

Abstract
Dengue viruses (DENV) are endemic pathogens of tropical and subtropical regions and cause significant morbidity and mortality worldwide. Although a partially effective vaccine is in use in several countries in which DENV are endemic, no antiviral therapeutics are approved for combating DENV-associated disease. Herein, we report the characterization of novel small molecule inhibitors of DENV replication, VGTI-A3 and VGTI-A3–03, as well as structure-activity relationship analysis of the molecules using a panel of chemical analogs. VGTI-A3 and VGTI-A3–03 are highly virus-specific, with greatest activity against DENV serotype 2. Further analysis revealed that treatment of infected cells with VGTI-A3–03 does not inhibit viral RNA replication or secretion of viral particles. Rather, the infectivity of secreted particles from A3–03 treated cells is significantly diminished compared to particles secreted from control cells. Elicitation of VGTI-A3–03-resistant mutants demonstrated a clear binding pocket in the capsid molecule at the dimerization interface. Additionally, we show that VGTI-A3–03 is incorporated into virus particles released from infected cells. In summary, these data provide detailed analysis of a potentially useful class of anti-DENV inhibitors and further identify a region of the viral capsid protein as a druggable target for other therapeutic approaches.
1. Introduction
Mosquito-borne flaviviruses, which include dengue viruses (DENV), West Nile virus (WNV), and the recently emerging Zika virus (ZIKV), are a significant cause of morbidity and mortality worldwide. DENV are comprised of four antigenically distinct serotypes (DENV1–4) and are estimated to cause ∼100 million symptomatic infections per year (Bhatt et al., 2013; Halasa et al., 2012). The range and threat of mosquito-borne flaviviruses is expanding, highlighting the critical need for a clearer understanding of the life cycles of this group of pathogens. Yet, there are currently no antivirals in use against DENV, while the vaccine currently licensed in some countries offers less than 100% protection (Capeding et al., 2014; Villar et al., 2015).
Development of DENV antivirals has been challenging, with many compounds demonstrating great potential in vitro but ultimately failing in clinical trials. Antiviral compounds designed to target host functions required for viral replication have been both developed specifically for DENV or repurposed (reviewed in (Acosta and Bartenschlager, 2016)). Inhibitors of viral entry and early events, such as ligands for putative DENV receptors or inhibitors of viral endocytosis, represent attractive targets. However, the timing of administration of such inhibitors in vivo is problematic, and limited clinical trial data has demonstrated no efficacy in humans for drugs targeting early events (Borges et al., 2013; Tricou et al., 2010). Host-targeted compounds directed towards later events in the viral life cycle have also been explored, such as inhibitors of cholesterol/lipid biosynthesis (Whitehorn et al., 2012, 2016), inhibitors of nucleotide biosynthesis (Carocci et al., 2015; Fischer et al., 2013; Fraser et al., 2014) and molecules that target the ubiquitin-proteasome pathway (Choy et al., 2015; Nag and Finley, 2012). The most advanced host-targeting DENV antiviral, Celgosivir, inhibits DENV by targeting glycosylation events of the viral proteins, envelope (E), pre-membrane (prM), and nonstructural protein 1 (NS1), that are critical for viral replication and formation of mature viral particles (Courageot et al., 2000; Rathore et al., 2011; Watanabe et al., 2012, 2016). Although limited efficacy was observed in clinical trials, attempts to optimize dosing and administration protocols are currently underway for further human studies (Low et al., 2014; Sung et al., 2016).
Direct targeting of viral proteins has proven a successful approach for identifying antivirals directed against other virus families, such as human immunodeficiency virus, hepatitis C virus, and influenza virus (reviewed in (Lou et al., 2014)). Extensive research has been conducted to discover flavivirus-targeting compounds using a variety of approaches, including in vitro enzymatic screens, displacement assays, and protein-protein interaction studies. High throughput screens have been developed and conducted to identify compounds that specifically inhibit the enzymatic activities of viral proteins, including the protease (nonstructural (NS) 2B/3), helicase (NS3), methyltransferase (NS5), and polymerase (NS5). Additionally, cell-based and replicon-based approaches have successfully identified viral protein targeting compounds as well (Lim et al., 2013). Unfortunately, to date, no compounds targeting DENV proteins have demonstrated efficacy in human trials, mostly due to pharmacokinetic obstacles. Additionally, viral resistance is always a concern when developing virus-targeted drugs.
Previous research from our group described a high content cell-based screen that identified inhibitors of dengue viral replication (Shum et al., 2010). Follow-up studies on several hits from this screen have demonstrated that inhibition by these compounds occurs through inhibition of host-targeted functions–pyrimidine biosynthesis (Fischer et al., 2013) and MAP kinase signaling (Smith et al., 2014). In this study, we describe the characterization of a potent inhibitor of DENV2 replication, which we have named VGTI-A3 (PubChem ID: 4259739). Through extensive medicinal chemistry and structural-activity relationship (SAR) analysis, we have identified an analog—VGTI-A3–03—with increased anti-DENV2 potency and solubility. We demonstrate that this inhibitor acts through direct targeting of the DENV2 capsid (C) protein and perform a detailed characterization of the binding region. Finally, we demonstrate that VGTI-A3–03 is directly incorporated into viral particles through this interaction with C. During the course of this investigation, a compound with similar structure was described, and its activity was also attributed to direct C binding (Byrd et al., 2013; Scaturro et al., 2014). Our study adds to this research by providing extensive SAR analysis of approximately 40 analogs, identifying several resistance-conferring mutations, thorough characterization of the region of the viral capsid required for VGTI-A3–03 inhibition, and demonstration of incorporation into the DENV virion.
2. Results
Previous research from our group described a high content immunofluorescence based screen for identification of small molecule inhibitors of DENV2 replication (Shum et al., 2010). One of the most potent compounds detected was 3-amino-6-phenyl-N- (4-phenyl-1,3-thiazol-2-yl)- 5,6,7,8- tetrahydrothieno[2,3-b]quinoline- 2-carboxamide (PubChem ID: 4259739)– from hereon referred to as “VGTI-A3” (Fig. 1A). This small molecule potently inhibited production of infectious progeny from DENV-2 infected HEK293 cells, causing a maximum viral load reduction (VLR) of approximately three logs, with an inhibitory concentration resulting in 90% reduction of titer (IC90) of 112 nM (Fig. 1D). Although VGTI-A3 displayed potent antiviral activity, low solubility presented an obstacle to development of an antiviral drug. Therefore, a large library of analogs was synthesized and tested in an attempt to identify molecules with more favorable physical and antiviral properties. All analogs were tested by quantitation of virus in cell culture supernatants by focus forming assays (ffa) to determine IC90 and maximum viral load reduction (VLR; Supplemental Table S1). Calculated logP values (ChemDraw Professional Version 15.0) were also consulted as a measure of solubility (low logP = high solubility). The goal of the structure activity relationship (SAR) study was to optimize the solubility of VGTI-A3 while maintaining its in vitro potency. The phenyl ring on the left-hand side of the molecule was replaced with smaller substituents (such as Me and H) and with pyridyl heterocycles. The bicyclic system attached via the peptide bond on the right-hand side of the molecule was both simplified with alkyl substituents and modified with various heterocyclic systems. Of the 39 compounds assayed, one was selected (VGTI-A3–03; Fig. 1B) for follow-up studies because of its increased antiviral activity (IC90 = 25 nM, VLR = 3.8), as well as its predicted improved solubility (Calc logP = 5.6 vs 4.7). VGTI A3–03 also lacks the undesirable chiral center (marked with an asterisk) at the saturated ring atom connected to the phenyl group. We next determined susceptibility of other DENV serotypes, DENV1, 3, and 4, and the related flavivirus, WNV, to VGTI-A3 or VGTI-A3–03 (Fig. 1E). Interestingly, while the antiviral activity of VGTI-A3 appeared to be limited to only DENV serotype 2 (grey bars), VGTI-A3–03 also demonstrated some inhibitory activity against DENV1, DENV4, and WNV (black bars). Anti-DENV activity of a related compound (ST-148) has been previously described (Fig. 1C; Supplemental Table 1: VGTI-A3–32) (Byrd et al., 2013; Scaturro et al., 2014). Because of the similar structures of our original compound (VGTI-A3), the improved analog (VGTI-A3–03), and the previously identified compound (ST-148), it is highly likely that the anti-DENV mechanism(s) are shared among these three small molecules.
Fig. 1. Identification of a potent anti-DENV compound and an analog with increased activity.
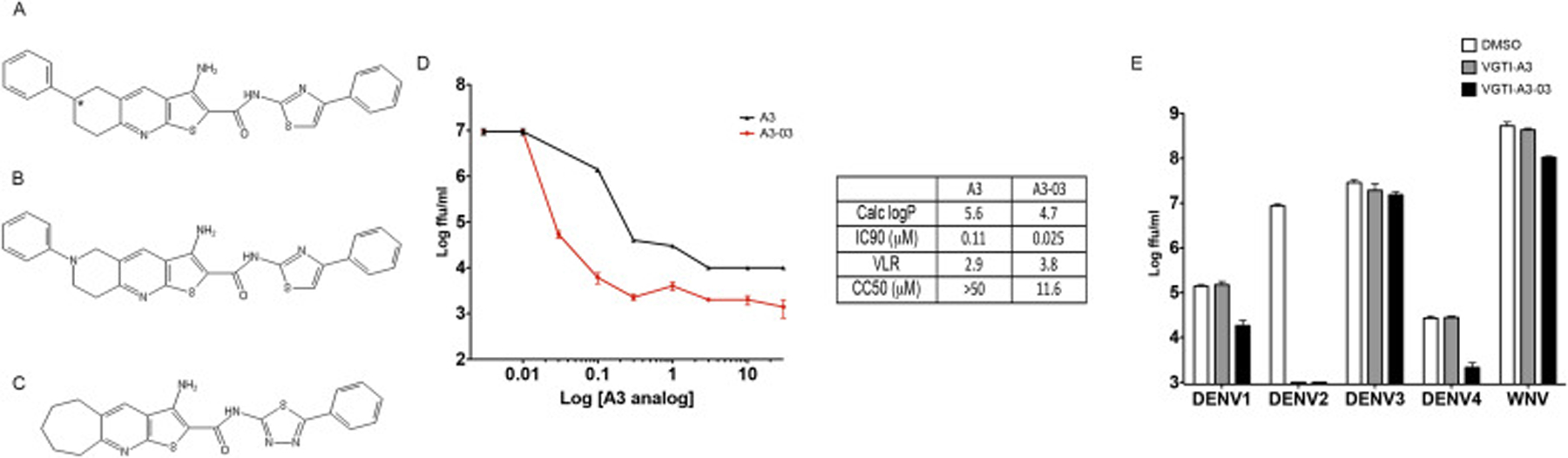
(A) Structure of VGTI-A3 and its analog, VGTI-A3–03 (B) and a previously identified anti-DENV compound, ST-148 (C). (D) HEK293 cells were infected with DENV at MOI = 0.1 ffu/cell + increasing concentrations of VGTI-A3 or VGTI-A3–03 (DMSO, 30 nM, 100 nM, 300 nM, 1uM, 3uM, 10uM, 30uM). Supernatants were collected at 3d pi, and assayed for infectious virus by focus forming assay (ffa). Calc LogP, IC90, VLR, and CC50 were calculated as described in Materials and Methods. (E) HEK293 cells were infected with indicated viruses at MOi = 0.1 ffu/cell in the presence of DMSO (white) or 1uM VGTI-A3 (grey) or VGTI-A3–03 (black). At 3 days pi, supernatants were collected and assayed by focus forming assay. Data are presented as mean ± SEM in triplicate.
To identify the time post infection at which A3–03 blocks DENV2 replication, we added the drug at different times pi (up to 24 h pi) and quantified supernatant virus at 48 h pi. Only a modest drop in antiviral activity was observed when VGTI-A3–03 was added at 4–24 h post-infection, as compared to addition at the time of infection and addition as late as 24 h pi still resulted in significant inhibition of viral replication (Fig. 2A). These results suggest that early events (i.e., entry, particle uncoating, or initiation of viral genome translation) in the viral life cycle are minimally affected by VGTI-A3–03 treatment, and that inhibition likely occurs during later steps of the viral life cycle (i.e., genome replication or viral particle assembly). To assess the affect of VGTI-A3–03 treatment on viral particle production, supernatants from cells infected at high multiplicity of infection (MOI = 3 ffu/cell) were assayed at 24 h pi for both infectious particles by focus forming assay and viral genome levels by reverse transcriptase quantitative PCR (RT-pPCR; Fig. 2B). Interestingly, although a significant decrease in infectious particle production was observed, levels of viral genomes were not reduced by VGTI-A3–03 treatment. These results suggest that VGTI-A3–03 does not inhibit production of genome-containing particles but instead promotes the formation and release of noninfectious virions.
Fig. 2. VGTI-A3–03 inhibits DENV during assembly of viral particles.
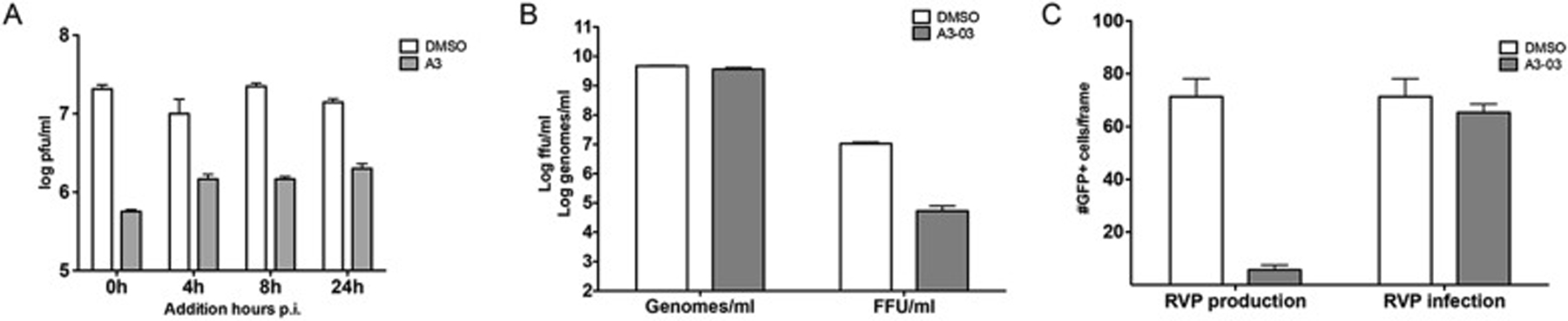
(A) HEK293 cells were infected with DENV at MOI = 3 ffu/cell. At indicated times p.i., media was removed wand replaced with media containing DMSO or 1uM VGTI-A3. Supernatants collected at 48 h p.i. were assayed by ffa. (B) Supernatants were collected from HEK293 cells infected at 3 ffu/cell ± 1uM A3 at 48 h p.i. Genomes per ml were quantified by RT-qPCR, and infectious virus was assayed by ffa. (C) Reporter virus particles (RVPs) were either produced in the presence of 1uM A3 (left, ‘RVP production’) or used to infect cells in 1uM A3 (right, ‘RVP infection’). The number of GFP + cells per frame (6 frames per sample) was quantified using ImageJ software. Data are presented as mean ± SEM in triplicate.
To further dissect the step during the viral life cycle where VGTI-A3–03 is acting, we utilized a powerful reporter viral particle (RVP) system (Ansarah-Sobrinho et al., 2008), in which replicon-bearing cells that produce the nonstructural region of the DENV genome (NS1-NS2A-NS2B-NS3-NS4A-NS4B-NS5) plus a GFP reporter are transfected with a plasmid encoding the DENV structural genes (C, pre-membrane/membrane (prM/M), and envelope (E)). Supernatants collected from these transfections contain RVPs carrying the reporter replicon that are able to enter naïve cells and initiate translation and replication of the viral replicon, but are unable to produce a new generation of viral particles, due to the lack of the C, prM, and E proteins. Using this assay, one can distinguish clearly between effects on early viral life cycle events (entry, initiation of viral translation) versus late (assembly, maturation, and release of viral particles). To determine whether VGTI-A3–03 affects particle production or assembly, we assessed the effects of adding compound either during production of RVPs by addition of compound post-transfection of the structural plasmid or during infection of naïve cells with RVPs. Addition of compound during RVP production had a profound inhibitory effect, while addition during RVP infection displayed no inhibition of GFP production (Fig. 2C). These results demonstrate that VGTI-A3–03 is acting against DENV infection by affecting the proper assembly of virus particles.
Another approach to identify antiviral mechanisms of action is to passage virus in the presence of compound to promote the formation of resistant viruses, followed by sequencing of resistant viruses to identify the viral factor being directly or indirectly targeted by the inhibitory compound. Production of resistant viruses by passage in the presence of VGTI-A3 was surprisingly easy, and resistant viruses started to appear as early as passage 3, with virtually the entire viral population of passage 4 displaying total resistance to VGTI-A3 treatment (Fig. 3A). Sequencing of multiple independently-derived resistant populations revealed a hot spot of mutations between residues 20–30 of the C protein. Fig. 3B indicates the mutations that were identified in resistant populations (threonine-25-isoleucine, leucine-35-proline, leucine-38-methionine; red) and the corresponding C regions of related flaviviruses. The L35P mutation was the most common residue change identified. Interestingly, Byrd et al. (2013) have reported resistant mutant formation against ST-148 at residue 34 of the C protein (serine-34-leucine), providing further evidence that these compounds similarly inhibit DENV2 infection.
Fig. 3. Mutations in the DENV capsid confer resistance to VGTI-A3.
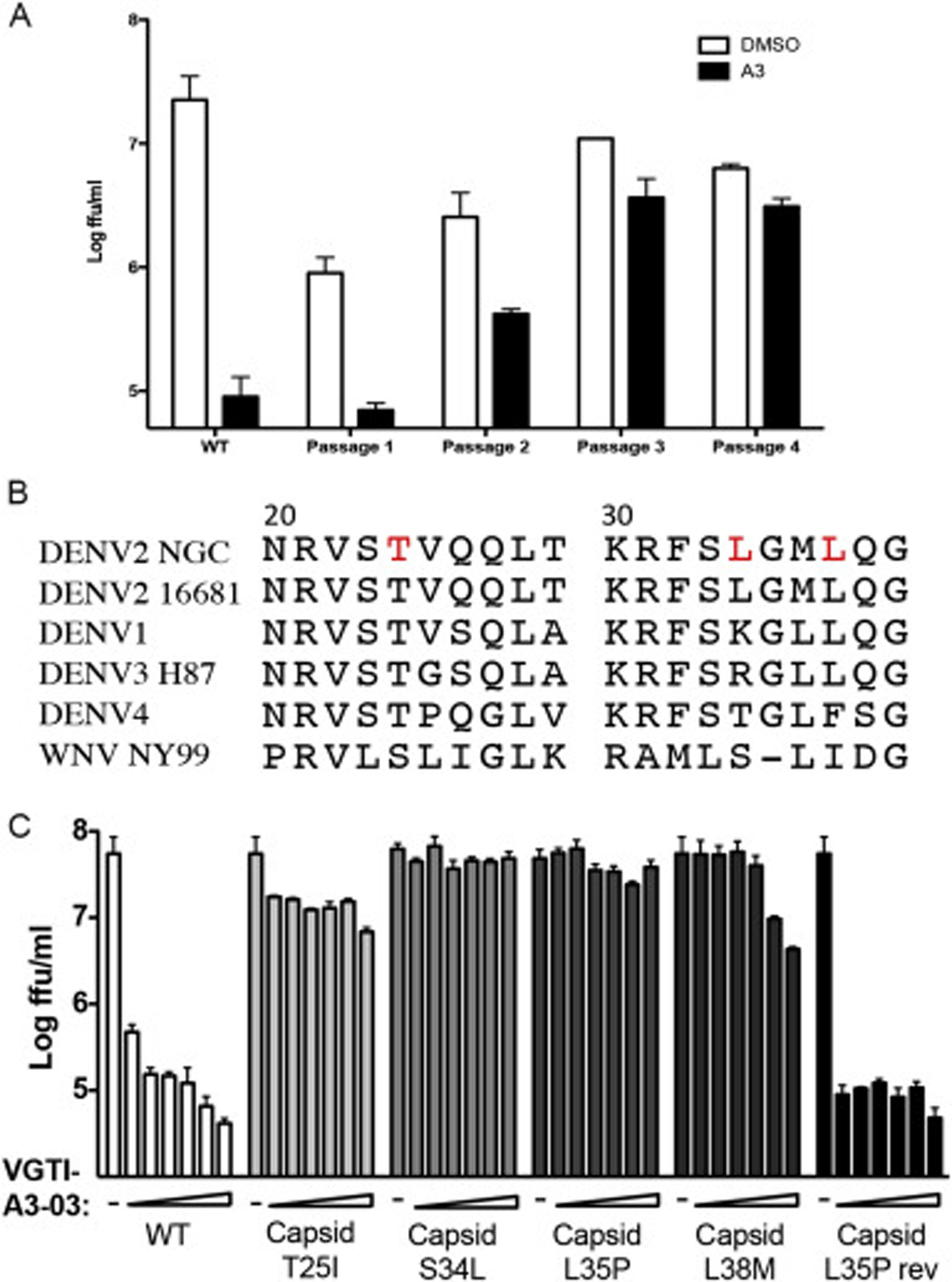
(A) Virus was passaged 4 times in the presence of 1uM VGTI-A3 at MOI = 0.1ffu/cell on HEK293 cells. Virus was assayed for resistance at each passage, and RNA isolated from p4 virus was sequenced by Sanger sequencing. (B) Region of the DENV2 C protein (residues 20–40) with mutations (red) found in resistant virus populations and corresponding region of related flaviviruses, DENV1–4 and WNV. (C) Mutations were introduced into the DENV2 16681 infectious clone, and resultant viruses were assayed for resistance by infection of HEK293 at MOI = 0.1 ffu/cell in the presence of 30 nM-10μM VGTI-A3. Infectious virus present in the supernatants collected at 3 days p.i. was quantitated by focus forming assay. Data are presented as mean ± SEM in triplicate. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
To confirm the role of these identified mutations in resistance to VGTI-A3–03 treatment, the three changes (T25I, L35P, and L38M) identified by our group, as well as the change reported by Byrd et al. (S34L) were engineered into the DENV2 16681 infectious cDNA clone (Huang et al., 2010). In dose-response assays, wild type virus displayed high susceptibility to VGTI-A3–03 treatment, as expected, while all of the mutant viruses demonstrated resistance. A revertant virus, in which the L35P residue was mutated back to the wild type amino acid within the infectious cDNA clone, exhibited susceptibility to VGTI-A3–03 treatment (Fig. 3C). These results confirm the role of this region of the C protein in the antiviral mechanism of VGTI-A3/VGTI-A3–03/ST-148, and support the notion of direct binding of these small molecules to the viral capsid.
To finely map the region of C dictating VGTI-A3–03 antiviral activity, the RVP system was again utilized. Introduction of the resistance-conferring mutations (L35P, S34L), as well as mutation of amino acids 23–39 of the C protein to alanine (A), in the RVP structural plasmid was assessed for ability to confer resistance to VGTI-A3–03 treatment during particle formation. Susceptibility of wild type RVPs and resistance of S34L or L35P RVPs was confirmed (Fig. 4A and B). The results of the alanine scan over the C 23–39 region are presented in Fig. 4C. Interestingly, while mutation of residues 34 and 35 to leucine and proline, respectively, was tolerated for RVP formation and conferred resistance as expected, mutation of either of these residues to alanine completely eliminated particle formation. After consultation of the published structure of DENV C dimer (PBD: 1R6R (Ma et al., 2004);), as well as the binding site of ST148 predicted by Byrd et al. on the C dimer, a number of other residues were selected for addition to the alanine scan study (F47, G64, R68, T71, and I72), due to their putative proximity to the predicted VGTI-A3–03 binding site. Ribbon diagrams (front and side views) of the C dimer highlighting residues that confer resistance (red), or are irrelevant to (green), VGTI-A3–03 inhibition are shown in Fig. 4D. These results suggest direct binding within a pocket formed between the α1 and α3’ helices of the C dimer. This pocket is in a distinct region from the of RNA and lipid binding motifs (Ma et al., 2004) of the C dimer and corresponds to the predicted binding site of ST148 by Byrd et al., who propose that ST-148 binding within this region promotes the formation of dimer-dimer interactions that support the higher order structure formation of the viral particle (Byrd et al., 2013; Mateo et al., 2015; Scaturro et al., 2014).
Fig. 4. Discovery of residues within compound binding pocket that define susceptibility to VGTI-A3–03.
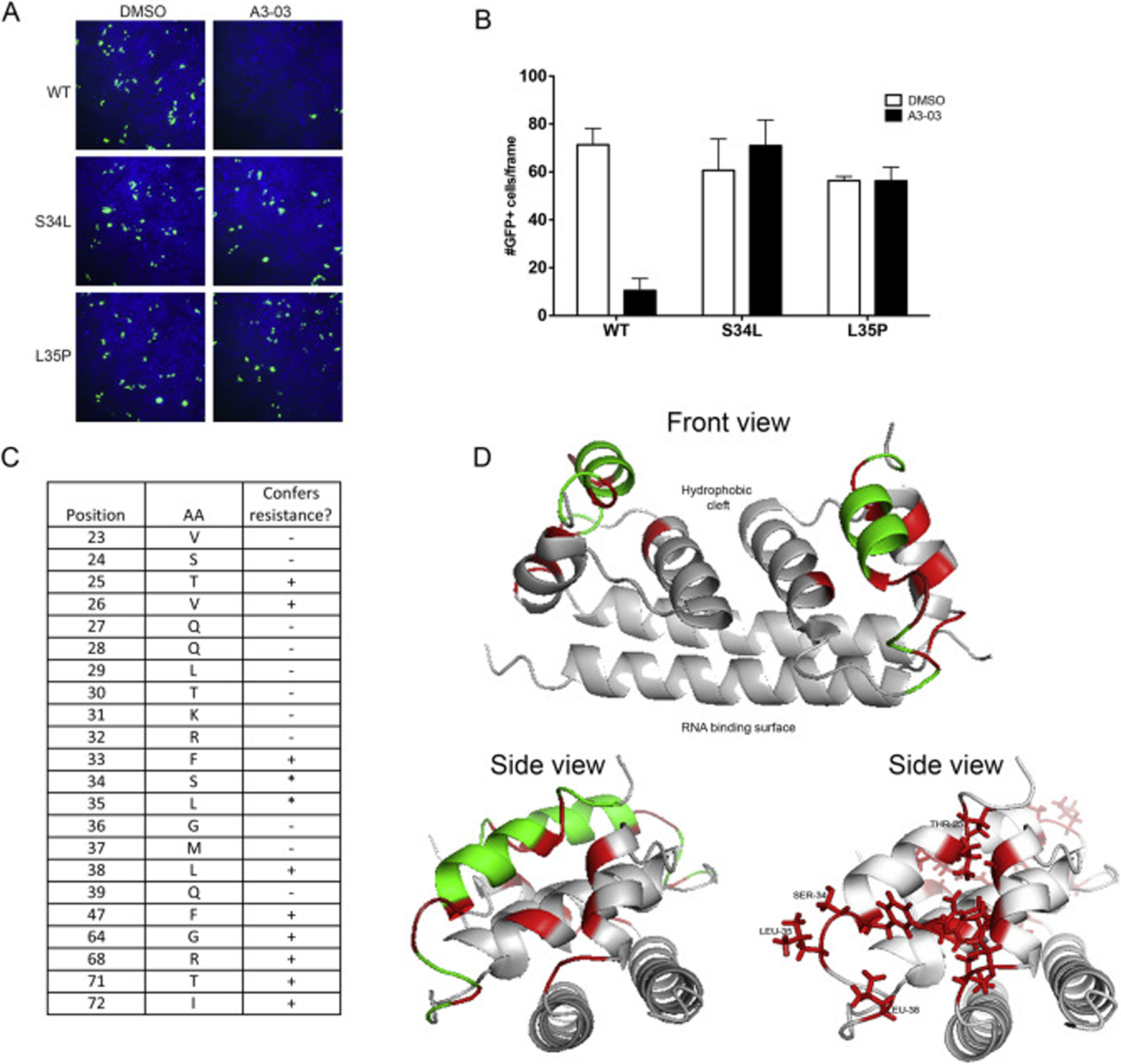
(A) DENV2 reporter virus particles (RVPs) expressing GFP were produced by transfection of DRep2A-expressing cells with a plasmid encoding DENV2 structural proteins (wild type or containing the C S34L or L35P mutations). At 1 day p.t., media was removed and replaced with 1uM VGTI-A3–03 or DMSO control. Supernatants containing RVPs were collected at 4 days p.t., purified over a sorbitol cushion, and used to infect naïve HEK293 cells at MOI = 5 IU/ml. GFP + cells were assayed at 3 days p.i. (B) GFP + cells were quantitated from 6 independent frames using ImageJ software. (C) RVPs constructed with indicated C residues mutated to alanines were tested for resistance conference to A3–03 as above. Residues that did not tolerate mutation to alanine, as indicated by no RVP production, are indicated with an asterisk. (D) Front and side views of MacPyMol depiction of the C dimer (PBD: 1R6R) with resistance conferring C mutations (red) or resistance neutral (green). The hydrophobic cleft, where the ER membrane is predicted to interact, and the putative RNA binding surface are indicated on front view. Data are presented as mean ± SEM in triplicate. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
To investigate the possibility that VGTI-A3–03 is directly binding to C dimers in viral particles and causing production of malformed virions, we examined the effect of VGTI-A3–03 treatment during virus production on the mobility of resultant particles in a sucrose density gradient. Supernatants collected at 24 h pi (+/− 1uM VGTI-A3–03) were purified over a sorbitol cushion, then centrifuged through a discontinuous sucrose gradient. Fractions were collected, and infectious particles, viral genomes, and envelope were measured by focus forming assay, RT-qPCR (Fig. 5A), and western blot (Fig. 5B). VGTI-A3–03 treatment did not cause a significant change in mobility of genome- or envelopeGTI-A3–03 treatment did not cause a significant change in mobility of genome- or econtaining particles over the sucrose gradient, suggesting that the noninfectious particles produced in the presence of VGTI-A3–03 have similar physical properties.
Fig. 5. VGTI-A3–03 is incorporated into DENV particles.
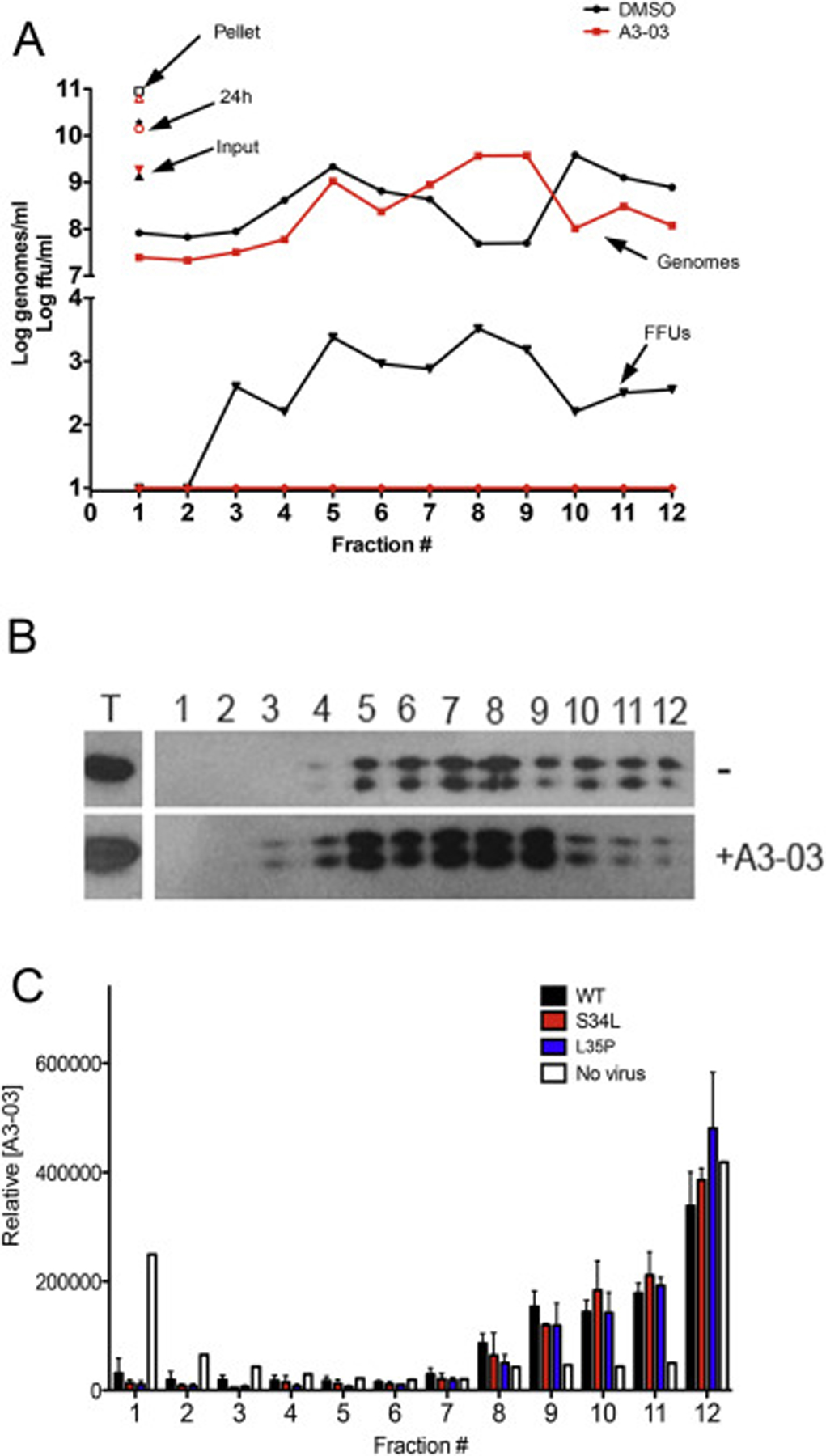
(A) Supernatants collected from HEK293 cells infected with DENV2 (MOI = 3 ffu/ml) for 24 h in the presence of 1uM VGTI-A3–03 or DMSO control were first pelleted over a sorbitol cushion, then loaded on top of a discontinuous (15/35/50%) sucrose gradient. Samples were centrifuged at 40,000 rpm for 4 h 1 ml fractions were collected and assayed for infectious particles by focus forming assay (FFUs) or viral genome equivalents by RT-qPCR or envelope by western blot (B). (C) Pelleted supernatants from no infection control, wild type or S34L-, L35P-mutated virus were centrifuged over a sucrose gradient as above. Fractions were assayed for relative VGTI-A3–03 levels by mass spectroscopy.
The possibility that VGTI-A3–03 is directly incorporated within released viral particles was also investigated. Gradients were prepared as above in the absence or presence of virus infection, and concentration of VGTI-A3–03 was assessed using mass spectrometry (Fig. 5C). In the absence of viral particles (white bars), the majority of compound fractionated to the lightest (23%) and heaviest fractions (40%; most likely a result of precipitation). Conversely, in the presence of wild type viral particle release (black bars), approximately 50% of the compound was found in fractions #8–11, the same fractions where particles were observed (Fig. 5A and B). These results demonstrate that VGTI-A3–03 is incorporated into the viral particle, presumably through interaction with C protein dimers during virion assembly. Assessment of VGTI-A3–03 fractionation in the presence of S34L (red) or L35P (blue) mutant viruses revealed similar patterns, suggesting that compound is incorporated into mutant virions with equal efficiency. These data are consistent with previous results that demonstrate ST-148 binding to wild type and S34L-mutated recombinant C proteins occurs with similar affinity (Byrd et al., 2013). Taken together, these results suggest that this class of compounds binds to C dimers during viral assembly and promotes dimer-dimer interactions, becoming incorporated into the virion. Resultant VGTI-A3–03 containing particles then display a defect in infection of naïve cells.
3. Discussion
In this study, a molecular compound with potent anti-DENV activity was identified, VGTI-A3, and, through extensive SAR analysis, a novel analog, VGTI-A3–03, was developed with improved solubility and antiviral characteristics. Previous identification of a molecule with a similar structure, ST-148 (Byrd et al., 2013; Scaturro et al., 2014), demonstrated direct binding of ST-148 to the DENV C protein. Data presented here suggests a similar mechanism of VGTI-A3–03, but extends this research to thoroughly characterize the residues of the C protein that direct this interaction. Interestingly, although Scaturro et al. observe inhibition of DENV entry, as well as impaired formation of infectious particles by ST-148, our data suggest that VGTI-A3–03 acts exclusively during virion morphogenesis. One explanation may be that ST-148 is able to penetrate the virion and bind C dimers during the initial entry more efficiently, while VGTI-A3–03 can only be incorporated during assembly of new particles. Whether this difference is due to the different structures of the two compounds or to the model systems employed still remains to be elucidated. Nevertheless, this class of compounds appears to be exerting its antiviral effects through direct binding in a pocket formed by the α1/α3′-helices of the DENV C dimer. Binding during virion formation results in VGTI-A3–03 incorporation into the viral particle, which results in the formation of noninfectious particles. Although VGTI-A3–03-containing virions display no discernible differences in density, as demonstrated by sedimentation patterns on a sucrose gradient, they are unable to initiate new infections in a naïve cell. These data are consistent with the mechanism proposed by Scaturro et al. that compound/capsid binding stabilizes dimer-dimer interactions within the virion, hindering particle disassembly following entry into a naïve cell.
Viral capsid/core proteins represent an attractive target for antiviral development due to the specificity and decreased likelihood of off-target effects resulting from binding to host factors, as compared to nucleoside analog based drug therapies, for example. Additionally, because these drugs can act during the later stages of infection, the time of effectiveness of such compounds may be extended in relation to drugs that rely on inhibition of entry or genome replication. A number of capsid targeting compounds have shown promise as antivirals against other enveloped viruses, including hepatitis B virus (Delaney et al., 2002; Deres et al., 2003; King et al., 1998) and human immunodeficiency virus (Blair et al., 2010; Fader et al., 2011; Tremblay et al., 2012). Of note, the mechanism of action of these capsid-targeting compounds is often related to stabilization of higher order structures as well.
In addition to the thorough SAR analysis of VGTI-A3 and its analogs and identification of VGTI-A3–03 as an analog with increased potency and solubility, we have also presented here an in-depth investigation into the specific residues within the VGTI-A3–03 binding pocket that dictate susceptibility to this class of compounds. Through this analysis, we are able to more comprehensively understand not only the C interaction properties of these compounds, but also the residues at the C dimer interface contributing to interdimer contact required for proper virion formation. Additionally, by demonstrating that virions assembled in the presence of VGTI-A3–03 do not display any apparent physical differences, but that they are indeed packaging compound within mature particles, our data add further support to the hypothesis that this class of compounds exerts its effects through promoting stronger dimer-dimer interactions and preventing viral genome un-coating.
Discovery and development of flavivirus-targeting antivirals has been challenging, compelling researchers to consider less traditional targeting strategies when the conventional approaches, such as nucleoside analogs, protease inhibitors, and entry-directed compounds, have failed. The class of compounds described here, which includes VGTI-A3, VGTI-A3–03, and the previously described ST-148 (Byrd et al., 2013), exert their antiviral effects by targeting the viral C proteins, a strategy that, although has been shown to work well against other enveloped viruses, has not yet been fully explored in the context of flavivirus infections. This study extends our understanding of the specific C-targeting mechanism of these compounds, provides some insight about the C interdimer interface, and highlights the value of exploring the therapeutic potential of C-targeting antivirals.
4. Materials and Methods
Cell culture and reagents
HEK293, C6/36, and Vero cells were grown in modified Eagle medium (MEM; Gibco) supplemented with 10% fetal bovine serum (Hyclone), 2 mM l-glutamine (Invitrogen), 100 U/ml penicillin G sodium, 100 μg/ml streptomycin sulfate (Invitrogen) and 1× nonessential amino acids (Gibco).
VGTI-A3 series synthesis
A detailed description of the synthesis methods is provided in Supplemental Material, including a diagram of VGTI-A3–03 synthesis strategy (Fig. S1).
IC90 and VLR assays
Infections of HEK293 cells were performed at MOI = 0.1 ffu/cell in the presence of 30uM, 10uM, 3uM, 1uM, 300 nM, 100 nM, 30 nM compound or DMSO in triplicate. At 3d pi, supernatants were collected and titrated by focus forming assay. IC90 values were determined by fitting data points to a nonlinear regression model using GraphPad Prism software. Viral load reduction (VLR) was calculated as VLR = log10(average(DMSO values))−log10 log10(average(30μM values)).
Virus strains
DENV1 (strain TH-Sman), DENV2 (New Guinea C), DENV3 (H87), and DENV4 (H241) were obtained from ATCC. West Nile virus (385–99) has been previously described (Xiao et al., 2001). Stocks were passaged one time on C6/36 cells, and purified stocks were produced by growth for 7 days on C6/36 cells. Supernatants were pelleted over a sorbitol cushion and resuspended in DMEM/0.1%FBS. Stock titers were determined by focus forming assay, as previously described (Shum et al., 2010).
Toxicity assays
HEK293 cells were incubated in cell media containing increasing concentrations of compounds in constant 1%DMSO (vol/vol). At 72 h, XTT reagent (ATCC: XTT Cell Proliferation Assay Kit; cat no 30–1011 K) was added, and absorbance was measured according to the manufacturer’s instructions. CC50 values were calculated using GraphPad Prism software.
Time-of-addition assay
HEK293 cells were infected with DENV2 at MOI = 3 ffu/cell in low volume with rocking for 1 h at 37 °C. Cells were washed and refed with regular growth media. At indicated times post-infection, media was removed and replaced with 1uM VGTI-A3 or DMSO diluted in media. Supernatants were collected at 48 h post-infection and quantitated by focus forming assay.
RNA replication assay
HEK293 cells were infected with DENV2 at MOI = 3 ffu/cell +1uM VGTI-A3–03 or DMSO control. At 24 h post-infection, RNA was isolated from supernatants and analyzed by RT-qPCR using a universal primer/probe set designed to amplify regions of the DENV genome within the 3’ untranslated region (UTR) (Gurukumar et al., 2009).
Reporter virus particle (RVP) production
The DENV replicon (pDENV2-reo-GZ) and structural (pDENV-CprME) plasmids were a kind gift of T. Pierson (NIH; (Ansarah-Sobrinho et al., 2008; Mukherjee et al., 2014). HEK293 cell lines stably maintaining pDENV2-reo-GZ were constructed by transfection and selection at 3d post-transfection with 300ug/ml zeocin. Clonal populations were isolated by limited dilution. To produce RVPs, DRep2A-293 cells were transfected with 6ug of pDENV-CprME in a 100 mm dish using Lipofectamine 3000 (Life Technologies), media was changed the next day, and supernatants were collected at 3d post-transfection. RVPs were pelleted at 30,000 rpm for 2 h over a 20% sorbitol cushion to purify and concentrate. Infectious units were quantified by TCID50 as previously described (Ansarah-Sobrinho et al., 2008). RVP treatments were carried out at MOI = 5 IU/cell, and number of GFP + cells/frame (6 independent frames) were counted at 3d post-infection using ImageJ software.
Resistance mutant generation
HEK293 cells were infected at MOI = 0.1 ffu/cell in the presence of 1uM VGTI-A3. Supernatants were collected at 3 days p.i. and passaged onto naïve HEK293 cells+ 1uM VGTI-A3. Virus collected at each passage was assayed for resistance to VGTI-A3 treatment and sequenced by Sanger sequencing. Results are reflective of more than 5 independent derivations of resistant viruses.
Infectious clone
The DENV2 cDNA infectious clone (16681 strain) was a kind gift of C. Huang (CDC Fort Collins, CO). Infectious clone-derived virus was produced as previously described (Huang et al., 2010). Briefly, plasmid was digested with XbaI to linearize, precipitated, then used as a template for capped in vitro transcription (ivt) using the mMessage mMachine T7 capped ivt kit (Ambion #AM1340). Ivt reactions were electroporated into 1 × 107 Vero cells. Media was changed the following day. At 7d post-transfection, supernatants were collected, cleared, then passaged on to naïve Vero cells at a 1:5 dilution for 7 days, before passage on to C6/36 cells. Supernatants were collected at 7d post-infection, cleared, and purified over a 20% sorbitol cushion. Susceptibility to VGTI-A3 was determined by infection at MOI = 0.1 ffu/cell +30 nM→→10μM and quantification of infectious virus at 3d pi by ffa. Introduction of mutations into the 16681 infectious clone or pDENV-CprME plasmids was carried out by site-directed mutagenesis using the QuikChange protocol with Phusion Taq mix (Thermofisher F530). Successful mutagenesis was confirmed by Sanger sequencing.
RVP resistance assay
RVPs were produced by transfection of pDENV-CprME (wild-type or mutated) into pDRep2rep-gz-HEK293 cells. At 24 h pt, media was removed and replaced with DMSO or 1uM VGTI-A3–03 in normal growth media. At 3d pt, supernatant were collected, cleared, and purified over a sorbitol cushion. Purified RVPs were used to infect naïve HEK293 cells, and GFP-positivity was assayed at 3d p.i. Susceptibility to VGTI-A3–03 was identified as > 50% reduction of RVPs produced in the presence of VGTI-A3–03 versus DMSO.
Sucrose gradients
Supernatants from infected HEK293 cells (+1uM VGTI-A3–03 or DMSO, 48 h) were purified over a sorbitol cushion, then spun over a discontinuous sucrose (15/35/50%) gradient (40,000 rpm, 4 h). 1 ml fractions were collected and infectious virus, genome levels, and envelope levels were determined by ffa, RT-qPCR, and western blot, respectively. Western blots were carried out using mouse anti-envelope (4G2 (Henchal et al., 1982);). The hybridoma for production of the pan-flavivirus anti-E antibody 4G2 was obtained from the ATCC. The hybridoma was maintained and antibody purified from culture supernatant by the Vaccine and Gene Therapy Institute monoclonal antibody core facility (OHSU).
LC-MS/MS Analysis of A3–03 in gradient fractions:
Frozen fractions were thawed and vortex mixed for 1 min then centrifuged for 1 min at 10,000 g. A portion of the supernatant was removed, placed in an autosampler vial and A3–03 content was analyzed in a 25 μL aliquot using a 4000 QTRAP hybrid/triple quadrupole linear ion trap mass spectrometer (SCIEX, Foster City, CA) with electrospray ionization (ESI) in positive mode. The mass spectrometer was interfaced to a Shimadzu (Columbia, MD) SIL-20AC XR auto-sampler followed by 2 LC-20AD XR LC pumps. HPLC separation consisted of a gradient mobile phase was delivered at a flow rate of 0.50 ml/min and consisted of two solvents, 10 mM ammonium formate in water (A) and 10 mM ammonium formate in 90% acetonitrile and 10% water (B). Initial concentration of B was 50% and increased to 95% B by 9 min held at 95% for 1 min, decreasing to 50% again over 0.5 min, followed by re-equilibration for 2.5 min. The column used was a Luna C8(2) 100 Å, 5u, 50 × 3 mm column maintained at 35 °C using a Shimadzu CTO-20AC column oven. The instrument was operated with the following settings: source voltage 5000 kV, GS1 50, GS2 50, CUR 20, TEM 600 and CAD gas Medium. A3–03 was detected using multiple reaction monitoring with the following transitions optimized by direct infusion of the pure compound. Each transition was monitored with a 50 ms dwell time: m/z = 484 → 282 and m/z = 484 → 308. Data were acquired using Analyst 1.6.2 software and the area of the A3 peak integrated using Multiquant 3.0.1 software. The area was used for the relative comparison of the content of A3 in each fraction.
Supplementary Material
Acknowledgements
Analytical support was provided by the Bioanalytical Shared Resource/Pharmacokinetics Core Facility that is part of the University Shared Resource Program at Oregon Health and Sciences University. This work was supported by the NIAID Pacific Northwest Regional Center of Excellence for Biodefense and Emerging Infectious Diseases Research (grant U54 AI 081680), the Antiviral Drug Discovery and Development Center (a NIAID Center of Excellence for Translational Research, grant U19 AI 109680), and 60° Pharmaceuticals, LLC.
Footnotes
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.antiviral.2018.04.019.
References
- Acosta EG, Bartenschlager R, 2016. The quest for host targets to combat dengue virus infections. Curr. Opin. Virol 20, 47–54. [DOI] [PubMed] [Google Scholar]
- Ansarah-Sobrinho C, Nelson S, Jost CA, Whitehead SS, Pierson TC, 2008. Temperature-dependent production of pseudo infectious dengue reporter virus particles by complementation. Virology 381, 67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, Drake JM, Brownstein JS, Hoen AG, Sankoh O, Myers MF, George DB, Jaenisch T, Wint GR, Simmons CP, Scott TW, Farrar JJ, Hay SI, 2013. The global distribution and burden of dengue. Nature 496, 504–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair WS, Pickford C, Irving SL, Brown DG, Anderson M, Bazin R, Cao J, Ciaramella G, Isaacson J, Jackson L, Hunt R, Kjerrstrom A, Nieman JA, Patick AK, Perros M, Scott AD, Whitby K, Wu H, Butler SL, 2010. HIV capsid is a tractable target for small molecule therapeutic intervention. PLoS Pathog 6, e1001220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges MC, Castro LA, Fonseca BA, 2013. Chloroquine use improves dengue-related symptoms. Memorias do Inst. Oswaldo Cruz 108, 596–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd CM, Dai D, Grosenbach DW, Berhanu A, Jones KF, Cardwell KB, Schneider C, Wineinger KA, Page JM, Harver C, Stavale E, Tyavanagimatt S, Stone MA, Bartenschlager R, Scaturro P, Hruby DE, Jordan R, 2013. A novel inhibitor of dengue virus replication that targets the capsid protein. Antimicrob. agents Chemother 57, 15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capeding MR, Tran NH, Hadinegoro SR, Ismail HI, Chotpitayasunondh T, Chua MN, Luong CQ, Rusmil K, Wirawan DN, Nallusamy R, Pitisuttithum P, Thisyakorn U, Yoon IK, van der Vliet D, Langevin E, Laot T, Hutagalung Y, Frago C, Boaz M, Wartel TA, Tornieporth NG, Saville M, Bouckenooghe A, Group CYDS, 2014. Clinical efficacy and safety of a novel tetravalent dengue vaccine in healthy children in Asia: a phase 3, randomised, observer-masked, placebo-controlled trial. Lancet 384, 1358–1365. [DOI] [PubMed] [Google Scholar]
- Carocci M, Hinshaw SM, Rodgers MA, Villareal VA, Burri DJ, Pilankatta R, Maharaj NP, Gack MU, Stavale EJ, Warfield KL, Yang PL, 2015. The bioactive lipid 4-hydroxyphenyl retinamide inhibits flavivirus replication. Antimicrob. agents Chemother 59, 85–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy MM, Zhang SL, Costa VV, Tan HC, Horrevorts S, Ooi EE, 2015. Proteasome inhibition suppresses dengue virus egress in antibody dependent infection. PLoS Negl. Trop. Dis 9, e0004058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courageot MP, Frenkiel MP, Dos Santos CD, Deubel V, Despres P, 2000. Alphaglucosidase inhibitors reduce dengue virus production by affecting the initial steps of virion morphogenesis in the endoplasmic reticulum. J. Virol 74, 564–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney W.E.t., Edwards R, Colledge D, Shaw T, Furman P, Painter G, Locarnini S, 2002. Phenylpropenamide derivatives AT-61 and AT-130 inhibit replication of wild-type and lamivudine-resistant strains of hepatitis B virus in vitro. Antimicrob. agents Chemother 46, 3057–3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deres K, Schroder CH, Paessens A, Goldmann S, Hacker HJ, Weber O, Kramer T, Niewohner U, Pleiss U, Stoltefuss J, Graef E, Koletzki D, Masantschek RN, Reimann A, Jaeger R, Gross R, Beckermann B, Schlemmer KH, Haebich D, Rubsamen-Waigmann H, 2003. Inhibition of hepatitis B virus replication by drug-induced depletion of nucleocapsids. Science 299, 893–896. [DOI] [PubMed] [Google Scholar]
- Fader LD, Bethell R, Bonneau P, Bos M, Bousquet Y, Cordingley MG, Coulombe R, Deroy P, Faucher AM, Gagnon A, Goudreau N, Grand-Maitre C, Guse I, Hucke O, Kawai SH, Lacoste JE, Landry S, Lemke CT, Malenfant E, Mason S, Morin S, O’Meara J, Simoneau B, Titolo S, Yoakim C, 2011. Discovery of a 1,5-dihydrobenzo[b][1,4]diazepine-2,4-dione series of inhibitors of HIV-1 capsid assembly. Bioorg. Med. Chem. Lett 21, 398–404. [DOI] [PubMed] [Google Scholar]
- Fischer MA, Smith JL, Shum D, Stein DA, Parkins C, Bhinder B, Radu C, Hirsch AJ, Djaballah H, Nelson JA, Fruh K, 2013. Flaviviruses are sensitive to inhibition of thymidine synthesis pathways. J. Virol 87, 9411–9419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser JE, Watanabe S, Wang C, Chan WK, Maher B, Lopez-Denman A, Hick C, Wagstaff KM, Mackenzie JM, Sexton PM, Vasudevan SG, Jans DA, 2014. A nuclear transport inhibitor that modulates the unfolded protein response and provides in vivo protection against lethal dengue virus infection. J. Infect. Dis 210, 1780–1791. [DOI] [PubMed] [Google Scholar]
- Gurukumar KR, Priyadarshini D, Patil JA, Bhagat A, Singh A, Shah PS, Cecilia D, 2009. Development of real time PCR for detection and quantitation of Dengue Viruses. Virol. J 6, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halasa YA, Shepard DS, Zeng W, 2012. Economic cost of dengue in Puerto Rico. Am. J. Trop. Med. Hyg 86, 745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henchal EA, Gentry MK, McCown JM, Brandt WE, 1982. Dengue virus-specific and flavivirus group determinants identified with monoclonal antibodies by indirect immunofluorescence. Am. J. Trop. Med. Hyg 31, 830–836. [DOI] [PubMed] [Google Scholar]
- Huang CY, Butrapet S, Moss KJ, Childers T, Erb SM, Calvert AE, Silengo SJ, Kinney RM, Blair CD, Roehrig JT, 2010. The dengue virus type 2 envelope protein fusion peptide is essential for membrane fusion. Virology 396, 305–315. [DOI] [PubMed] [Google Scholar]
- King RW, Ladner SK, Miller TJ, Zaifert K, Perni RB, Conway SC, Otto MJ, 1998. Inhibition of human hepatitis B virus replication by AT-61, a phenylpropenamide derivative, alone and in combination with (−)beta-L-2’,3’-dideoxy-3’-thiacytidine. Antimicrob. agents Chemother 42, 3179–3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim SP, Wang QY, Noble CG, Chen YL, Dong H, Zou B, Yokokawa F, Nilar S, Smith P, Beer D, Lescar J, Shi PY, 2013. Ten years of dengue drug discovery: progress and prospects. Antivir. Res 100, 500–519. [DOI] [PubMed] [Google Scholar]
- Lou Z, Sun Y, Rao Z, 2014. Current progress in antiviral strategies. Trends Pharmacol. Sci 35, 86–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low JG, Sung C, Wijaya L, Wei Y, Rathore AP, Watanabe S, Tan BH, Toh L, Chua LT, Hou Y, Chow A, Howe S, Chan WK, Tan KH, Chung JS, Cherng BP, Lye DC, Tambayah PA, Ng LC, Connolly J, Hibberd ML, Leo YS, Cheung YB, Ooi EE, Vasudevan SG, 2014. Efficacy and safety of celgosivir in patients with dengue fever (CELADEN): a phase 1b, randomised, double-blind, placebo-controlled, proof-of-concept trial. Lancet Infect. Dis 14, 706–715. [DOI] [PubMed] [Google Scholar]
- Ma L, Jones CT, Groesch TD, Kuhn RJ, Post CB, 2004. Solution structure of dengue virus capsid protein reveals another fold. Proc. Natl. Acad. Sci. U. S. A 101, 3414–3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateo R, Nagamine CM, Kirkegaard K, 2015. Suppression of drug resistance in dengue virus. mBio 6 e01960–01915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S, Pierson TC, Dowd KA, 2014. Pseudo-infectious reporter virus particles for measuring antibody-mediated neutralization and enhancement of dengue virus infection. Methods Mol. Biol 1138, 75–97. [DOI] [PubMed] [Google Scholar]
- Nag DK, Finley D, 2012. A small-molecule inhibitor of deubiquitinating enzyme USP14 inhibits Dengue virus replication. Virus Res 165, 103–106. [DOI] [PubMed] [Google Scholar]
- Rathore AP, Paradkar PN, Watanabe S, Tan KH, Sung C, Connolly JE, Low J, Ooi EE, Vasudevan SG, 2011. Celgosivir treatment misfolds dengue virus NS1 protein, induces cellular pro-survival genes and protects against lethal challenge mouse model. Antivir. Res 92, 453–460. [DOI] [PubMed] [Google Scholar]
- Scaturro P, Trist IM, Paul D, Kumar A, Acosta EG, Byrd CM, Jordan R, Brancale A, Bartenschlager R, 2014. Characterization of the mode of action of a potent dengue virus capsid inhibitor. J. Virol 88, 11540–11555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shum D, Smith JL, Hirsch AJ, Bhinder B, Radu C, Stein DA, Nelson JA, Fruh K, Djaballah H, 2010. High-content assay to identify inhibitors of dengue virus infection. Assay drug Dev. Technol 8, 553–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JL, Stein DA, Shum D, Fischer MA, Radu C, Bhinder B, Djaballah H, Nelson JA, Fruh K, Hirsch AJ, 2014. Inhibition of dengue virus replication by a class of small-molecule compounds that antagonize dopamine receptor d4 and downstream mitogen-activated protein kinase signaling. J. Virol 88, 5533–5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung C, Wei Y, Watanabe S, Lee HS, Khoo YM, Fan L, Rathore AP, Chan KW, Choy MM, Kamaraj US, Sessions OM, Aw P, de Sessions PF, Lee B, Connolly JE, Hibberd ML, Vijaykrishna D, Wijaya L, Ooi EE, Low JG, Vasudevan SG, 2016. Extended evaluation of virological, immunological and pharmacokinetic endpoints of CELADEN: a randomized, placebo-controlled trial of celgosivir in dengue fever patients. PLoS Negl. Trop. Dis 10, e0004851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay M, Bonneau P, Bousquet Y, DeRoy P, Duan J, Duplessis M, Gagnon A, Garneau M, Goudreau N, Guse I, Hucke O, Kawai SH, Lemke CT, Mason SW, Simoneau B, Surprenant S, Titolo S, Yoakim C, 2012. Inhibition of HIV-1 capsid assembly: optimization of the antiviral potency by site selective modifications at N1, C2 and C16 of a 5-(5-furan-2-yl-pyrazol-1-yl)-1H-benzimidazole scaffold. Bioorg. Med. Chem. Lett 22, 7512–7517. [DOI] [PubMed] [Google Scholar]
- Tricou V, Minh NN, Van TP, Lee SJ, Farrar J, Wills B, Tran HT, Simmons CP, 2010. A randomized controlled trial of chloroquine for the treatment of dengue in Vietnamese adults. PLoS Negl. Trop. Dis 4, e785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villar L, Dayan GH, Arredondo-Garcia JL, Rivera DM, Cunha R, Deseda C, Reynales H, Costa MS, Morales-Ramirez JO, Carrasquilla G, Rey LC, Dietze R, Luz K, Rivas E, Miranda Montoya MC, Cortes Supelano M, Zambrano B, Langevin E, Boaz M, Tornieporth N, Saville M, Noriega F, Group CYDS, 2015. Efficacy of a tetravalent dengue vaccine in children in Latin America. N. Engl. J. Med 372, 113–123. [DOI] [PubMed] [Google Scholar]
- Watanabe S, Chan KW, Dow G, Ooi EE, Low JG, Vasudevan SG, 2016. Optimizing celgosivir therapy in mouse models of dengue virus infection of serotypes 1 and 2: the search for a window for potential therapeutic efficacy. Antivir. Res 127, 10–19. [DOI] [PubMed] [Google Scholar]
- Watanabe S, Rathore AP, Sung C, Lu F, Khoo YM, Connolly J, Low J, Ooi EE, Lee HS, Vasudevan SG, 2012. Dose- and schedule-dependent protective efficacy of celgosivir in a lethal mouse model for dengue virus infection informs dosing regimen for a proof of concept clinical trial. Antivir. Res 96, 32–35. [DOI] [PubMed] [Google Scholar]
- Whitehorn J, Nguyen CV, Khanh LP, Kien DT, Quyen NT, Tran NT, Hang NT, Truong NT, Hue Tai LT, Cam Huong NT, Nhon VT, Van Tram T, Farrar J, Wolbers M, Simmons CP, Wills B, 2016. Lovastatin for the treatment of adult patients with dengue: a randomized, double-blind, placebo-controlled trial. Clin. Infect. Dis. Offi. Publ. Infect. Dis. Soc. Am 62, 468–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehorn J, Van Vinh Chau N, Truong NT, Tai LT, Van Hao N, Hien TT, Wolbers M, Merson L, Dung NT, Peeling R, Simmons C, Wills B, Farrar J, 2012. Lovastatin for adult patients with dengue: protocol for a randomised controlled trial. Trials 13, 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao SY, Guzman H, Zhang H, Travassos da Rosa AP, Tesh RB, 2001. West Nile virus infection in the golden hamster (Mesocricetus auratus): a model for West Nile encephalitis. Emerg. Infect. Dis 7, 714–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.