

Abstract
Background
ROS1 tyrosine kinase inhibitors (TKIs) have demonstrated significant clinical benefit for ROS1+ NSCLC patients. However, TKI resistance inevitably develops through ROS1 kinase domain (KD) modification or another kinase driving bypass signaling. While multiple TKIs have been designed to target ROS1 KD mutations, less is known about bypass signaling in TKI‐resistant ROS1+ lung cancers.
Methods
Utilizing a primary, patient‐derived TPM3‐ROS1 cell line (CUTO28), we derived an entrectinib‐resistant line (CUTO28‐ER). We evaluated proliferation and signaling responses to TKIs, and utilized RNA sequencing, whole exome sequencing, and fluorescence in situ hybridization to detect transcriptional, mutational, and copy number alterations, respectively. We substantiated in vitro findings using a CD74‐ROS1 NSCLC patient's tumor samples. Last, we analyzed circulating tumor DNA (ctDNA) from ROS1+ NSCLC patients in the STARTRK‐2 entrectinib trial to determine the prevalence of MET amplification.
Results
CUTO28‐ER cells did not exhibit ROS1 KD mutations. MET TKIs inhibited proliferation and downstream signaling and MET transcription was elevated in CUTO28‐ER cells. CUTO28‐ER cells displayed extrachromosomal (ecDNA) MET amplification without MET activating mutations, exon 14 skipping, or fusions. The CD74‐ROS1 patient samples illustrated MET amplification while receiving ROS1 TKI. Finally, two of 105 (1.9%) entrectinib‐resistant ROS1+ NSCLC STARTRK‐2 patients with ctDNA analysis at enrollment and disease progression displayed MET amplification.
Conclusions
Treatment with ROS1‐selective inhibitors may lead to MET‐mediated resistance. The discovery of ecDNA MET amplification is noteworthy, as ecDNA is associated with more aggressive cancers. Following progression on ROS1‐selective inhibitors, MET gene testing and treatments targeting MET should be explored to overcome MET‐driven resistance.
Keywords: drug resistance, entrectinib, MET, NSCLC, ROS1
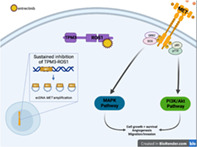
Sustained inhibition of TPM3‐ROS1 with entrectinib drives pharmacological selection of tumor cells which have evolved extrachromosomal MET gene amplification. MET amplification results in increased MET transcript and protein expression, as well as aberrant MET activation. MET activation propagates downstream signaling via the PI3K/Akt and RAS/MAPK signaling pathways, driving cancer cell proliferation and survival, and thus resistance to entrectinib therapy.
INTRODUCTION
The c‐ros oncogene 1, receptor tyrosine kinase (ROS1) was first discovered through characterization of the oncogenic product of avian sarcoma virus UR2 and is thought to be activated primarily in the patterning of epithelial tissues during development. 1 , 2 , 3 However, chromosomal rearrangement events—which fuse a 5′ gene partner to the 3′ kinase domain of ROS1—stimulate aberrant ROS1 tyrosine kinase activity, leading to sustained proliferative and survival signaling and subsequent malignant cellular transformation. 4 , 5 , 6 These chromosomal rearrangements leading to oncogenic ROS1 fusion proteins were first discovered in glioblastoma in 2003, and in non‐small cell lung cancer (NSCLC) in 2007. 4 , 7 ROS1 fusions have since been identified in approximately 2% of all NSCLC cases. 6
Two ROS1‐targeted tyrosine kinase inhibitors (TKIs) are currently approved by the U.S. Food and Drug Administration (FDA) to treat ROS1 fusion‐positive (ROS1+) lung adenocarcinoma – crizotinib and entrectinib. 6 , 8 Since its 2019 approval, pan‐TRK/ROS1/ALK TKI entrectinib has been utilized for ROS1+ NSCLC due to its greater ability to cross the blood–brain barrier and therefore treat or delay brain metastases associated with ROS1+ NSCLC. 9 , 10 , 11
However, while entrectinib and crizotinib have significantly improved outcomes for patients, drug resistance to TKIs inevitably develops, leading to disease progression. Approximately one‐third of ROS1+ NSCLC patients resistant to entrectinib demonstrate on‐target ROS1 kinase domain (KD) mutations. 12 Repotrectinib is being developed for inhibition of ROS1 resistance mutations. 13 However, the mechanism of resistance in the majority of patients still remains unknown or poorly characterized. The purpose of this study was to identify and characterize these less well understood mechanisms of acquired resistance to entrectinib. While several studies have characterized crizotinib resistance in ROS1 positive cancer utilizing both patient samples and in vitro analysis, 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 there is little data regarding mechanisms underlying entrectinib resistance, which we hypothesized may be different from crizotinib as both are multikinase inhibitors with different spectrums of activity.
Using previously described methods by our laboratory, 23 we sought to create an in vitro model of acquired resistance to entrectinib utilizing a primary, patient‐derived cell line harboring a TPM3‐ROS1 fusion. We demonstrated that this entrectinib‐resistant cell line was dependent on MET expression and activation for survival. We investigated the mechanism by which MET was upregulated, and found it occurred via extrachromosomal (ecDNA) MET gene amplification. Although bypass signaling driven by MET activation has been documented clinically in EGFR‐mutant and ALK+ NSCLC, 24 , 25 , 26 as well as lorlatinib‐refractory ROS1+ NSCLC, 22 to our knowledge, this is the first in vitro description of MET gene amplification to drive bypass signaling in entrectinib‐refractory ROS1+ NSCLC.
METHODS
Cell lines
CUTO28 cell line derivation was performed as previously described following Institutional Review Board approved informed consent of the patient. 20 All cell lines were cultured in RPMI1640 (ThermoFisher) with 10% FBS (ThermoFisher) at 37°C with 5% CO2. The CUTO28‐ER cell line was derived in vitro following sustained exposure of CUTO28 cells to increasing doses of entrectinib. CUTO28 cells were authenticated short tandem repeat (STR) analysis by the Barbara Davis Center Molecular Biology Service Center at the University of Colorado supported by Award Number P30DK116073 from the National Institute of Diabetes And Digestive And Kidney Diseases.
Sequencing
DNA and RNA were isolated from replicate biological samples using the QIAamp DNA Kit and RNEasy Kit (Qiagen) and submitted for sequencing and annotation by Novogene. 27 Differential gene expression analysis was done by DEseq2 R package. 28 The volcano plot was created using EnhancedVolcano R package (https://github.com/kevinblighe/EnhancedVolcano). Patient ctDNA samples from the STARTRK‐2 trial were sequenced utilizing Foundation Medicine's FoundationOne Liquid CDx test.
Fluorescence in situ hybridization (FISH)
MET amplification FISH was performed using probes for MET and chromosome 7 centromere (CEP7, as an internal control). Sample preparation and analysis were performed as previously described. 5 , 29 To determine whether observed MET locus amplification was occurring extrachromosomally, metaphase FISH was performed as previously described. 30
Reagents
Entrectinib, crizotinib, capmatinib, and afatinib were purchased from Selleck Chemicals. Antibodies to total AKT, AKT pS473, total ERK, ERK pT202/Y204, total ROS1, ROS1 pY2274, total MET, MET pY1234/1235, total AXL, total EGFR, EGFR pY1068, EGFR pY1086, EGFR pY1173, total HER2, HER2 pY1196, HER2 pY1221/1222, HER3 pY1289 were purchased from Cell Signaling Technologies. Antibody to GAPDH was purchased from Santa Cruz Biotechnologies Inc. MET and CEP7 interphase FISH probes were purchased from Abbott Molecular Inc. MET, CEP7, MYC, and CEP8 metaphase FISH probes were purchased from Empire Genomics.
Proliferation assays
Cells were seeded 1000 cells/well and treated 24 h after seeding at the drug concentrations described on each graph. CUTO28‐ER cells were seeded with entrectinib, and media changed just prior to treatment at 24 h. Following 72 h of treatment, CellTiter 96 AQueous One Solution (Promega) was added and absorbance values were read using Gen5 software. Each assay was performed in triplicate in at least 3 independent biological replicates. Data were plotted and IC50 values calculated using GraphPad Prism software.
Immunoblotting
Immunoblotting was performed as previously described. 29 Cells were treated at the indicated doses for 2–3 h, then lysed in RIPA buffer with Halt protease and phosphatase inhibitor cocktail (Thermo‐Scientific) and diluted in loading buffer (LI‐COR Biosciences). Membranes were scanned using the Odyssey Imaging System and software (LI‐COR). All western blot images are representative of at least three independent experiments.
STARTRK‐2 patient recruitment
Patients were recruited to the open‐label, multicenter, global phase 2 basket study of entrectinib if they had locally advanced or metastatic solid tumors driven by NTRK1/2/3, ALK, or ROS1 gene rearrangements. Only those patients with ROS1 rearrangements were analyzed for MET amplification in this study.
RESULTS
Derivation of CUTO28 and CUTO28‐ER cell lines
The patient‐derived CUTO28 cell line was generated from the pleural effusion of a patient with ROS1+ NSCLC. The patient had clinical testing using only ROS1 FISH at diagnosis. In the CUTO28 cell line a ROS1 gene rearrangement was confirmed and the 5′ partner gene was determined to be TPM3 via genome alignment of RNA sequencing. While the CUTO28 cell line was derived from a pleural effusion following sustained crizotinib treatment, suggestive of clinical tumor progression, this cell line demonstrates exquisite sensitivity to ROS1 inhibitors entrectinib and crizotinib (IC50 12 nM and 18 nM, respectively). Notably, our laboratory routinely observes TKI sensitivity in cell lines derived from patients while on TKI; this observed phenomenon may indicate outgrowth of a TKI‐sensitive subclone, particularly when cell lines, such as the case here, are not derived in the presence of the TKI.
To generate the CUTO28 entrectinib‐resistant (CUTO28‐ER) cell line, CUTO28 cells were passaged in the presence of increasing doses of entrectinib, administered in the media every 3–5 days, and dose escalation was ceased once CUTO28‐ER cells were stably proliferating at 500 nM entrectinib (approximately 40 times the IC50 of parental CUTO28). Whole exome sequencing, validated by RT‐PCR with Sanger direct DNA sequencing, did not reveal any mutation in the kinase domain of ROS1, and expression of TPM3‐ROS1 mRNA by RNA Seq was not significantly different between parental and CUTO28‐ER cells (not shown).
CUTO28‐ER resistance to entrectinib is driven by MET
CUTO28‐ER cells exhibited an IC50 to entrectinib of over 2 μM, approximately 157‐fold that of parental CUTO28 cells (Figure 1a, b). However, in response to dual ROS1/MET inhibitor crizotinib, CUTO28‐ER cells exhibited greater sensitivity than CUTO28 cells (IC50 of 14 nM vs 24 nM, respectively), suggesting that MET might be driving proliferation. To validate this potential MET sensitivity, we treated cells with capmatinib to determine relative sensitivity to a MET‐selective TKI that does not target ROS1. While parental CUTO28 cells were completely resistant to capmatinib (IC50 > 10 μM), CUTO28‐ER cells displayed marked single agent sensitivity (IC50 202 nM).
FIGURE 1.
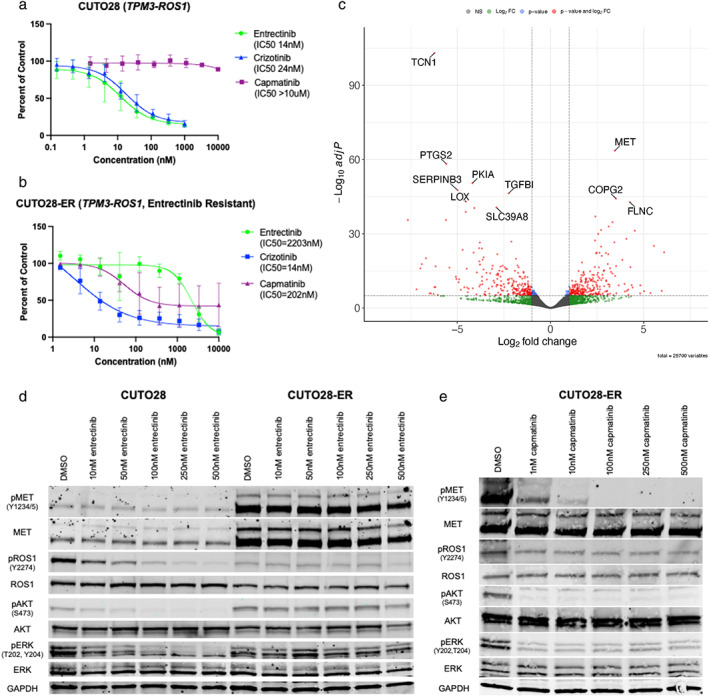
MET inhibitors regulate cell proliferation and survival in entrectinib‐resistant cells. (a, b) Cell survival assay of CUTO28 parental (a) and CUTO28‐ER (b) cells treated with the indicated doses of entrectinib, crizotinib, or capmatinib for 72 h. Cell survival was assayed by CellTiter AQueous one solution colorimetric proliferation assay. (c) Volcano plot depicting genes significantly up‐ or downregulated at the mRNA transcript level in CUTO28‐ER cells as compared to CUTO28 parental cells. Significantly regulated genes (|Log2FoldChage| > 1, adjusted p‐value <1.0 × 10−5) are shown as red dots. The top 10 significantly up‐ or downregulated genes are labeled. (d, e) CUTO28 and CUTO28‐ER cells were treated with the indicated inhibitors for 2 h. Lysates were probed with antibodies directed against the specific proteins labeled
While activation of ROS1 (pROS1) was abrogated in parental CUTO28 cells with low dose entrectinib, pROS1 levels in CUTO28‐ER cells were consistently low in the absence of entrectinib treatment and across all tested doses of entrectinib, despite similar levels of ROS1 total protein (Figure 1d). While signals of both activated Akt (pAkt) and activated ERK1/2 (pERK1/2) were decreased by entrectinib in a dose‐dependent manner in CUTO28 parental cells, pAkt and pERK1/2 were not inhibited by entrectinib in CUTO28‐ER cells, even at 500 nM (Figure 1d). Additionally, total MET and activated MET (pMET) were markedly increased in CUTO28‐ER cells compared to parental. Taken together, these data indicate CUTO28‐ER cells no longer rely on ROS1 activation for growth and survival, and are upregulating production and activation of MET. To this point, pAkt and pERK signals were strongly decreased in a dose‐dependent manner in CUTO28‐ER cells treated with the MET‐selective inhibitor capmatinib for 2 h, as was pMET (Figure 1e). Inhibition of MET alone was as effective at inhibiting downstream signaling as dual inhibition of ROS1 and MET in CUTO28‐ER cells, and inhibition of MET had no effect on signaling in CUTO28 parental cells (Figure S1a).
RNA sequencing reveals differential expression of MET
Analysis of CUTO28 and CUTO28‐ER RNA samples by bulk RNA sequencing indicated expression of MET was significantly upregulated in CUTO28‐ER cells as compared to parental (Figure 1c; Log2FoldChange of 3.48, adjusted p‐value of 2.78 × 10−64). Pertinently, we did not see any increase in hepatocyte growth factor (HGF) production, as the ligand for MET receptor. Previously our laboratory has shown that EGFR or other ERBB family members can drive ROS1 TKI resistance, but we observed no evidence of upregulation of ERBB family of receptors or their ligands. 20 , 23 Consistent with these data, the pan‐ERBB TKI afatinib did not have any effect on downstream signaling in CUTO28‐ER cells (Figure S1a) nor on proliferation (data not shown). Additionally, no upregulation or alterations were observed in common kinase drivers of bypass signaling (FGFR family, Src family, KIT, PI3K, MYC) or other proteins downstream in this signaling cascade (ERK, AKT, STAT3, RAS, etc).
Amplification of the MET locus drives MET overexpression in CUTO28‐ER cells
We next sought to identify the mechanism(s) by which CUTO28‐ER cells had significantly upregulated the expression and activation of MET. Given the increased RNA and protein expression observed in CUTO28‐ER compared to the parental CUTO28, we evaluated for copy number amplification (CNA) using fluorescence in situ hybridization (FISH) with probes specific to both the MET locus and centrosome 7 chromosome enumeration probe 7 (CEP7). While parental CUTO28 cells displayed polyploidy typical of cancer cells, with four hybridized probe signals each of MET and chromosome 7 per cell on average (a MET:CEP7 ratio of 1.0), CUTO28‐ER cells harbored 17.1 hybridized probe signals for MET and 4.1 hybridized probe signals for chromosome 7 per cell on average, indicating a MET:CEP7 ratio of 4.2 (Figure 2a).
FIGURE 2.
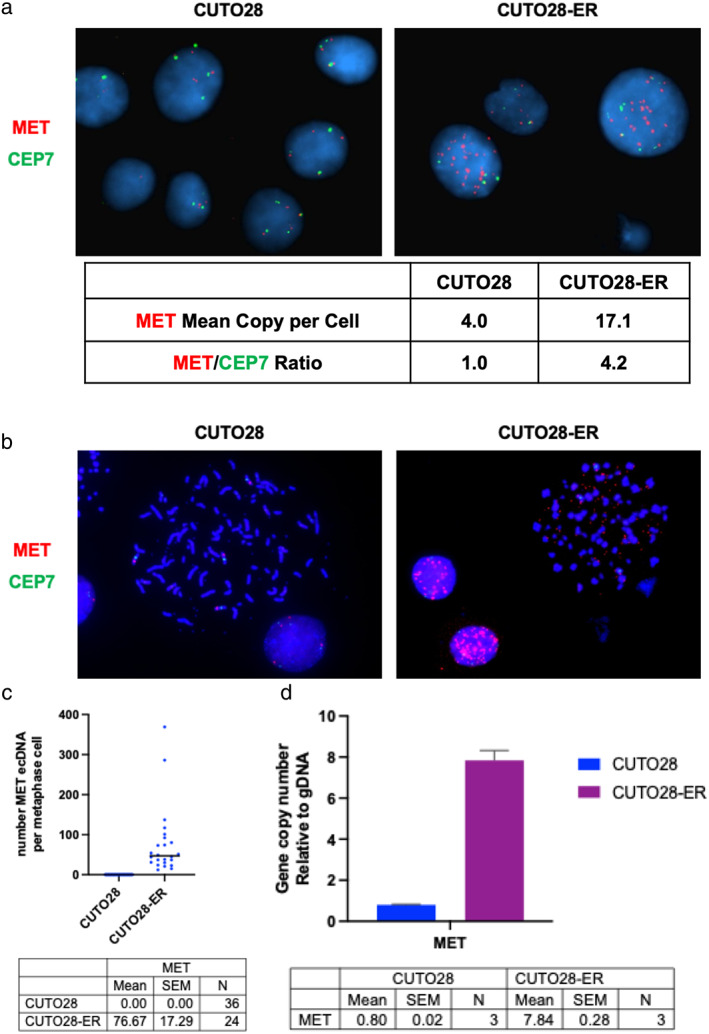
Amplification of the MET locus drives MET overexpression in CUTO28‐ER cells. (a) Interphase florescence in situ hybridization (FISH) imaging using probes which label the MET gene (red) and its corresponding chromosome's centrosome, CEP7 (green). The ratio of MET/CEP7 copies is used as a clinical measure of focal MET gene amplification. The number of MET and CEP7 hybridization signals per cell, as well as MET/CEP7 ratio, are quantified by cell line. Images taken at 100x magnification. (b) Metaphase FISH was performed to visualize how much of this amplification was occurring outside of the genomic DNA. (c) The number of extrachromosomal MET probe hybridization signals per metaphase‐halted cell represented in panel B are quantified by cell line. (d) The ecDNA MET copy number gain relative to genomic DNA is quantified by cell line via genomic qPCR
To determine if the observed MET locus amplification was occurring intrachromosomally and/or extrachromosomally, we performed metaphase FISH and used Keyence digital microscopy to localize amplified MET in the genome. Here, we discovered that MET locus amplification in CUTO28‐ER cells was achieved solely through ecDNA amplification (Figure 2b). The mean number of ecDNA MET probe signals observed was 76.67 (SEM ±17.29) in the CUTO28‐ER cells, compared to 0.00 (SEM ±0.00) in parental CUTO28 cells (Figure 2c). Genomic qPCR was used to obtain the ecDNA gene copy number relative to genomic DNA; here, we found ecDNA expression of the MET locus was 7.84‐fold that genomic DNA in CUTO28‐ER cells (Figure 2d). We also observed ecDNA amplification of the MYC locus in CUTO28 parental cells, which is conserved in CUTO28‐ER cells (Figure S1b–d). This increase in the MET gene copy number by ecDNA amplification explains the high total MET protein level in CUTO28‐ER.
We also investigated known mechanisms of increased/sustained activation of MET protein. One mechanism through which MET protein activation is increased is by MET exon 14 skipping or Y1003 mutation, which leads to loss of Cbl binding and thus increased protein expression. 31 , 32 , 33 , 34 , 35 , 36 Cells can also affect constitutive activation of MET via activating point mutations in the MET kinase domain or through oncogenic fusion with a 5′ gene partner. 37 , 38 , 39 , 40 , 41 , 42 , 43 , 44 CUTO28‐ER cells were subjected to both whole exome and RNA sequencing, and validated with RT‐PCR and direct Sanger sequencing. We found no evidence of mutations leading to MET exon 14 skipping or a Y1003 mutation, nor any activating point mutations in the MET kinase domain (data not shown). Lastly, and importantly for both MET expression and activation, no MET gene fusions were identified by RNA seq (data not shown).
MET drives bypass signaling via CNA in ROS1+ NSCLC patients resistant to ROS1 TKI
A 58‐year‐old white male never smoker had a diagnostic right upper lobe wedge biopsy demonstrating adenocarcinoma of the lung with molecular testing showing a CD74‐ROS1 rearrangement. First line therapy crizotinib for metastatic disease was not well tolerated due to elevated transaminases and was therefore discontinued after approximately 5 months. He was next enrolled in the STARTRK‐2 basket trial for entrectinib treatment. Following 18 months on entrectinib, computed tomography (CT) scan determined disease progression. At this time, a cell line was derived from his resultant pleural effusion (CUTO38). CUTO38 retained sensitivity to entrectinib in vitro and showed a MET:CEP7 ratio of 1.1 (7.2 FISH probe signals for MET, 6.6 FISH probe signals for CEP7; Figure 3). Following progression on entrectinib, the patient received carboplatin/pemetrexed chemotherapy for 3 months, followed by pemetrexed/pembrolizumab maintenance therapy for approximately 16 months. A positron emission technology (PET)‐CT scan revealed new bone metastases. The patient was next enrolled in a clinical trial for repotrectinib and remained on treatment for 2 weeks before admission to the emergency department for drainage of a large left pleural effusion. It was from this effusion that the first formalin‐fixed paraffin‐embedded (FFPE) tissue sample was obtained; FISH analysis revealed a MET:CEP7 ratio of 0.9 (mean of 3.4 probe signals for MET per cell, 3.9 probe signals for CEP7 per cell; Figure 3). Due to progressive shortness of breath and persistent worsening of swelling in the lower extremities, repotrectinib treatment was discontinued. Following repotrectinib discontinuation, a CT scan showed disease progression in the form of diffuse lesions in both lungs. The patient next initiated cabozantinib therapy, which was well tolerated and improved both the shortness of breath and swelling. After 2 months on cabozantinib, CT scan showed lung metastases that were significantly decreased in size, but that cardiophrenic lymph nodes had increased in size and worsening ascites, from which the second FFPE sample was obtained. FISH performed on this second sample determined a MET:CEP7 ratio of 2.5 (mean of 9.5 probe signals for MET per cell, 3.8 probe signals for CEP7 per cell; Figure 3), a measurable CNA from the 0.9 MET:CEP7 ratio detected in the previous sample. Cabozantinib treatment was stopped due to disease progression and the patient died in hospice care shortly after this. Because of the positive response of the patient's lung lesions to cabozantinib which is a dual ROS1/MET inhibitor, it is possible that a MET‐amplified subpopulation gained an unspecified secondary resistance mechanism, driving the ascites fluid accumulation wherein cells with MET amplification were detected.
FIGURE 3.
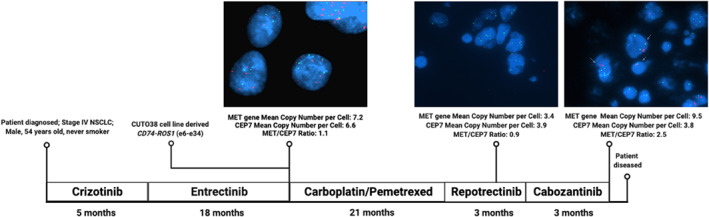
FISH performed on ROS1+ NSCLC patient pleural fluid/ascites samples reveals MET gene amplification at progression on ROS1 TKI. A patient harboring a CD74‐ROS1 fusion attempted a series of ROS1 TKIs due to unmanageable side effects. From both the CUTO38 cell line (derived April 2018 following progression on crizotinib) and a sample obtained during repotrectinib treatments (January 2020), MET FISH revealed a MET/CEP7 ratio of 1.1 or 0.9, respectively. When the patient returned to clinic in May 2020 with recurrence, a sample collected from ascites fluid and subjected to FISH revealed a MET/CEP7 ratio of 2.5. All images were taken at 100x magnification
To gain a better understanding of the prevalence of MET amplification as a mechanism of resistance to entrectinib in ROS1+ NSCLC, we evaluated circulating tumor DNA (ctDNA) of ROS1+ STARTRK‐2 trial participants. 45 Of 105 ROS1+ NSCLC patients with successfully sequenced ctDNA both at study enrollment and at progression on entrectinib, 2 (1.9%) samples displayed CNA of MET via FoundationOne Liquid CDx. Of these two patients, one patient had no detectible CNA at study enrollment but displayed MET amplification by day 166 of entrectinib therapy. The second patient had MET CNA at study enrollment but demonstrated disease progression as best response only 28 days later where the MET CNA was maintained (Table 1). Both patients received three lines of therapy prior to entrectinib, none of which targeted ROS1 or MET. Because the FoundationOne sample multiplexed ctDNA sequencing assay calculates CNA ratio based upon number of reads for a gene in the test sample compared to a process‐matched control sample from the International HapMap Project, gene amplifications are reported on a Yes/No basis in the case of a focal amplification of at least six copies. 46
TABLE 1.
ctDNA reveals MET amplification in two STRTRK‐2 ROS1+ patients receiving entrectinib
| Patient 1 | Patient 2 | |
|---|---|---|
| ROS1 fusion | CD74‐ROS1 | SLC34A2‐ROS1 |
| MET copy number alteration (CNA) at baseline | Not amplified | Amplified |
| MET CNA at detection | Amplified | Amplified |
| Days receiving entrectinib until MET CNA detected | 166 | 28 |
| Demographics | ||
| Age | 53 | 68 |
| Gender | Male | Male |
| Ethnicity | Asian | White |
| Prior lines of therapy | ||
| Cisplatin/pemetrexed | Cisplatin/pemetrexed | |
| Docetaxel | Carboplatin/pemetrexed | |
| Nivolumab | Pemetrexed |
Two of 105 patients (1.9%) positive for MET CNA as captured by circulating tumor DNA (ctDNA) in STARTRK‐2 clinical trial. Denominator of 105 determined by number of patients with successfully sequenced ctDNA at both C1D1 and progression. Successful detection rate of ROS1 fusion via ctDNA in ROS1+ NSCLC patients is ~65%, and CNAs are more challenging than ROS1 fusions to detect in plasma due to lower event frequency and wild‐type DNA interference, thus there is reason to believe the rate of MET amplification could be significantly greater than detected in this study population. CNA = copy number amplification.
DISCUSSION
MET gene amplification has previously been described as a bypass resistance mechanism to TKIs, first discovered in EGFR mutant NSCLC resistant to gefitinib 47 and subsequently observed in ALK rearranged and RET rearranged NSCLC. 26 , 48 , 49 , 50 , 51 , 52 , 53 , 54 , 55 More recently, MET gene amplification has been observed in ROS1+ lung cancers that progressed on lorlatinib, a next‐generation ROS1 TKI utilized to target some ROS1 mutations driven by first‐generation ROS1 TKIs. 22 To our knowledge, this represents the first case of MET‐mediated resistance to entrectinib in ROS1+ NSCLC, and importantly shows a novel mechanism involving ecDNA MET gene amplification. Our in vitro and patient data presented herein make the case for monitoring for MET amplification and other activating MET gene alterations at the time of progression on entrectinib.
In CUTO28‐ER, pROS1 levels were low in the absence of ROS1 TKI treatment indicative of loss of ROS1 signaling. We have noted this phenomenon in previously derived TKI resistant cell lines. In the first instance, the SLC34A2‐ROS1 cell line HCC78 lost a copy of the ROS1 gene fusion at TKI resistance leading to loss of expression of the ROS1 fusion protein. 23 Interestingly, a different study of ROS1 TKI resistance in this same cell line also demonstrated loss of ROS1 protein expression. 56 In the second instance, the CCDC6‐RET cell line LC‐2/Ad maintained the genomic RET fusion and RET fusion protein expression, but lost phosphorylation of RET. 57 This latter case is similar to CUTO28‐ER cells. We can speculate that in some instances of bypass signaling, there may be a selective advantage to reduce signaling from the original oncogene and this may lead to a more complete dependence on the bypass signaling pathway. This oncogenic switch was observed with CUTO28‐ER with near complete switch in dependence from ROS1 to MET.
A dose‐dependent decrease in both Akt and ERK1/2 activation by the MET selective TKI capmatinib in CUTO28‐ER cells suggested MET was driving a bypass signaling program through both the PI3K/Akt and RAS/RAF/MAPK pathways. We saw no evidence of MET exon 14 skipping, activating point mutations, or MET gene fusions, but rather only CNA via ecDNA. The nascent ecDNA amplification of MYC may indicate parental CUTO28 cells are predisposed to ecDNA amplification. This is noteworthy, as ecDNA is associated with more aggressive cancers. 58
MET CNA observed by ctDNA analysis in entrectinib‐refractory ROS1+ NSCLC patients confirms the clinical significance of these in vitro findings. Successful detection of ROS1 fusion via ctDNA in this trial was approximately 65%, consistent with known sensitivity issues for the detection of gene fusions. 59 Similarly, given the challenges surrounding CNV detection in ctDNA due to low sensitivity, 60 there is reason to believe the rate of MET amplification could be significantly greater than detected in this study population using only ctDNA analysis. Additionally, the MET CNA observed by MET interphase FISH within a patient tumor cell sample from ascites fluid—but not in another sample from pleural fluid 4 months prior, or in a tumor‐derived primary cell line derived 2 years prior—highlights not only the temporal but also spatial heterogeneity which may have yielded differential MET copy numbers due to selection pressure from different treatments and/or different metastatic sites. The possibility that a MET‐amplified subpopulation gained an unspecified secondary resistance mechanism was not ruled out prior to the patient's death.
There are only two prior in vitro studies of entrectinib‐mediated drug resistance in ROS1+ NSCLC. One study detected KRAS G12C mutation, KRAS amplification, and/or FGF3 amplification in entrectinib‐resistant HCC78 cells, 56 and in another ROS1F2004C was observed as a recurrent on‐target resistance mutation to entrectinib in a mutagenesis screen of Ba/F3 cells driven by either CD74‐ROS1 or EZR‐ROS1 fusions. 61 Our discovery of MET‐mediated bypass signaling in entrectinib‐refractory ROS1+ NSCLC is important both because of the novel mechanism of ecDNA gene amplification but also because it is readily targetable with US FDA approved drugs such as crizotinib, capmatinib, or tepotinib. While there are no clinical reports of MET‐mediated entrectinib‐resistance in NSCLC, MET amplification has been detected in two TKI‐refractory ROS1+ NSCLC patients via interphase FISH. 22 MET has been documented as an important driver of bypass signaling across many cancers, and across many NSCLCs with disparate primary RTK drivers. There is currently a planned basket trial for NSCLC patients receiving a TKI for a driver mutation (e.g., ROS1, ALK, RET, etc.) who demonstrate MET‐mediated resistance upon progression via next generation sequencing and/or FISH. This study will evaluate the efficacy and safety of addition of the MET inhibitor tepotinib to their current TKI (NCT04739358). We speculate that continued advancement of ctDNA testing and consistent monitoring for MET alterations such as gene amplification will capture a larger incidence of MET‐mediated resistance and has the potential to improve patient outcomes.
CONFLICT OF INTEREST
LCT, NC, HN, LB, AZD: No disclosures to announce; ATL: Patent with Abbott Molecular, licensing of CUTO cell lines; TRW, DC, BS: Employed by Genentech, Inc. and have equity in Roche; KMT, DP, SK, JC: Previously employed by Boundless Bio, Inc.; RCD is currently an employee and shareholder of Rain Therapeutics. RCD has received licensing fees from Takeda, BMS, ThermoFisher, Genentech, Black Diamond, Pearl River, Voronoi, Scorpion Therapeutics, Foundation Medicine, Ignyta, Chugai, Blueprint Medicines, Abbott Molecular and Rain Therapeutics. RCD has received consulting fees from Genentech/Roche, Ignyta, Pfizer, Takeda, AstraZeneca, Blueprint Medicines, Pfizer, Green Peptide, Anchiano, Bayer, Guardant, Ariad, and Rain Therapeutics.
Supporting information
Figure S1. CUTO28 cells do not respond to MET inhibition and both CUTO28 and CUTO28‐ER cells display ecDNA amplification of the MYC locus. (a) CUTO28 and CUTO28‐ER cells were treated with the indicated inhibitors for 2 h. Lysates were probed with antibodies directed against the specific proteins labeled. (b) Metaphase FISH was performed using probes which label the MYC gene (red) and its corresponding chromosome's centrosome, CEP8 (green). (c) The number of extrachromosomal MYC probe hybridization signals per metaphase‐halted cell represented in Panel B are quantified by cell line. (e) The ecDNA MYC copy number gain relative to genomic DNA is quantified by cell line via genomic qPCR
Tyler LC, Le AT, Chen N, Nijmeh H, Bao L, Wilson TR, et al. MET gene amplification is a mechanism of resistance to entrectinib in ROS1+ NSCLC . Thorac Cancer. 2022;13(21):3032–3041. 10.1111/1759-7714.14656
Funding information Bonnie J. Addario Lung Cancer Foundation
REFERENCES
- 1. Matsushime H, Wang L‐H, Shibuya M. Human c‐ros‐1 gene homologous to the v‐ros sequence of UR2 sarcoma virus encodes for a transmembrane receptorlike molecule. Mol Cell Biol. 1986;6(8):3000–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Birchmeier C, Birnbaum D, Waitches G, Fasano O, Wigler M. Characterization of an activated human ros gene. Mol Cell Biol. 1986;6(9):3109–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Acquaviva J, Wong R, Charest A. The multifaceted roles of the receptor tyrosine kinase ROS in development and cancer. Biochim Biophys Acta. 2009;1795:37–52. [DOI] [PubMed] [Google Scholar]
- 4. Charest A, Kheifets V, Park J, Lane K, McMahon K, Nutt CL, et al. Oncogenic targeting of an activated tyrosine kinase to the Golgi apparatus in a glioblastoma. Proc Natl Acad Sci. 2003;100(3):916–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Davies JD, Le AT, Theodoro MF, Skokan MC, Aisner DL, Berge EM, et al. Identifying and targeting ROS1 gene fusions in non‐small cell lung cancer. Clin Cancer Res. 2012;18(17):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lin JJ, Shaw AT. Recent advances in targeting ROS1 in lung cancer. J Thorac Oncol. 2017;12(11):1611–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131:1190–203. [DOI] [PubMed] [Google Scholar]
- 8. Administration, U.S.F.D , FDA Approves Entrectinib for NTRK Solid Tumors and ROS‐1 NSCLC. 2019.
- 9. Ardini E, Menichincheri M, Banfi P, Bosotti R, De Ponti C, Pulci R, et al. Entrectinib, a pan‐TRK, ROS1, and ALK inhibitor with activity in multiple molecularly defined cancer indications. Mol Cancer Ther. 2016;15(4):628–39. [DOI] [PubMed] [Google Scholar]
- 10. Patil T, Smith DE, Bunn PA, Aisner DL, Le AT, Hancock M, et al. The incidence of brain metastases in stage IV ROS1‐rearranged non‐small cell lung cancer and rate of central nervous system progression on crizotinib. J Thorac Oncol. 2018;13(11):1717–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Doebele RC, Perez L, Trinh H, Martinec M, Martina R, Riehl T, et al. Comparative effectiveness analysis between entrectinib clinical trial and crizotinib real‐world data in ROS1+ NSCLC. J Comp Eff Res. 2021;10(17):1271–82. [DOI] [PubMed] [Google Scholar]
- 12. Doebele RC, Drilon A, Shaw AT, Wolf J, Farago AF, et al. Genomic landscape of entrectinib resistance from ctDNA analysis in STARTRK‐2. Ann Oncol. 2019;30(5):v865. [Google Scholar]
- 13. Yun MR, Kim DH, Kim S‐Y, Joo H‐S, Lee YW, Choi HM, et al. Repotrectinib exhibits potent antitumor activity in treatment‐naive and solvent‐front‐mutant ROS1‐rearranged non‐small cell lung cancer. Clin Cancer Res. 2020;26(13):3287–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Awad MM, Katayama R, McTigue M, Liu W, Deng Y‐L, Brooun A, et al. Acquired resistance to crizotinib from a mutation in CD74‐ROS1. New Engl J Med. 2013;368:2395–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Drilon A, Somwar R, Wagner JP, Vellore NA, Eide CA, Zabriskie MS, et al. A novel crizotinib‐resistant solvent‐front mutation responsive to cabozantinib therapy in a patient with ROS1‐rearranged lung cancer. Clin Cancer Res. 2015;22(10):2351–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zou HY, Li Q, Engstrom LD, West M, Appleman V, Wong KA, et al. PF‐06463922 is a potent and selective next‐generation ROS1/ALK inhibitor capable of blocking crizotinib‐resistant ROS1 mutations. Proc Natl Acad Sci. 2015;112(11):3493–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dziadziuszko R, Le AT, Wrona A, Jassem J, Camidge DR, Varella‐Garcia M, et al. An activating KIT mutation induces crizotinib resistance in ROS1‐positive lung cancer. J Thorac Oncol. 2016;11(8):1273–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Facchinetti F, Loriot Y, Kuo M‐S, Mahjoubi L, Lacroix L, Planchard D, et al. Crizotinib‐resistant ROS1 mutations reveal a predictive kinase inhibitor sensitivity model for ROS1‐ and ALK‐rearranged lung cancers. Clin Cancer Res. 2016;22(24):5983–91. [DOI] [PubMed] [Google Scholar]
- 19. Gainor JF, Friboulet L, Yoda S, Alghalandis LD, Farago AF, Logan J, et al. Frequency and spectrum of ROS1 resistance mutations in ROS1‐positive lung cancer patients progressing on crizotinib. J Clin Oncol. 2016;34(15):9072. [Google Scholar]
- 20. McCoach CE, Le AT, Gowan K, Jones K, Schubert L, Doak A, et al. Resistance mechanisms to targeted therapies in ROS1+ and ALK+ non‐small cell lung cancer. Clin Cancer Res. 2018;24(14):3334–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dagogo‐Jack I, Rooney M, Nagy RJ, Lin JJ, Chin E, Ferris LA, et al. Molecular analysis of plasma from patients with ROS1‐positive NSCLC. J Thorac Oncol. 2019;14(5):816–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lin JJ, Yoda S, Zhu VW, Johnson TW, Sakhtemani R, et al. Spectrum of mechanisms of resistance to crizotinib and lorlatinib in ROS1 fusion‐positive lung cancer. Clin Cancer Res. 2021;27:2899–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Davies KD, Mahale S, Astling DP, Aisner DL, Le AT, Hinz TK, et al. Resistance to ROS1 inhibition mediated by EGFR pathway activation in non‐small cell lung cancer. PLoS One. 2013;8(12):e82236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Piotrowska Z, Isozaki H, Lennerz JK, Gainor JF, Lennes IT, Zhu VW, et al. Landscape of acquired resistance to osimertinib in EGFR‐mutant NSCLC and clinical validation of combined EGFR and RET inhibition with osimertinib and BLU‐667 for acquired RET fusion. Cancer Discovery. 2018;8:1529–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR‐TKI therapy in 155 patients with EGFR‐mutant lung cancers. Clin Cancer Res. 2013;19:2240–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dagogo‐Jack I, Yoda S, Lennerz JK, Langenbucher A, Lin JJ, Rooney MM, et al. MET alterations are a recurring and actionable resistance mechanism in ALK‐positive lung cancer. Clin Cancer Res. 2020;26(11):2535–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li F, Li Y, Liang H, Xu T, Kong Y, Huang M, et al. HECTD3 mediates TRAF3 polyubiquitination and type I interferon induction during bacterial infection. J Clin Investig. 2018;128(9):4148–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, Weickhardt AJ, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non‐small cell lung cancer. Clin Cancer Res. 2012;18(5):1472–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wu STK, Nguyen N, Raviram R, Erb M, Santini J, et al. Circular ecDNA promotes accessible chromatin and high oncogene expression. Nature. 2019;575(7784):699–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lee JK, Madison R, Classon A, Gjoerup O, Rosenzweig M, Frampton GM, et al. Characterization of non‐small‐cell lung cancers with MET exon 14 skipping alterations detected in tissue or liquid: clinicogenomics and real‐world treatment patterns. J Clin Oncol. 2021;5:1354–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kong‐Beltran M, Seshagiri S, Zha J, Zhu W, Bhawe K, Mendoza N, et al. Somatic mutations lead to an oncogenic deletion of MET in lung cancer. Cancer Res. 2006;66(1):283–9. [DOI] [PubMed] [Google Scholar]
- 33. Frampton GM, Ali SM, Rosenzweig M, Chmielecki J, Lu Z, Bauer TM, et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov. 2015;5(8):850–9. [DOI] [PubMed] [Google Scholar]
- 34. Lee J‐H, Gao CF, Lee CC, Kim MD, Vande Woude GF. An alternatively spliced form of MET receptor is tumorigenic. Exp Mol Med. 2006;38(5):565–73. [DOI] [PubMed] [Google Scholar]
- 35. Abella JV, Peschard P, Naujokas MA, Lin T, Saucier C, Urbe S, et al. MET/hepatocyte growth factor receptor ubiquitination suppresses transformation and is required for Hrs phosphorylation. Mol Cell Biol. 2005;25(21):9632–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Peschard P, Fournier TM, Lamorte L, Naujokas MA, Band H, Langdon WY, et al. Mutation of the c‐Cbl TKB domain binding site on the MET receptor tyrosine kinase converts it into a transforming protein. Mol Cell. 2001;8(5):995–1004. [DOI] [PubMed] [Google Scholar]
- 37. Davies KD, Ng TL, Estrada‐Bernal A, Le AT, Ennever PR, Camidge DR, et al. Dramatic response to crizotinib in a patient with lung cancer positive for an HLA‐DRB1‐MET gene fusion. J Clin Oncol. 2017;1:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schmidt L, Duh F‐M, Chen F, Kishida T, Glenn G, Choyke P, et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto‐oncogene in papillary renal carcinomas. Nat Genet. 1997;16(1):68–73. [DOI] [PubMed] [Google Scholar]
- 39. Albiges L, Guegan J, Le Formal A, Verkarre V, Rioux‐Leclercq N, Sibony M, et al. MET is a potential target across all papillary renal cell carcinomas: result from a large molecular study of pRCC with CGH array and matching gene expression array. Clin Cancer Res. 2014;20(13):3411–21. [DOI] [PubMed] [Google Scholar]
- 40. Graveel C, Su Y, Koeman J, Wang L‐M, Tessarollo L, Fiscella M, et al. Activating MET mutations produce unique tumor profiles in mice with selective duplication of the mutant allele. Proc Natl Acad Sci. 2004;101(49):17198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jeffers M, Schmidt L, Nakaigawa N, Webb CP, Weirich G, Kishida T, et al. Activating mutations for the MET tyrosine kinase receptor in human cancer. Proc Natl Acad Sci. 1997;94(21):11445–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lubensky IA, Schmidt L, Zhuang Z, Weirich G, Pack S, Zambrano N, et al. Hereditary and sporadic papillary renal carcinomas with c‐MET mutations share a distinct morphological phenotype. Am J Pathol. 1999;155(2):517–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schmidt L, Junker K, Nakaigawa N, Kinjerski T, Weirich G, Miller M, et al. Novel mutations of the MET proto‐oncogene in papillary renal carcinomas. Oncogene. 1999;18(14):2343–50. [DOI] [PubMed] [Google Scholar]
- 44. Network CGAR, Linehan WM, Spellman PT, Ricketts CJ, Creighton CJ, Fei SS, et al. Comprehensive molecular characterization of papillary renal‐cell carcinoma. New Engl J Med. 2016;374(2):135–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dziadziuszko R, Hung T, Wang K, Choeurng V, Drilon A, Doebele RC, et al. Pre‐ and post‐treatment blood‐based genomic landscape of patients with ROS1 or NTRK fusion‐positive solid tumours treated with entrectinib. Mol Oncol. 2022;16:2000–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31(11):1023–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Engelman JA, Zejnullahu K, Mitzudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316(5827):1039–43. [DOI] [PubMed] [Google Scholar]
- 48. Harada D, Isozaki H, Kozuki T, Yokoyama T, Yoshioka H, Bessho A, et al. Crizotinib for recurring non‐small‐cell lung cancer with EML4‐ALK fusion genes previously treated with alectinib: a phase II trial. Thorac Cancer. 2021;12(5):643–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ji J, Mitra A, Camidge DR, Riess JW. Early alectinib resistance from MET amplification in ALK‐rearranged NSCLC: response to crizotinib with re‐response to alectinib and crizotinib. Clin Lung Cancer. 2021;22(6):e851–5. [DOI] [PubMed] [Google Scholar]
- 50. Li M, An Z, Tang Q, Ma Y, Yan J, Chen S, et al. Mixed responses to first‐line alectinib in non‐small cell lung cancer patients with rare ALK gene fusions: a case series and literature review. J Cell Mol Med. 2021;25(19):9476–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sakakibara‐Konishi J, Kitai H, Ikezawa Y, Hatanaka Y, Sasaki T, Yoshida R, et al. Response to crizotinib re‐administration after progression on lorlatinib in a patient with ALK‐rearranged non‐small‐cell lung cancer. Clin Lung Cancer. 2019;20(5):e555–9. [DOI] [PubMed] [Google Scholar]
- 52. Shi R, Filho SNM, Li M, Fares A, Weiss J, Pham N‐A, et al. BRAF V600E mutation and MET amplification as resistance pathways of the second‐generation anaplastic lymphoma kinase (ALK) inhibitor alectinib in lung cancer. Lung Cancer. 2020;146:78–85. [DOI] [PubMed] [Google Scholar]
- 53. Lin JJ, Liu SV, McCoach CE, Zhu VW, Tan A‐C, Yoda S, et al. Mechanisms of resistance to selective RET tyrosine kinase inhibitors in RET fusion‐positive non‐small‐cell lung cancer. Ann Oncol. 2020;31(12):1725–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lin Y‐T, Chiang C‐L, Hung J‐Y, Lee M‐H, Su W‐C, Wu S‐Y, et al. Resistance profiles of anaplastic lymphoma kinase tyrosine kinase inhibitors in advanced non‐small‐cell lung cancer: a multicenter study using targeted next‐generation sequencing. Eur J Cancer. 2021;156:1–11. [DOI] [PubMed] [Google Scholar]
- 55. Rosen EY, Johnson ML, Clifford SE, Somwar R, Kherani JF, Son J, et al. Overcoming MET‐dependent resistance to selective RET inhibition in patients with RET fusion‐positive lung cancer by combining selpercatinib with crizotinib. Clin Cancer Res. 2020;27(1):34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ku BM, Bae YH, Lee KY, Sun J‐M, Lee S‐H, Ahn JS, et al. Entrectinib resistance mechanisms in ROS1‐rearranged non‐small cell lung cancer. Invest New Drugs. 2020;38:360–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nelson‐Taylor SK, Le AT, Yoo M, Schubert L, Mishall KM, Doak A, et al. Resistance to RET‐inhibition in RET‐rearranged NSCLC is mediated by reactivation of RAS/MAPK signaling. Mol Cancer Ther. 2017;16(8):1623–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kim H, Nguyen N‐P, Turner K, Wu S, Gujar AD, Luebeck J, et al. Extrachromosomal DNA is associated with oncogene amplification and poor outcome across multiple cancers. Nat Genet. 2020;52:891–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. McCoach CE, Blakely CM, Banks KC, Levy B, Chue BM, Raymond VM, et al. Clinical utility of cell‐free DNA for the detection of ALK fusions and genomic mechanisms of ALK inhibitor resistance in non‐small cell lung cancer. Clin Cancer Res. 2018;24(12):2758–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Peng H, Lu L, Zhou Z, Liu J, Zhang D, Nan K, et al. CNV detection from circulating tumor DNA in late stage non‐small cell lung cancer patients. Genes. 2019;10:926–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Keddy C, Shinde P, Jones K, Kaech S, Somwar R, Shinde U, et al. Resistance profile and structural modeling of next generation ROS1 tyrosine kinase inhibitors. Mol Cancer Ther. 2021;11:336–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. CUTO28 cells do not respond to MET inhibition and both CUTO28 and CUTO28‐ER cells display ecDNA amplification of the MYC locus. (a) CUTO28 and CUTO28‐ER cells were treated with the indicated inhibitors for 2 h. Lysates were probed with antibodies directed against the specific proteins labeled. (b) Metaphase FISH was performed using probes which label the MYC gene (red) and its corresponding chromosome's centrosome, CEP8 (green). (c) The number of extrachromosomal MYC probe hybridization signals per metaphase‐halted cell represented in Panel B are quantified by cell line. (e) The ecDNA MYC copy number gain relative to genomic DNA is quantified by cell line via genomic qPCR