

Abstract
Synthetic supramolecular structures constructed through the cooperative action of numerous non-covalent forces are highly desirable as models to unravel and understand the complexity of systems created in nature via self-assembly. Taking advantage of the low cost of 2,4,6-trichloro-1,3,5-triazine (cyanuric chloride) and the sequential nucleophilic substitution reactions with almost all types of nucleophiles, a series of six structurally related novel s-triazine derivatives 1–6 were synthesized and structurally characterized based on their physical, spectral and crystallographic data. The solid-state structures of all the six compounds showed intriguing and unique molecular duplexes featuring NH···N, CH···O and CH···π interactions. Careful analysis of different geometric parameters of the involved H-bonds indicates that they are linear, significant and are therefore responsible for guiding the three-dimensional structure of these compounds in the solid state. The prevalence of sextuple hydrogen bond array-driven molecular duplexes and the possibility of structural modifications on the s-triazine ring render these novel triazine derivatives 1–6 attractive as a platform to create heteroduplex constructs and their subsequent utility in the field of supramolecular chemistry and crystal engineering.
Keywords: triazine derivatives, self-assembly, molecular duplexes, CH···O interactions, CH···π interactions, supramolecular synthons
1. Introduction
Material properties are frequently dictated by the interactions and hence the way in which their constituent entities are assembled [1]. Consequently, molecular self-assembly has turned out to be an important 'bottom-up' approach to create intriguing materials of nano- and micro-structures, starting from simple components [2–6]. However, even with the best present-day synthetic approaches, accurate prediction of structures and the preparation of complementary nanostructures having efficiency equivalent to natural systems still remain a challenge and the ultimate goal of crystal engineers and supramolecular chemists [2–6]. The main reason is inadequate knowledge of intermolecular forces and poor understanding of the principles involved [7]. So far, numerous artificial self-assembly systems have been developed, employing a variety of supramolecular forces [8–11]. The most interesting are the systems involving arrays of multiple parallel or near-parallel H-bonds [12–18], developed primarily mimicking the DNA duplex having highly predictable intermolecular interactions and programmable sequence specificity.
Importance of weak hydrogen bonds such as CH···O and CH···π has recently been recognized in both natural and synthetic systems [19–24]. Although C–H groups are generally much weaker H-bond donors than hydrogens bound to heteroatoms, the CH···O/π interactions have been found to play important roles in physical, chemical and biological properties of a variety of substances [19–24]. Interestingly, the CH···O interactions are reported to constitute 20–25% of the total number of hydrogen bonds in proteins, playing key role in stabilizing the structures of proteins [25–30]. Similarly, the CH···π interactions have been found to be robust enough to stabilize a particular conformation of molecules for their higher order self-assembly [19,20]. These are documented as the main supramolecular forces in protein folding that stabilizes their secondary and tertiary structures [31,32], in addition to their role in arranging alkyl chains towards the phenyl group of amino acid residues and binding of proteins with cofactors and carbohydrates [33]. Owing to the recognition of their importance in natural systems, the CH···O/π interactions are now considered as vital supramolecular forces in various synthetic self-assembly processes, molecular and anion recognition events, and in crystal engineering field [34–38].
To date, a variety of self-assembled duplex systems involving arrays of conventional H-bonds have been recognized and subsequently used in the field of supramolecular chemistry [12–18]. However, the similar duplex systems involving weak hydrogen bonds such as CH···O/N/π are rarely identified and explored [39]. Keeping in view the importance of weak hydrogen bonds such as CH···O/N/π (vide supra), the duplex systems involving CH···O/N/π interactions are extremely important to be identified to meet the nature's selectivity in artificial systems. Stimulated by these observations and as continuation of our research interests in non-covalent interactions [40–46], herein we report the synthesis and structures of six novel triazine derivatives having AAADDD type H-bond acceptors and donor sites (π, O and N acceptors, and CH, CH and NH donors, respectively). Owing to the special structural features (figure 1), the formation of unique molecular duplexes featuring NH···N, CH···O and CH···π has been observed in the solid-state self-assembly of all the synthesized compounds.
Figure 1.

Proposed general structural features of synthesized triazine derivatives. Note: in cases where n = 0, neighbouring aryl CH may act as weak H-bond donor.
2. Results and discussion
The s-triazine derivatives 1–6 were synthesized in three steps starting from readily available and inexpensive cyanuric chloride (scheme 1). In the first step, cyanuric chloride was reacted with a variety of phenols in dry tetrahydrofuran containing anhydrous potassium carbonate to obtain 2-chloro-4,6-diaryloxy-1,3,5-triazines A which were then treated with hydrazine monohydrate in chloroform solvent at room temperature in the second step to afford 2-hydrazinyl-4,6-diaryloxy-1,3,5-triazines B. Finally, the intermediates B were reacted with different aromatic aldehydes in refluxing dry ethanol containing the catalytic amounts of sodium hydrogen sulphite to furnish s-triazine derivatives 1–6 in good to excellent yields (scheme 1). All the synthesized triazine derivatives were at first characterized by their physical and spectral (FT-IR, 1H and 13C-NMR) data, and later by their single crystal X-ray diffraction analysis. The single crystals suitable for X-ray diffraction analysis of all the derivatives (table 1) were obtained by slow evaporation of their solution in ethanol solvent.
Scheme 1.

Synthesis of s-triazine derivatives 1–6.
Table 1.
X-ray crystallographic data of 1–6.
| crystal data | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| CCDC | 2 155 046 | 2 155 047 | 2 155 048 | 2 155 049 | 2 155 050 | 2 155 051 |
| chemical formula | C24H21N5O2 | C22H17N5O3 | C22H15Br2N5O3 | C24H20Cl2N6O2 | C26H23N5O2 | C24H17Cl2N5O2 |
| Mr | 411.46 | 399.40 | 557.21 | 495.36 | 437.49 | 478.32 |
| crystal system, space group | monoclinic, P21/c | triclinic, P̄1 | monoclinic, P21 | triclinic, P̄1 | triclinic, P̄1 | monoclinic, P21/c |
| temperature (K) | 296 | 296 | 296 | 296 | 296 | 296 |
| a, b, c (Å) | 11.585 (2), 8.1214 (11), 22.565 (3) | 11.9832 (12), 12.5490 (11), 13.8420 (11) | 9.4582 (16), 22.182 (4), 10.407 (2) | 11.8120 (11), 14.0117 (14), 16.0983 (14) | 12.9628 (11), 13.0656 (12), 15.2100 (13) | 16.525 (9), 8.180 (7), 18.726 (13) |
| α, β, γ (°) | 90.391 (10) | 106.918 (4), 93.462 (5), 99.538 (5) | 90.648 (13) | 99.163 (3), 108.034 (4), 104.982 (6) | 68.900 (4), 79.785 (4), 77.004 (4) | 109.48 (3) |
| V (Å3) | 2123.1 (6) | 1950.9 (3) | 2183.3 (7) | 2362.9 (4) | 2328.6 (4) | 2386 (3) |
| Z | 4 | 4 | 4 | 4 | 4 | 4 |
| radiation type | Mo Kα | Mo Kα | Mo Kα | Mo Kα | Mo Kα | Mo Kα |
| µ (mm−1) | 0.09 | 0.09 | 3.75 | 0.31 | 0.08 | 0.30 |
| crystal size (mm) | 0.44 × 0.22 × 0.16 | 0.37 × 0.33 × 0.22 | 0.38 × 0.26 × 0.22 | 0.38 × 0.32 × 0.24 | 0.44 × 0.35 × 0.28 | 0.38 × 0.34 × 0.28 |
| data collection | ||||||
| diffractometer | Bruker Kappa APEXII CCD | Bruker Kappa APEXII CCD | Bruker Kappa APEXII CCD | Bruker Kappa APEXII CCD | Bruker Kappa APEXII CCD | Bruker Kappa APEXII CCD |
| absorption correction | multi-scan (SADABS; Bruker, 2005) | multi-scan (SADABS; Bruker, 2005) | multi-scan (SADABS; Bruker, 2005) | multi-scan (SADABS; Bruker, 2005) | multi-scan (SADABS; Bruker, 2005) | multi-scan (SADABS; Bruker, 2005) |
| Tmin, Tmax | 0.675, 0.746 | 0.675, 0.746 | 0.345, 0.550 | 0.675, 0.746 | 0.675, 0.746 | 0.675, 0.746 |
| no. of measured, independent and observed [I > 2σ(I)] reflections | 13 303, 4617, 2420 | 22 828, 7656, 3849 | 12 636, 7647, 3970 | 26 688, 8743, 4532 | 25 415, 8613, 4140 | 14 629, 5204, 2763 |
| Rint | 0.056 | 0.073 | 0.058 | 0.071 | 0.062 | 0.058 |
| (sin θ/λ)max (Å−1) | 0.639 | 0.617 | 0.606 | 0.606 | 0.606 | 0.639 |
| refinement | ||||||
| R[F2 > 2σ(F2)], wR(F2), S | 0.056, 0.151, 1.00 | 0.054, 0.146, 0.96 | 0.062, 0.150, 0.96 | 0.058, 0.153, 0.98 | 0.057, 0.169, 0.98 | 0.056, 0.178, 1.01 |
| no. of reflections | 4617 | 7656 | 7647 | 8743 | 8613 | 5204 |
| no. of parameters | 282 | 543 | 521 | 617 | 600 | 298 |
| no. of restraints | — | — | 1 | — | — | — |
| H-atom treatment | H-atom parameters constrained | H-atom parameters constrained | H atoms treated by a mixture of independent and constrained refinement | H-atom parameters constrained | H-atom parameters constrained | H-atom parameters constrained |
| Δ>max, Δ>min (e Å−3) | 0.19, −0.20 | 0.15, −0.18 | 0.96, −0.53 | 0.28, −0.32 | 0.24, −0.22 | 0.46, −0.41 |
The molecular structures of triazine derivatives 1–6 along with crystallographic numbering schemes are illustrated in figure 2. Interestingly, the compounds 1, 3 and 6 crystallize in monoclinic system with space group P21/c, P21 and P21/c, respectively, whereas compounds 2, 4 and 5 crystallize in triclinic system, all having P̄1 space group. Despite having almost the same molecular core, the structures of 1–6 are not isomorphous, reflecting the impact of the substituents on the molecular packing (table 1). The compounds 1 and 6 have one molecule in the asymmetric unit whereas, two different/independent molecules/conformers having slightly different bond lengths, dihedral angles and torsions angles were observed in the unit cell of compounds 2–5 (figure 2). Selected geometric parameters for all the six compounds are presented in the electronic supplementary material, table S1. The magnitude of the standard uncertainty values linked with these geometric parameters prohibits description of any definitive trends across the series. The central core comprising the imino group of hydrazone and triazinyloxy moiety is essentially planar in the crystal structures of all six compounds. This planarity can be attributed to the partial double bond character of both C-N and C-O bonds involving triazine ring due to significant delocalization of nitrogen and oxygen lone pairs towards the triazine ring. This is clearly indicated by the shorter bond lengths of C-N and C-O bonds [N4-C2 1.347(2) Å and O2-C3 1.342(2) Å in 1, N4-C3 1.341(3) Å, N9-C25 1.339(3) Å and O1-C1 1.355(3) Å, O5-C24 1.344(3) Å in 2, N4-C3 1.331(16) Å, N9-C25 1.348(15) Å and O1-C1 1.364(13) Å, O5-C24 1.343(15) Å in 3, N4-C3 1.336(4) Å, N9-C25 1.344(3)Å and O1-C1 1.357(3) Å, O3-C25 1.360(3) Å in 4, N4-C2 1.340(3) Å, N9-C28 1.340(3) Å and O1-C1 1.348(3) Å, O3-C27 1.348(3) Å in 5, N4-C2 1.352(3) Å and O1-C3 1.360(3) Å in 6] (electronic supplementary material, table S1). The aryl ring attached to the oxygen of central/planar core is tilted/twisted and in some cases present nearly at right angle compared with the triazine plane [C3-O2-C11-C12 93.7(3)° in 1, C1-O1-C4-C5 −121.9(3)° and C24-O5-C32-C33 123.5(3)° in 2, C1-O1-C4-C5 46.9(13)° and C24-O5-C32-C33 141.3(10)° in 3, C1-O1-C4-C5 −143.8(3)° and C25-O3-C28-C29 −87.1(4)° in 4, C1-O1-C4-C5 132.4(2)° and C27-O3-C30-C31 119.4(3)° in 5, C3-O1-C4-C9 −123.6(3)° in 6]. The deviation of aryl ring plane from the right-angled geometry can be attributed to the varying degree of conjugation of oxygen with neighbouring (hetero)aromatics (figure 1). By contrast, the other aryl ring attached to the central/planar core in all the compounds except 1 is almost present in the same plane as triazine ring owing to the extended conjugation [N5-C18-C19-C20 165.5(2)° in 1, N5-C16-C17-C22 174.3(3)°, N10-C38-C39-C44 173.0(3)° in 2, N5-C16-C17-C22 176.4(10)°, N10-C38-C39-C44 −173.7(14)° in 3, N5-C16-C17-C22 2.3(5)°, N11-C40-C41-C42 1.9(5)° in 4, C19-C20-C21-C22 170.3(3)°, C45-C46-C47-C52 176.5(3)° in 5 and C17-C18-C19-C24 176.6(3)° in 6]. The slightly more tilt observed in the case of compound 1 may be attributed to the packing effect. This particular arrangement of central core and the two attached aryl rings offers three H-bond donors [NH, CH (imine) and CH (aryl/sp2)] and three H-bond acceptors [N (triazine ring), O (bridged between (hetero)aromatic rings, π system (aryl)]. Interestingly, the presence of sp2-hybridized nitrogen atom in the central core increases acidity of both nearby NH and CH groups, making them better H-bond donors. As the strength of any hydrogen bond is more dependent on donor acidity than acceptor basicity [22], an expected consequence of this particular arrangement and increased acidity of NH and CH groups is the facile formation of molecular duplexes featuring NH···N, CH···O and CH···π interactions (vide infra).
Figure 2.

The molecular structures (Oak Ridge thermal ellipsoid plot (ORTEP) diagram) of triazine derivatives 1–6. Displacement ellipsoids are drawn at 50% probability level.
As shown in figure 3, all the six solid-state structures feature a molecular duplex driven by NH···N, CH···O and CH···π interactions, and this clearly is the predominant supramolecular synthon operating in each of the crystal structures; geometric parameters associated with this synthon are listed in table 2. In addition to observation of a centrosymmetric molecular duplex in the solid-state structure of compounds 1 (1.1) and 6 (6.6) that has one molecule in their asymmetric units, a centrosymmetric molecular duplex has also been observed for compound 2 having two independent molecules in the crystal structure. In this compound, two different centrosymmetric duplexes [(2.2) and (2′.2′)] have been observed. However, all other compounds of the series i.e. 3, 4 and 5 having two different conformers/independent molecules in their unit cells showed the formation of duplexes between two different conformers [duplexes of type (3.3′), (4.4′) and (5.5′), respectively] (figure 3).
Figure 3.

Molecular duplexes observed in the solid-state structures of triazine derivatives 1–6.
Table 2.
Geometric parameters associated with the hydrogen bonds of molecular duplexes in 1–6.
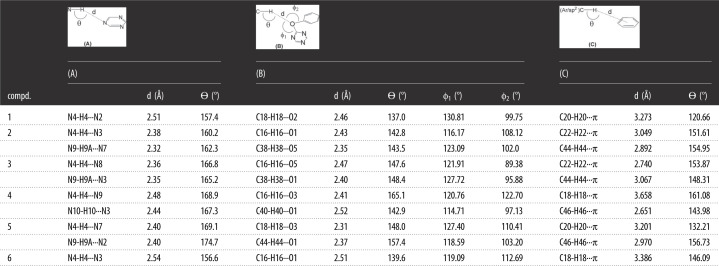 |
Over and beyond the NH···N, CH···O and CH···π interactions involved in the formation of molecular duplexes, the crystal packing of 1 (R = 4-CH3, R1 = H, n = 0) is dominated by π···π, CH···π and CH···N interactions. The self-complementary molecular duplex of 1 interacts with a neighbouring duplex by means of an antiparallel π···π [C5⋯C6 3.337 Å] stacking and a CH···π [C24-H24···C6 2.828 Å] interaction, resulting in the formation of one-dimensional tapes (electronic supplementary material, figure S1). These tapes then connect to the neighbouring tapes by means of C-H···π [C12-H12···C14 2.769 Å] and CH···N [C9-H9···N2 2.692 Å] interactions, making an overall three-dimensional network structure (electronic supplementary material, figure S2). Similar one-dimensional tapes are observed in compound 2 (R = H, R1 = 3-OH, n = 0) and 3 (R = 4-Br, R1 = 3-OH, n = 0), both having a strong H-bonding OH group. Owing to the presence of two different/independent molecules/conformers in the unit cell of 2, two self-complementary molecular duplexes, (2.2) and (2′.2′), interact with neighbouring duplexes by means of OH···N [O3-H3···N2 2.221 Å and O6-H6A···N8 2.137 Å] and CH···O [C18-H18···O3 2.399 Å, C20-H20···O2 2.458 Å and C40-H40···O6 2.473 Å, C42-H42···O4 2.448 Å] contacts. These tapes of 2 joins with the neighbouring tapes with the help of π···π [C2···C30 3.363 Å] and CH···π [C33-H33···C13 2.891 Å] interactions, making an overall three-dimensional network structure of 2 (electronic supplementary material, figure S2). In contrast with the duplexes of 2 and as pointed out above, a molecular duplex of 3 featuring two independent conformers (3.3′) is formed despite having two different/independent molecules/conformers in its unit cell. These duplexes interact with each other by means of a bifurcated OH···N and OH···O [O3-H3···N7 2.218 Å and O3-H3···O4 2.603 Å] and two CH···O [C20-H20···O4 2.491 Å, C40-H40···O3 2.489 Å] interactions forming one-dimensional tapes (electronic supplementary material, figure S1). These tapes connect with the neighbouring tapes by CH···π [C6-H6···O6 2.648 Å], CH···Br [C33-H33···Br 2.878 Å] and a lone pair···π [Br4···C7 3.518 Å] interactions to extend a three-dimensional network structure of 3 (electronic supplementary material, figure S2). Interestingly, the tape structures observed for 1–3 are not observed for compound 4. In compound 4, each molecular duplex featuring two independent conformers (3.3′) stack on neighbouring duplex by means of CH···N [C8-H8···N8 2.610 Å] and CH···π [C9-H9···C42 2.885 Å] interactions (electronic supplementary material, figure S1). These stacked duplexes attach to the neighbouring stacked duplexes with the help of CH···Cl [C19-H19···Cl3 2.903 Å], CH···O [C23-H23B···O1 2.651 Å] and a centrosymmetric lone pair···π [Cl2···C6 3.428 Å] interactions providing three-dimensional network structure of 4 (electronic supplementary material, figure S2). Supramolecular tapes featuring molecular duplexes (5.5′) are also formed for 5 by means of C-H···N [C49-H49···N5 2.675 Å] and C-H···O [C50-H50···O2 2.598 Å] interactions (electronic supplementary material, figure S1). These tapes are interconnected by means of C-H···π [C41-H41···C49 2.878 Å] and CH···N [C34-H34···N2 2.699 Å] interactions in the three-dimensional network structure of 5 (electronic supplementary material, figure S1). The self-complementary molecular duplexes observed for 6, however, form one-dimensional zigzag chains by means of CH···N [C6-H6···N5 2.806 Å], a bifurcated CH···N/O [C5-H5···N2 2.836 Å and C5-H5···O2 2.560 Å] and C-H···O [C11-H11···O2 2.785 Å] interactions (electronic supplementary material, figure S1). These chains connect to the neighbouring chains by C-H···π [C21-H21···C24 2.991 Å, C15-H15···C5 2.804 Å] and CH···Cl [C22-H22···Cl1 3.010 Å] interactions, furnishing three-dimensional network structure of 6 (electronic supplementary material, figure S2).
Directionality is considered as one of the most important features of a hydrogen bond to distinguish it from the mere London dispersion forces. Generally it is believed that the linear bonds (150° < ϴ < 180°) are structurally more significant due to the dipole–monopole and dipole–dipole contribution to the electrostatic energy which is a maximum at ϴ = 180° and zero at ϴ = 90° [22]. Careful analysis of the bond angles ϴ in table 2 demonstrates that the NH···N hydrogen bonds involved in the formation of molecular duplexes are relatively linear and the most significant. However, the same angle ϴ associated with C-H···O and C-H···π interactions involved in the formation of molecular duplexes is slightly more bent as compared with ϴ associated with NH···N hydrogen bonds, although it is still in the range of significant interactions. Interestingly, most of the CH···O distances are comparable to NH···N distances (please see d (Å) in table 2). As compared with the distances of NH···N and CH···O, the distances of CH···π [CH (aryl/sp2) to centre of aryl ring of neighbouring molecule] interactions are slightly longer, although falls within the limits [47]. However, the angle associated with these interactions clearly indicates the significant role of these interactions, as many CH···π interactions are clustered around a distance of C–π centre approximately 4–6 Å and angle ϴ approximately 120°–150° [47]. It is important to mention here that most of the CH···π interactions having greater angle ϴ have shorter distances, although whole data prohibits description of any definitive trends across the series (See d (Å) under column C in table 2). The angle ϕ is usually measured to see O/N-atom lone pair directionality. The angles ϕ1 and ϕ2 in table 2 have been observed in the range of 114.71–130.81° and 89.38–122.70° indicating the sidewise approach of the H-bond donors [48]. The variation of angles ϕ in these compounds also indicates the formation of various conjugation systems of oxygen with the adjacent aromatic rings. It can finally be inferred from the data gathered in table 2 that the cooperativity of NH···N, CH···O and CH···π interactions observed in triazine derivatives 1–6 make the otherwise weak CH···O and CH···π interactions strong and significant. Owing to this cooperativity, the molecular duplexes are observed even in compounds having strong competing H-bonding interactions, especially those involving OH groups. Therefore, the observed molecular duplexes are robust and are responsible for guiding the three-dimensional structures of these compounds in the solid state.
3. Conclusion
In summary, we have synthesized and crystallographically characterized six new structurally similar s-triazine derivatives 1–6 in order to see the formation of molecular duplexes through the cooperative action of NH···N, CH···O and CH···π interactions. Fascinatingly, the solid-state structures of all the six compounds showed the formation of expected molecular duplexes. The analysis of different geometric parameters clearly indicates the linear and significant nature of the involved non-covalent interactions highlighting their cooperative and interdependent nature, mutually influencing the strength of each other. Owing to the cooperative role of NH···N, CH···O and CH···π interactions, the observed molecular duplexes are robust supramolecular synthons, responsible for guiding the three-dimensional structures of synthesized triazine derivatives. Exploring the detailed structural and energetic impact of this cooperativity and the formation of duplexes will be interesting in host-guest chemistry, crystal engineering and other fields of supramolecular chemistry, especially when there is a possibility of structural modifications on the s-triazine ring (figure 1) keeping all the observed AAADDD type H-bond acceptors and donor sites intact.
4. Experimental section
4.1. General procedure for the synthesis 2-hydrazinyl-4,6-substituted-diphenoxy-1,3,5-triazines (B1–B4)
The 2-chloro-4,6-diaryloxy-1,3,5-triazine (A1–A4) (0.0066 mol) was dissolved in 30 ml of chloroform and added dropwise to the solution of hydrazine monohydrate (2 ml, 0.326 mol) in chloroform with constant stirring over a period of 16 h at room temperature. The reaction mixture was further stirred for another 6–8 h. After the completion of reaction as indicated by TLC, the mixture was washed with 3 × 30 ml of water in order to remove excess of hydrazine hydrate. The chloroform layer was then collected and dried over anhydrous sodium sulfate. The solid obtained after evaporation of chloroform under vacuum was recrystallized from ethanol to get respective pure product (B1–B4).
4.1.1. 2-Hydrazinyl-4,6-bis(4-tolyloxy)–1,3,5-triazine (B1)
Yield: 68%; m.p.: 185–187°C; Rf: 0.62 (chloroform : methanol; 9 : 1); 1H-NMR (300 MHz, DMSO-d6): δ (ppm) 9.19 (s, 1H, NH), 7.22–7.19 (4H, m, H-6,6′,10,10′), 7.12–7.09 (2H, d, J = 8.4 Hz, H-5,9), 7.10 (2H, d, J = 8.4 Hz, H-5′,9′), 4.34 (2H, s, NH2), 2.31, 2.29 (6H, s, 2 × CH3); 13C-NMR (75 MHz, DMSO-d6):δ (ppm) 172.5 (C-1), 171.6, 169.5 (C-2,3),150.1, 150.0 (C-4,8), 135.0, 134.9 (C-7,11), 130.3, 130.1 (C-6,6′,10,10′), 122.2, 121.8 (C-5,5′,9,9′), 20.8, 20.8 (2 × CH3)
4.1.2. 2-Hydrazinyl-4,6-diphenoxy-1,3,5-triazine (B2)
Yield: 67%; m.p.: 106–108°C [Lit. 107] [49]; Rf: 0.51 (chloroform : methanol; 9 : 1); 1H-NMR (300 MHz, DMSO-d6): δ (ppm) 9.22 (s, 1H, NH), 7.44–7.36 (4H, m, H-6,6′,10,10′), 7.27–7.15 (6H, m, H-5,5′,9,9′,7,11), 4.34 (2H, d, J = 2.7 Hz, NH2), 13C-NMR (75 M Hz, DMSO-d6): δ (ppm) 172.3 (C-1) 171.4, 169.6 (C-2,3), 152.2, 152.2 (C-4,8) 129.9, 129.8 (C-6,6′,10,10′), 125.9 (C-7,11) 122.1(C-5,5′,9,9).
4.1.3. 2,4-bis(4-Bromophenoxy)-6-hydrazinyl-1,3,5-triazine (B3)
Yield: 66%; m.p.: 183–185°C; Rf: 0.45 (chloroform : methanol; 9 : 1); 1H-NMR (300 MHz, DMSO-d6) (300 MHz, DMSO-d6): δ (ppm) 9.27 (s, 1H, NH), 7.61–7.55 (4H, m, H-6,6′,10,10′), 7.24–7.13 (4H, m, H-5,5′,9,9′), 4.37 (2H, s, NH2); 13C-NMR (75 MHz, DMSO-d6): δ (ppm) 172.0 (C-1), 171.2, 169.4 (C-2,3), 151.5,151.4 (C-4,8), 132.7, 132.6 (C-6,6′,10,10′), 124.6, 124.5 (C-5,5′,9,9′), 118.2, 118.1 (C-7,11).
4.1.4. 2,4-bis(4-Chlorophenoxy)-6-hydrazinyl-1,3,5-triazine (B4)
Yield: 60%; m.p.: 166–168°C; Rf: 0.49 (chloroform : methanol; 9 : 1); 1H-NMR (300 MHz, DMSO-d6) (300 MHz, DMSO-d6): δ (ppm) 9.27 (s, 1H, NH), 7.47–7.43 (4H, m, H-6,6′,10,10′), 7.27 (2H, d, J = 9.0 Hz, H-5,9), 7.21 (2H, d, J = 9.0 Hz, H-5′,9′), 4.37 (2H, s, NH2); 13C-NMR (75 M Hz, DMSO-d6): δ (ppm) 172.1 (C-1), 171.2, 169.4 (C-2,3), 151.0, 150.9 (C-4,8), 130.1, 130.0 (C-6,6′,10,10′), 129.8, 129.7 (C-7,11) 124.1, 124.1 (C-5,5′,9,9′).
4.2. General procedure for the synthesis 2-(substituted-benzylidenehydrazinyl)-4,6-substituted-diphenoxy-1,3,5-triazine (1–6)
The 2-hydrazinyl-4,6-diphenoxy-1,3,5-triazine (B1–B4) (0.01 mol) was added to a 20 ml of dry ethanol and the mixture was stirred for 15 min at room temperature, followed by the addition of 0.012 mol of substituted aldehydes and catalytic amount of sodium hydrogen sulphite. The whole reaction mixture was then refluxed for 6–12 h, which resulted in the appearance of a solid product. After completion of the reaction as indicated by TLC, the solid product was filtered, washed with cold ethanol and recrystallized from a mixture of acetonitrile and ethanol to afford respective pure product (1–6).
4.2.1. 2-(2-Benzylidenehydrazinyl)-4,6-bis(p-tolyloxy)-1,3,5-triazine (1)
Yield: 70%; m.p.: 218–219°C; Rf: 0.70 (n-Hexane : ethyl acetate; 6 : 4); IR (ATR, ῡ): cm−1 3243 (N-H stretch.), 3030 (Csp2—H stretch.), 2918 (Csp3—H stretch.), 1604 (C=N stretch.), 1570, 1504 (2 × C=C stretch.), 1359, 1199 (2 × C-O stretch.); 1H-NMR (300 MHz, DMSO-d6): δ (ppm) 11.79 (1H, s, NH), 8.16 (1H, s, H-12), 7.62 (2H, dd, J = 7.5, 4.2 Hz, H-14,18), 7.43–7.40 (3H, m, H-15,16,17), 7.26–7.20 (4H, m, H-6,6′,10,10′), 6.71–7.08 (4H, m,H-5,5′,9,9′), 2.34, 2.31 (6H, s, 2 × CH3); 13C-NMR (75 MHz, DMSO-d6): δ (ppm) 172.9 (C-1), 172.1, 166.9 (C-2,3), 150.0 (C-4,8), 146.2 (C-12), 135.3, 135.1 (C-7,11), 134.7 (C-13), 130.4, 130.1 (C-6,6′,10,10′), 129.2 (C-14,16,18), 127.3 (C-15,17), 121.9 (C-5,5′,9,9′), 20.8(2 × CH3).
4.2.2. 3-((2-(4,6-Diphenoxy-1,3,5-triazin-2-yl)hydrazono)methyl)phenol (2)
Yield: 89%; m.p.: 258–260°C; Rf : 0.50 (chloroform : methanol; 9 : 1); IR (ATR, ῡ): cm−1 3400–3000 (broad, O-H stretch.), 3232 (N-H stretch.), 3062 (Csp2—H stretch.), 1612 (C=N stretch.), 1571, 1544 (2 × C=C stretch.), 1368, 1191 (2 × C-O stretch.); 1H-NMR (300 MHz, DMSO-d6): δ (ppm) 11.79 (1H, s, NH), 9.63 (1H, s, OH) 7.44 (1H, s, H-12), 7.44–7.42 (4H, m, H-6,6′,10,10′), 7.27–7.14 (8H, m, H-5,5′,9,9′,7,11,14,17), 7.00 (1H, d, J = 6.6 Hz, H-18), 6.80 (1H, d, J = 6.6 Hz, H-16); 13C-NMR (75 MHz, DMSO-d6): δ (ppm) 172.8 (C-1), 171.9, 166.9 (C-2,3), 158.1 (C-15), 152.2 (C-4,8), 146.5 (C-12), 135.9 (C-15), 130.3 (C-17), 130.0, 129.8 (C-6,6′,10,10′) 126.1 (C-7,11), 122.2 (C-5,5′,9,9′), 119.0 (C-18), 117.7 (C-16), 112.9 (C-14).
4.2.3. 3-((2-(4, 6-bis(4-Bromophenoxy)-1,3,5-triazinyl)hydrazono)methyl) phenol (3)
Yield: 71%; m.p.: 254°C; Rf: 0.60 (chloroform : methanol; 9 : 1); IR (ATR, ῡ): cm−1 3500–2500 (broad, O-H stretch.), 3223 (N-H stretch.), 3124 (Csp2—H stretch.), 1607 (C=N stretch.), 1565, 1480 (2 × C=C stretch.), 1367, 1195 (2 × C-O stretch.); 1H-NMR (300 MHz, DMSO-d6): δ (ppm) 11.81 (1H, s, NH), 9.61 (1H, s, OH), 8.08 (1H, s, H-12), 7.62 (4H, m, H-6,6′,10,10′), 7.27–7.19 (5H, m, H-5,5′,9,9′,17), 7.13 (1H, bs, H-14), 7.00 (1H, d, J = 7.2 Hz, H-18), 6.80 (1H, dd, J = 8.1, 2.4 Hz, H-16); 13C-NMR (75 MHz, DMSO-d6): δ (ppm) 172.5 (C-1), 171.7, 166.8 (C-2,3), 158.1 (C-15), 151.4 (C-4,8), 135.8 (C-13), 132.8, 132.6 (C-6,6′,10,10′), 130.3 (C-17), 124.6 (C-5,5′,9,9′), 119.0 (C-18), 118.5, 118.3 (C-7,11), 117.8 (C-16), 113.0 (C-14).
4.2.4. 4-((2-(4,6-bis(4-Chlorophenoxy)-1,3,5-triazin-2-yl)hydrazono)methyl-N,N dimethylaniline (4)
Yield: 93%; m.p.: 226–228°C; Rf: 0.80 (chloroform : methanol; 9 : 1); IR (ATR, ῡ): cm−1 3243 (N-H stretch.), 3140 (Csp2—H stretch.), 2895 (Csp3—H stretch.), 1599 (C=N stretch.), 1572, 1483 (2 × C=C stretch.), 1366, 1193 (2 × C-O stretch.), 1084 (C-Cl); 1H-NMR (300 MHz, DMSO-d6): δ (ppm) 11.56 (1H, s, NH), 8.03 (1H, s, H-12), 7.50–7.42 (6H, m, H-6,6′,10,10′,14,18), 7.31–7.24 (4H, m, H-5,5′,9,9′), 6.71 (2H, d, J = 8.7 Hz, H-15,17), 2.95 (6H, s, N(CH3)2); 13NMR(75 MHz, DMSO-d6): (ppm) 172.6 (C-1), 171.6, 166.3 (C-2,3), 151.9 (C-16), 151.0 (C-4,8), 147.7 (C-12), 130.3, 130.1 (C-7,11), 129.9, 129.7 (C-6,6′,10,10′), 128.8 (C-14,18), 124.2 (C-5,5′,9,9′), 121.8 (C-13), 112.2 (C-15,14).
4.2.5. 2-((E)-2-((E)-3-phenylallylidene)hydrazinyl)-4,6-bis(p-tolyloxy)-1,3,5-triazine (5)
Yield: 77%; m.p.: 225–227°C; Rf: 0.69 (chloroform : methanol; 9 : 1); IR (ATR, ῡ): cm−1 3235 (N-H stretch.), 3124 (Csp2—H stretch.), 2916 (Csp3—H stretch.), 1625 (C = N stretch.), 1567, 1503 (2 × C = C stretch), 1366, 1193 (2 × C-O stretch); 1H-NMR (300 MHz, DMSO-d6): δ (ppm) 11.67 (1H, s, NH), 7.98 (1H, dd, J = 6.6, 1.8 Hz, H-12), 7.59 (2H, d, J = 6.9 Hz, H-14,18), 7.40–7.30 (3H, m, H-15,16,17), 7.24–7.20 (4H, m, H-6,6′,10,10′), 7.13–7.08 (4H, m, H-5,5′,9,9′), 6.97 (2H, d, J = 6.6 Hz, H-X,Y), 2.32, 2.31 (6H, s, 2 × CH3); 13C-NMR (75 MHz, DMSO-d6): δ (ppm); 172.9 (C-1), 172.1, 166.7 (C-2,3), 150.0 (C-4,8), 148.8 (C-12), 139.2 (C-13), 136.3 (C-X), 135.3, 135.1 (C-7,11), 130.4, 130.3 (C-6,6′,10,10′), 129.2 (C-14,15,17,18), 127.5 (C-16), 125.8 (C-Y), 121.87 (C-5,5′,9,9′), 20.8 (2 × CH3).
4.2.6. 2,4-bis(4-Chlorophenoxy)-6-(2-((E)-3-phenylallylidene)hydrazinyl)-1,3,5-triazine (6)
Yield: 87%; m.p: 248–250°C; Rf: 0.73 (chloroform : methanol; 9 : 1); IR (ATR, ῡ): cm−1 3240 (N-H stretch.), 3132 (Csp2—H stretch.), 1626 (C=N stretch.), 1574, 1485 (2 × C=C stretch.), 1366, 1209 (2 × C-O stretch.), 1085 (C-Cl); 1H-NMR (300 MHz, DMSO-d6): δ (ppm) 11.76 (1H, s, NH), 7.99 (1H, dd, J = 5.1, 3.0 Hz, H-12), 7.61–7.47 (6H, m, H-6,6′,10,10′,H-15,17), 7.39–7.26 (7H, m, H-5,5′,9,9′,14,16,18), 6.99–6.97 (2H, m, H-19,20); 13C-NMR (75 MHz, DMSO-d6): δ (ppm); 172.6 (C-1), 171.8, 166.6 (C-2,3), 151.0 (C-4,8), 149.2 (C-12), 139.5 (C-13), 136.3 (C-20), 130.4, 130.2 (C-7,11), 130.0, 129.8 (C-6,6′,10,10′), 129.3 (C-14,16,18), 127.6 (C-15,17), 125.7 (C-19), 124.2, 124.1 (C-5,5′,9,9′).
4.3. Crystallographic data collection and structural refinement
Single crystals of 1–6 were mounted on a thin glass fibre at room temperature and the reflection data were collected on a Bruker kappa APE XII CCD diffractometer equipped with graphite mono-chromated MoKα radiation (λ = 0.71073 Å). The data were also corrected for Lorentz and polarization effects. The structure was solved using SHELXS-97. Final refinement on F2 was carried out by full-matrix least-squares techniques using SHELXL-97 [50]. The crystal data of 1–6 (CCDC: 2155046-2155051) and refinement values are summarized in table 1.
Contributor Information
Shahid Hameed, Email: shameed@qau.edu.pk.
Muhammad Moazzam Naseer, Email: moazzam@qau.edu.pk.
Ethics
It is not relevant to our work
Data accessibility
Data available as electronic supplementary material [51]. The crystallographic data can also be obtained from https://www.ccdc.cam.ac.uk/data_request/cif.
CCDC: 2155046-2155051
Authors' contributions
S.A.: data curation, formal analysis and methodology; S.H.: conceptualization, funding acquisition, project administration, resources, supervision, validation and writing—review and editing; M.N.T.: data curation, formal analysis and software; M.M.N.: conceptualization, funding acquisition, project administration, supervision, validation, writing—original draft and writing—review and editing.
All authors gave final approval for publication and agreed to be held accountable for the work performed therein.
Conflict of interest declaration
We declare we have no competing interests.
Funding
We are highly grateful to the Higher Education Commission (HEC), Govt. of Pakistan for financial support.
References
- 1.Pochan D, Scherman O. 2021. Introduction: molecular self-assembly. Chem. Rev. 121, 13 699-13 700. ( 10.1021/acschemrev.1c00884) [DOI] [PubMed] [Google Scholar]
- 2.Li T, Lu XM, Zhang MR, Hu K, Li Z. 2022. Peptide-based nanomaterials: self-assembly, properties and applications. Bioact. Mater. 11, 268-282. ( 10.1016/j.bioactmat.2021.09.029) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ariga K, Nishikawa M, Mori T, Takeya J, Shrestha LK, Hill JP. 2019. Self-assembly as a key player for materials nanoarchitectonics. Sci. Technol. Adv. Mater. 20, 51-95. ( 10.1080/14686996.2018.1553108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sevim S, Sorrenti A, Franco C, Furukawa S, Pané S, Demello AJ, Puigmartí-Luis J. 2018. Self-assembled materials and supramolecular chemistry within microfluidic environments: from common thermodynamic states to non-equilibrium structures. Chem. Soc. Rev. 47, 3788-3803. ( 10.1039/C8CS00025E) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Talapin DV, Engel M, Braun PV. 2020. Functional materials and devices by self-assembly. MRS Bull. 45, 799-806. ( 10.1557/mrs.2020.252) [DOI] [Google Scholar]
- 6.Sang Y, Liu M. 2022. Hierarchical self-assembly into chiral nanostructures. Chem. Sci. 13, 633-656. ( 10.1039/D2SC00111J) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pavc D, Sebastian N, Spindler L, Drevenšek-Olenik I, Podboršek GK, Plavec J, Šket P. 2022. Understanding self-assembly at molecular level enables controlled design of DNA G-wires of different properties. Nat. commun. 13, 1-11. ( 10.1038/s41467-022-28726-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Conn MM, Rebek J. 1997. Self-assembling capsules. Chem. Rev. 97, 1647-1668. ( 10.1021/cr9603800) [DOI] [PubMed] [Google Scholar]
- 9.Li Y, Wang Y, Huang G, Gao J. 2018. Cooperativity principles in self-assembled nanomedicine. Chem. Rev. 118, 5359-5391. ( 10.1021/acs.chemrev.8b00195) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vriezema DM, Comellas Aragonès M, Elemans JA, Cornelissen JJ, Rowan AE, Nolte RJ. 2005. Self-assembled nanoreactors. Chem. Rev. 105, 1445-1490. ( 10.1021/cr0300688) [DOI] [PubMed] [Google Scholar]
- 11.Sinha NJ, Langenstein MG, Pochan DJ, Kloxin CJ, Saven JG. 2021. Peptide design and self-assembly into targeted nanostructure and functional materials. Chem. Rev. 121, 13 915-13 935. ( 10.1021/acs.chemrev.1c00712) [DOI] [PubMed] [Google Scholar]
- 12.Baruah PK, Khan S. 2013. Self-complementary quadruple hydrogen bonding motifs: from design to function. RSC Adv. 3, 21 202-21 217. ( 10.1039/C3RA43814G) [DOI] [Google Scholar]
- 13.Ligthart GB, Guo D, Spek AL, Kooijman H, Zuilhof H, Sijbesma RP. 2008. Ureidobenzotriazine multiple H-bonding arrays: the importance of geometrical details on the stability of H-bonds. J. Org. Chem. 73, 111-117. ( 10.1021/jo7019338) [DOI] [PubMed] [Google Scholar]
- 14.Cafferty BJ, Musetti C, Kim K, Horowitz ED, Krishnamurthy R, Hud NV. 2016. Small molecule-mediated duplex formation of nucleic acids with ‘incompatible' backbones. Chem. Commun. 52, 5436-5439. ( 10.1039/C6CC00779A) [DOI] [PubMed] [Google Scholar]
- 15.Zhang Y, et al. 2018. Hydrogen-bonded duplexes with lengthened linkers. Org. lett. 20, 1555-1558. ( 10.1021/acs.orglett.8b00283) [DOI] [PubMed] [Google Scholar]
- 16.Sun Y, Gu J, Wang H, Sessler JL, Thordarson P, Lin YJ, Gong H. 2019. AAAA–DDDD quadruple H-bond-assisted ionic interactions: robust bis (guanidinium)/dicarboxylate heteroduplexes in water. J. Am. Chem. Soc. 141, 20 146-20 154. ( 10.1021/jacs.9b09503) [DOI] [PubMed] [Google Scholar]
- 17.Zeng H, Miller RS, Flowers RA, Gong B. 2000. A highly stable, six-hydrogen-bonded molecular duplex. J. Am. Chem. Soc. 122, 2635-2644. ( 10.1021/ja9942742) [DOI] [Google Scholar]
- 18.Liu J, Wang Y, Lei G, Peng J, Huang Y, Cao Y, Xie M, Pu X, Lu Z. 2009. A sextuple hydrogen bonding molecular duplex bearing 1,8-naphthalimide moieties and polymer light-emitting diode based on it. J. Mater. Chem. 19, 7753-7758. ( 10.1039/B910045H) [DOI] [Google Scholar]
- 19.Desiraju GR. 1999. T. Steiner in the weak hydrogen bond. London, UK: Oxford University Press. [Google Scholar]
- 20.Nishio M, Hirota M, Umezawa Y. 1998. The CH/π interaction: evidence, nature, and consequences. New York, NY: John Wiley & Sons. [Google Scholar]
- 21.Melandri S. 2011. ‘Union is strength’: how weak hydrogen bonds become stronger. Phys. Chem. Chem. Phys. 13, 13 901-13 911. ( 10.1039/C1CP20824A) [DOI] [PubMed] [Google Scholar]
- 22.Desiraju GR. 1996. The C–H….O hydrogen bond: structural implications and supramolecular design. Acc. Chem. Res. 29, 441-449. ( 10.1021/ar950135n) [DOI] [PubMed] [Google Scholar]
- 23.Nishio M, Umezawa Y, Fantini J, Weiss MS, Chakrabarti P. 2014. CH–π hydrogen bonds in biological macromolecules. Phys. Chem. Chem. Phys. 16, 12 648-12 683. ( 10.1039/C4CP00099D) [DOI] [PubMed] [Google Scholar]
- 24.Veljković DŽ, Janjić GV, Zarić SD. 2011. Are C–H….O interactions linear? The case of aromatic CH donors. CrystEngComm 13, 5005-5010. ( 10.1039/C1CE05065F) [DOI] [Google Scholar]
- 25.Castellano RK. 2004. Progress toward understanding the nature and function of CH…O interactions. Curr. Org. Chem. 8, 845-865. ( 10.2174/1385272043370384) [DOI] [Google Scholar]
- 26.Scheiner S, Kar T. 2005. Effect of solvent upon CH….O hydrogen bonds with implications for protein folding. J. Phys. Chem. B 109, 3681-3689. ( 10.1021/jp0446736) [DOI] [PubMed] [Google Scholar]
- 27.Chakrabarti P, Bhattacharyya R. 2007. Geometry of nonbonded interactions involving planar groups in proteins. Prog. Biophys. Mol. Biol. 95, 83-137. ( 10.1016/j.pbiomolbio.2007.03.016) [DOI] [PubMed] [Google Scholar]
- 28.Scheiner S. 2006. Contributions of NH….O and C….O hydrogen bonds to the stability of β-sheets in proteins. J. Phys. Chem. B 110, 18 670-18 679. ( 10.1021/jp063225q) [DOI] [PubMed] [Google Scholar]
- 29.Park H, Yoon J, Seok C. 2008. Strength of Cα– H….OC hydrogen bonds in transmembrane proteins. J. Phys. Chem. B 112, 1041-1048. ( 10.1021/jp077285n) [DOI] [PubMed] [Google Scholar]
- 30.Manikandan K, Ramakumar S. 2004. The occurrence of C-H….O hydrogen bonds in α-helices and helix termini in globular proteins. Proteins Struct. Funct. Bioinf. 56, 768-781. ( 10.1002/prot.20152) [DOI] [PubMed] [Google Scholar]
- 31.Plevin MJ, Bryce DL, Boisbouvier J. 2010. Direct detection of CH/π interactions in proteins. Nat. Chem. 2, 466-471. ( 10.1038/nchem.6500) [DOI] [PubMed] [Google Scholar]
- 32.Sacksteder CA, Bender SL, Barry BA. 2005. Role for bound water and CH–π aromatic interactions in photosynthetic electron transfer. J. Am. Chem. Soc. 127, 7879-7890. ( 10.1021/ja050659a) [DOI] [PubMed] [Google Scholar]
- 33.Brandl M, Weiss MS, Jabs A, Sühnel J, Hilgenfeld R. 2001. C-H⋯π-interactions in proteins. J. Mol. Biol. 307, 357-377. ( 10.1006/jmbi.2000.4473) [DOI] [PubMed] [Google Scholar]
- 34.Yu L, Zhang M, Lou D, Li J, Wang X, Bai M. 2021. CH/π-interaction-driven self-assembly of tetraphenylethylene derivatives into the face to face arrangement. RSC Adv. 11, 2377-2382. ( 10.1039/D0RA10572D) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Radhakrishnan KV, Anas S, Suresh E, Koga N, Suresh CH. 2007. Molecular recognition in an organic host–guest complex: CH…O and CH…π interactions completely control the crystal packing and the host–guest complexation. Bull. Chem. Soc. Jpn. 80, 484-490. ( 10.1246/bcsj.80.484) [DOI] [Google Scholar]
- 36.da Silva JF, Ferreira AP, Marques MM, Harjivan SG, da Piedade MF, Duarte MT. 2011. Effect of C–H…X interactions (X=O, S, π) in the supramolecular arrangements of 3-ferrocenyl-methoxybenzo [b] thiophene isomers. CrystEngComm 13, 1638-1645. ( 10.1039/C0CE00434K) [DOI] [Google Scholar]
- 37.Aburaya K, Nakano K, Sada K, Yoswathananont N, Shigesato M, Hisaki I, Tohnai N, Miyata M. 2008. Importance of weak hydrogen bonds in the formation of cholamide inclusion crystals with aromatic guests. Cryst. Growth Des. 8, 1013-1022. ( 10.1021/cg701011r) [DOI] [Google Scholar]
- 38.Desiraju GR. 2005. C–H⋯O and other weak hydrogen bonds. From crystal engineering to virtual screening. Chem. Commun. 24, 2995-3001. ( 10.1039/B504372G) [DOI] [PubMed] [Google Scholar]
- 39.Leigh DA, Robertson CC, Slawin AM, Thomson PI. 2013. AAAA-DDDD quadruple hydrogen-bond arrays featuring NH···N and CH···N hydrogen bonds. J. Am. Chem. Soc. 135, 9939-9943. ( 10.1021/ja404504m) [DOI] [PubMed] [Google Scholar]
- 40.Mehreen S, Zia M, Khan A, Hussain J, Ullah S, Anwar MU, Al-Harrasi A, Naseer MM. 2022. Phenoxy pendant isatins as potent α-glucosidase inhibitors: reciprocal carbonyl⋯carbonyl interactions, antiparallel π⋯ π stacking driven solid state self-assembly and biological evaluation. RSC Adv. 12, 20 919-20 928. ( 10.1039/D2RA03307K) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zia M, Hameed S, Frontera A, Irran E, Naseer MM. 2021. Understanding the planar conformations in diarylsubstituted heteroarenes: structural and theoretical insights. CrystEngComm 23, 3144-3151. ( 10.1039/D1CE00354B) [DOI] [Google Scholar]
- 42.Naseer MM, Hussain M, Bauzá A, Lo KM, Frontera A. 2018. Intramolecular noncovalent carbon bonding interaction stabilizes the cis conformation in acylhydrazones. ChemPlusChem 83, 881-885. ( 10.1002/cplu.201800329) [DOI] [PubMed] [Google Scholar]
- 43.Naseer MM, Bauzá A, Alnasr H, Jurkschat K, Frontera A. 2018. Lone pair–π vs. σ-hole–π interactions in bromine head-containing oxacalix[2]arene[2]triazines. CrystEngComm 20, 3251-3257. ( 10.1039/C8CE00666K) [DOI] [Google Scholar]
- 44.Hussain M, Bauzá A, Frontera A, Lo KM, Naseer MM. 2018. Structure guided or structure guiding? Mixed carbon/hydrogen bonding in a bis-Schiff base of N-allyl isatin. CrystEngComm 20, 150-154. ( 10.1039/C7CE01697B) [DOI] [Google Scholar]
- 45.Abbas A, Flores-Holguin N, Naseer MM. 2015. Structure-fluorescence relationship: interplay of non-covalent interactions in homologous 1,3,5-triaryl-2-pyrazolines. New J. Chem. 39, 4359-4367. ( 10.1039/C5NJ00179J) [DOI] [Google Scholar]
- 46.Jawaria R, Hussain M, Shafiq Z, Ahmad HB, Tahir MN, Shad HA, Naseer MM. 2015. Robustness of thioamide dimer synthon, carbon bonding and thioamide–thioamide stacking in ferrocene-based thiosemicarbazones. CrystEngComm 17, 2553-2561. ( 10.1039/C4CE02566K) [DOI] [Google Scholar]
- 47.Wang J, Yao L. 2019. Dissecting C–H···π and N–H···π interactions in two proteins using a combined experimental and computational approach. Sci. Rep. 9, 1-9. ( 10.1038/s41598-019-56607-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Custelcean R, Engle NL, Bonnesen PV. 2007. Crystalline hydrogen-bonded nanocolumns of cyclic thiourea octamers. CrystEngComm 9, 452-455. ( 10.1039/B701570D) [DOI] [Google Scholar]
- 49.Federico S, Ciancetta A, Porta N, Redenti S, Pastorin G, Cacciari B, Klotz KN, Moro S, Spalluto G. 2016. 5,7-disubstituted-[1,2,4] triazolo[1,5-a][1,3,5]triazines as pharmacological tools to explore the antagonist selectivity profiles toward adenosine receptors. Eur. J. Med. Chem. 108, 529-541. ( 10.1016/j.ejmech.2015.12.019) [DOI] [PubMed] [Google Scholar]
- 50.Sheldrick GM. 2008. A short history of SHELX. Acta Crystallogr. Sec. A, Found. Crystallogr. 64, 112-122. ( 10.1107/S0108767307043930) [DOI] [PubMed] [Google Scholar]
- 51.Asghar S, Hameed S, Tahir MN, Naseer MM. 2022. Molecular duplexes featuring NH···N, CH···O and CH···π interactions in solid-state self-assembly of triazine-based compounds. Figshare. ( 10.6084/m9.figshare.c.6251577) [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Asghar S, Hameed S, Tahir MN, Naseer MM. 2022. Molecular duplexes featuring NH···N, CH···O and CH···π interactions in solid-state self-assembly of triazine-based compounds. Figshare. ( 10.6084/m9.figshare.c.6251577) [DOI] [PMC free article] [PubMed]
Data Availability Statement
Data available as electronic supplementary material [51]. The crystallographic data can also be obtained from https://www.ccdc.cam.ac.uk/data_request/cif.
CCDC: 2155046-2155051