl, which were not detected in either cluster 3 or cluster 0 (Figure 4K, L). Analysis of the DAM signature among clusters 0, 3 and 6 indicated that cluster 3 occupies a transitional state between homeostatic microglia and DAM (Figure 4M). Gene set enrichment analysis of differentially expressed genes in TM and DAM clusters from SykiΔMG:5×FAD and Sykfl/fl:5×FAD microglia showed that metabolic pathways were highly affected by SYK deficiency (Figure S6H, I). We conclude that SYK deficiency and the related metabolic defect prevent the acquisition of a complete DAM phenotype in response to Aβ pathology. However, SYK deficiency does not restrain microglia proliferation and allows accumulation of a microglial population expressing intermediate levels of both homeostatic and DAM genes that is prodromal to DAM.
SYK- and TREM2-deficient microglia exhibit distinct DAM trajectory defects
SYK is a critical component of TREM2 signaling, but is also required by other receptors (Hamerman et al., 2009; Mócsai et al., 2006). Therefore, we sought to examine how SYK-deficiency impacts microglia activation in comparison with TREM2-deficiency. To do so, we re-clustered our scRNA-seq data of SykiΔMG:5×FAD and Sykfl/fl:5×FAD microglia together with newly generated scRNA-seq data from Trem2+/+-5×FAD and Trem2−/−-5×FAD microglia. We identified 10 clusters recapitulating the same populations described above (Figure 5A, B). Because the MHC-II cluster (cluster 9) contained only 167 cells, this population was excluded from further analyses. We next analyzed the representation of the remaining microglial clusters in each genotype. Compared to their respective controls, both SykiΔMG and Trem2−/− microglia evinced a decrease in DAM (cluster 4), IFN-R (cluster 5), and TM2 (cluster 6, a DAM prodromal stage), whereas some homeostatic populations were expanded (particularly cluster 2). However, an increase in the TM1 transitional stage (cluster 3) was only evident in SykiΔMG microglia (Figure 5C). Moreover, cycling microglia (cluster 8) was depleted Trem2r−/−, but not in SykiΔMG microglia (Figure 5C). Pseudotime trajectory analysis corroborated that the homeostatic population differentiated into three terminal stages (DAM, IFN-R and cycling microglia). TM1 and TM2 stages appeared to precede DAM, but not IFN-R and cycling clusters (Figure 5D). We next considered the distribution of Trem2−/− and SykiΔMG microglia along these three trajectories. Trem2−/− microglia were associated with a complete blockade of all fates. By contrast, SykiΔMG microglia were partially inhibited along the DAM trajectory with an arrest at the transitional stage TM1, the IFN-R trajectory was moderately impaired, and no defect was observed in the cycling trajectory (Figure 5E). Thus, unlike Trem2−/− microglia, SykiΔMG microglia can proliferate and initiate a DAM differentiation program, but are unable to reach a DAM terminal stage, resulting in the accumulation of the prodromal stage TM1 (Figure 5F).
Figure 5. scRNA-seq reveals distinct defects in the DAM trajectories of SYK- and TREM2-deficient microglia.
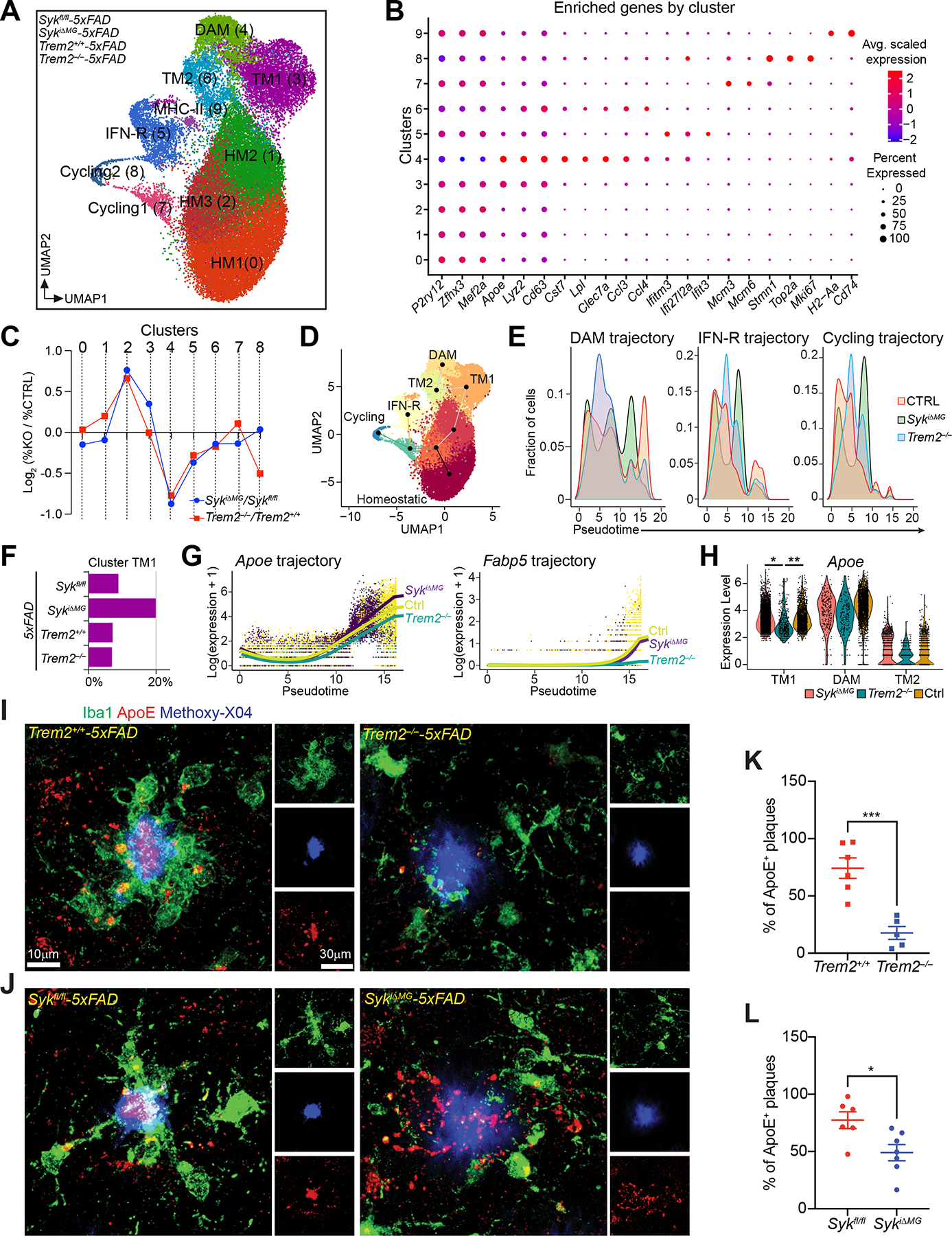
(A) UMAP plots of microglia from each genotype (n = 2–3 mice/genotype).
(B) Average scaled expression levels of selected signature genes per cluster.
(C) Log2 fold-change of cluster sizes in SykiΔMG-5×FAD and Trem2−/−-5×FAD respective to controls (Sykfl/fl-5×FAD and Trem2+/+-5×FAD).
(D) UMAP plot showing the projection of the three identified phenotypic trajectories.
(E) Pseudotime distribution of microglia from each genotype along DAM, IFN-R, and cycling trajectories. CTRL group is a pool of microglia from Sykfl/fl-5×FAD and Trem2+/+-5×FAD mice.
(F) Relative abundance of cluster TM1 for each genotype.
(G) Expression levels of Apoe and Fabp5 along the DAM trajectory for indicated genotypes. CTRL group is a pool of microglia from Sykfl/fl-5×FAD and Trem2+/+-5×FAD mice.
(H) Violin plots comparing Apoe expression among indicated genotypes in selected clusters (TM1, TM2 and DAM). CTRL group is a pool of microglia from Sykfl/fl-5×FAD and Trem2+/+-5×FAD mice.
(I and J) Representative confocal images of methoxy-X04, Iba-1 and ApoE staining the cortex of 6-month-old Trem2+/+-5×FAD, Trem2−/−-5×FAD, Sykfl/fl-5×FAD and SykiΔMG-5×FAD mice.
(K and L) Percentage of ApoE-positive plaques in each genotype. Data points represent the average of two technical repeats (one experiment; n = 5–7 mice/genotype).
*, P < 0.05; **, P < 0.01; ***, P < 0.001 by two-tailed unpaired Student’s t test (K and L), or mixed model (H) (see METHOD DETAILS). Data are presented as mean ± SEM.
We then assessed the DEGs between these two genotypes along the DAM trajectory. We identified 16 DEGs, including the DAM signature genes Apoe and Fabp5, which were both reduced in Trem2−/− microglia compared to SykiΔMG and control microglia (Figure 5G). Focusing specifically on the TM1 cluster, Apoe was sharply enriched in SykiΔMG microglia, compared to Trem2−/− microglia (Figure 5H). Collectively, these data suggest that induction of Apoe is TREM2-dependent but SYK-independent, at least in part. Yet, absence of SYK impedes the acquisition of a complete DAM signature. To further corroborate this conclusion, we performed immunofluorescence staining for ApoE on brain sections from all analyzed groups (Figure 5I, J). Both Trem2−/−-5×FAD and SykiΔMG- 5×FAD mice had a reduced percentage of ApoE+ plaques compared to respective controls, which is consistent with the significant reduction of DAMs in both genotypes. However, reduction in ApoE+ plaques was more significant in Trem2−/−-5×FAD mice than in SykiΔMG-5×FAD mice (−76.2% vs −36.7% respectively) (Figure 5K, L). Moreover, the staining pattern of plaque-associated ApoE in SykiΔMG-5×FAD mice appeared more diffuse than in Sykfl/fl-5×FAD mice (Figure 5J). This may be due to the defective clustering of SykiΔMG microglia and/or tempered ApoE production. We conclude that Aβ pathology may elicit a TREM2-dependent, yet SYK-independent, pathway in microglia that is sufficient to support both proliferation and advancement to a prodromal stage of DAM, which is demarcated by simultaneous expression of homeostatic genes and Apoe.
DAP10 activates microglia independently of SYK
We sought to determine how TREM2 activates microglia independently of SYK. It has been shown that the TREM2-DAP12 complex modulates ERK activation through a pathway independent of SYK (Peng et al., 2010). We asked whether this pathway could induce the large Apoe+ TM1 cluster observed in SYK-deficient microglia. To test this, we used DAP12 knock-in mice (known as Dap12KΔY75), which harbor a deletion in the cytoplasmic motif of DAP12 that disrupts signaling without affecting pairing with TREM2 and trafficking to the cell membrane (Tomasello et al., 2000). We then crossed Dap12KΔY75 with 5×FAD mice and analyzed microglia from 6-month-old Dap12KΔY75-5×FAD and 5×FAD mice by scRNA-seq. Dap12KΔY75:5×FAD microglia evinced a phenotype mirroring that of microglia in SykiΔMG:5×FAD mice - i.e. preserved cycling microglia and accumulation of Apoe+ TM1 population (Figure 6A, B). Biochemical analysis of bone marrow-derived macrophages (BMDM) from Dap12KΔY75, Trem2−/− and SykcΔMG mice showed that lack of either TREM2 or DAP12, but not SYK, impacted ERK activation (Figure 6C), corroborating that the microglia phenotype induced by SYK deficiency is independent of any signal dependent on the TREM2-DAP12 axis.
Figure 6. DAP10 activates microglia independently of SYK.
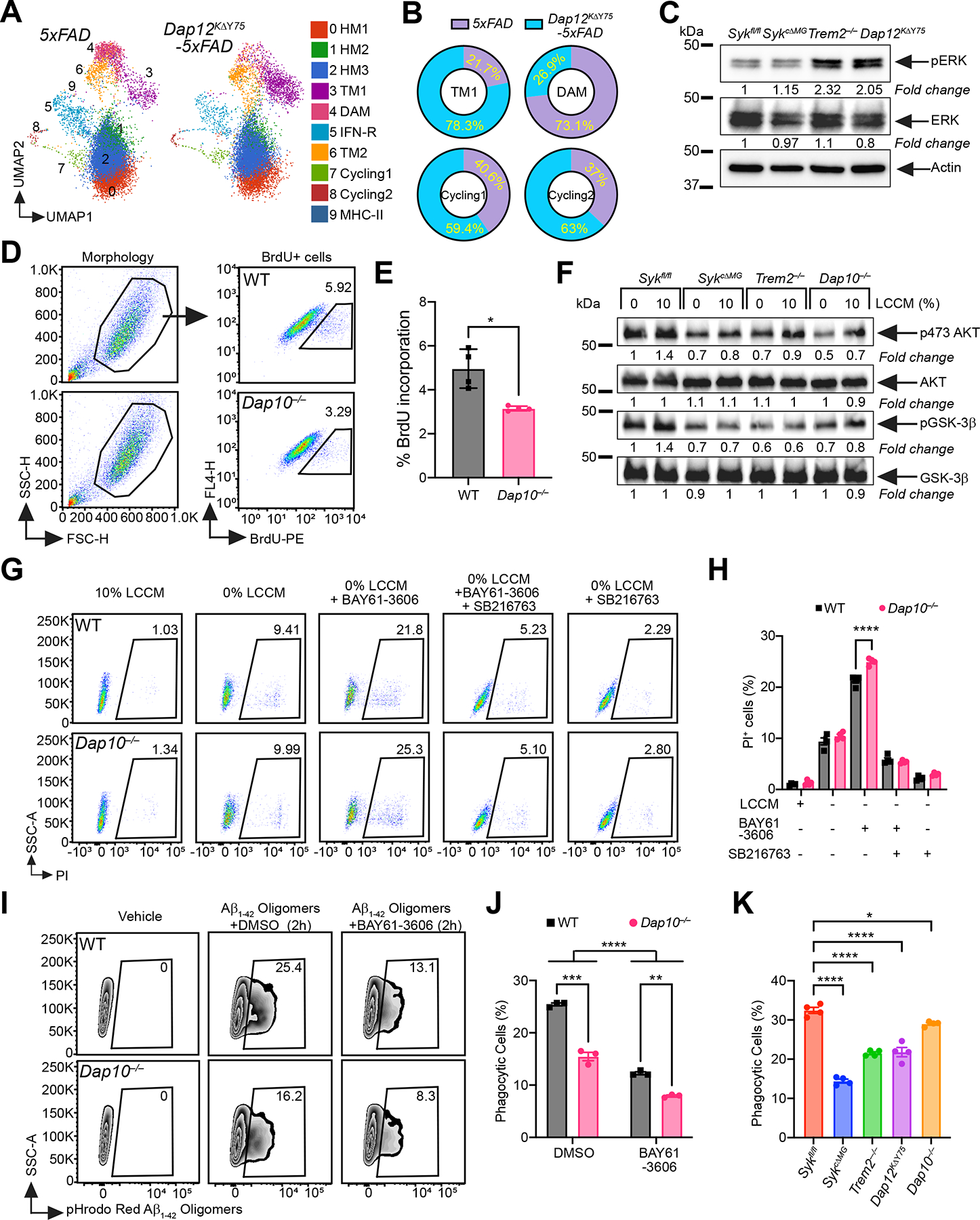
(A) UMAP plots of microglia from each genotype (n = 3–4 mice/genotype). Cluster annotation based on expression of signature genes shown in Figure 5B and S5.
(B) Proportional contribution of each genotype to designated clusters.
(C) Immunoblots of ERK and pERK on BMDM from indicated genotypes. Representative of two independent experiments.
(D) Representative FACS plots of BrdU staining on WT and Dap10−/− primary microglia.
(E) Percentage of BrdU+ microglia in each genotype. Representative of three independent experiments (n = 4 technical repeats/genotype).
(F) Immunoblots of AKT, pSer473 AKT, GSK3-β and pGSK3-β on cultured primary microglia from indicated genotypes, with or without 10% LCCM stimulation. Representative of two independent experiments.
(G) Representative FACS plots of PI staining on WT and Dap10−/− BMDM, cultured 48 hours under five different conditions: 1) 10% LCCM; 2) 0% LCCM; 3) 0% LCCM + BAY61–3606 (SYK inhibitor); 4) 0% LCCM + BAY61–3606 + SB216763 (GSK-3β inhibitor); 5) 0% LCCM + SB216763.
(H) Percentage of dead BMDMs in each genotype, at the indicated conditions. Representative of three independent experiments (n = 4 technical repeats/group).
(I) Representative FACS plots of WT and Dap10−/− BMDMs cultured with pHrodo-Red labeled Aβ1–42 for 2 hours, with or without SYK inhibitor.
(J) Quantification of Aβ1–42 uptake by BMDMs from each genotype, with or without SYK inhibitor. Representative of three independent experiments (n = 3 technical repeats/group).
(K) Same experiment as in (I and J), on BMDMs of indicated genotypes. Representative of three independent experiments (n = 4 technical repeats/genotype).
*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 by two-tailed unpaired Student’s t test (E), two-way ANOVA with Sidak’s multiple comparisons test (H and J), or one-way ANOVA with Tukey’s multiple comparisons test (K). Data are presented as mean ± SEM.
In addition to DAP12, TREM2 associates with the transmembrane adapter DAP10 (Hcst) in BMDM (Peng et al., 2010). DAP10 is similar to DAP12 but lacks a canonical cytoplasmic ITAM. In fact, it contains a cytoplasmic Y-x-N-M motif that directly recruits PI3K and the adapter GRB2 (Lanier, 2009). Thus, we asked whether TREM2-dependent SYK-independent microglia activation requires DAP10. We cultured microglia from Dap10−/− and wild-type neonatal mice pulsed with BrdU and showed that lack of DAP10 impaired microglia proliferation (Figure 6D, E). Biochemical analysis of microglia cultures from Dap10−/−, Trem2−/−, SykcΔMG and Sykfl/fl mice showed that DAP10-deficiency affected phosphorylation of AKT and its target GSK-3β (Figure 6F). Additionally, Dap10−/− BMDM were more sensitive to the pro-apoptotic effect of the SYK inhibitor BAY61–3606 than wild-type BMDM. The survival defect was completely reversed by a pharmacological inhibitor of GSK-3β, promoting cell survival and proliferation (Eldar-Finkelman, 2002) (Figure 6G, H). Additionally, Dap10−/− BMDM cultured with Aβ oligomers appeared less phagocytic than wild-type BMDM, while SYK inhibition reduced phagocytic capacity in both groups (Figure 6I, J). Comparing Dap10−/−, Trem2−/−, Dap12KΔY75, SykcΔMG and Sykfl/fl BMDM, we confirmed that DAP10 deficiency curtailed phagocytosis, albeit to a lesser extent than did lack of either SYK, TREM2, or DAP12 signaling (Figure 6K). These data demonstrate that DAP10 signaling sustains microglia proliferation, metabolic activation, survival and Aβ phagocytosis independently of SYK, while both pathways are required for full microglia activation.
Anti-CLEC7A improves microglia activation in 5×FAD mice carrying a TREM2 hypofunctional variant
We previously described a reduced microglia response to Aβ pathology in 5×FAD mice carrying the human TREM2R47H variant rather than the common variant (TREM2CV) (Song et al., 2018). Since the TREM2R47H variant is unable bind ligands and activate SYK, we attempted to rescue SYK activation in this mouse model by engaging an alternative surface receptor that recruits SYK. We chose CLEC7A (DECTIN1), a member of the C-type lectin receptor family that is expressed in neutrophils, dendritic cells, alveolar and peritoneal macrophages (Brown et al., 2002), and is upregulated in microglia during neurodegeneration (Herre et al., 2004; Krasemann et al., 2017; Mass et al., 2017). CLEC7A recognizes β-glucan, a component of the fungal wall, promoting antifungal responses in macrophages (Brown et al., 2002; Underhill et al., 2005). The CLEC7A cytoplasmic domain contains a hemi-ITAM domain that directly recruits and activates SYK upon dimerization (Herre et al., 2004; Rogers et al., 2005; Underhill et al., 2005). To engage CLEC7A in microglia during Aβ pathology, we initially evaluated a recombinant Fc-mutated variant of the anti-CLEC7A monoclonal antibody (mAb) 2A11 (Brown et al., 2002): mAb 2A11 specifically stained microglia cultured from neonatal mice (Figure 7A); moreover, 2A11 mAb-mediated cross-linking of CLEC7A in cultured microglia induced phosphorylation of SYK and S6K (Figure 7B), indicating that this mAb activates CLEC7A in microglia. To test whether CLEC7A engagement can enhance Aβ phagocytosis, TREM2R47H-5×FAD mice were injected i.p. with a single dose of 2A11 mAb or isotype control. Twenty-four hours after treatment, mice were injected with methoxy-X04 and microglia were analyzed by flow cytometry. More methoxy-X04+ microglia were found in the 2A11 mAb- than in the control mAb-treated group, indicating increased Aβ uptake (Figure 7C, D). We next mimicked short-term treatment by injecting TREM2R47H-5×FAD mice i.p. with mAb 2A11 or isotype control for 10 days. Twenty-four hours after the final injection, mice were analyzed by imaging (Figure 7E). Mice receiving the 2A11 mAb exhibited a significant increase of Iba1 (Figure 7F, G) and CD11c staining (Figure 7H, I), indicating more robust microglia activation. Increased expression of Itgax (encoding CD11c) was validated by RT-PCR in brain lysates (Figure 7J). Although short-term treatment did not affect the overall Aβ1–42 load in cortex and hippocampus (Figure S7A, B), 2A11 mAb -treated mice had fewer filamentous plaques and more dynamic and inert plaques than did control mice (Figure S7C, D). Thus, CLEC7A engagement in microglia with hypofunctional TREM2 can, at least, partially rescue microglial responses to Aβ (Figure 7K). Lastly, we mined published snRNA-seq data of human AD and age matched controls for expression of SYK and CLEC7A mRNA using TREM2 for comparison (Zhou et al., 2020). We confirmed that SYK and CLEC7A are highly expressed in human microglia from both AD and control subjects (Figure 7L). Thus, the CLEC7A-SYK pathway in microglia might be a potential therapeutic target for AD.
Figure 7. Acute treatment with anti-CLEC7A induces microglial activation in TREM2R47H-5×FAD mice.
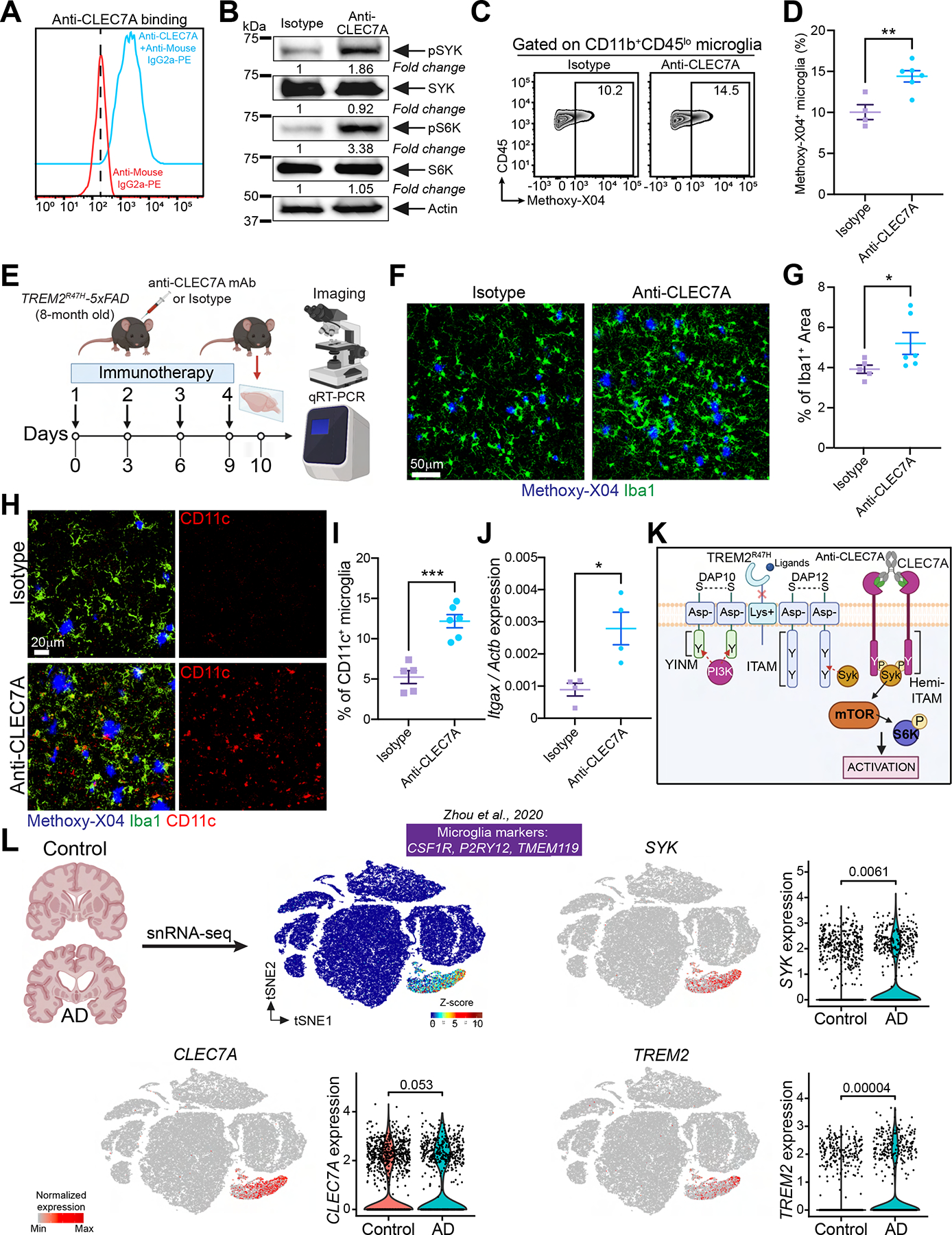
(A) Cultured WT primary microglia from P4 neonates stained with anti-CLEC7A. Cells stained only with secondary Ab served as negative control. Representative of three independent experiments.
(B) Immunoblot of SYK, pSYK, S6K and pS6K on primary microglia treated with either isotype control or anti-CLEC7A. Representative of two independent experiments.
(C) Representative FACS plots of methoxy-X04+ microglia from TREM2R47H-5×FAD mice, injected ip. with either isotype control or anti-CLEC7A. WT mice served as a negative control to set the methoxy-X04+ gate.
(D) Percentage of methoxy-X04+ microglia in the tested groups (two independent experiments; n = 4–6 mice/group).
(E) Schematic of the experimental design.
(F) Representative confocal images of Iba1 and methoxy-X04 staining in the cortex of TREM2R47H-5×FAD mice injected with either isotype control or anti-CLEC7A.
(G) Percentage of Iba1+ area in the tested groups. Data points represent the average of two technical repeats (one experiment; n = 5–6 mice/group).
(H) Representative confocal images of methoxy-X04, Iba-1 and CD11c in the cortex of TREM2R47H-5×FAD mice injected with either isotype control or anti-CLEC7A.
(I) Percentage of CD11c+ microglia in the tested groups. Data points represent the average of two technical repeats (one experiment; n = 5–6 mice/group).
(J) Itgax mRNA levels assessed by qRT-PCR in whole cortical tissue from TREM2R47H-5×FAD mice injected with either isotype control or anti-CLEC7A (one experiment; n = 4 mice/group).
(K) Cartoon summarizing the hypothetical pharmacodynamics of anti-CLEC7A in microglia.
(L) tSNE plots of human brain cells from AD patients and control subjects analyzed by snRNA-seq. Expression of SYK, CLEC7A and TREM2 is displayed.
*, P < 0.05, **, P < 0.01, ***, P < 0.001 by two-tailed unpaired Student’s t test (D, G, I and J), or Wilcoxon rank-sum test (L). Data are presented as mean ± SEM.
See also Figure S7 in the supplemental information.
DISCUSSION
This study demonstrates that SYK signaling is a central node in microglia responses to Aβ pathology. Conditional deletion of SYK in microglia prior to or after disease onset dramatically impaired the ability of microglia to encase Aβ plaques, resulting in augmented Aβ deposition, increased neuronal damage, and worsened behavioral deficits, suggesting that SYK is required for generation and maintenance of microglia responses to Aβ. Mechanistically, SYK deficiency impaired the activation of the PI3K-AKT-GSK-3β-mTOR axis. In parallel, SYK deficient microglia escalated autophagy, which may provide an alternative energetic pathway. Using scRNA-seq, we resolved intermediate and terminal stages of the microglial response to Aβ, showing that SYK-deficient microglia fail to acquire DAM profiles. SYK function in the microglial response to Aβ strikingly mimics that of Drosophila Shark, an analogous non-receptor tyrosine kinase that is recruited to the phagocytic receptor Draper through an ITAM and is required for phagocytosis of neuronal cell corpses by glia (Ziegenfuss et al., 2008). This similarity epitomizes a highly conserved mechanism to clear protein aggregates and cellular debris.
We revealed a notable discrepancy between TREM2-deficient and SYK-deficient microglia at the transcriptomic level. While both SYK and TREM2 deficiencies did impede the generation of DAM, SYK deficiency led to the expansion of a microglia cluster with an intermediate phenotype prodromal to DAM. This suggests that TREM2 activates microglia through a SYK-dependent and a yet unexplored SYK-independent pathway. We provided evidence that TREM2 can sustain SYK-independent microglia activation through DAP10, which directly recruits PI3K (Peng et al., 2010). Accordingly, DAP10 deficiency affected phosphorylation of AKT and GSK-3β and moderated microglia proliferation, survival and phagocytosis.
Lastly, we demonstrated that systemic administration of an agonistic anti-CLEC7A antibody to 5×FAD mice carrying the hypofunctional TREM2R47H variant promotes SYK signaling and improves microglia activation. Since both CLEC7A and SYK are expressed in human microglia, CLEC7A emerges as a potential therapeutic target to boost microglia activation in AD. Thus, therapeutic activation of microglia in AD may be achieved by using agonists for the broad range of receptors that signal through SYK.
Limitations of the study
Our study of SYK-deficient microglia is limited to a prototypic mouse model of Aβ accumulation. While we showed for the first time an impact of SYK and DAP10 signaling on microglia responses to amyloid pathology, how these pathways orchestrate microglia functions remains unclear. Finally, it will be important to determine therapeutic and side effects of the anti-CLEC7A mAb dosing regimen.
STAR + METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Marco Colonna (mcolonna@wustl.edu).
Materials Availability
Materials and reagents used in this study are listed in the Key Resources Table. Reagents generated in our laboratory in this study or previous studies are available upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD45-PE (clone 30-F11) | BioLegend | Cat #103106 |
| CD45-APCCy7 (clone 30-F11) | BioLegend | Cat #103116 |
| CD11b-APC (clone M1/70) | BioLegend | Cat #101212 |
| Ly6C-APCCy7 (clone HK1.4) | BioLegend | Cat #128026 |
| Ly6G-FITC (clone 1A8) | BioLegend | Cat #127606 |
| P2RY12-BV421 (clone S16001E) | BioLegend | Cat #392106 |
| Syk-PE (clone 5F5) | BioLegend | Cat #646004 |
| BrdU-PE (Bu20a) | BioLegend | Cat #339812 |
| anti-CLEC7A antibody-Fc mutated (clone 2A11) | This paper | N/A |
| anti-human ILT1-Fc mutated (clone 135.5) | In house | (Molgora et al., 2020) |
| anti-mouse IgG2a-PE | SouthernBiotech | Cat #1080-09 |
| anti-phospho Syk (rabbit monoclonal) | Cell Signaling Technology | Cat #2717 |
| anti-Syk (rabbit monoclonal) | Cell Signaling Technology | Cat #13198 |
| anti-phospho Akt 473 (rabbit monoclonal) | Cell Signaling Technology | Cat #4060 |
| anti-Akt (rabbit monoclonal) | Cell Signaling Technology | Cat #4691 |
| anti-phospho NDRG1 (rabbit polyclonal) | Cell Signaling Technology | Cat #3217 |
| anti-NDRG1 (rabbit polyclonal) | Cell Signaling Technology | Cat #5196 |
| anti-phospho GSK3-β (rabbit polyclonal) | Cell Signaling Technology | Cat #9336 |
| anti-GSK3-β (rabbit monoclonal) | Cell Signaling Technology | Cat #12456 |
| anti-phospho ERK (rabbit polyclonal) | Cell Signaling Technology | Cat #9101 |
| anti-ERK (rabbit monoclonal) | Cell Signaling Technology | Cat #4695 |
| anti-phospho S6K (rabbit monoclonal) | Cell Signaling Technology | Cat #9234 |
| anti-S6K (rabbit polyclonal) | Cell Signaling Technology | Cat #9202 |
| anti-LC3 (rabbit polyclonal) | Cell Signaling Technology | Cat #2775 |
| anti-beta-actin-HRP (mouse monoclonal) | Santa Cruz Biotechnology | Cat #sc-47778 HRP |
| anti-rabbit IgG, HRP | Cell Signaling Technology | Cat #7074 |
| anti-Iba1 (rabbit monoclonal) | Cell Signaling Technology | Cat #17198 |
| anti-Iba1 (goat polyclonal) | Abcam | Cat #5076 |
| anti-CD68 (rat monoclonal) | BioLegend | Cat #137002 |
| anti-Pu.1 (rabbit monoclonal) | Cell Signaling Technology | Cat #2258 |
| anti-Aβ1–42 (rabbit recombinant monoclonal) | Thermo Fisher Scientific | Cat #700254 |
| anti-Aβ1–16 (6E10) | BioLegend | Cat #803013 |
| anti-Lamp1 (rat Monoclonal) | BioLegend | Cat #121602 |
| anti-APP (22C11 mouse mAb) | Millipore | Cat #MAB348 |
| anti-LC3-Alexa Fluor® 488 (rabbit monoclonal) | Cell Signaling Technology | Cat #13082 |
| anti-CD11c (rabbit monoclonal) | Cell Signaling Technology | Cat #97585 |
| anti-CD74-Alexa Fluor® 647 (rat monoclonal) | BioLegend | Cat # 151004 |
| anti-Ki67 (rabbit polyclonal) | Abcam | Cat #15580 |
| anti-ApoE (biotinylated HJ6.3) | Holtzman Lab | (Xiong et al., 2021) |
| anti-Trem2 (polyclonal sheep) | R&D Systems | Cat #AF1729 |
| anti-Tmem119 (rabbit monoclonal) | Cell Signaling Technology | Cat #90840 |
| anti-Clec7a (rat monoclonal) | InvivoGene | Cat #mabg-mmcl |
| anti-goat IgG Alexa-Fluor®488 (donkey polyclonal) | Abcam | Cat #150129 |
| anti-rabbit IgG Alexa Fluor® 555 (donkey polyclonal | Abcam | Cat #150074 |
| anti-goat IgG Alexa Fluor® 647 (donkey polyclonal) | Abcam | Cat #150131 |
| anti-rabbit IgG Alexa Fluor® 647 (goat recombinant polyclonal) | Invitrogen | Cat #A-21244 |
| anti-rat IgG Alexa Fluor®647 (chicken polyclonal) | Invitrogen | Cat # A-21472 |
| anti-sheep IgG Alexa Fluor® 555 (donkey polyclonal) | Abcam | Cat #150178 |
| Streptavidin-Alexa Fluor® 647 | BioLegend | Cat #405237 |
| goat F(ab’)2 anti-mouse IgG(H+L) (polyclonal) | SouthernBiotech | Cat #1032–01 |
| Bacterial and virus strains | ||
| pFUSEmIgG2A-Fc1 vector expressing a DNA fragment encoding heavy chain variable and CH1 regions of anti-CLEC7A antibody (clone 2A11) | This paper | N/A |
| Fc-null pFUSE-mIgG2A-Fc1 vector expressing a DNA fragment encoding the light chain of anti-CLEC7A antibody (clone 2A11) | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| MitoTracker Green | Invitrogen | Cat #M7514 |
| Aqua fluorescent reactive dye | Life technologies | Cat #L34966 |
| Methoxy-X04 | Tocris Biosciences | Cat #4920 |
| Propidium Iodide | Invitrogen | Cat # P1304MP |
| BrdU | Invitrogen | Cat # B23151 |
| β-amyloid (1–42) peptide - unlabeled | Anaspec | Cat #AS-20276 |
| SuperSignal West Pico PLUS Chemiluminescent substrate | Thermo Fisher Scientific | Cat #34579 |
| TO-PRO-3 iodide | Life Technologies | Cat #T3605 |
| BAY61–3606 | Selleckchem | Cat #S7006 |
| SB216763 | Tocris Biosciences | Cat #1616 |
| Critical commercial assays | ||
| Foxp3/Transcription Factor Staining Buffer Set | Invitrogen | Cat #00-5523-00 |
| BD Pharmingen BrdU Flow Kit | BD Biosciences | Cat #557892 |
| pHRodo iFL Labeling kit | Invitrogen | Cat #P36014 |
| miRNeasy Mini Kit | QIAGEN | Cat #217084 |
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat #4368814 |
| SYBR Green real-time PCR master mix | Applied Biosystems | Cat # 4309155 |
| Deposited data | ||
| 10x Single Cell RNA-seq: Sykfl/fl, Sykfl/fl-Cx3cr1CreERT2, Sykfl/fl-5×FAD, 5×FAD transgenic mice, Dap12KΔY75-5×FAD and Trem2−/−-5×FAD | This paper | GSE210258 |
| Experimental models: Cell lines | ||
| Primary murine microglia | This paper | N/A |
| Primary murine BMDM | This paper | N/A |
| Expi293F Cells | Thermo Fisher Scientific | Cat #A14527 |
| Experimental models: Organisms/strains | ||
| C57BL6/J | Jackson Laboratory | Cat #000664 |
| 5×FAD transgenic mice | Jackson Laboratory | Cat #034840-JAX |
| Cx3cr1Cre | Jackson Laboratory | Cat #025524 |
| Cx3cr1CreERT2 | Jackson Laboratory | Cat #021160 |
| Sykfl/fl | Jackson Laboratory | Cat #017309 |
| Sykfl/fl-5×FAD | This paper | N/A |
| Sykfl/fl-Cx3cr1Cre | This paper | N/A |
| Sykfl/fl-Cx3cr1CreERT2 | This paper | N/A |
| Sykfl/fl-Cx3cr1 CreERT2-5×FAD | This paper | N/A |
| Dap10−/− | In house | (Gilfillan et al., 2002) |
| Dap12KΔY75 | In house | (Tomasello et al., 2000) |
| Dap12KΔY75-5×FAD | This paper | N/A |
| Trem2−/− | In house | (Wang et al., 2015) |
| Trem2−/−-5×FAD | In house | (Wang et al., 2015) |
| Trem2R47H-5×FAD | In house | (Song et al., 2018) |
| Oligonucleotides | ||
| Itgax qPCR FW: CTGGATAGCCTTTCTTCTGCTG RV: GCACACTGTGTCCGAACTCA |
This paper | N/A |
| Actin qPCR FW: GGAGGGGGTTGAGGTGTT RV: TGTGCACTTTTATTGGTCTCAA |
This paper | N/A |
| Recombinant DNA | ||
| anti-CLEC7A antibody-Fc mutated plasmids (clone 2A11) | This paper | N/A |
| anti-human ILT1-Fc mutated plasmids (clone 135.5) | In house | (Molgora et al., 2020) |
| Software and algorithms | ||
| MATLAB | MathWorks | N/A |
| ImageJ | National Institutes of Health | N/A |
| Bio-Rad ChemiDoc MP imaging system | Bio-Rad Laboratories | N/A |
| AMT Image Capture Engine V602 software | Advanced Microscopy Techniques | N/A |
| FlowJo | FlowJo | www.flowjo.com/ |
| Imaris 7.7 | Bitplane | N/A |
| Cell Ranger Software Suite (v6.0.0) | 10x Genomics | N/A |
| Seurat (v4.0) | (Hao et al., 2021) | N/A |
| R (v4.1.0) | R Core Team (2017). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL http://www.R-project.org/. | N/A |
| BBrowser v2.7.5 | BioTuring Inc. | www.bioturing.com/bbrowser |
| slingshot (v2.1.0) | (Street et al., 2018) | N/A |
| tradeSeq (v1.6) | (Van den Berge et al., 2020) | N/A |
| GraphPad Prism 9 | GraphPad | www.graphpad.com |
| Other | ||
| TAM Diet (500, 2016) | Envigo | TD.130857 |
Data and Code Availability
The accession number for the data reported in this paper is GE0:GSE210258.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Mice were of mixed sexes. Mice within experiments were age and sex matched. Sykfl/fl- and SykiΔMG-5×FAD mice were generated by crossing the Sykfl/fl-Cx3cr1CreERT2/+ mice with Sykfl/fl-5×FAD. Sykfl/fl mice were also crossed with Cx3cr1Cre mice to ensure constitutive Syk deletion, named SykcΔMG in contrast to TAM-inducible Sykfl/fl-Cx3cr1CreERT2 mice referred to as Syki΄MG mice, that were used for in vitro experiments. Trem2−/−, Dap12KΔY75, Dap10−/− and Trem2R47H-5×FAD mice were previously described (Gilfillan et al., 2002; Song et al., 2018; Tomasello et al., 2000; Wang et al., 2015). For generation of bone marrow-derived macrophages (BMDM) 6–12 weeks old mice were used. Primary microglia for culture were isolated from pups at postnatal day 4. Mice used in this study were sacrificed at indicated time points. Mice were housed in the animal facilities of Washington University in St. Louis. All animal experiments were conducted in compliance with Institutional regulations, under authorized protocols # 19–0981 approved by the Institutional Animal Care and Use Committee.
Primary cells
Primary microglia cultures of high purity were generated as previously described (Wang et al., 2015) for Aβ phagocytosis assay, immunoblotting, BrdU proliferation assay and antibody binding assay. In brief, the neonatal mouse brain at P4 was dissected and dissociated by Trypsin (1.67 mg/mL) with Dnase (0.33 mg/mL). The mixed glial culture was resuspended in the complete DMEM medium (supplemented with 1 × GlutaMAX™ supplement, 100 U/mL penicillin-streptomycin, 1 mM sodium pyruvate) containing 10% heat-inactivated fetal bovine serum (FBS) and seeded on PLL-coated T25 flask. From day 5, floating cells (primary microglia) were collected and seeded in 24-well plates at the density of 2 × 105 cells per well in the complete DMEM medium containing 10% heat-inactivated FBS and 10% of L-929 cell conditioned medium (LCCM) for further experiments.
To prepare BMDM for immunoblotting, survival and Aβ phagocytosis assays, bone marrow was collected from femurs and tibias and cells were plated at 5 × 106 cells/100-mm Petri dish in complete RPMI medium (supplemented with 1 × GlutaMAX™ supplement, 100 U/mL penicillin-streptomycin, 1 mM sodium pyruvate) with 10% heat-inactivated FBS and 10% LCCM. Cells were cultured for 5 days before use.
METHOD DETAILS
Mice
For generation of Sykfl/fl- and SykiΔMG-5×FAD mice, 1- or 4-month-old littermates were fed a tamoxifen (TAM) diet for 4 weeks followed by replacement with regular chow. Treated mice were sacrificed at 6-, 9- or 5-, 9-months of age as indicated in the RESULTS. To determine fibrillar Aβ phagocytosis by microglia in vivo, 6-month-old Sykfl/fl- and SykiΔMG-5×FAD mice were intraperitoneally injected with methoxy-X04 (Tocris) at 10 mg/kg in a PBS/DMSO mixture. Mice were sacrificed after 3 hours of injection and brains were made into single-cell suspension. For the short term of anti-CLEC7A antibody treatment, 8-month-old Trem2R47H-5×FAD mice received intraperitoneal injection of anti-CLEC7A antibody (2A11), or anti-human ILT1 antibody (Fc mutated, clone 135.5) (Molgora et al., 2020) as a control, for 4 times within 10 days. Mice were sacrificed within 24 hours after the last injection for assessment. For testing the in vivo effect of 2A11 treatment on fibrillar Aβ phagocytosis by microglia Trem2R47H-5×FAD mice were first injected ip. with a single dose of 2A11 followed by ip. injection of methoxy-X04 after 24 hours.
Flow cytometry
Cells were incubated with Fc block prior to staining. The following antibodies were used in the study: CD45-PE or -APCCy7 (clone 30-F11), CD11b-APC (clone M1/70), Ly6C-APCCy7 (clone HK1.4), Ly6G-FITC (clone 1A8); P2RY12-BV421 (clone S16001E), Syk-PE (clone 5F5), BrdU-PE (Bu20a). Cells, stained with indicated antibodies for 30 min on ice, were acquired (LSR FORTESSA, Canto II, Calibur 3, BD Bioscience) and analyzed using FlowJo software. Cellular mitochondrial mass was assessed by using MitoTracker Green. For antibody binding assay, cells were stained with anti-CLEC7A (2A11) for 30 min on ice followed by a secondary anti-mouse IgG2a-PE staining for 30 min at 4°C. Cells stained by anti-mouse IgG2a-PE only were used as a control. Cell viability was determined by Aqua LIVE/Dead-405 nm staining (Invitrogen) or DAPI, and negative cells were considered viable. Propidium iodide (PI) was used for survival assay. Foxp3/Transcription Factor Staining Buffer Set (eBioscience) was used for intracellular staining.
Cell sorting
Cells from brain were isolated from the indicated animals as previously described (Wang et al., 2015). CD45loCD11b+ microglia or CD45+ cells in the brain were fluorescence-activated cell-sorted (FACS Aria II, BD Bioscience) directly into 2%FBS in PBS for TEM or immunoblot lysates or 0.04% bovine serum albumin (BSA) for single-cell RNAseq.
Immunoblotting
Sorted microglia, cultured primary microglia, or BMDM were lysed in cell lysis buffer (Cell Signaling Technology) for 20–30 min on ice. Cell extract was centrifuged at 12000 rpm for 15 min and supernatant was collected and subjected to protein estimation. Equal amount of protein from each sample mixed in 4 × Laemmli sample buffer was loaded on 4–20% mini protein TGX gels from Bio-Rad and run using Tris-Glycine-SDS buffer. Resolved proteins were transferred on PVDF membranes. Membranes were washed using TBST (Tris-buffered-saline with 0.1% tween 20), blocked using 5% BSA or non-fat dried milk and incubated in primary antibody overnight at 4°C. The following primary antibodies were used in the study: anti-phospho Syk (1:500, rabbit monoclonal, Cell Signaling Technology), anti-Syk (1:1000, rabbit monoclonal, Cell Signaling Technology), anti-phospho Akt 473 (1:1000, rabbit monoclonal, Cell Signaling Technology), anti-Akt (1:1000, rabbit monoclonal, Cell Signaling Technology), anti-phospho NDRG1 (1:1000, rabbit polyclonal, Cell Signaling Technologies), anti-NDRG1 (1:1000, rabbit polyclonal, Cell Signaling Technology), anti-phospho GSK3- β (1:1000, rabbit polyclonal, Cell Signaling Technology), anti-GSK3-β (1:1000, rabbit monoclonal, Cell Signaling Technology), anti-phospho ERK (1:1000, rabbit polyclonal, Cell Signaling Technology), anti-ERK (1:1000, rabbit monoclonal, Cell Signaling Technology), anti-phospho S6K (1:1000, rabbit monoclonal, Cell Signaling Technology), anti-S6K (1:1000, rabbit polyclonal, Cell signaling Technology), anti-LC3 (1:1000, rabbit polyclonal, Cell Signaling Technology), anti-beta-actin-HRP (1:4,000, mouse monoclonal, Santa Cruz Biotechnology). After incubation in primary antibody, membranes were washed and incubated in HRP conjugated secondary antibody for 1 hour at room temperature (RT), washed again and developed using SuperSignal West Pico Plus Chemiluminescent substrate inside Bio-Rad ChemiDoc MP imaging system. Quantification of immunoblotting was performed by ImageJ (National Institutes of Health).
Sample preparation for TEM
Sorted microglia were fixed in 2% paraformaldehyde (PFA)/2.5% glutaraldehyde (Ted Pella Inc.) in 100 mM sodium cacodylate buffer pH 7.2 for 1 hour at RT. Following washes with sodium cacodylate buffer, cells were embedded in 2.5% agarose and post-fixed with 1% osmium tetroxide (Ted Pella Inc.) for 1 hour. After three washes in dH2O, samples were en bloc stained in 1% aqueous uranyl acetate (Electron Microscopy Sciences) for 1 hour. Samples were washed with dH2O, dehydrated in an ethanol series, and infiltrated with Eponate 12 resin (Ted Pella Inc.). Ultrathin sections (95 nm) were cut with a diamond knife using a Leica Ultracut UCT ultramicrotome (Leica Microsystems Inc.) and counterstained with uranyl acetate and lead citrate. Samples were analyzed and imaged on a JEOL 1200 EX II transmission electron microscope (JEOL USA Inc.) equipped with an AMT 8-megapixel digital camera and AMT Image Capture Engine V602 software (Advanced Microscopy Techniques). Quantitative evaluation was performed by taking random images of 30 cells of comparable size and sectioned through the nucleus (indicating center of cell) and measuring the cross-sectional area using ImageJ. Data was expressed as the total number of a multivesicular/multilamellar structures or lipid droplets per cross-sectional area of cytosol.
Immunofluorescence staining of brain samples
Confocal microscopy analysis was performed as previously described (Wang et al., 2020). Briefly, after transcardial perfusion with ice-cold PBS containing 1 U/ml of heparin, brains were fixed in 4% PFA overnight followed by incubating in 30% sucrose overnight at 4°C before embedding in a 2:1 mixture of 30% sucrose and optimal cutting temperature compound. 40-μm serial coronal sections were cut on a cryo-sliding microtome. Sections were blocked with PBS+5% BSA and permeabilized with 0.5% Triton-X 100 in blocking solution. Primary antibodies were added at indicated combinations overnight at a dilution of 1:1,000 (rabbit monoclonal, Cell Signaling Technology) or 1:500 (goat polyclonal, Abcam) for Iba1, 1:1,000 for CD68 (rat monoclonal, BioLegend), 1:100 for Pu.1 (rabbit monoclonal, Cell Signaling Technology), 1:500 for Aβ1–42 (rabbit recombinant monoclonal, Thermo Fisher Scientific) or 1:1,000 for Ap1–16 (6E10, BioLegend), 1:1,000 for Lamp1 (rat monoclonal, BioLegend), 1:100 for APP (22C11 mouse mAb, Millipore), 1:50 for LC3-Alexa Fluor® 488 (rabbit monoclonal, Cell Signaling Technology), 1:100 for CD11c (rabbit monoclonal, Cell Signaling Technology), 1:300 for CD74-Alexa Fluor® 647 (rat monoclonal, BioLegend), 1:100 for Ki67 (rabbit polyclonal, Abcam), 1:300 for ApoE (biotinylated HJ6.3, (Xiong et al., 2021), 1:50 for Trem2 (polyclonal sheep, R&D Systems), 1:100 for Tmem119 (rabbit monoclonal, Cell Signaling Technology), 1:50 for Clec7a (rat monoclonal, InvivoGene). Secondary antibodies were added as follows: anti-goat IgG Alexa-Fluor®488 1:1,000 (donkey polyclonal, Abcam), anti-rabbit IgG Alexa Fluor® 555 1:1,000 (donkey polyclonal, Abcam), anti-goat IgG Alexa Fluor® 647 1:1,000 (donkey polyclonal, Abcam), anti-rabbit IgG Alexa Fluor® 647 1:1,000 (goat recombinant polyclonal, Invitrogen), anti-rat IgG Alexa Fluor®647 1:1,000 (chicken polyclonal, Invitrogen), Streptavidin-Alexa Fluor® 647 1:500 (BioLegend), anti-sheep IgG Alexa Fluor®555 1:1,000 (donkey polyclonal, Abcam) for 1.5 hours at RT. TO-PRO-3 iodide (300 nM; Thermo Fisher Scientific) was used to counterstain for nuclei, and Aβ plaques were labeled with methoxy-X04 (3 μg/ml; Tocris). The respective areas were captured using a conventional fluorescence microscope (Nikon Eclipse 80i microscope) or confocal microscopes (Nikon A1Rsi+ or Zeiss LSM880 airyscan), or a Zeiss Axio Scan 7 Brightfield / Fluorescence Slide Scanner. Images were then processed with Imaris 7.7 (Bitplane).
Microglia clustering analysis
The density of microglial clustering around Aβ plaques within z stacks was determined by using the Spots and Surface function of Imaris followed by running an automated scripts in MATLAB as described before (Ulland et al., 2017). Briefly, the Spots function was used to identify microglia within the Iba1 and TO-PRO-3 colocalized channel and the volume as well as location of Aβ plaques was reported by Surface function on methoxy-X04 image data. The density of microglia within 15-μm of plaque surfaces was estimated in MATLAB mathematically. The volumes of dystrophic neurites around plaques were calculated by a similar approach in which the Surfaces function was used to identify the volumes and location of methoxy-X04 as well as Lamp1 or APP positive signals within z stacks. Microglial coverage around Aβ plaques was calculated as the total Iba1+methoxy-X04+ volume divided by the total methoxy-X04+ volume. Iba-1 intensity profile was generated by Surface function and the number of microglia was determined by the Spots function of Imaris on Pu.1 image data. Three-dimensional reconstruction of methoxy-X04 and CD68 positive signals was performed by Surfaces function for visualization of Aβ engulfed by microglial CD68+ phagosome on the images.
Quantification of Aβ plaques pathology
The measurement of total methoxy-X04 and Aβ1–42 area coverage was performed as described before (Wang et al., 2020). Briefly, cortex and hippocampus regions or a similar regions encompassing portions of cortex were drawn in ImageJ from individual image and a threshold in which a fixed intensity was much greater than the mean for the selected region was used for calculating the percentage of area coverage automatically by batch processing in ImageJ. Two or three brain sections per mouse were used for quantification. The plaque load for each mouse was shown as the average of two or three sections. Quantification of Iba1, Tmem119, or Clec7a coverage in the cortex was performed by a similar procedure, with different threshold settings. The conformation of individual plaque in each condition was determined as described before (Wang et al., 2020). Briefly, we evaluated: 1) “inert plaques” made of fibrillar Aβ mostly stained by methoxy-X04; 2) “dynamic plaques” exhibiting a core of methoxy-X04+ fibrillar Aβ surrounded by a halo of 6E10+ non-fibrillar Aβ; 3) “filamentous plaques” with little content of β-sheet structure and branched amyloid fibrils mainly stained with the 6E10 antibody. The proportion of distinct plaque morphology was calculated by normalizing the total number of plaques from each z-stack image.
Quantification of microglial activation
For measurement of CD11c+ pixels within Iba1+ pixels, the Spots function on z-stack image data was used to conduct the Iba1+ channel. The thresholds for CD11c+ within Iba1+ pixels were determined by visual inspection and used for all images. For each z-stack image, the percentages of CD11c+ pixels within Iba1+ pixels were calculated. A similar procedure was used for measurement of percentage of LC3+ or CD74+ microglia over total microglia population. The mean intensity of the TREM2 channel within Iba1+ pixels was exported to Microsoft Excel. A threshold of mean TREM2 intensity was set and simply averaged to obtain the final mean intensity. Sholl analysis of non-plaque associated microglia was perform by ImageJ. Five individuals Tmem119+Iba1+non-plaque associated microglia in each brain image were analyzed and data from individual cell was averaged to determine the number of intersections in each brain section. Two brain sections from each mouse were calculated and the final results were shown as the average of the number of intersections in each mouse of two genotypes.
Aβ phagocytosis assay in vitro
Human Aβ1–42 peptides were aggregated and labeled by pHrodo Red and diluted to a final concentration of 100 μM. 5 × 104 cultured primary microglia or BMDM were seeded overnight into a 96-well plate before adding pHrodo Red labeled Aβ1–42 oligomers. Uptake of Aβ1–42 aggregates was then captured in a period of 1 or 2 hours by FACS after removing Aβ1–42 oligomers from culture media.
In vitro BrdU incorporation
Cultured primary microglia were carefully washed of growth factors and re-plated in a non-tissue culture-treated 96-well plate at 1× 105 cells per well. After 48 hours cells were pulsed with 10 μM BrdU for additional 48 hours. Cells were detached with 1mM EDTA and BrdU incorporation measured as instructed by the manufacturer (BD Pharmingen BrdU Flow Kit).
Survival assay
To assess cell survival, WT or Dap10−/− BMDM were seeded into 96-well plate at 5 × 104/well and cultured with following conditions: 1) 10% LLCM; 2) 0% LLCM; 3) 0% LLCM with 1 μM BAY61–3606 (Syk inhibitor); 4) 0% LLCM with 1 μM BAY61–3606 (Syk inhibitor) and 10 μM SB216763 (GSK-3β inhibitor) 5) 0% LLCM with 10 μM SB216763 (GSK-3β inhibitor) for 48 hours. Frequencies of viable cells were determined by FACS with PI staining.
General design of behavioral tests
All behavioral testing was conducted during the light cycle, by a female experimenter blinded to experimental group. Equipment was cleaned with 2% chlorhexidine diacetate between animals. The order of tests conducted for each cohort were as follows: 1 hour locomotor activity, elevated plus maze, visible platform trials of the water maze, submerged platform trials, and water maze probe trial.
One-hour locomotor activity and open field test.
To evaluate general activity levels mice were evaluated over a 1-hour period in transparent (47.6 × 25.4 × 20.6 cm high) polystyrene enclosures. Each cage was surrounded by a frame containing a 4 × 8 matrix of photocell pairs, the output of which was fed to an on-line computer (Hamilton-Kinder). The system software (Hamilton-Kinder) was used to define a 33 × 11 cm central zone and a peripheral or surrounding zone that was 5.5 cm wide with the sides of the cage being the outermost boundary. This peripheral area extended along the entire perimeter of the cage. Variables that were analyzed included the total number of ambulations and rearing on hindlimbs, as well as the number of entries, the time spent, and the distance traveled in the center area as well as the distance traveled in the periphery surrounding the center.
Elevated Plus Maze (EPM).
EPM was conducted as previously described (Wozniak et al., 2013). Briefly, a 5 min trial was conducted in a dimly lit room using a standard mouse elevated plus maze from Kinder Scientific (Poway). The maze consisted of a black acrylic surface measuring 5×5 cm and elevated 63 cm above the floor equipped with photo beam pairs. Four arms (35 cm long, 5 cm wide; two open and two with 15 cm high walls) extended from a central area. The MotorMonitor software (Kinder Scientific) quantified beam breaks as duration, distance traveled, entries, and time at rest in each zone (closed arms, open arms and center area).
Morris water maze (MWM).
MWM was conducted as previously described (Wozniak et al., 2004). Briefly, cued (visible platform), place (submerged platform), and probe trials were conducted in a galvanized steel pool, measuring 120 cm in diameter, and filled with opaque water (diluted nontoxic white tempera paint). The PVC escape platform measured 11.5 cm in diameter. A digital video camera connected to a PC computer and the computer software program ANY-maze (Stoelting Co) tracked the swimming pathway of the mouse to the escape platform and quantified path length, latency to find escape platform, and swimming speeds.
On two consecutive days, animals received four cued trials, separated by 1 hour, during which a red tennis ball atop a rod was attached to the escape platform and served as a visual cue. To prevent spatial learning, the escape platform was moved to a different quadrant location for each trial. The mouse was released from the quadrant opposite to the platform location and allowed 60 s to locate the platform. Once the mouse found the platform, it was allowed to remain there for 10 s before being returned to its home cage. Three days following visible platform testing, the cue was removed from the platform, and it was submerged 1 cm under the water for the hidden platform tests. Animals received two blocks of two consecutive trials on five consecutive days, with an inter-trial interval between 30–90 s and approximately 2 hour separating trial blocks. The escape platform remained in the same quadrant location for all trials and distal cues were placed on the walls of the room to support spatial learning. The mouse was released from a different location for each trial on each day. The mouse was allowed 60 s to find the escape platform and allowed to sit on it for 10 s before being returned to its home cage. Visible and submerged platform trials were combined into blocks of two or four trials for analyses, respectively. Following completion of submerged platform trials on the 5th day of training, the escape platform was removed from the pool and one 60 s probe trial was conducted to assess memory retention for the location of the platform.
Single-cell RNAseq
6-month-old of Sykfl/fl, SykiΔMG, Sykfl/fl-5×FAD and SykiΔMG-5×FAD, as well as Trem2+/+-5×FAD, Trem2−/−-5×FAD and DAP12KΔY75-5×FAD male mice were sacrificed for the sample preparation. After perfusion with cold PBS, cortices were dissociated, and CD45+ cells were sorted using FACS. All CD45+ cell libraries were prepared using the 10x Genomics Chromium Single Cell 3’ v3 Gene Expression Kit and sequenced on Illumina NovaSeq.
Anti-CLEC7A-Fc mutated antibody production
Anti-mouse CLEC7A monoclonal antibody (mAb) 2A11 was previously reported (Brown et al., 2002). A DNA fragment encoding heavy chain variable (VH) and CH1 regions was cloned into the pFUSEmIgG2A-Fc1 vector with L234A, L235A, P329G (LALA-PG) mutation, which blocks Fc binding. A DNA fragment encoding the light chain was cloned into Fc-null pFUSE-mIgG2A-Fc1 vector as previously described (Molgora et al., 2020). The heavy chain and light chain plasmids were co-transfected into Expi293F cells (Thermo Fisher Scientific) for expression at mass ratio 1:2. When cell viability was below 50% (5–7 days), the supernatant was collected and filtered with 0.22-μm filters. The antibody was purified with protein A agarose (GoldBio). Final preps were concentrated to about 10 mg/mL into PBS and stored in 80°C for future use.
Stimulation of cultured primary microglia by anti-CLEC7A
Before stimulation, primary microglia were starved for 3 hours in 1% serum RPMI. Cells were incubated on ice for 30 min with 10 μg/ml of anti-CLEC7A or anti-human ILT1 antibody (Fc mutated, clone 135.5) (Molgora et al., 2020) as a control. Cells were then washed 3 times with cold PBS at 4°C and incubated at 37°C for 5 min in the presence of purified goat F(ab’)2 anti-mouse IgG(H+L) (15 μg/ml, polyclonal, SouthernBiotech). After stimulation, cells were washed 3 times once again with cold PBS at 4°C and lysed with lysis buffer for the immunoblotting.
qRT-PCR analyses
Total RNA was extracted from cortices of the left-brain hemispheres using miRNeasy Mini Kit (QIAGEN). After cDNA synthesis using a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems), quantitative PCR was performed using SYBR Green real-time PCR master mix (Thermo Fisher Scientific) and LightCycler 96 detection system (Roche). The reference gene was actin and relative gene expression levels were determined by the ΔΔCt method. Gene expression was considered undetectable if Ct values were >35 cycles.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis
All graphs represent the mean of all samples in each group ± SEM as indicated in the figure legends. Statistical analysis was performed using GraphPad Prism software. P < 0.05 was considered significant difference between different conditions, determined by a two-tailed unpaired t test, one-way ANOVA with Tukey’s multiple comparisons test or two-way ANOVA with Sidak’s multiple comparisons test as indicated in the figure legends. P-values in violin plots shown in the Figure 7L were calculated using stat_compare_means() function of ggpubr package (0.4.0) in R, using Wilcox rank-sum test. Quantification of fluorescence microscopy, confocal microscopy and electron microscopy images were performed using Imaris, ImageJ and MATLAB.
Single-cell RNAseq analysis
Cell Ranger Software Suite (v6.0.0) from 10xGenomics was used for sample demultiplexing, barcode processing, and single-cell counting. Cellranger count was used to align samples to the reference genome GRCm39, quantify reads, and filter reads and barcodes. Only protein-coding genes, with the exception of Clec7a, were retained in the analysis. The Seurat (v4.0) package in R (v4.1.0) was used for downstream analysis (Hao et al., 2021). For quality control, cells with mitochondrial content more than 7.5% were removed. Cells with low UMI and gene number per cell were filtered out. Cutoffs for UMI and gene number were empirically determined on the basis of histograms showing cell density as a function of UMI per gene counts. Cutoffs of UMI greater than 2000 and less than 20000, and genes greater than 1000 were applied. Genes expressed in fewer than 10 cells were removed from the dataset. Microglia clusters were digitally isolated and further filtered for clusters containing doublets, stress genes, and apoptotic cells. After filtering, the dataset from Sykfl/fl, SykiΔMG, Sykfl/fl-5×FAD and SykiΔMG-5×FAD mice (n = 3 for each genotype) showed in Figure 4 and S5 (refer to as SYK ONLY) contained a total of 57,830 microglia cells with a median 5871 UMI and median 2537 genes and the integration dataset from Sykfl/fl-, SykiΔMG-, Trem2+/+- (n = 2) and Trem2−/−-5×FAD mice (n = 3) showed in Figure 5 (refer to as SYK-TREM2) contained a total of 38,404 microglia cells remained with a median 5483 UMI and median 2359 genes per cell. For 5×FAD (n = 3), DAP12KΔY75-5×FAD (n = 4) dataset, cells with mitochondrial content of more than 10% were removed. Cutoffs of UMI greater than 500 and less than 100,000, and genes greater than 500 and less than 10,000 were applied. Genes expressed in fewer than 10 cells were removed from the dataset. After quality control, 26,921 microglia cells with a median 6,369 UMI and median 2,663 genes were analyzed (refer to as DAP12 ONLY). Then microglia from the DAP12 ONLY dataset were reference mapped to the UMAP of integration SYK-TREM2 dataset using FindTransferAnchors () and MapQuery () functions in the Seurat package and the UMAP was shown in Figure 6A.
Clustering and differential expression analysis
For both SYK ONLY and SYK-TREM2 datasets, data were normalized using the SCTransform method regressed on mitochondrial gene percentage and integrated using FindIntegrationAnchors function and Canonical Correlation Analysis (CCA) (Hafemeister and Satija, 2019; Stuart et al., 2019). Principle component analysis was performed, and the top 30 principal components were selected for dimensionality reduction using the Uniform Approximation and Projection (UMAP) algorithm. Clustering was performed using the FindNeighbors and FindClusters functions using a resolution of 0.6 (SYK ONLY) or 0.4 (SYK-TREM2). Marker genes were identified by comparing each cluster against all other clusters using the FindAllMarkers function with default settings (log-fold change threshold of 0.25 and >10% cells expressing the gene). Cell clusters from each tissue were annotated based on marker gene expression. For data visualization and figure preparation, the scRNA-seq data was also analyzed with BBrowser version 2.7.5 (SingleCell). Differential gene expression testing was performed using the mixed model method “dream” from the muscat package (Crowell et al., 2020). Genes detected in fewer than 10 cells or with total count less than 10 were excluded from analysis. Gene set enrichment analysis was performed with GSEA (Daly et al., 2003; Subramanian et al., 2005). Fold changes were calculated by comparing SykiΔMG-5×FAD vs. Sykfl/fl-5×FAD on cells aggregated from TM1, TM2, and DAM clusters using FindMarkers with the MAST method and a nominal FDR threshold (q < 0.05). Genes were then ranked by log2 fold change and submitted for gene set enrichment analysis using GSEAPreranked and the hallmark gene sets from MSigDB.
Pseudotime trajectory analysis
Single-cell pseudotime trajectories were inferred using the R package slingshot (v2.1.0) (Street et al., 2018). Normalized values resulting from batch integration were used as input for the calculation of cell lineages and pseudotime. Genes that were significantly differentially expressed along pseudotime were identified using the R package tradeSeq (v1.6) (Van den Berge et al., 2020). A negative binomial generalized additive model was fit to the identified lineages using the fitGAM function using the top 3000 variable features. Genes that changed significantly along the DAM trajectory were identified using associationTest. Microglia from SykiΔMG-5×FAD were then compared against Trem2−/−-5×FAD microglia along the DAM trajectory using the conditionTest function. DE genes (FDR < 0.05) were further filtered by selecting those which overlapped with those that changed significantly along the DAM trajectory.
Supplementary Material
Microglia must activate SYK to restrain Aβ pathology
SYK sustains microglia fitness through the PI3K-AKT-GSK3β-mTOR pathway
TREM2 activates microglia through SYK and SYK-independent DAP10-dependent pathways
Direct activation of SYK by anti-CLEC7A rescues microglia with hypofunctional TREM2
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (RF1AG05148501, R21 AG059176, and RF1 AG059082) and Centene. We thank Richard Cho for generous gift of antibodies.
DECLARATION OF INTERESTS
M. Co. is a member of Vigil Neuro scientific advisory board (SAB), is consultant for Cell Signaling Technology and NGM Bio, has received research grants from Vigil Neuro during the conduct of the study and has a patent to TREM2 pending. DMH co-founded and is on the SAB of C2N Diagnostics, is on the SAB of Denali, Genentech, and Cajal Neuroscience, consults for Alector and is on the Advisory Board for Cell. No other disclosures were reported.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Askew K, Li K, Olmos-Alonso A, Garcia-Moreno F, Liang Y, Richardson P, Tipton T, Chapman MA, Riecken K, Beccari S, et al. (2017). Coupled Proliferation and Apoptosis Maintain the Rapid Turnover of Microglia in the Adult Brain. Cell Rep. 18, 391–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atagi Y, Liu CC, Painter MM, Chen XF, Verbeeck C, Zheng H, Li X, Rademakers R, Kang SS, Xu H, et al. (2015). Apolipoprotein E is a ligand for triggering receptor expressed on myeloid cells 2 (TREM2). J. Biol. Chem 290, 26043–26050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Berge K, Roux de Bézieux H, Street K, Saelens W, Cannoodt R, Saeys Y, Dudoit S, and Clement L (2020). Trajectory-based differential expression analysis for single-cell sequencing data. Nat. Commun 11, 1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GD, Taylor PR, Reid DM, Willment JA, Williams DL, Martinez-Pomares L, Wong SYC, and Gordon S (2002). Dectin-1 is a major β-glucan receptor on macrophages. J. Exp. Med 196, 407–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, and Colonna M (2021). Microglia in Alzheimer’s disease at single-cell level. Are there common patterns in humans and mice? J. Exp. Med 218, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu DH, Morita CT, and Weiss A (1998). The Syk family of protein tyrosine kinases in T-cell activation and development. Immunol. Rev 165, 167–180. [DOI] [PubMed] [Google Scholar]
- Claes C, Danhash EP, Hasselmann J, Chadarevian JP, Shabestari SK, England WE, Lim TE, Hidalgo JLS, Spitale RC, Davtyan H, et al. (2021). Plaque-associated human microglia accumulate lipid droplets in a chimeric model of Alzheimer’s disease. Mol. Neurodegener 16, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condello C, Yuan P, Schain A, and Grutzendler J (2015). Microglia constitute a barrier that prevents neurotoxic protofibrillar Aβ42 hotspots around plaques. Nat. Commun 6, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowell HL, Soneson C, Germain PL, Calini D, Collin L, Raposo C, Malhotra D, and Robinson MD (2020). Muscat Detects Subpopulation-Specific State Transitions From Multi-Sample Multi-Condition Single-Cell Transcriptomics Data. Nat. Commun 11, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, and Spiegelman B (2003). PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet 34, 267–273. [DOI] [PubMed] [Google Scholar]
- Eldar-Finkelman H (2002). Glycogen synthase kinase 3: An emerging therapeutic target. Trends Mol. Med 8, 126–132. [DOI] [PubMed] [Google Scholar]
- Ellwanger DC, Wang S, Brioschi S, Shao Z, Green L, Case R, Yoo D, Weishuhn D, Rathanaswami P, Bradley J, et al. (2021). Prior activation state shapes the microglia response to antihuman TREM2 in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman BA, Srinivasan K, Ayalon G, Meilandt WJ, Lin H, Huntley MA, Cao Y, Lee SH, Haddick PCG, Ngu H, et al. (2018). Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell Rep. 22, 832–847. [DOI] [PubMed] [Google Scholar]
- Gilfillan S, Ho EL, Cella M, Yokohama WM, and Colonna M (2002). NKG2D recriuts two distinct adapters to trigger NK cell activation and costimulation. Nat. Immunol 3, 1150–1155. [DOI] [PubMed] [Google Scholar]
- Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, et al. (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science (80-.). 330, 841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldmann T, Wieghofer P, Muller PF, Wolf Y, Varol D, Yona S, Brendecke SM, Kierdorf K, Staszewski O, Datta M, et al. (2013). A new type of microglia gene targeting shows TAK1 to be pivotal in CNS autoimmune inflammation. Nat. Neurosci 16, 1618–1626. [DOI] [PubMed] [Google Scholar]
- Goldmann T, Wieghofer P, Jordão MJC, Prutek F, Hagemeyer N, Frenzel K, Amann L, Staszewski O, Kierdorf K, Krueger M, et al. (2016). Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat. Immunol 17, 797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, Garner H, Trouillet C, De Bruijn MF, Geissmann F, et al. (2015). Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 518, 547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafemeister C, and Satija R (2019). Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. BioRxiv 576827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamerman JA, Ni M, Killebrew JR, Chu CL, and Lowell CA (2009). The expanding roles of ITAM adapters FcRγ and DAP12 in myeloid cells. Immunol. Rev 232, 42–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y, Hao S, Andersen-Nissen E, Mauck WM, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herre J, Marshall ASJ, Caron E, Edwards AD, Williams DL, Schweighoffer E, Tybulewicz V, Reis E Sousa C, Gordon S, and Brown GD (2004). Dectin-1 uses novel mechanisms for yeast phagocytosis in macrophages. Blood 104, 4038–4045. [DOI] [PubMed] [Google Scholar]
- Jay TR, Miller CM, Cheng PJ, Graham LC, Bemiller S, Broihier ML, Xu G, Margevicius D, Karlo JC, Sousa GL, et al. (2015). TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J. Exp. Med 212, 287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay TR, Hirsch AM, Broihier ML, Miller CM, Neilson LE, Ransohoff RM, Lamb BT, and Landreth GE (2017). Disease progression-dependent effects of TREM2 deficiency in a mouse model of Alzheimer’s disease. J. Neurosci 37, 637–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, et al. (2017). A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169, 1276–1290.e17. [DOI] [PubMed] [Google Scholar]
- Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, Beckers L, O’Loughlin E, Xu Y, Fanek Z, et al. (2017). The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 47, 566–581.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanier LL (2009). DAP10- and DAP12-associated receptors in innate immunity. Immunol. Rev 227, 150–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Meilandt WJ, Xie L, Gandham VD, Ngu H, Barck KH, Rezzonico MG, Imperio J, Lalehzadeh G, Huntley MA, et al. (2021). Trem2 restrains the enhancement of tau accumulation and neurodegeneration by β-amyloid pathology. Neuron 109, 1283–1301.e6. [DOI] [PubMed] [Google Scholar]
- Long JM, and Holtzman DM (2019). Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 1–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh SE, Walker AJ, Kamath T, Dissing-Olesen L, Hammond TR, de Soysa TY, Young AMH, Murphy S, Abdulraouf A, Nadaf N, et al. (2022). Dissection of artifactual and confounding glial signatures by single-cell sequencing of mouse and human brain. Nat. Neurosci 25, 306–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mass E, Jacome-Galarza CE, Blank T, Lazarov T, Durham BH, Ozkaya N, Pastore A, Schwabenland M, Chung YR, Rosenblum MK, et al. (2017). A somatic mutation in erythro-myeloid progenitors causes neurodegenerative disease. Nature 549, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters CL, and Selkoe DJ (2012). Biochemistry of amyloid β-protein and amyloid deposits in Alzheimer disease. Cold Spring Harb. Perspect. Med 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda T, Amann L, Monaco G, Sankowski R, Staszewski O, Krueger M, Del Gaudio F, He L, Paterson N, Nent E, et al. (2022). Specification of CNS macrophage subsets occurs postnatally in defined niches. Nature 604, 740–748. [DOI] [PubMed] [Google Scholar]
- Mathys H, Adaikkan C, Gao F, Young JZ, Manet E, Hemberg M, De Jager PL, Ransohoff RM, Regev A, and Tsai LH (2017). Temporal Tracking of Microglia Activation in Neurodegeneration at Single-Cell Resolution. Cell Rep. 21, 366–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mócsai A, Abram CL, Jakus Z, Hu Y, Lanier LL, and Lowell CA (2006). Integrin signaling in neutrophils and macrophages uses adaptors containing immunoreceptor tyrosine-based activation motifs. Nat. Immunol 7, 1326–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molgora M, Esaulova E, Vermi W, Hou J, Chen Y, Luo J, Brioschi S, Bugatti M, Omodei AS, Ricci B, et al. (2020). TREM2 Modulation Remodels the Tumor Myeloid Landscape Enhancing Anti-PD-1 Immunotherapy. Cell 182, 886–900.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L, and Selkoe DJ (2012). Neurotoxicity of amyloid β-protein: Synaptic and network dysfunction. Cold Spring Harb. Perspect. Med 2, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent AA, Lin K, van Lengerich B, Lianoglou S, Przybyla L, Davis SS, Llapashtica C, Wang J, Kim DJ, Xia D, et al. (2020). TREM2 Regulates Microglial Cholesterol Metabolism upon Chronic Phagocytic Challenge. Neuron 105, 837–854.e9. [DOI] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, et al. (2006). Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci 26, 10129–10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Q, Malhotra S, Torchia JA, Kerr WG, Coggeshall KM, and Humphrey MB (2010). TREM2- and DAP12-dependent activation of PI3K requires DAP10 and is inhibited by SHIP1. Sci. Signal 3, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz M, Masuda T, Wheeler MA, and Quintana FJ (2021). Microglia and Central Nervous System-Associated Macrophages mdash from Origin to Disease Modulation. Annu. Rev. Immunol 39, 251–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers NC, Slack EC, Edwards AD, Nolte MA, Schulz O, Schweighoffer E, Williams DL, Gordon S, Tybulewicz VL, Brown GD, et al. (2005). Syk-dependent cytokine induction by dectin-1 reveals a novel pattern recognition pathway for C type lectins. Immunity 22, 507–517. [DOI] [PubMed] [Google Scholar]
- Safaiyan S, Besson-Girard S, Kaya T, Cantuti-Castelvetri L, Liu L, Ji H, Schifferer M, Gouna G, Usifo F, Kannaiyan N, et al. (2021). White matter aging drives microglial diversity. Neuron 109, 1100–1117.e10. [DOI] [PubMed] [Google Scholar]
- Saijo K, Schmedt C, Su I hsin, Karasuyama H, Lowell CA, Reth M, Adachi T, Patke A, Santana A, and Tarakhovsky A (2003). Essential role of Src-family protein tyrosine kinases in NF-κB activation during B cell development. Nat. Immunol 4, 274–279. [DOI] [PubMed] [Google Scholar]
- Sala Frigerio C, Wolfs L, Fattorelli N, Thrupp N, Voytyuk I, Schmidt I, Mancuso R, Chen WT, Woodbury ME, Srivastava G, et al. (2019). The Major Risk Factors for Alzheimer’s Disease: Age, Sex, and Genes Modulate the Microglia Response to Aβ Plaques. Cell Rep. 27, 1293–1306.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz C, Perdiguero EG, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, Prinz M, Wu B, Jacobsen SEW, Pollard JW, et al. (2012). A lineage of myeloid cells independent of myb and hematopoietic stem cells. Science (80-.). 335, 86–90. [DOI] [PubMed] [Google Scholar]
- Song WM, Joshita S, Zhou Y, Ulland TK, Gilfillan S, and Colonna M (2018). Humanized TREM2 mice reveal microglia-intrinsic and -extrinsic effects of R47H polymorphism. J. Exp. Med 215, 745–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangenberg E, Severson PL, Hohsfield LA, Crapser J, Zhang J, Burton EA, Zhang Y, Spevak W, Lin J, Phan NY, et al. (2019). Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat. Commun 10, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Street K, Risso D, Fletcher RB, Das D, Ngai J, Yosef N, Purdom E, and Dudoit S (2018). Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics 19, 477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay TL, Mai D, Dautzenberg J, Fernández-Klett F, Lin G, Sagar S, Datta M, Drougard A, Stempfl T, Ardura-Fabregat A, et al. (2017). A new fate mapping system reveals context-dependent random or clonal expansion of microglia. Nat. Neurosci 20, 793–803. [DOI] [PubMed] [Google Scholar]
- Thion MS, Ginhoux F, and Garel S (2018). Microglia and early brain development: An intimate journey. Science (80-.). 362, 185–189. [DOI] [PubMed] [Google Scholar]
- Tomasello E, Desmoulins PO, Chemin K, Guia S, Cremer H, Ortaldo J, Love P, Kaiserlian D, and Vivier E (2000). Combined natural killer cell and dendritic cell functional deficiency in KARAP/DAP12 loss-of-function mutant mice. Immunity 13, 355–364. [DOI] [PubMed] [Google Scholar]
- Turner M, Schweighoffer E, Colucci F, Di Santo JP, and Tybulewicz VL (2000). Tyrosine kinase SYK: Essential functions for immunoreceptor signalling. Immunol. Today 21, 148–154. [DOI] [PubMed] [Google Scholar]
- Ulland TK, and Colonna M (2018). TREM2 — a key player in microglial biology and Alzheimer disease. Nat. Rev. Neurol 14, 667–675. [DOI] [PubMed] [Google Scholar]
- Ulland TK, Song WM, Huang SCC, Ulrich JD, Sergushichev A, Beatty WL, Loboda AA, Zhou Y, Cairns NJ, Kambal A, et al. (2017). TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell 170, 649–663.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underhill DM, Rossnagle E, Lowell CA, and Simmons RM (2005). Dectin-1 activates Syk tyrosine kinase in a dynamic subset of macrophages for reactive oxygen production. Blood 106, 2543–2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utz SG, See P, Mildenberger W, Thion MS, Silvin A, Lutz M, Ingelfinger F, Rayan NA, Lelios I, Buttgereit A, et al. (2020). Early Fate Defines Microglia and Non-parenchymal Brain Macrophage Development. Cell 181, 557–573.e18. [DOI] [PubMed] [Google Scholar]
- Wang S, Mustafa M, Yuede CM, Salazar SV, Kong P, Long H, Ward M, Siddiqui O, Paul R, Gilfillan S, et al. (2020). Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s disease model. J. Exp. Med 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Gilfillan S, Krishnan GM, Sudhakar S, Zinselmeyer BH, et al. (2015). TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 160, 1061–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ulland TK, Ulrich JD, Song W, Tzaferis JA, Hole JT, Yuan P, Mahan TE, Shi Y, Gilfillan S, et al. (2016). TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J. Exp. Med 213, 667–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wes PD, Sayed FA, Bard F, and Gan L (2016). Targeting microglia for the treatment of Alzheimer’s Disease. Glia 64, 1710–1732. [DOI] [PubMed] [Google Scholar]
- Wozniak DF, Hartman RE, Boyle MP, Vogt SK, Brooks AR, Tenkova T, Young C, Olney JW, and Muglia LJ (2004). Apoptotic neurodegeneration induced by ethanol in neonatal mice is associated with profound learning/memory deficits in juveniles followed by progressive functional recovery in adults. Neurobiol. Dis 17, 403–414. [DOI] [PubMed] [Google Scholar]
- Wozniak DF, Diggs-Andrews KA, Conyers S, Yuede CM, Dearborn JT, Brown JA, Tokuda K, Izumi Y, Zorumski CF, and Gutmann DH (2013). Motivational Disturbances and Effects of L-dopa Administration in Neurofibromatosis-1 Model Mice. PLoS One 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright GJ, Cherwinski H, Foster-Cuevas M, Brooke G, Puklavec MJ, Bigler M, Song Y, Jenmalm M, Gorman D, McClanahan T, et al. (2003). Characterization of the CD200 Receptor Family in Mice and Humans and Their Interactions with CD200. J. Immunol 171, 3034–3046. [DOI] [PubMed] [Google Scholar]
- Xiong M, Jiang H, Serrano JR, Gonzales ER, Wang C, Gratuze M, Hoyle R, Bien-Ly N, Silverman AP, Sullivan PM, et al. (2021). APOE immunotherapy reduces cerebral amyloid angiopathy and amyloid plaques while improving cerebrovascular function. Sci. Transl. Med 13, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanishi Y, Kitaura J, Izawa K, Matsuoka T, Oki T, Lu Y, Shibata F, Yamazaki S, Kumagai H, Nakajima H, et al. (2008). Analysis of mouse LMIR5/CLM-7 as an activating receptor: Differential regulation of LMIR5/CLM-7 in mouse versus human cells. Blood 111, 688–698. [DOI] [PubMed] [Google Scholar]
- Yao H, Coppola K, Schweig JE, Crawford F, Mullan M, and Paris D (2019). Distinct Signaling Pathways Regulate TREM2 Phagocytic and NFκB Antagonistic Activities. Front. Cell. Neurosci 13, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh FL, Wang Y, Tom I, Gonzalez LC, and Sheng M (2016). TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia. Neuron 91, 328–340. [DOI] [PubMed] [Google Scholar]
- Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, et al. (2013). Fate Mapping Reveals Origins and Dynamics of Monocytes and Tissue Macrophages under Homeostasis. Immunity 38, 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan P, Condello C, Keene CD, Wang Y, Bird TD, Paul SM, Luo W, Colonna M, Baddeley D, and Grutzendler J (2016). TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron 90, 724–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Wu X, Li X, Jiang LL, Gui X, Liu Y, Sun Y, Zhu B, Piña-Crespo JC, Zhang M, et al. (2018). TREM2 Is a Receptor for β-Amyloid that Mediates Microglial Function. Neuron 97, 1023–1031.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Song WM, Andhey PS, Swain A, Levy T, Miller KR, Poliani PL, Cominelli M, Grover S, Gilfillan S, et al. (2020). Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med 26, 131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegenfuss JS, Biswas R, Avery MA, Hong K, Sheehan AE, Yeung Y-G, Stanley ER, and Freeman MR (2008). Draper-dependent glial phagocytic activity is mediated by Src and Syk family kinase signalling. Nature 453, 935–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2021). 2021 Alzheimer’s disease facts and figures. Alzheimers. Dement 17, 327–406. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the data reported in this paper is GE0:GSE210258.