

Abstract
Detailed investigations of macrophage phagocytosis and killing of Aspergillus fumigatus conidia have been limited by technical difficulties in quantifying fungal uptake and viability. In order to study early events in cell pathogen ingestion and killing, we developed a new flow cytometry assay that utilizes the fungus-specific viability dye FUN-1. Metabolically active A. fumigatus conidia accumulate orange fluorescence in vacuoles, while dormant or dead conidia stain green. After incubation within THP-1 cells, recovered conidia are costained with propidium iodide (PI) to discriminate between dormant and dead cells. Flow cytometric measurements of FUN-1 metabolism and PI uptake provide indicators of conidial viability, dormancy, and death. Conidial phagocytosis and killing are also assessed by measurement of green and orange FUN-1 fluorescence within the THP-1 cell population. Compared to previously described methods, this assay has less error introduced by membrane permeability changes and serial dilution of filamentous fungal forms. Results suggest that the THP-1 cells kill conidia rapidly (within 6 h) after exposure. Conidia that are preexposed to human serum are ingested and killed more quickly than are nonopsonized conidia.
Aspergillus fumigatus is a common cause of invasive pulmonary infection in immunocompromised patients, especially in recipients of myeloablative chemotherapy or hematopoietic stem cell transplantation. In these patients, who have various degrees and durations of compromised phagocytic and cell-mediated immunity, inhaled conidia of A. fumigatus can mature into hyphae in the lungs, invade pulmonary vasculature, and disseminate to other critical organs. Attributable mortality approximates 60 to 95%, largely depending on the severity of the hosts' immune deficit and the stage of recognition of disease (6).
One of the first critical mechanisms of host defense is the pulmonary macrophage, which can ingest conidia, kill the organism, and coordinate defense (2). The mechanisms by which macrophages ingest and kill A. fumigatus conidia are not well described, in part because methods used to quantify conidial killing have not been standardized. Measurement of fungal viability using serial dilution quantification of CFU is limited by the error introduced by the morphological switch from conidia to hyphae, cellular clumping, and hyphal fragmentation. Recognition of these limitations has encouraged the development of other methods to quantify the burden of filamentous fungi in animal models, such as quantification of the cell wall component chitin (2). Unfortunately, these methods are not sensitive enough for evaluating early stages of fungal growth, such as conidial maturation.
Previous methods used to measure intracellular killing of conidia include quantification of CFU by serial dilution plating (13), measurement of intracellular conidial germination using light microscopy (14), and quantification of conidial death by flow cytometric analysis of propidium iodide (PI) uptake (4, 8). Studies using these methods have reported that macrophage killing (i) is variable, ranging from 15 to 75% efficacy; (ii) relies on nonoxidative mechanisms; and (iii) occurs late after phagocytosis, reaching peak values up to 18 h after conidial ingestion (7, 14). We hypothesized that much of the reported variability may be due to error in measurements that rely on serial dilution of filamentous forms. Also, the timing of killing cannot be reliably predicted based on conidial germination into hyphae, given the potential of long lag periods of microbial dormancy. Finally, PI uptake as a measure of cell death may be limited by nonspecific positivity introduced by changes in membrane permeability.
FUN-1 [2-chloro-4-(2,3-dihydro-3-methyl-(benzo-1,3-thiazol-2-yl)-methylidene)-1-phenylquinolinium iodide] stain (Molecular Probes, Eugene, Oreg.) has been used as an indicator of metabolic activity in yeasts (9) and A. fumigatus hyphae (5). In metabolically active cells, green fluorescence is converted into orange-red cylindrical intracellular structures. We report here the development of a flow cytometric assay that incorporates FUN-1 and PI to provide two parameters of conidial viability. Studies performed suggest that a macrophage like cell line (THP-1) kills A. fumigatus conidia efficiently and rapidly after phagocytosis, especially in the presence of serum components.
MATERIALS AND METHODS
Microorganisms and preparation of inoculum.
The A. fumigatus isolate used in these studies was obtained from a patient who developed proven pulmonary aspergillosis at the National Institutes of Health (B-5233) (17). Isolates were maintained on potato dextrose agar and plated fresh on RPMI 1640 agar prior to use. Conidial suspensions were harvested by flooding each colony with sterile RPMI 1640 (plus 0.025% Tween 20). An aliquot of cells was counted with a hemacytometer, and the inoculum was adjusted to a concentration of 106 cells/ml and stored at 4°C until use.
FUN-1 stain was obtained from Molecular Probes. Conidia were incubated with FUN-1 (final concentration of 5 mM) with gentle shaking at room temperature for 30 min in the dark (9, 19). Dye uptake and inoculum viability were documented by visualization through a Texas Red fluorescein filter, and cells were washed in phosphate-buffered saline (PBS) and stored at 4°C in RPMI 1640 until use. In control experiments, FUN-1 positivity was measured in live conidia over time, as well as in conidia killed with heat (85°C for 25 min), using the flow cytometry methods described below. For certain experiments, conidia were coincubated with RPMI 1640-HEPES containing 10% fresh human serum (Gemini Bio-Products, Woodland, Calif.) prior to exposure to macrophages.
THP-1 cells.
THP-1 cells (American Type Culture Collection) were maintained in complete medium containing RPMI 1640-HEPES (plus 10% fetal bovine serum) at 37°C (5% CO2), with passage every 3 days. THP-1 cells were plated onto six-well plates and allowed to differentiate in the presence of 0.01 M phorbol 12-myristate 13-acetate (Sigma). Cells that were adherent after 72 h were utilized in phagocytosis and killing experiments.
Measurement of conidial phagocytosis and killing. (i) Phagocytosis quantification.
Adherence of THP-1 cells was confirmed by light microscopy (usually >80% of cells), and nonadherent cells were removed by gentle washing with prewarmed (37°C) 1× PBS. To each well, FUN-1-treated conidia, suspended in fresh or serum-containing media, were added, and plates were centrifuged at 125 × g for 5 min to allow for even and rapid conidial exposure to macrophages. Control THP-1 cells were pretreated with 1 μg of cytochalasin B (Sigma) per ml for 2 h at 37°C; cytochalasin B specifically inhibits the polymerization of actin and prevents phagocytosis but not extracellular binding of conidia to macrophages. THP-1 cells were allowed to ingest conidia for 1, 5, 15, 30, and 60 min. Supernatants were discarded, and the wells were washed gently three times with ice-cold Hanks balanced salt solution (Gibco BRL, Gaithersburg, Md.). THP-1 cells were lifted from the wells with gentle pipetting with PBS containing 1 mM EDTA (pH 7.5) and 5 mM sodium azide. To discriminate between ingested and adherent conidia, THP-1 cells were further incubated with Calcofluor white M2R (Molecular Probes), which stains the fungal cell wall fluorescent blue. The cells were washed and resuspended in PBS containing 1% formaldehyde and analyzed using a FACScan cytometer (Becton Dickinson, San Jose, Calif.). Analyses were performed with Cellquest software (Becton Dickinson). The THP-1 cell population was gated based on light scatter characteristics. The population of cells which were single positive for FUN-1 orange were considered to have phagocytosed but not adherent conidia, cells positive for Calcofluor white fluorescence had adherent conidia, and double-positive cells had both internalized and adherent conidia.
(ii) Killing analysis by serial dilution.
To measure macrophage killing of conidia by serial dilution, THP-1 cells were allowed to ingest FUN-1-labeled conidia for 1 h. Medium containing nonadherent, nonphagocytosed conidia was removed, and wells were washed three times using warm 1× PBS. Macrophages were then allowed to kill conidia for 2 and 4 h before intracellular conidia were harvested. Plates were frozen at −70°C and rapidly thawed at 37°C to lyse the THP-1 cells and harvest conidia. Cellular lysis was confirmed by microscopy. The supernatant was mixed vigorously, and serial dilutions were performed in sterile RPMI 1640 and immediately plated on yeast extract agar containing Triton (10 g of yeast extract, 20 g of peptone, 20 g of dextrose, 20 g of agar, and 50 μl of Triton X-100 per liter). Plates were incubated at 30°C, and colonies were counted after 48 and 72 h of growth. CFU were compared in samples harvested at different time points. Controls were performed by measuring CFU from conidia treated in the same fashion in the absence of THP-1 cells.
(iii) Flow cytometric killing assays.
Conidia were harvested from macrophages using the freeze-thaw step described above. The suspension was then drawn through a pipette tip three times and transferred to Falcon tubes. Thirty microliters of DNase-RNase master mix (10 μl of DNase [10 U/ml], 10 μl of RNase [500 μg/ml], 1 M MgCl2, and 1 M CaCl2) was added to each tube and incubated at 37°C for 15 min. Six microliters of proteinase K (1 μg/ml) was added to all tubes, and incubation was continued for 15 min at 30°C. Tubes were stored at 4°C overnight, and PI was added to the reisolated conidia to achieve a final concentration of 25 μg/ml. After incubation for 20 min at room temperature in the dark, the samples were analyzed. Live conidia incubated with FUN-1 were used to set light scatter gates for conidial characteristics and FUN-1 positivity, and conidia killed with heat (85°C, 25 min) were used to set gates for PI positivity. Additional controls include FUN-1-stained THP-1 cells in the absence of Aspergillus conidia.
RESULTS
Conidial killing: serial dilution plating.
Initial experiments were performed to quantify THP-1 cell killing of A. fumigatus conidia using serial dilution plating. A. fumigatus conidia were incubated with activated THP-1 cells (105) at 1:1, 50:1, and 100:1 ratios, and macrophages were allowed to kill the organisms at 37°C for 2 and 4 h before conidial recovery. As controls, equivalent numbers of A. fumigatus conidia were allowed to germinate in the absence of THP-1 cells under the same conditions. Figure 1 shows that minimal killing occurred after 2 h (“2 hour kill”). THP-1 cells appeared to kill A. fumigatus after 4 h, as fewer CFU were recovered at all concentrations tested (“4 hour kill”). However, fewer CFU were recovered after A. fumigatus growth in the absence of phagocytic cells (“2 hour growth” versus “4 hour growth”). Further, the percentage of CFU recovered after exposure to THP-1 cells was always greater than or equal to the percentage of CFU recovered from the growth controls. We hypothesized that this might be secondary to conidial clumping during growth, resulting in a falsely decreased estimate of viability by CFU. Light microscopy documented that, at 4 h, conidia were swollen and starting to germinate, and clumping of cells occurred despite vigorous vortexing in the presence of detergent. Hence, conidial maturation and growth into hyphae, in the absence of macrophage killing, yielded fewer CFU upon serial dilution testing. As this decrease in CFU could potentially result in overestimation of macrophage conidial killing, new methods to measure macrophage killing were sought.
FIG. 1.

CFU recovered from THP-1 cells relative to inoculum (y axis) after 2- and 4-h incubations (“2 hour kill” and “4 hour kill”) and after 2- and 4-h growth in the absence of THP-1 cells (“2 hour growth” and “4 hour growth”). Data shown are from three experiments, each with triplicate plating per well.
Conidial killing: flow cytometry.
Previous studies evaluated conidial viability after recovery from lysed macrophages by cytofluorometric quantification of fluorescein isothiocyanate-labeled, PI-positive conidia (8). These methods relied on either streptolysin O or freezing-thawing to lyse macrophages (4). We were unable to efficiently lyse THP-1 cells using streptolysin O. Freeze-thawing resulted in lysis of >80% of THP-1 cells, with control experiments documenting minimal (<10%) killing of A. fumigatus conidia (data not shown). Initial experiments performed to measure macrophage killing thus incorporated freeze-thaw lysis and PI staining. As shown in Fig. 2, this method allowed for identification of a PI-positive population, and the proportion increased in a population of heat-killed cells. However, a significant proportion of live conidia appeared PI positive. As this occurred in cells with light scatter characteristics indicating significant size (outlined in Fig. 2b), in the absence of autofluorescence (Fig. 2a), it is likely that cell wall and membrane changes that occur during conidial maturation result in PI uptake. A second problem encountered with this method is a large amount background staining of macrophage cell debris (data not shown).
FIG. 2.

Forward scatter characteristics (FSC-H, y axis) are plotted against PI uptake (x axis) for live, naked A. fumigatus conidia (a); live, PI-exposed A. fumigatus conidia (b); and heat-killed, PI-exposed conidia (c). Large PI-positive cells within the live population are apparent (b) despite the absence of significant autofluorescence (a). Heat killing (verified using serial dilution plating) resulted in a high proportion of PI-positive cells (c).
In order to more accurately discriminate between conidia and cellular debris and to provide an additional measure of conidial viability after recovery from macrophages, we first stained A. fumigatus conidia with the viability dye FUN-1. Maturing A. fumigatus conidia process the FUN-1 dye such that metabolically active cells accumulate fluorescent orange-red vacuoles, while dormant or dead cells exhibit a diffuse green cytoplasm (Fig. 3a). The heterogeneous population of A. fumigatus conidia in Fig. 3a is indicative of asynchronous growth. Measurement of FUN-1 dye green-to-orange metabolism over time shows that almost the entire population of A. fumigatus conidia (87%) becomes intensely positive within 60 min, with the log fluorescence intensity also increasing over time (Fig. 3b). There was no difference in FUN-1 metabolism of two A. fumigatus isolates examined, and FUN-1 orange fluorescence is stable within conidia, continuing to increase as organisms mature into hyphae.
FIG. 3.
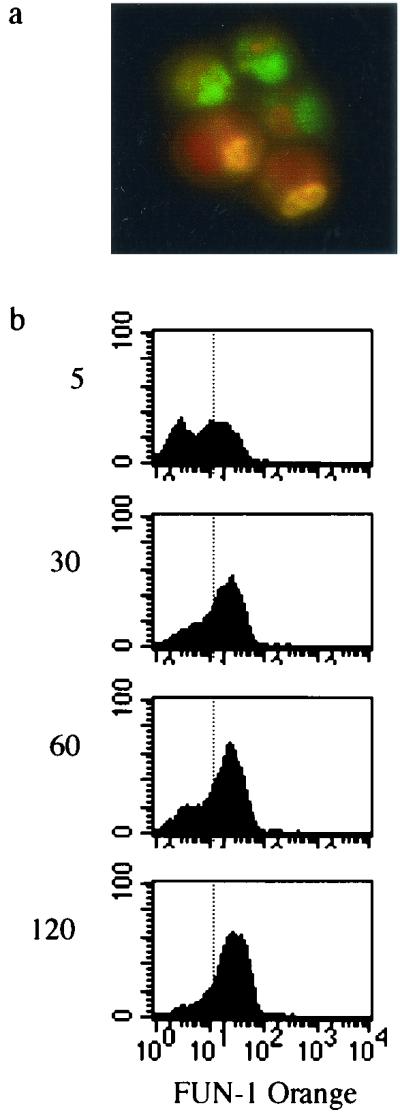
(a) FUN-1-stained green and orange A. fumigatus conidia. (b) FUN-1 metabolism of live A. fumigatus conidia over time. After indicated incubations at 30°C, conidial metabolism was halted by rapid exposure to 4°C and cells were fixed, as described in Materials and Methods. The fluorescence-activated cell sorting profiles shown were produced by gating upon the entire cellular population. Log fluorescence intensity is shown on the x axis, and the relative cell number is shown on the y axis, for each time point indicated (5, 30, 60, and 120 min).
However, by this method alone, dormant conidia might not be distinguishable from dead conidia, as both would fluoresce green. To distinguish between dormant and dead cells and to minimize nonspecific PI positivity, we colabeled the recovered conidial population with PI. Fungal cells were analyzed by gating conidia based on appropriate light scatter characteristics and examining PI fluorescence within the FUN-1 orange-negative population (Fig. 4). In the live control in Fig. 4, the majority of the cells (91%) fluoresce orange. Within the orange-negative population, 26% are dead (PI positive), while the majority (74%) are dormant (PI negative). Orange fluorescence of FUN-1 is no longer apparent after FUN-1-stained cells are exposed to heat. The majority of green cells (99%) are dead, with strong PI positivity. Combining measurements of FUN-1 metabolism and PI positivity thus provides measures of cellular metabolic activity, death, and dormancy.
FIG. 4.

Control experiments for FUN-1 metabolism (top) and PI uptake within the FUN-1 orange-negative population (bottom) in freshly harvested live cells (left) and heat-killed conidia (right).
Cellular killing of conidia.
THP-1 cell killing of A. fumigatus conidia was measured using both parameters (Fig. 5). Histograms measuring FUN-1 orange metabolism (Fig. 5a) and PI uptake within the FUN-1 orange-negative population (Fig. 5b) are shown in a control live population and after 2- and 6-h incubations in THP-1 cells. FUN-1 metabolism of conidia recovered after 2-h incubations in THP-1 cells is higher than that after 6-h incubations within macrophages (bold histogram). The proportion of dead cells within the FUN-1 orange-negative population increases over time, with 65% of cells harvested from macrophages after 6 h dead (PI positive).
FIG. 5.

(a) Histogram plot of FUN-1 metabolism of live A. fumigatus conidia (dotted line) and conidia harvested from macrophages after 2-h (solid line) and 6-h (bold line) incubations. Log fluorescence intensity is shown on the x axis, and the relative cell number is on the y axis. (b) PI uptake in live conidia and conidia recovered from THP-1 cells after 2 and 6 h. FUN-1 green-to-orange metabolism is measured within the entire population of recovered conidia, while PI uptake is measured only within the FUN-1 orange-negative population. Shown are the results of one experiment, which is representative of at least six different experiments.
These results suggest that THP-1 cells kill the majority of A. fumigatus conidia within 6 h of incubation but that the process is not complete. We hypothesized that another method to determine conidial killing would be to measure intracellular FUN-1 green and orange fluorescence within intact THP-1 cells. To quantify intracellular fluorescence, THP-1 cells, exposed to FUN-1-fluorescent conidia, were washed and lifted off wells, as described in Materials and Methods. Green and orange fluorescence was measured within the gate containing intact THP-1 cells. To ensure that measured fluorescence represented internalized and not externally adherent conidia or nonspecific staining of THP-1 cells, fluorescence was also measured within a population of sodium azide-pretreated THP-1 cells. Figure 6 shows increased green (Fig. 6a) and orange (Fig. 6b) fluorescence in THP-1 cells exposed to conidia for 2 h (bold histogram) compared to those exposed for 5 min and sodium azide controls (solid histograms). Upon microscopic examination, orange fluorescence of internalized conidia was detected. However, there was a large amount of nonspecific background staining with FUN-1 green, explaining the positive control population in Fig. 6a. Measurement of FUN-1 orange within the intact THP-1 cells thus provides an additional measure of intracellular conidial viability.
FIG. 6.

Histogram plots of green fluorescence (a) and orange fluorescence (b) within intact THP-1 cells after 5-min (solid line) and 2-h (bold line) incubations, compared to intracellular staining of sodium azide-treated controls after 2 h (solid gray area).
These experiments demonstrate two methods to measure macrophage ingestion and killing of conidia. Quantification of conidial FUN-1 green-to-orange metabolism and PI staining within non-metabolically active conidia reflect organism viability and death after recovery from macrophages. As previous reports suggest that serum components may mediate responses of phagocytes to A. fumigatus (15, 18), we utilized this new method to measure phagocytosis and killing of opsonized and nonopsonized conidia. Figure 7 shows that a large proportion of serum-naïve conidia (solid histogram) harvested from THP-1 cells after 2 h remain metabolically active, while few serum-exposed conidia (open histogram) are viable. The percentages of viable and dead conidia among the recovered population are shown in Fig. 7b. Fewer viable and more dead opsonized conidia were recovered after 2 h, although killing levels appear equivalent after 6-h incubations. However, our ability to estimate killing of opsonized conidia after 6 h may be limited by the recovery of relatively few (<1,000) intact conidia. The same results were demonstrated after conidia were opsonized with heat-killed human serum (data not shown). Importantly, exposure to serum components did not alter FUN-1 labeling of conidia, when measured by either percentage of positive cells within the population or mean fluorescence intensity over time (data not shown).
FIG. 7.

(a) Histogram plots of intracellular orange fluorescence 2 h after exposure to nonopsonized A. fumigatus conidia (solid gray area) and conidia preexposed to non-heat-inactivated human serum (solid line). Counts are shown on the y axis. (b) Percent conidia viable (FUN-1)orange) and dead (PI positive) after recovery under the conditions indicated. Data from triplicate measures in three different experiments.
As free-floating conidia were removed from wells after 1 h of incubation in both settings, these data suggest that fewer viable A. fumigatus conidia remain attached to or within macrophages after 2 h if the conidia are first exposed to serum. Serum components could thus alter either internalization or killing or potentially both. To determine if the increased killing is explained by faster conidial phagocytosis, internalization of opsonized conidia and that of nonopsonized conidia were compared. The percentage of THP-1 cells containing internalized conidia that were preexposed to serum was higher than that for cells exposed to nonopsonized conidia (Fig. 8), although internalization equilibrated with longer incubations. As expected, cytochalasin-treated THP-1 cell controls did not internalize A. fumigatus conidia; instead, adherent conidia provided a large amount of blue fluorescence (data not shown).
FIG. 8.

Percentage of THP-1 cells with internalized conidia (FUN-1 orange, y axis) after indicated incubations (x axis). Percentages of cells with internalized naked A. fumigatus conidia and conidia preexposed to fresh serum are shown. Results were obtained from triplicate measurements in two experiments.
DISCUSSION
Detailed study of macrophage interactions with filamentous fungi has been limited by difficulties in measuring early stages of cellular growth. We have developed a sensitive assay to measure cellular conidial killing, using the viability dye FUN-1. Compared to previously described methods, this assay has less error introduced by membrane permeability changes and serial dilution of filamentous fungal forms. Results suggest that the macrophage-like cell line THP-1 kills conidia rapidly after exposure. Opsonization of conidia with serum components results in more rapid ingestion and killing.
Serial dilution methods have long been the “gold standard” to quantify the growth of organisms after exposure to antimicrobial compounds or phagocytic cells. These methods perform particularly well in quantifying growth of organisms that reproduce as single CFU but are a less accurate reflection of growth for filamentous fungi, which change morphology between early (conidial) and late (hyphal) forms and tend to cluster in both cellular states (as seen in Fig. 3a). The studies presented here document that growth of the organism could result in decreased numbers of CFU upon serial dilution, which likely occurs from clumping of both conidia and early hyphal forms. Although light microscopic detection of conidial germination within macrophages has also been used to measure efficacy of conidial killing, this method is limited by the possibility that the intracellular environment suppresses germination without killing the organism.
Flow cytometry as a method to measure conidial viability appears useful, particularly if evaluating early stages of cell growth, as conidia of various sizes can be evaluated within a population by accounting for light scatter characteristics. This has been accomplished previously using PI (4): however, the studies here demonstrate that limitations of this method include nonspecific PI positivity in metabolically active conidia and background staining of cellular debris. As conidial maturation is marked by sloughing of a chitinous cell wall with swelling resulting from exposure of a permeable cell membrane (6), it is likely that PI uptake occurs because of the changes in cell membrane permeability. The recently reported observation that the detergent deoxycholate results in increased PI uptake is consistent with this explanation (4).
We included in our assay the viability dye FUN-1, which has been shown elsewhere to be useful in measuring the metabolic state of yeasts by accumulation of orange fluorescent intracellular vacuoles (9). This dye, which has an excitation wavelength of 508 nm and an emission wavelength of 525 to 590 nm, is distinguishable from PI, which has excitation and emission wavelengths of 536 and 625 nm, respectively (1). Using the two dyes together, one can measure both conidial viability (FUN-1 orange fluorescence) and conidial death (PI positivity). Measurement of PI positivity within the FUN-1 green population of cells minimizes the error introduced by changes in membrane permeability, as such metabolically active cells fluoresce FUN-1 orange, and enables discrimination of PI-positive cellular debris. Finally, this technique has the added advantage of enabling measurement of conidial ingestion and viability within phagocytic cells.
Results of experiments presented here suggest that the macrophage cell line THP-1 ingests and kills A. fumigatus rapidly, with the majority of conidia killed within 6 h after exposure. These results are in contrast to those of experiments that measured alveolar macrophage conidial killing by enumeration of intracellular conidial germination, which suggest that there is a lag of 3 to 6 h between ingestion and killing and that maximal killing does not occur until 30 h after conidial ingestion (14). Also, previous studies found that murine alveolar macrophages kill conidia with a mean efficacy of only 71% after 18 h (7). As enumeration of intracellular conidial germination is a measure both of cellular killing and of the metabolic state of conidia, it is possible that the discrepancy results from a lag period of conidial growth introduced by the intracellular environment. Consistent with this is the observation that conidia that are preincubated (swollen) before exposure to murine alveolar macrophages are killed more rapidly (7). Although this observation was interpreted as a sign of increased susceptibility to antimicrobial peptides, it is also possible that this difference represents a more accurate measurement of killing, by eliminating the lag period introduced by delayed intracellular conidial maturation. Finally, one cannot exclude the possibility that phorbol 12-myristate 13-acetate-activated THP-1 cells kill conidia more effectively than do other cells. Previous studies have indicated variation in killing efficacy depending on the specific cellular population studied (14).
THP-1 cells killed conidia that were preexposed to serum components more rapidly than they killed naked conidia, with almost complete killing occurring by 2 h after exposure. Measurement of intracellular FUN-1 orange demonstrated that THP-1 cells internalize opsonized conidia more effectively than they internalize nonopsonized conidia. This finding is consistent with previous studies demonstrating increased neutrophil internalization of plasma-treated conidia (16). Although this has been attributed in part to the effects of complement binding, numerous serum components, such as immunoglobulin, mannose binding lectins, or soluble CD14, may mediate ingestion through specific pathogen recognition receptors on macrophages (10–12, 18). Mannose binding lectin, which has been previously shown to bind to A. fumigatus (11), enables amplification of complement activation and phagocyte recognition (3). The method described here might prove valuable for further detailed investigations of conidium-phagocyte interactions.
In conclusion, we have developed sensitive assays to measure both conidial phagocytosis and killing, minimizing multiple potential sources of error introduced by previously employed techniques. The results of studies using these methods indicate that preopsonization of A. fumigatus conidia results in more rapid ingestion, coupled with faster intracellular killing. Further studies are necessary to explain specific mechanisms of conidial phagocytosis and killing.
ACKNOWLEDGMENTS
These studies were supported by grants from the National Institutes of Health (K08-AI01571) and a Research Grant from the American Lung Association (K.A.M.).
We thank Janet Staab and Theodore White for helpful suggestions and manuscript review.
REFERENCES
- 1.Alvarez-Barrientos A, Arroyo J, Canton R, Nombela C, Sanchez-Perez M. Applications of flow cytometry to clinical microbiology. Clin Microbiol Rev. 2000;13:167–195. doi: 10.1128/cmr.13.2.167-195.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.deRepentigny L, Petitbois S, Boushira M, Michaliszyn E, Senechal S, Gendrom N, Montplaiser S. Acquired immunity in experimental murine aspergillosis is mediated by macrophages. Infect Immun. 1993;61:3791–3802. doi: 10.1128/iai.61.9.3791-3802.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Epstein J, Eichbaum Q, Sheriff S, Ezekowitz R A. The collectins in innate immunity. Curr Opin Immunol. 1996;8:29–35. doi: 10.1016/s0952-7915(96)80101-4. [DOI] [PubMed] [Google Scholar]
- 4.Jahn B, Rampp A, Dick C, Jahn A, Palmer M, Bhakdi S. Accumulation of amphotericin B in human macrophages enhances activity against Aspergillus fumigatus conidia: quantification of conidial kill at the single-cell level. Antimicrob Agents Chemother. 1998;42:2569–2575. doi: 10.1128/aac.42.10.2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lass-Florl C, Nagl M, Speth C, Ulmer H, Dierich M P, Wurzner R. Studies of in vitro activities of voriconazole and itraconazole against Aspergillus hyphae using viability staining. Antimicrob Agents Chemother. 2001;45:124–128. doi: 10.1128/AAC.45.1.124-128.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Latge J. Aspergillus fumigatus and aspergillosis. Clin Microbiol Rev. 1999;12:310–350. doi: 10.1128/cmr.12.2.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levitz S, Selsted M, Ganz T, Lehrer R, Diamond R. In vitro killing of spores and hyphae of Aspergillus fumigatus and Rhizopus oryzae by rabbit neutrophil cationic peptides and bronchoalveolar macrophages. J Infect Dis. 1986;154:483–489. doi: 10.1093/infdis/154.3.483. [DOI] [PubMed] [Google Scholar]
- 8.Michaliszyn E, Senechal S, Martel P, deRepentigny L. Lack of involvement of nitric oxide in killing of Aspergillus fumigatus conidia by pulmonary alveolar macrophages. Infect Immun. 1995;63:2075–2078. doi: 10.1128/iai.63.5.2075-2078.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Millard P, Roth B, Thi H, Yue S, Haugland R. Development of the FUN-1 family of fluorescent probes for vacuole labeling and viability testing of yeasts. Appl Environ Microbiol. 1997;63:2897–2905. doi: 10.1128/aem.63.7.2897-2905.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mold C, Gresham H, DuClos T. Serum amyloid P component and C-reactive protein mediate phagocytosis through murine Fc gamma receptors. J Immunol. 2001;166:1200–1205. doi: 10.4049/jimmunol.166.2.1200. [DOI] [PubMed] [Google Scholar]
- 11.Neth O, Jack D, Dodds A, Holzel H, Klein N, Turner M. Mannose-binding lectin binds to a range of clinically relevant microorganisms and promotes complement deposition. Infect Immun. 2000;68:688–693. doi: 10.1128/iai.68.2.688-693.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olson E, Standing J, Griego-Harper N, Hoffman O, Limper A. Fungal β-glucan interacts with vitronectin and stimulates tumor necrosis factor alpha release from macrophages. Infect Immun. 1996;64:3548–3554. doi: 10.1128/iai.64.9.3548-3554.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robertson M, Kerr K, Seaton A. Killing of Aspergillus fumigatus spores by human lung macrophages: a paradoxical effect of heat-labile serum components. J Med Vet Mycol. 1989;27:295–302. doi: 10.1080/02681218980000401. [DOI] [PubMed] [Google Scholar]
- 14.Schaffner A, Douglas H, Braude A I, Davis C E. Killing of Aspergillus spores depends on the anatomical source of the macrophage. Infect Immun. 1983;42:1109–1115. doi: 10.1128/iai.42.3.1109-1115.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sturtevant J, Latge J P. Participation of complement in the phagocytosis of the conidia of Aspergillus fumigatus by human polymorphonuclear cells. J Infect Dis. 1992;166:580–586. doi: 10.1093/infdis/166.3.580. [DOI] [PubMed] [Google Scholar]
- 16.Sturtevant J E, Latge J P. Interactions between conidia of Aspergillus fumigatus and human complement component C3. Infect Immun. 1992;60:1913–1918. doi: 10.1128/iai.60.5.1913-1918.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsai H, Wheeler M, Chang Y, Kwon-Chung K. A developmentally regulated gene cluster involved in conidial biosynthesis in Aspergillus fumigatus. J Bacteriol. 1999;181:6469–6477. doi: 10.1128/jb.181.20.6469-6477.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang J E, Warris A, Ellingsen E A, Jorgensen P F, Flo T H, Espevik T, Solberg R, Verweij P E, Aasen A O. Involvement of CD14 and Toll-like receptors in activation of human monocytes by Aspergillus fumigatus hyphae. Infect Immun. 2001;69:2402–2406. doi: 10.1128/IAI.69.4.2402-2406.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wenisch C, Linnau K, Parschalk B, Zedtwitz-Liebenstein K, Georgopoulos A. Rapid susceptibility testing of fungi by flow cytometry using vital staining. J Clin Microbiol. 1997;35:5–10. doi: 10.1128/jcm.35.1.5-10.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]