

Abstract
Streptococcus defectivus is one of the nutritionally variant streptococci, a class of viridans group streptococci first isolated from patients with endocarditis and otitis media. In previous studies, NVS-47, a clinical isolate of S. defectivus, was shown to bind to the extracellular matrix. A high-molecular-weight surface protein was identified and proposed to be responsible for mediating this binding. In the present study, the gene encoding this protein was identified by transposon mutagenesis and characterized. The gene (emb) was found to be larger than 14 kb and was partially sequenced. It encodes a protein containing at least 50 repeats of 77 amino acids predicted to assume an alternating coiled-coil conformation. The domain responsible for extracellular matrix binding was mapped to the N terminus of the protein. From sequence analysis, Emb is proposed to be the prototype of a new family of streptococcal fibrillar proteins.
Nutritionally variant streptococci (NVS) are a class of viridans group streptococci first isolated by Frenkel and Hirsch in 1961 (7) as satellite colonies of Haemophilus species from patients with endocarditis and otitis media. In the absence of helper bacteria, they are unable to grow in most common laboratory media unless supplemented with pyridoxal hydrochloride or l-cysteine (5, 22). van de Rijn and George (30), studying a large collection of NVS, identified three serotype antigens not found in other streptococci. The distribution of these antigens allowed the delineation of three serotypes, with serotypes I and II comprising 97% of the isolates. Later, serotypes I and II were placed in two new streptococcal species, Streptococcus defectivus and Streptococcus adjacens, respectively (4). More recently, based on 16S RNA sequences, Kawamura et al. (10) proposed that S. defectivus and S. adjacens be moved to a new genus called Abiotrophia.
NVS are part of the normal oral flora (8), but they can also act as pathogens both in humans and in nonhuman animals (22). The most important NVS infection in humans is subacute bacterial endocarditis. NVS are responsible for 5 to 10% of all streptococcal endocarditis cases and are probably an important cause of the blood culture-negative form of the disease (20). Patients with NVS endocarditis have higher relapse and mortality rates than those with endocarditis caused by enterococci and the other viridans group streptococci (26). The clinical severity of NVS endocarditis has been correlated with both a delay in bacteriological diagnosis and an intrinsic resistance of NVS to antibiotics (2). The slow growth rate and metabolic defect of NVS often lead to negative blood cultures and seem to induce an in vivo antibiotic-tolerant status (3).
The pathogenesis of bacterial endocarditis begins with bacterial adhesion to a damaged heart valve. The most important candidates for the target of this adhesion are the platelet-fibrin matrix and the extracellular matrix (ECM) components exposed on the damaged tissue (9). In previous studies, S. defectivus was not able to bind to the platelet-fibrin matrix (31), but 13 of 15 strains analyzed were shown to bind to the ECM (28). By chemical mutagenesis, Tart and van de Rijn (29) obtained an S. defectivus mutant lacking the ability to bind ECM and identified a high-molecular-weight surface protein which they proposed was responsible for mediating such binding.
In this report, we describe the localization by transposon mutagenesis and the characterization of the gene encoding this ECM binding protein (Emb). The gene was found to be larger than 14 kb and was partially sequenced. It encodes a protein containing at least 50 repeats of 77 amino acids predicted to assume a coiled-coil conformation. The domain responsible for binding to ECM was mapped to the N terminus of the protein. Emb is proposed to be the prototype of a new family of streptococcal fibrillar proteins.
MATERIALS AND METHODS
Bacteria, media, and growth conditions.
S. defectivus NVS-47, NVS-59, NVS-52, and NVS-100 and S. adjacens NVS-61 and NVS-63 were from our collection. Bacteria were grown in semisynthetic medium (CDMT) (5) or in Todd-Hewitt broth supplemented with 0.2% yeast extract plus pyridoxal hydrochloride (0.5 mg/ml) (THYP) at 37°C. When bacteria were grown on solid media, sheep blood (3%) was added and plates were incubated in a 5% CO2 atmosphere. Escherichia coli JM109 and BL21(DE3) were grown in Luria broth or TYPG medium (16 g of tryptone, 16 g of yeast extract, 5 g of NaCl, 2.5 g of K2HPO4, and 5 g of glucose/liter of distilled H2O) at 37°C with agitation. Antibiotics, when required, were added at the following concentrations: kanamycin, 500 μg/ml for S. defectivus and 50 μg/ml for E. coli; ampicillin, 50 μg/ml.
DNA preparation.
A culture (500 ml) of NVS-47 was grown in THYP overnight, washed once in saline solution (50 ml), and then resuspended in cold TE–sucrose (25%, 10 ml) containing lysozyme (9.3 × 104 U/ml) and mutanolysin (12 U/ml). After incubation at 65°C for 1 h, 20% sodium sarcosyl (0.5 ml) and RNase to a final concentration of 0.3 mg/ml were added. The sample was further incubated for 30 min at 37°C. DNA was then extracted three times with phenol-chloroform (vol/vol) and twice with chloroform-isoamyl alcohol (24:1, vol/vol) and precipitated with ethanol. DNA was finally washed with ethanol (70%), dried, and resuspended in TE (200 μl).
Plasmid DNA from E. coli was prepared with a Qiagen (Santa Clarita, Calif.) plasmid kit in accordance with the manufacturer’s suggestions.
DNA manipulations.
All recombinant DNA techniques were performed by standard procedures (1) with E. coli JM109 as a host. DNA restriction enzymes were obtained from Promega Corp., Madison, Wis., and used in accordance with the manufacturer’s suggestions. DNA amplification (PCR) was performed with a model 480 DNA Thermal Cycler (Perkin-Elmer-Cetus, Norwalk, Conn.) and Pwo DNA polymerase (Boehringer Mannheim Corp., Indianapolis, Ind.) in accordance with the manufacturers’ directions. Southern blotting was performed by the standard procedure (1); probes were labeled and detected with a Genius 1 DNA labeling and detection kit (Boehringer).
pRC6 and pRC10 construction.
pRC6 was constructed by replacing the 1.15-kb BamHI fragment of pGh9:ISS1 (14), containing the erm gene, with a 1.16-kb BglII PCR product containing the aphIII gene from pKM1 (11), conferring resistance to kanamycin. The resulting plasmid replicates in both gram-positive and gram-negative bacteria at 28°C but is unable to replicate at 37°C. It carries the lactococcal insertion sequence ISS1. After ISS1 replicative transposition, the entire plasmid flanked by duplicated ISS1 sequences is integrated in the recipient chromosome.
pRC10 was constructed by replacing the ISS1-containing EcoRI-ClaI fragment of pRC6 with the 1.4-kb fragment of emb immediately upstream of the target of pRC6 integration (from base 2955 to base 4359) obtained by PCR.
DNA sequencing and sequence analysis.
Plasmid DNA and PCR products were prepared for chain termination sequencing (24) as previously described (15). Samples were sequenced with a Sequenase 2.0 kit (U.S. Biochemical Corp., Cleveland, Ohio) and [32P]dATP (ICN Biochemicals, Costa Mesa, Calif.). Sequencing reactions were resolved on 6% polyacrylamide gels, which were subsequently dried and exposed at −65°C to Kodak X-OMAT AR film. When PCR products were sequenced, in order to correct the mutations that might have occurred during amplification, each strand was sequenced from DNA obtained from a different PCR.
DNA sequence data were analyzed with the Genetics Computer Group software package (6). The signal peptide cleavage site in the Emb sequence was identified by the method of Nielsen et al. (16). Analysis for the prediction of coiled-coil structures was performed with COILS 2.2, available at the Internet address http://ulrec3.unil.ch/software/COILS_form.html/ (13). The Streptococcus pyogenes genome has been made available by the Streptococcal Genome Sequencing Project at the Internet address http://www.genome.ou.edu/strep.html.
Preparation and screening of enriched libraries.
The chromosomal DNA of NVS-47 was digested with the appropriate enzyme(s) and separated on agarose gels. Fragments to be cloned in the range of the predicted size (±1 kb) were purified from the gels with a QIAquick gel extraction kit (Qiagen) and ligated to pACYC177 (21). The ligation mixture was used to transform JM109, and the colonies were analyzed by colony blotting (23).
Preparation of frozen electrocompetent cells and electroporation procedure.
Frozen electrocompetent cells were prepared essentially by the procedure previously described for Streptococcus agalactiae (19). Briefly, bacterial cells were grown in THYP (500 ml) to the midexponential phase, harvested by centrifugation, resuspended, and then washed four times in 1 volume of glycerol (10%). For the final wash, cells were resuspended in a 1/100 volume, distributed in 1.5-ml test tubes, and centrifuged for 5 min at 13,000 × g. After calculation of its volume, the pellet was resuspended in 1 volume of glycerol (20%) and frozen at −70°C.
For electroporation, frozen electrocompetent cells were thawed on ice and diluted 1:2 in ice-cold glycerol (10%). An aliquot of 50 μl was mixed with the transforming DNA and loaded into a prechilled Gene Pulser cuvette (0.1-cm electrode gap) (Bio-Rad, Hercules, Calif.). The cuvette was exposed to a single pulse with a Bio-Rad Gene Pulser apparatus (capacitance, 25 μF; voltage, 20 kV/cm; resistance, 200 Ω). Immediately after the pulse, cells were diluted in THYP (1 ml), incubated for 3 h at 28°C, and then plated on selective plates.
Transposon mutagenesis.
pRC6 was introduced into NVS-47 by electroporation. Kanamycin-resistant transformants were selected at the permissive temperature (28°C) to allow plasmid replication. Seven transformants were grown overnight at 28°C in medium without kanamycin, diluted 1:10, and incubated for 4 h at the nonpermissive temperature (37°C) to allow plasmid curing. Finally, the cultures were plated on kanamycin at 37°C to select the transposants. The same procedure was used with pRC10 to select its insertion into the emb gene.
ECM adherence assay.
Bacterial adherence to baby hamster kidney cell-secreted ECM was detected by an enzyme-linked immunosorbent assay as previously described (28). Briefly, bacteria were grown overnight in CDMT, washed, and resuspended in ice-cold phosphate-buffered saline (PBS; 0.02 M sodium phosphate, 0.15 M NaCl [pH 7.4]) to an optical density of 0.5 at 530 nm (18-mm cells; Spectronic 20D spectrophotometer; Bausch & Lomb, Inc., Rochester, N.Y.). Bacteria were then mixed with an equal volume of blocking solution (3% casein–0.05% Tween 20 in 0.01 M Tris–0.1 M NaCl [pH 7.4]). After appropriate washes, specific bacterial antibody and then horseradish peroxidase-labeled goat anti-rabbit immunoglobulin were used to detect ECM-adherent organisms. The A620 was measured after tetramethylbenzidine dihydrochloride color substrate development. In each assay, the control plastic binding value was determined and subtracted from the mean ECM binding values to calculate specific ECM adherence. For inhibition studies, bacteria were incubated with antibodies for 1 h under rotation at room temperature prior to the ECM adherence assay. The total reaction volume was kept constant by resuspending the bacteria in a one-half volume of PBS in order to maintain a 1:1 ratio of sample to blocking solution with the addition of the antibodies.
Serum adsorption for the enrichment of adhesin-specific antibodies.
Enrichment of adhesin-specific antibodies was performed as previously described (29). Briefly, antiserum Rb342 (40 μl, generated by hyperimmunization of New Zealand White rabbits with strain NVS-47) was diluted 1:25 in PBS and then adsorbed with transposon insertion mutant strain WFemb1 from a culture grown overnight (100 ml) for 1 h at 4°C. After the cells were sedimented by centrifugation, the serum was filtered and the incubation was repeated with the parent strain (NVS-47, 100 ml). Antibody-coated bacteria were recovered by centrifugation and washed three times in PBS to remove unbound antibodies. Finally, bacteria were resuspended in 0.1 M glycine containing bovine serum albumin (0.5%) at pH 2.5 and incubated for 1 h at 4°C to release surface-bound antibodies. Bacteria were then centrifuged, and the eluted antibodies were neutralized with 1 M Tris (pH 9.0) and filtered through a 0.22-μm-pore-size membrane (Millipore Corp., Bedford, Mass.).
Preparation of solubilized cell wall extracts.
Solubilized cell wall extracts were prepared as described by Tart and van de Rijn (29). Briefly, bacteria were grown in 100 ml of CDMT to the mid-exponential phase, at which time penicillin (16.7 U/ml) was added. Incubation was continued for another 6 h. Bacteria were then centrifuged, washed twice in PBS, and resuspended in 30% raffinose–0.04 M Tris (pH 8, 2.8 ml) to which mutanolysin was added (200 μl; 2 mg/ml in 0.1 M KPO4 [pH 6.2]). Protoplast formation was carried out for 18 h at 37°C with rotation. Protoplasts were then pelleted, and the resulting supernatant was dialyzed against 0.01 M NaPO4 (pH 7.0) and filtered through a 0.22-μm-pore-size membrane (Millipore).
SDS-PAGE and Western blot analysis.
Proteins from bacterial samples and biotinylated molecular weight markers (Bio-Rad) were separated by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) by the method of Laemmli (12) and transferred (18 h, 80 mA) to nitrocellulose (NitroBind; Micron Separation Inc., Westborough, Mass.). Blots were blocked with 5% powdered milk–Tris-buffered saline (0.10 M Tris [pH 7.4], 0.15 M NaCl) and then incubated with enriched antiadhesin antibody (described above) at room temperature for 45 min. Next, the membranes were washed three times in Tris-buffered saline, incubated with horseradish peroxidase-labeled goat anti-rabbit immunoglobulin (Bio-Rad) and avidin-horseradish peroxidase conjugate (Bio-Rad) at room temperature for 45 min, and washed as before. Finally, the blots were developed with 3.4 mM 4-chloro-1-naphthol in CH3OH (1 part)–0.01 M imidazole in PBS (5 parts)-H2O2 (0.1%) for 30 min.
Overexpression of Emb fragments in E. coli.
PCR-amplified fragments of the emb gene (Table 1) were separated by agarose gel electrophoresis, recovered with a QIAquick gel extraction kit, and cloned in the SmaI site of pBluescript (Stratagene, LaJolla, Calif.). Cloned inserts were subsequently excised with restriction enzyme sites introduced by means of the oligonucleotide primers and ligated into expression vector pT77 or pET11a (27) (Table 1), and the ligation mixture was transformed in BL21(DE3). TYPG medium was inoculated with a single microcolony from the above transformation, and the culture was grown at 37°C until the optical density at 600 nm reached 0.8. One milliliter of the culture was removed as a preinduction sample; isopropyl-β-d-thiogalactopyranoside (IPTG) (1 mM final concentration) was added to the remaining culture, which was incubated at 37°C for an additional 3 h. Cells were washed in PBS, lysed by two passes through a French press at 20,000 lb/in2, and centrifuged for 30 min at 20,000 × g to remove cellular debris.
TABLE 1.
Recombinant polypeptide fragments of Emb and oligonucleotide primers used for PCR amplification
| Fragment | Amino acid residues | kDaa | Position of oligonucleotide primerb
|
|
|---|---|---|---|---|
| 5′ | 3′ (complement) | |||
| Emb-A | 503–963 | 48.0 | (NdeI) 1754–1770 | 3135–3117 (BamHI) |
| Emb-B | 928–139 | 48.5 | (NdeI) 3029–3045 | 4414–4399 (BamHI) |
| Emb-C | 50–530 | 51.8 | (EcoRI) 349–412 | 1837–1821 (BamHI) |
| Emb-D | 50–661 | 65.6 | (EcoRI) 349–412 | 2230–2212 (BamHI) |
For the Emb-derived portion of the fused protein only. Recombinant fragments C and D included a six-residue N-terminal sequence encoded by the plasmid.
PCR primers were designed to include the nucleotide sequence of the emb gene as shown. Each primer included a unique restriction enzyme site (indicated in parentheses). Additional nucleotides were added to maintain the reading frame (5′ primer) or to introduce a stop codon (3′ primer).
Affinity-purified antibody preparation.
Overexpressed fragments of the Emb protein were separated by SDS-PAGE and transferred to nitrocellulose. The blot was stained with Ponceau red (0.2% in 3% trichloroacetic acid–3% 5-sulfosalicylic acid), and strips containing the overexpressed polypeptides as well as those containing the comigrating proteins from uninduced E. coli cultures were isolated from the blot. The nitrocellulose strips were blocked with 5% powdered milk–Tris-saline-azide solution (0.05 M Tris [pH 7.4], 0.15 M NaCl, 0.03 M NaN3) overnight at 4°C. Rb342 serum was added at a 1:10 dilution and incubated with the strips for 2 h at 4°C. The strips were washed five times in Tris-saline-azide solution before incubation in 0.1 M glycine–0.05% bovine serum albumin (pH 2.5) for 1 h at 4°C to elute bound antibodies. The eluted antibodies were neutralized with 1 M Tris (pH 9.0) and filtered through a 0.22-μm-pore-size membrane (Millipore).
Nucleotide sequence accession number.
The nucleotide sequence of the emb gene has been deposited in GenBank under accession no. AF067776.
RESULTS
Transposon mutagenesis of NVS-47.
In order to produce an adhesin-negative mutant, the ISS1-containing thermosensitive plasmid pRC6 was introduced into NVS-47 by electroporation. Transformants were selected at 28°C to allow plasmid replication. Seven strains containing the plasmid were subjected to a temperature shift (see Materials and Methods) and plated on selective plates at 37°C to recover the transposants. Seven of 2,168 transposants tested were shown to be deficient in ECM adherence. One representative mutant, WFemb1, was chosen for further characterization.
To determine the effect on surface protein expression of the insertion of the transposon, enriched antiadhesin antibodies (prepared as described in Materials and Methods) were used to probe parent and mutant S. defectivus cell wall-associated proteins by Western blotting. Four high-molecular-weight bands (Fig. 1) were recognized in NVS-47 (lane a), while these proteins were absent in the corresponding transposon insertion mutant strain, WFemb1 (lane b).
FIG. 1.
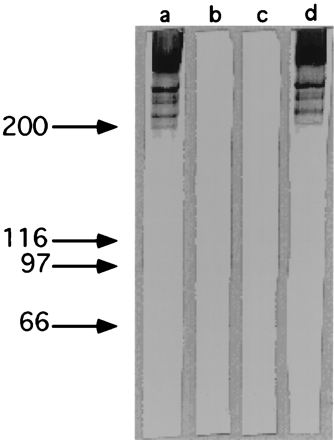
Western blot analysis of S. defectivus parent and mutant strain cell wall-associated proteins. Samples were separated by SDS-PAGE (8% polyacrylamide gel) and then transferred to nitrocellulose. The nitrocellulose blots were probed with adhesin-enriched antibodies (a to c) or anti-Emb-A antibodies (d). Lanes a and d, NVS-47 parent strain; lane b, WFemb1 mutant strain; lane c, WFemb2 mutant strain. The band present at the top of lanes a and d represents material that did not enter the separating gel. Molecular mass standards are shown on the left (kilodaltons).
Cloning and characterization of the emb gene.
The region surrounding pRC6 integration was mapped by Southern blotting (data not shown) to develop a cloning strategy. Seven overlapping DNA fragments (Fig. 2) covering 6,413 bp were amplified by inverted PCR and sequenced. Sequence analysis showed a single open reading frame (ORF) (emb) starting at base 248. After the first 1.9 kb, the ORF assumed a repeat structure made up of 231-bp tandem repeats. Four additional overlapping fragments of 2.7, 3, 5.5, and 6.6 kb (Fig. 2) covering an additional 8 kb downstream from the sequenced region were subsequently cloned in pACYC177. The distal regions of each fragment were sequenced. All the resulting sequences showed the same repeat structure of 231-bp tandem repeats previously found in the first part of the gene (data not shown), suggesting either that the ORF continues in all of them (making the size of the gene more than 14 kb and the number of repeats almost 50) or the presence of a second gene homologous to emb and in tandem with it. Potential −10, −35, and Shine-Dalgarno sequences were identified in the region 5′ to the coding sequence (Fig. 3).
FIG. 2.
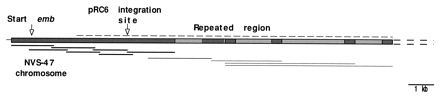
Sequencing strategy for the emb gene. Thick solid lines indicate fragments obtained by inverse PCR and completely sequenced; thin solid lines indicate fragments cloned by enriched libraries and partially sequenced; thin dotted line indicates repeated region. The sequenced regions are shown in dark cross-hatching. Arrows indicate the predicted start codon of emb and the pRC6 integration site in WFemb1.
FIG. 3.

Sequence of the region upstream of emb. Putative −10, −35, and Shine-Dalgarno (SD) sequences are shown, as is the putative start site for Emb translation.
A strain with a disruption in the emb gene was constructed to confirm that the deficiency in ECM adherence shown by WFemb1 was due to the emb disruption caused by pRC6 insertion and not by some other mutation. For this purpose, temperature-sensitive plasmid pRC10 into which an internal fragment of the gene had been cloned was introduced into strain NVS-47. After the temperature shift, five NVS-47 derivative strains with pRC10 integrated into the chromosome were isolated. Southern blot analysis (data not shown) showed that in all of the recombinants, the emb gene was disrupted. When tested for adherence, none of the five mutants was able to bind to ECM (data not shown), adding further evidence that the emb gene encodes an adhesin molecule. The same four polypeptide bands were found to be absent when cell wall-associated proteins of one of these mutants (WFemb2) were compared to those of the transposon insertion mutant WFemb1 by Western blot analysis (Fig. 1, lane c).
Characterization of the bands missing in the cell wall-associated protein profiles of WFemb1 and WFemb2.
As demonstrated above, four high-molecular-weight bands that were absent from the mutants were recognized by enriched antiadhesin antibodies in NVS-47 cell wall-associated protein profiles (Fig. 1). While these bands are probably the result of proteolytic degradation of Emb and/or different multimeric forms of the protein, it is possible that one or more of them represent another protein whose expression or stability in the cell wall depends on the presence of Emb. To address this issue, antibodies to a purified fragment of Emb overexpressed in E. coli (see below) were affinity purified and used to analyze NVS-47 cell wall-associated proteins. As shown in Fig. 1, lane d, these antibodies also recognized the four bands present in the wild type and missing in the two mutants. This result suggests that the four bands represent either degradation products of Emb or different multimeric forms of the protein.
Structure of Emb.
The sequenced part of the gene encodes a polypeptide of 2,055 amino acids (Fig. 4). The first 48 residues have the properties of a bacterial signal peptide, with a charged region followed by a hydrophobic sequence. The most likely site for signal peptide cleavage, as determined by the method of Nielsen et al. (16), is Ala-48. The predicted molecular mass of this portion of the protein after signal peptide processing is 210 kDa. At the N terminus of the predicted mature protein, between Ala-77 and Gln-128, is a region of 52 amino acids with 29 serine residues (55.8%). The region from Pro-633 to the end of the polypeptide is rich in alanine (25.4%) and includes 17 tandem repeats of 77 residues. The first eight repeats have an average identity of about 37% (similarity, 68%), while the last nine repeats have an average identity of 88% (similarity, 92%). The two repeats encoded by the more distal partially sequenced part of the gene are 82% identical to each other. Compared with the consensus sequence calculated for the nine highly conserved repeats, the identity is 50% (similarity, 74%). The sequence LTXEEK, at the beginning of each repeat, is conserved throughout the sequence.
FIG. 4.
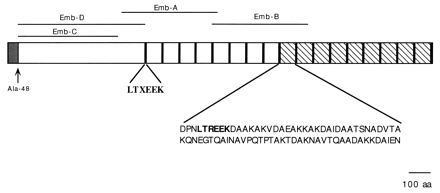
Structure of the first 2,055 amino acids (aa) of Emb. The putative leader peptide is shown as a cross-hatched box. The 18 LTXEEK conserved sequences at the beginning of each repeat are shown as black boxes. The nine more highly conserved repeats are shown as hatched boxes; their consensus sequence is also shown. Emb-A, Emb-B, Emb-C, and Emb-D represent the fragments of Emb overexpressed in E. coli.
When the sequence of Emb is compared with sequences in protein databases, only two significant matches are found: the EF* protein of Streptococcus suis (1,822 amino acids) (25) and an uncharacterized protein of S. pyogenes (2,060 amino acids). Both proteins have a highly repeated structure, and the homology with Emb is restricted to the repeated regions (Fig. 5A). The consensus sequence of the Emb repeats (77 amino acids) is 51% identical (70% similar) to that of the EF* repeats (76 amino acids) and 34% identical (46% similar) to that of the S. pyogenes protein repeats (81 amino acids) (Fig. 5B). The N terminus of each of the three proteins does not show any significant match with the databases.
FIG. 5.
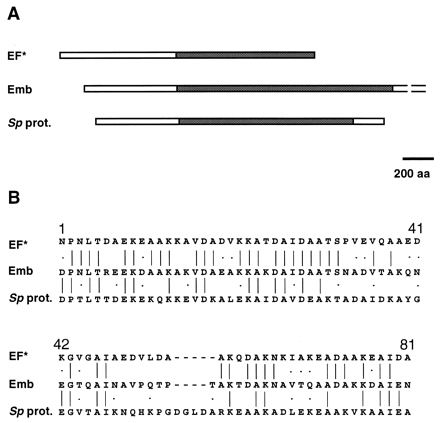
Homology of Emb with EF* of S. suis and the uncharacterized protein of S. pyogenes (Sp prot.). (A) Graphic map of the proteins. Homologous regions are shown as cross-hatched boxes. aa, amino acids. (B) Alignment of the consensus sequences of the three proteins. Identical amino acids are joined by a line; similar amino acids are indicated by a dot. The consensus sequence for Emb repeats (77 aa) is 51% identical (70% similar) to that for the repeats of EF* (76 aa) and 34% identical (46% similar) to that for the repeats of the uncharacterized protein of S. pyogenes (81 aa).
Localization of the ECM binding domain of Emb.
In order to show the location of the adhesin activity of the Emb protein, four overlapping fragments of the emb gene were overexpressed in E. coli. Table 1 shows the sizes and molecular masses of the various fragments. Affinity-purified antibodies to the overexpressed protein fragments and to comigrating proteins of uninduced E. coli were prepared from anti-NVS-47 serum by adsorption and subsequent elution from the nitrocellulose blot. Affinity-purified antibodies or whole antiserum was incubated with NVS-47, which was then tested for its ability to bind to ECM. Table 2 shows the complete inhibition of binding in the presence of whole antiserum. Bacteria incubated with either anti-Emb-C or anti-Emb-D antibodies were inhibited 92 and 90%, respectively. Binding was inhibited 8 and 15% when bacteria were incubated with anti-Emb-A and anti-Emb-B antibodies. No inhibition was observed with affinity-purified antibodies to proteins of uninduced E. coli.
TABLE 2.
Inhibition of S. defectivus adherence to ECM by affinity-purified antibodies to Emb fragments
| Inhibitor | A260a | % Inhibition |
|---|---|---|
| PBS | 0.38 | |
| Rb342b | 0.01 | 96 |
| Affinity-purified antibodiesc | ||
| Emb-A | 0.35 | 8 |
| Emb-B | 0.32 | 15 |
| Emb-C | 0.02 | 92 |
| Emb-D | 0.07 | 90 |
| Emb-A, Emb-B, and Emb-C com. prot. | 0.41 | |
| Emb-D com. prot. | 0.40 |
Except for values for Emb-C, all values represent the mean of two separate experiments.
Whole antiserum.
Affinity-purified antibodies to different fragments of the Emb protein after its overexpression in E. coli BL21(DE3) and affinity-purified antibodies to uninduced E. coli proteins comigrating (com. prot.) with Emb-A, Emb-B, Emb-C, or Emb-D.
Presence of the emb gene in clinical isolates of NVS.
In order to determine whether the emb gene was found in other clinical isolates of NVS, total DNA from four S. defectivus strains (NVS-47, NVS-59, NVS-52, and NVS-100) and two S. adjacens strains (NVS-61 and NVS-63) was tested by Southern blotting. The chromosomal DNA of the six strains was digested with KpnI, separated on an agarose gel, transferred to a filter, and probed at a low stringency with the 1.4-kb fragment of emb cloned in pRC10. All four DNA preparations from the S. defectivus strains reacted with the emb gene probe, whereas no reaction was observed with the S. adjacens DNA (data not shown). The patterns of bands recognized by the probe were different in the four S. defectivus strains: three bands were present in the hybridization profiles of NVS-47 and NVS-100 (8.8, 3.5, and 1.6 kb and 12.6, 4.0, and 0.9 kb, respectively), whereas only two bands were present in the hybridization profiles of NVS-59 and NVS-52 (9.0 and 5.0 kb and 12.6 and 3.5 kb, respectively) (data not shown).
DISCUSSION
In this paper, we describe the partial cloning and characterization of emb, the gene encoding the major adhesin of S. defectivus. After isolation of a mutant by transposon mutagenesis, 6.4 kb of the region flanking the transposon insertion was cloned by inverse PCR and sequenced. This region was shown to contain a partial ORF 6.1 kb long (emb) and its putative promoter region. The last 4.5 kb of the fragment contained 17 tandem repeats of 231 bp. In order to clone the rest of the gene, five additional overlapping fragments covering a total of 8 kb of DNA downstream of the first fragment were subsequently cloned, and their termini were sequenced. All of the sequences contained repeats of 231 bp homologous to those found in the first fragment, suggesting that the emb ORF continues through the additional 8 kb without interruption. Although we cannot rule out the existence in this region of a second gene homologous to emb and in tandem with it, this possibility is unlikely, since cutting in this region with restriction enzymes whose cutting sites are represented in the repeats produced only fragments of the size of the repeat or a multiple of it (data not shown). The highly repeated structure of the additional 8 kb made it impossible to design internal primers for both PCR and sequencing, leaving the construction of a library of nested deletions the most suitable strategy to complete the sequence. Since to map and to sequence by use of nested deletions a DNA fragment of 8 kb containing about 36 repeats is a major project, we decided to focus on the characterization of the ECM binding domain of Emb, leaving cloning and sequencing of the rest of the gene to a future project.
DNA hybridization studies showed that homologs of the emb gene of NVS-47 are present in three other clinical isolates of S. defectivus previously shown to be able to bind to ECM (28) but are absent in two isolates of the closely related organism S. adjacens previously shown to be unable to bind to ECM (28). The pattern of bands recognized by the probe, based on the repeated portion of emb, showed heterogeneity among the genes present in the different strains, as expected for genes containing tandem repeats.
Western blot analysis of cell wall extracts probed with affinity-purified antibodies to an overexpressed fragment of Emb showed four bands in the wild-type strain that were missing in the mutant strains, the smallest migrating at 200 kDa. Since S. defectivus peptidoglycan is partially resistant to both lysozyme and mutanolysin, bacteria were incubated with these two enzymes at 37°C for 18 h to release cell wall-associated proteins. Under these conditions, Emb might undergo some degradation, which could explain the presence of four bands. An alternative explanation is that the four bands might represent multimers of Emb, although this explanation is unlikely, since treatment with reducing agents did not affect the protein profile (data not shown) and the denaturing conditions of the gel should have been able to resolve multimeric structures not tightly bound by disulfide bridges. The considerable amount of material that did not enter the separating gel probably represents Emb molecules still associated with partially degraded peptidoglycan fragments.
In order to map the Emb domain responsible for ECM binding, antibodies affinity purified by binding to individual overexpressed portions of the protein were assayed for their ability to inhibit the binding of NVS-47 to ECM. Only affinity-purified antibodies to the overlapping fragments Emb-C and Emb-D were able to cause significant inhibition, thereby mapping the binding site in the N-terminal 480 residues of the predicted mature protein. Attempts were made to show direct binding of the overexpressed Emb-C and Emb-D polypeptides to ECM and their direct inhibition of NVS-47 binding to ECM, but these were unsuccessful, suggesting that the folding of the binding site of the protein fragments was incorrect after overexpression in E. coli (data not shown).
The repeated region of Emb shows homology to two proteins present in protein databases: EF* of S. suis type 2 and an uncharacterized protein of S. pyogenes. EF* is a secreted protein that is 1,822 amino acids long; its function is still unknown, but there are indications that it could be involved in virulence (25). It is noteworthy that the homology between EF* and Emb is restricted to the repeated region, while the N-terminal domains are completely unrelated. Although Emb is localized on the cell wall, EF* is secreted (25). However, after the stop codon of the gene encoding EF* and in frame with it, there is a sequence encoding a classical C-terminal anchor domain, suggesting EF* evolution from a cell wall protein. The uncharacterized protein of S. pyogenes is a cell wall protein of 2,060 amino acids that has a classical C-terminal anchor domain. The homology between this protein and Emb is restricted to the repeated region.
The secondary structure of the consensus sequence for the Emb repeats is predicted to be α helical, with one interruption in the region where the residues Pro-51 and Pro-54 are located. Also, in the junctions between the repeats, the helical structure is predicted to be interrupted by the residue Pro-2. The presence of repeats suggests that the protein may adopt some form of repeated helical structure. An analysis of the Emb repeats with COILS 2.2 divided each repeat into two regions which were predicted to assume a coiled-coil conformation (with the first having the highest probability) and two regions which were not predicted to assume such a structure. As shown in Fig. 6, a similar conformation is predicted for the repeats of EF* and of the uncharacterized protein of S. pyogenes, although the probability that the second regions of the repeats assume a coiled-coil conformation is much higher in these proteins than in Emb. Usually in fibrillar coiled-coil proteins, such as M6 of S. pyogenes, the coiled-coil structure is conserved for the entire length of the molecule (18). Further studies are necessary to understand the unusual structure of Emb and related proteins in which segments predicted to assume a coiled-coil conformation alternate with segments predicted to have a different structure.
FIG. 6.
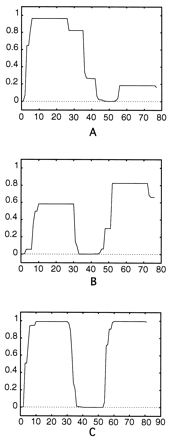
Coiled-coil prediction in the consensus sequence for the repeats of Emb (A), EF* (B), and the uncharacterized protein of S. pyogenes (C). The consensus sequence for the various repeats was analyzed with COILS 2.2 to predict the probability that they could assume a coiled-coil conformation. The amino acids number is shown on the x axis; the probability that a region might assume a coiled-coil conformation is shown on the y axis. The calculation was done with the MTIDK matrix, considering a 21-residue scanning window (13).
The data suggest that these three proteins belong to a new family of long, repeated, exported proteins of streptococci. It is interesting that only the repeated portion of the molecules is conserved throughout the members of the family, while the N termini, which in Emb carry the binding site for ECM, are completely unrelated. The peculiar structure of these proteins suggests that their repeated regions could function as a conserved partially coiled-coil mechanical module used by different streptococci to expose different functional domains on their surfaces.
In this study, we began the characterization of Emb, a long fibrillar adhesin that enables S. defectivus to bind to ECM, and proposed that it belongs to a new family of streptococcal proteins. Further studies are required to identify the ECM component(s) bound by the protein, the eventual accessory functions that could be carried out by such a long protein, and the structure of the repeated domain. Such information will provide a basis both for a better understanding of S. defectivus pathogenicity and for the development of vaccines and antiadhesion therapy (17) against this fastidious opportunistic pathogen.
ACKNOWLEDGMENTS
We are indebted to Kelley Bellomo for outstanding technical assistance. We also thank I. Blomfield for helpful advice; E. Garret for help in screening the emb mutants; and D. Dubnau, E. Dubnau, and I. Smith for carefully reading the manuscript.
This work was supported in part by Public Health Service grant AI37320 from the National Institutes of Health to I.V.D.R. and the Oligonucleotide Core Laboratory of the Comprehensive Cancer Center of Wake Forest University (grant CA12107).
REFERENCES
- 1.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K. Current protocols in molecular biology. New York, N.Y: John Wiley & Sons, Inc.; 1995. [Google Scholar]
- 2.Bouvet, A. 1995. Human endocarditis due to nutritionally variant streptococci: Streptococcus adjacens and Streptococcus defectivus. Eur. Heart J. 16(Suppl. B):24–27. [DOI] [PubMed]
- 3.Bouvet A, Cremieux A C, Contrepois A, Vallois J, Lamesch C, Carbon C. Comparison of penicillin and vancomycin, individually and in combination with gentamicin and amikacin, in the treatment of experimental endocarditis induced by nutritionally variant streptococci. Antimicrob Agents Chemother. 1985;28:607–611. doi: 10.1128/aac.28.5.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bouvet A, Grimont F, Grimont P A D. Streptococcus defectivus sp. nov. and Streptococcus adjacens sp. nov., nutritionally variant streptococci from human clinical specimens. Int J Syst Bacteriol. 1989;39:290–294. doi: 10.1099/00207713-41-4-483. [DOI] [PubMed] [Google Scholar]
- 5.Bouvet A, van de Rijn I, McCarty M. Nutritionally variant streptococci from patients with endocarditis: growth parameters in semisynthetic medium and demonstration of a chromophore. J Bacteriol. 1981;146:1075–1082. doi: 10.1128/jb.146.3.1075-1082.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Devereux J, Haeberly P, Smithies O. A comprehensive set of sequence analysis programs for the VAX. Nucleic Acids Res. 1984;12:387–395. doi: 10.1093/nar/12.1part1.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frenkel A, Hirsch W. Spontaneous development of L forms of streptococci requiring secretions of other bacteria or sulphydryl compounds for normal growth. Nature. 1961;191:728–730. doi: 10.1038/191728a0. [DOI] [PubMed] [Google Scholar]
- 8.George R H. The isolation of symbiotic streptococci. J Med Microbiol. 1974;7:77–83. doi: 10.1099/00222615-7-1-77. [DOI] [PubMed] [Google Scholar]
- 9.Herzberg M C. Platelet-streptococcal interactions in endocarditis. Crit Rev Oral Biol Med. 1996;7:222–236. doi: 10.1177/10454411960070030201. [DOI] [PubMed] [Google Scholar]
- 10.Kawamura, Y., X. Hou, F. Sultana, S. Liu, H. Yamamoto, and H. Ezaki. Transfer of Streptococcus adjacens and Streptococcus defectivus to Abiotrophia gen. nov. as Abiotrophia adjacens comb. nov. and Abiotrophia defectiva comb. nov., respectively. Int. J. Syst. Bacteriol. 45:798–803. [DOI] [PubMed]
- 11.Kiel J A K W, Vossen J P M J, Venema G. A general method for the construction of Escherichia coli mutants by homologous recombination and plasmid segregation. Mol Gen Genet. 1987;207:294–301. doi: 10.1007/BF00331592. [DOI] [PubMed] [Google Scholar]
- 12.Laemmli U K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 13.Lupas A. Prediction and analysis of coiled-coil structures. Methods Enzymol. 1996;266:513–525. doi: 10.1016/s0076-6879(96)66032-7. [DOI] [PubMed] [Google Scholar]
- 14.Maguin E, Prevost H, Ehrlich D, Gruss A. Efficient insertional mutagenesis in lactococci and other gram-positive bacteria. J Bacteriol. 1996;178:931–935. doi: 10.1128/jb.178.3.931-935.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Manganelli R, Ricci S, Pozzi G. The joint of Tn916 circular intermediates is a homoduplex in Enterococcus faecalis. Plasmid. 1997;38:71–78. doi: 10.1006/plas.1997.1300. [DOI] [PubMed] [Google Scholar]
- 16.Nielsen H, Engelbrecht J, Brunak S, von Heijne G. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 1997;10:1–6. doi: 10.1093/protein/10.1.1. http://www.cbs.dtu.dk . ( http://www.cbs.dtu.dk) ) [DOI] [PubMed] [Google Scholar]
- 17.Ofek I, Kahane I, Sharon N. Toward anti-adhesion therapy for microbial diseases. Trends Microbiol. 1996;4:297–299. doi: 10.1016/0966-842x(96)30023-1. [DOI] [PubMed] [Google Scholar]
- 18.Phillips G N, Flicker P F, Cohen C, Manjula B N, Fischetti V A. Streptococcal M protein: α-helical coiled-coil structure and arrangement on the cell surface. Proc Natl Acad Sci USA. 1981;78:4689–4693. doi: 10.1073/pnas.78.8.4689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ricci M L, Manganelli R, Berneri C, Orefici G, Pozzi G. Electrotransformation of Streptococcus agalactiae with plasmid DNA. FEMS Microbiol Lett. 1994;119:47–52. doi: 10.1111/j.1574-6968.1994.tb06865.x. [DOI] [PubMed] [Google Scholar]
- 20.Roberts R B, Krieger A K, Schiller N L, Gross K G. Viridans streptococcal endocarditis: the role of various species, including pyridoxal-dependent streptococci. Rev Infect Dis. 1979;11:955–966. doi: 10.1093/clinids/1.6.955. [DOI] [PubMed] [Google Scholar]
- 21.Rose R E. The nucleotide sequence of pACYC184. Nucleic Acids Res. 1988;16:355. doi: 10.1093/nar/16.1.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ruoff K L. Nutritionally variant streptococci. Clin Microbiol Rev. 1991;4:184–190. doi: 10.1128/cmr.4.2.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 24.Sanger F, Nicklen S, Coulson A R. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith H E, Reek F H, Vecht U, Gielkens A L J, Smits M A. Repeats in an extracellular protein of weakly pathogenic strains of Streptococcus suis type 2 are absent in pathogenic strains. Infect Immun. 1993;61:3318–3326. doi: 10.1128/iai.61.8.3318-3326.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stein D S, Nelson K E. Endocarditis due to nutritionally variant streptococci: therapeutic dilemma. Rev Infect Dis. 1987;9:908–916. doi: 10.1093/clinids/9.5.908. [DOI] [PubMed] [Google Scholar]
- 27.Studier F W, Rosenberg A H, Dunn J J, Dubendorff J W. Use of T7 polymerase to direct expression of cloned genes. Methods Enzymol. 1990;185:60–98. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- 28.Tart R C, van de Rijn I. Analysis of adherence of Streptococcus defectivus and endocarditis-associated streptococci to extracellular matrix. Infect Immun. 1991;59:857–862. doi: 10.1128/iai.59.3.857-862.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tart R C, van de Rijn I. Identification of the surface component of Streptococcus defectivus that mediates extracellular matrix adherence. Infect Immun. 1993;61:4994–5000. doi: 10.1128/iai.61.12.4994-5000.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van de Rijn I, George M. Immunochemical study of nutritionally variant streptococci. J Immunol. 1984;133:2220–2225. [PubMed] [Google Scholar]
- 31.van de Rijn I, Tart R C. Interaction of nutritionally variant streptococci with extracellular matrix. In: Orefici G, editor. New perspectives on streptococci and streptococcal infections. Stuggart, Germany: Gustav Fischer Verlag; 1992. pp. 133–136. [Google Scholar]