

Graphical abstract

Keywords: SARS CoV-2 main protease, monomer–dimer equilibrium, conformational stability, inhibitor binding, room-temperature X-ray crystallography
Abstract
We recently demonstrated that inhibitor binding reorganizes the oxyanion loop of a monomeric catalytic domain of SARS CoV-2 main protease (MPro) from an unwound (E) to a wound (active, E*) conformation, independent of dimerization. Here we assess the effect of the flanking N-terminal residues, to imitate the MPro precursor prior to its autoprocessing, on conformational equilibria rendering stability and inhibitor binding. Thermal denaturation (Tm) of C145A mutant, unlike H41A, increases by 6.8 °C, relative to wild-type mature dimer. An inactivating H41A mutation to maintain a miniprecursor containing TSAVL[Q or E] of the flanking nsp4 sequence in an intact form [(-6)MProH41A and (-6*)MProH41A, respectively], and its corresponding mature MProH41A were systematically examined. While the H41A mutation exerts negligible effect on Tm and dimer dissociation constant (Kdimer) of MProH41A, relative to the wild type MPro, both miniprecursors show a 4–5 °C decrease in Tm and > 85-fold increase in Kdimer as compared to MProH41A. The Kd for the binding of the covalent inhibitor GC373 to (-6*)MProH41A increases ∼12-fold, relative to MProH41A, concomitant with its dimerization. While the inhibitor-free dimer exhibits a state in transit from E to E* with a conformational asymmetry of the protomers’ oxyanion loops and helical domains, inhibitor binding restores the asymmetry to mature-like oxyanion loop conformations (E*) but not of the helical domains. Disorder of the terminal residues 1–2 and 302–306 observed in both structures suggest that N-terminal autoprocessing is tightly coupled to the E-E* equilibrium and stable dimer formation.
Introduction
SARS CoV-2, a positive-sense single stranded RNA virus, encodes its proteins for its maturation and propagation in at least 12 open reading frames.1, 2 Two-thirds of the ∼30 kb genome is translated as 2 large polyproteins, pp1a and pp1ab.3, 4, 5 Polyproteins pp1a and pp1ab span the non-structural proteins (nsp) nsp1-nsp10 and nsp1-nsp16, respectively.3 A single copy of the main protease (MPro) is synthesized as part (nsp5) of these polyproteins proximal to membrane spanning segments within nsp4 and nsp6.6 It catalyzes its own release (termed autoprocessing) promoting cleavages at its termini and the various sites in the polyproteins between nsp6 and nsp16.7, 8 Together with a papain-like protease, encoded within nsp3, which cleaves the polyproteins to release nsp1-nsp3, the mature nsps assemble to form the replication/transcription complex essential to produce the mature progeny virus.3
Mature MPro, also known as 3C-like protease, is a homodimeric cysteine protease.7 Each protomer (306 amino acids) comprises 3 domains. Domains 1 (residues 8–101) and II (residues 102–184) together form the catalytic region exhibiting a chymotrypsin-like fold. This region is connected to domain III (residues 201–306), which encompasses 5 helices, through a long loop region (residues 185–200).6, 9 Each protomer includes the non-canonical active site dyad, C145 and H41 residues, with a characteristic oxyanion loop conformation.6 The active sites of the dimer are catalytically equivalent.10
Since the emergence of the SARS CoV-2 pandemic, MPro has been a renewed target for developing potent oral inhibitors for the treatment of COVID-19.11, 12 Developing such inhibitors is even more urgent considering emerging variants that escape the current vaccine treatments. The chemical structure and specificity of inhibitors mimic the substrate making critical contacts with the binding pockets of the enzyme. They include various chemically reactive groups such as a carbonyl or nitrile, which form a covalent bond with the active site C145 residue.13 Known inhibitors include nirmatrelvir, which is currently in use for the treatment of COVID-19 under an emergency use authorization by the FDA,14 and the recently described covalent and non-covalent inhibitors.13, 15, 16, 17, 18, 19, 20, 21 It is noteworthy that the majority of these inhibitors are designed to act on the mature MPro and only limited knowledge exists regarding the efficacy of binding of such inhibitors to the active site of MPro precursor prior to its maturation at its termini or to the monomeric form of mature MPro in vivo.22, 23
In vitro experiments to determine the order of cleavage at the termini of MPro require intact model precursors containing the nsp4 and nsp6 sequences flanking the MPro. However, lack of accumulation of such precursors due to their rapid autoprocessing of MPro upon expression in E. coli precludes their accumulation and isolation.8, 10 Insolubility of the constructs also limits such endeavors (our unpublished results). A precise mechanism by which an MPro protomer or a dimer catalyzes cleavages at its termini, either through an intra- or intermolecular mechanism, is not fully understood.24, 25, 26 Moreover, few such studies pertain only to the previous SARS CoV isolate and not to the recent SARS CoV-2 MPro.22, 23 A recent study aiming to address the conformation of the flanking C-terminal residues of nsp4 (SAVLQ) appended to MProC145S resulted in observing only the cleaved product SAVLQ bound to the dimeric MProC145S in the crystal structure.27 Structures of the dimeric MPro bearing a C145A mutation displaying the C-terminal residues of one dimer bound to the active site of a second dimer have led to the proposal that C-terminal autoprocessing occurs via an intermolecular process.27, 28, 29
From a handful of previous studies of SARS CoV MPro, a consensus emerges suggesting that cleavage at the N-terminus precedes the C-terminal cleavage and that the N-terminal cleavage is accompanied by the conformational reorganization of 1) the active site oxyanion loop and 2) the interface promoted by the free N-terminal residues through intra- and inter-protomer contacts with domains II and III in conjunction with a distinctive reorientation of the latter.8, 23, 27 These rearrangements lead to a significant decrease in the dimer dissociation constant and appearance of mature-like catalytic activity.8, 10 Thus, mutational, kinetics and structural studies have focused mostly on the previous SARS CoV isolate examining the relationship of catalytic activity to conformational rearrangements/dimerization of the mature MPro and not of the MPro precursor.22, 23
It is evident from various investigations that substrate or inhibitor binding reorganizes the active site oxyanion loop from an inactive (E) to an active (E*) state that is typical of the catalytically active conformation of the mature dimeric MPro.10, 23, 30 This transition is concomitant with the monomer–dimer equilibrium shifting to the dimer form. Furthermore, by using a construct that is exclusively monomeric consisting of only the catalytic domain and loop region (residues 1–199, MPro1-199), we recently showed that inhibitor binding transitions the E state to E*I state independent of dimerization.30 Consistent with this observation, a miniprecursor of MPro1-199 containing the N-terminal flanking region undergoes autoprocessing suggesting that precursor processing may also be governed by the binding of the N-terminal cleavage site sequence modulating the E-E* equilibrium.30 The above result is in accordance with earlier observations showing that monomeric model precursors, in which the N-terminal flanking residues of nsp4 are appended to the full-length MPro E290A/R298A mutant, also undergo N-terminal autoprocessing, albeit at a slower rate than the wild-type.8, 10, 30
It is evident from the above observations that biochemical and structural studies of model precursors of MPro are feasible only through either mutating the active site or cleavage site residues to permit their accumulation and isolation for in vitro studies. Here we report the results of a systematic study towards understanding the effect of active site mutations and the presence of N-terminal flanking region sequence, to mimic an MPro precursor prior to the N-terminal cleavage, on the dimer dissociation constant, conformational stability, and thermodynamics of inhibitor binding. As monomers did not yield crystals and crystal growth was selective only for dimers, two dimeric structures of (-6*)MProH41A, with and without GC373 (the reactive aldehyde form of GC376),10 are described and compared with the corresponding mature MPro structures.
Results
Active site mutations and the N-terminal flanking sequence TSAVLQ alter the conformational stability and dimer dissociation constant of MPro
Differential scanning fluorimetry (DSF) was utilized to assess the effect of active site mutations on the overall conformational stability of MPro. Proteins were subjected to DSF at 10 µM in buffer B (pH 7) containing 0.1 % DMSO. Tm values along with the estimated dimer dissociation constants (Kdimer) of the various constructs are listed in Table 1 (Figures 1 , 2 and S2). MProC145A and MProH41A are mainly dimeric at 10 µM (25 °C) based on the apparent Kdimer by SV-AUC and SEC-MALS analyses resembling that of MProWT (Figure 1(A), (B) and (D)).6, 10 The mid-point (Tm) for the two-state unfolding transition of MProWT is 53.9 °C. This Tm is consistent with the value reported by differential scanning calorimetry (DSC) for SARS CoV MProWT, which is 96 % identical in sequence to SARS CoV-2 MProWT.31
Table 1.
Thermal denaturation (Tm) and relationship to Kdimer of various MPro constructs in the absence and presence of GC373.
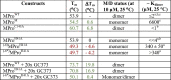 |
Tm was determined by DSF at a concentration of 10 µM in buffer B. M/D denotes monomer/dimer. a estimated by SV-AUC. b monitored by SEC-MALS. c cited from references.6, 10 DSF scans are shown in Figure 2. Green and red lettering denote the increase and decrease, respectively, of Tm and ΔTm, relative to the corresponding mature MPro construct.
Figure 1.

SEC-MALS and SV-AUC analyses of MProWT and its analogues. (A-C) An injection volume of 125 µl (ranging from 7.5 to 60 µM) on Superose-12 column (1 × 30 cm) in buffer A. Calculated mass of the constructs are shown in Figure S1. Eluting peak apex concentration and mass (Da) are indicated. (C) Analysis of (-6*)MProH41A (60 µM) in the absence (black trace) and presence (red trace) of GC373 at 5x the concentration of protein. (D-F) Observed absorbance distributions by SV-AUC. Estimated Kdimer are listed in Table 1. M, D denote the monomer and dimer, respectively, and M/D the equilibrium boundary. The estimated Kdimer for (-6*)MProH41A in the presence of 2-fold molar excess of GC373 is 38 ± 5 µM (Figure S3).
Figure 2.

DSF profiles of MProWT and its active site mutants and miniprecursor. Unfolding of the proteins as a function of temperature was carried out in buffer B. The gray dashed line indicates the midpoint for the transition for MProWT (black trace) and serves as a reference to compare the rest of the constructs. Some traces are repeated for ease of comparison. Estimated Tm’s are shown in parentheses beside the construct designation and in Table 1.
Circular dichroism (CD) profiles of dimeric MProWT, MProH41A and MProC145A at 10 µM concentration in buffer B at 25 °C are similar (Figure 3(A)). Surprisingly, the Tm for MProC145A is 6.8 °C higher than that observed for MProH41A, which displays a nearly identical Tm as that of MProWT (Table 1 and Figure 2). Consistent with the DSF data, thermal denaturation CD profiles also return a Tm difference (ΔTm) of 6.8 °C between MProWT (or MProH41A) and MProC145A (Figure 3 (B)). To ascertain, if this two-state behavior is exclusive to a dimer form as well as evaluate the contribution of the dimer interface to the overall conformational stability, a previously described construct, MProM was analyzed. MProM is predominantly monomeric, with a Kdimer of 6600 µM,10 because of two critical interface mutations E290A and R298A, leading to MPro dimer dissociation.10 The observed Tm of MProM is almost the same (Table 1 and Figure 2(A)) as MProWT. This suggests that the Tm signifies the unfolding transition of the monomer fold of MProWT and MProH41A. In this context, it is worth noting that the secondary structure CD profiles as well as the tertiary fold of monomer and dimer forms are similar.8, 10, 32, 33
Figure 3.

Comparison of the CD profiles of MProWT and its active site mutants. CD spectra acquired using 10 µM protein in buffer B at 25 °C (A) and at 222 nm as a function of increasing temperature (B). Values in parentheses indicate Tm in °C.
As H41A mutation does not alter the Tm or the Kdimer, relative to the wild type, but is expected to inactivate the enzyme, MProH41A was chosen to engineer a construct containing 6 amino acids of the flanking nsp4 sequence TSAVLQ to mimic a miniprecursor of MPro prior to its cleavage at its N-terminus, termed (-6)MProH41A (Figure S1). Appending these residues decreases the Tm by 4.6 °C and increases the Kdimer drastically by > 85-fold (Table 1 and Figure 1(E) and S2). However, when subjecting this construct to prolonged incubation at ∼300 µM for > 2 weeks at ambient temperature, it exhibits very slow cleavage at the nsp4/nsp5 junction. A product corresponding to mature MProH41A was identified by mass spectrometry indicative of this cleavage at the nsp4/nsp5 junction. The crystal structure of MproWT determined by neutron diffraction shows that both the side chains of C145 and H41 are ionized and form an ion pair.34 The substitution of the basic H41 residue could result in a change in the ionization state of the sulfur atom of C145 from thiolate to thiol which is a less effective nucleophile and thus, accounts for the observed low catalytic activity. To further limit autoprocessing, the conserved Q residue in the P1 position of the nsp4/nsp5 junction was substituted to E26, 35 (denoted by an asterisk), the resulting construct was termed (-6*)MProH41A. The Tm of (-6*)MProH41A is about the same as (-6)MProH41A, but the Kdimer is predicted to be much higher than 340 µM as no dimer was detectable by SV-AUC at 40 µM (compare Figure 1(E) and (F)). There is evidence for very weak self-association at 80 µM, unlike (-6)MProH41A, which clearly exhibits a species indicative of a monomer/dimer equilibrium boundary at a similar concentration of 90 µM and a Kdimer of 340 ± 50 µM (Figure 1(E) and S2). Thus, the influence of the presence of the N-terminal flanking sequence is reflected in a decreased Tm of 4–5 °C for (-6)MProH41A and (-6*)MProH41A. Of significance is the observation that the P1 Q to E mutation results in an even larger increase in Kdimer. As expected, no N-terminal autoprocessing of (-6*)MProH41A was observed after prolonged periods of incubation at high concentrations enabling the determination of crystal structures described below.
GC373 exhibits weaker binding to the miniprecursor mimetic relative to its mature counterpart
The reversible covalent inhibitor GC373 binds to MProWT with a dissociation constant (Kd) of 0.15 µM (Table 2 ).10, 21 This Kd equates to a Tm of 70 °C with twofold (2x) molar excess of GC373, matching the same protein to GC373 ratio at the completion of the ITC experiment, and a Tm increase (ΔTm) of 16.1 °C relative to the inhibitor-free MProWT (Table 1). This suggests that inhibitor binding with high affinity enhances the conformational stability of the dimer. It is well-known that increased thermal stability correlates with inhibitor binding affinity.36, 37 A gradual increase in Tm is observed with increasing GC373 concentration (Figure 2(C)), and at 20x GC373, ΔTm increases to 19.8 °C (Table 1). At 50 µM MProC145A, no thermal response was observed when titrating with GC373 indicative of its weak binding. Even though, H41 lacks close contacts with GC373, the Kd for the binding of GC373 to MProH41A increases by ∼9-fold (Kd = 1.4 µM, Table 2 and Figure 4 ) due to the lower reactivity of the protonated sulfur of C145 compared to the unprotonated of the wild type. This relates to a Tm of 70.8 °C, which is a difference in Tm of 2.9 °C and 16.9 °C compared to MProWT + 20x GC373 and inhibitor-free MProH41A, respectively.
Table 2.
Estimated thermodynamic parameters of GC373 binding to MProWT, its active site H41A mutant and miniprecursor as determined by ITC.
| Construct | N | Kd = Ki (µM) | ΔH (kcal/mol) | ΔS (cal/mol/K) | ΔG (kcal/mol) |
|---|---|---|---|---|---|
| MProWT | 0.99 ± 0.01 | 0.15 ± 0.03 | −6.7 ± 0.1 | 9.1 | −9.4 |
| MProC145A | no response | ||||
| MProH41A | 0.97 ± 0.03 | 1.4 ± 0.5 | −5.6 ± 0.2 | 8.3 | −8.1 |
| (-6*)MProH41A | 0.94 ± 0.04 | 16.7 ± 4.2 | −0.8 ± 0.06 | 19.2 | −6.6 |
Titrations were performed in buffer C with 30–50 µM protein in the cell and inhibitor in the syringe at 10 times the concentration of the protein. As no thermal response was observed for (-6*)MProH41A at 50 µM, as was also the case with MProC145A, the concentration of (-6*)MProH41A was increased to 150 µM protein and 1.5 mM GC373 to attain meaningful thermal response and thermodynamic parameters. MProWT data is listed here from reference10 for ease of comparison.
Figure 4.

Binding isotherm of GC373 to MProH41A and its miniprecursor. Titrations were carried out in buffer C at 28 °C. The thermodynamic parameters are listed in Table 2.
In contrast to the binding of GC373 to the MProH41A dimer, even when the Kd increases by 9-fold with only a small decrease in Tm (ΔTm) of ∼3 °C relative to MProWT + 20x GC373, the binding of GC373 to (-6*)MProH41A results only in a very small Tm increase of 0.4 °C as compared to inhibitor-free (-6*)MProH41A. This result parallels an even weaker binding of GC373 to (-6*)MProH41A. The Kd for GC373 binding to (-6*)MProH41A is ∼12 times larger than MProH41A (Table 2 and Figure 4). Notably, addition of GC373 promotes dimerization of (-6*)MProH41A with an ∼50:50 distribution of dimer to monomer population when 60 µM (-6*)MProH41A was mixed and incubated for 60 min with GC373 at 5-times the concentration of protein prior to SEC-MALS as shown in Figure 1C. The estimated Kdimer for (-6*)MProH41A with GC373 is 38 ± 5 µM (Figure S3). In the absence of GC373, (-6*)MProH41A is mainly monomeric with a Kdimer much higher than 340 µM. Thus, the single binding isotherm observed for the titration of GC373 to (-6*)MProH41A reflects the two processes of binding concomitant with dimerization, evident from the slow thermal response (Figure 4) and the large increase in entropy (ΔS) which offsets the decrease in ΔH (Table 2). Here the ΔH likely signifies the weaker interaction of GC373 with the active site and ΔS, the conformational and hydration changes associated with dimer formation. Of these two processes, namely inhibitor binding and shift in the M-D equilibrium to the dimer, P1-Q to E mutation largely accounts for the increase in Kdimer, which indirectly suggests that P1 Q plays a role in enhancing dimer stability. We have recently shown that the active site oxyanion loop exists mainly in the unwound (inactive, E) state in the monomeric form and that it switches to the wound/mature-like (active, E*) state upon inhibitor binding, the latter resembling the loop conformation of the mature dimer. The E-E* loop equilibrium being mainly in the unwound E state, as expected to occur in the monomeric (-6*)MProH41A miniprecursor, is likely to account for the increased Kd (∼12-fold) of GC373 as compared to its mature counterpart, MProH41A.
Revelations of the minor distorted and asymmetric conformations of the MPro miniprecursor dimer in the absence and presence of GC373
Because (-6*)MProH41A is monomeric in solution up to a concentration of 340 μM (∼11 mg/mL), we were interested in obtaining its crystal structure in the inhibitor-free form. The room-temperature structure was obtained at 1.90 Å resolution. (-6*)MProH41A crystallizes in the P21 space group. Unexpectedly, we found a homodimer instead of a monomer in the asymmetric unit, with the dimer’s quaternary structure resembling that of the inhibitor-free mature MProWT dimer (Figure 5 (A)). We modeled residues 3–301 in (-6*)MProH41A structure, whereas the rest of the N-terminal and C-terminal residues are disordered and are not visible in the electron density map.
Figure 5.

Room-temperature structure of (-6*)MProH41A and comparison with MProWT. (A) Cartoon representation of the homodimeric (-6*)MProH41A. Catalytic C145 and the site of H41A mutation are shown as spheres. Helical and catalytic domains are colored in light violet and light orange, respectively. (B and C) Superposition of (-6*)MProH41A (colored like in A) and MProWT (light grey). Movement of the helical domain of protomer B in (-6*)MProH41A is shown in panel C by the increased distances between corresponding Ala285 residues.
To evaluate how similar the inhibitor-free (-6*)MProH41A and MProWT dimers are, we superimposed each protomer molecule of (-6*)MProH41A with the monomer of MProWT (PDB ID 7JUN,.34 Of note, MProWT crystallizes with one protomer in the asymmetric unit, and the homodimer is created through a crystallographic twofold symmetry axis. In the MProWT dimer, the protomers A and B have identical conformations. The root mean square deviations (RMSDs) on the main-chain atoms are 0.53 Å and 1.34 Å, respectively, when protomers A and B of (-6*)MProH41A are each aligned with an MProWT protomer. This result demonstrates the structural similarity of protomer A but points to a significant difference between the tertiary structures of protomer B in (-6*)MProH41A compared to MProWT. Notably, it also signifies asymmetry in the quaternary structure of the (-6*)MProH41A dimer. This is demonstrated in Figure 5(B), which shows the superposition of inhibitor-free (-6*)MProH41A and MProWT, in which protomer A of the miniprecursor was aligned with one of the protomers of the wild-type mature enzyme. There is a major conformational shift of the helical domain of protomer B in (-6*)MProH41A, where some helices move by 5–7 Å from their positions in the native MProWT structure. As a whole, the helical domain of protomer B moves away from the corresponding domain of protomer A by ∼2 Å adopting a more open conformation relative to MProWT (Figure 5(C)). At the helical domains interface the closest residues Ala285 are separated by 7.3 Å in (-6*)MProH41A versus 5.2 Å in MProWT. A similar opening of the helical domains interface was previously observed in a MPro construct with non-native Gly-Ala-Met residues fused to the N-terminal Ser1, termed IMT MPro (MProIMT hereafter, PDB ID 7KFI).27 In MProIMT, however, the helical domains move away from each other symmetrically, making the distance between the interfacing Ala285 residues of about 10 Å. Therefore, (-6*)MProH41A conformation can be considered ‘semi-open’, whereas that found in MProIMT can be viewed as ‘open’. In addition, the presence of non-native residues at the N-terminus of MProIMT results in major structural alterations in the oxyanion hole and subsites S1, S2 and S4 caused by the cascade effect to avoid steric clashes by the N-terminal residues with Phe140, Glu166 and Pro168. In contrast, there is no substantial reshaping of the oxyanion loop or the substrate-binding subsites in (-6*)MProH41A protomer A (Figure 6 A and (B)). A significant change, however, occurs for the conformation of Glu166 in protomer B of (-6*)MProH41A where it flips down to form a new hydrogen bond of 3.1 Å with His163, which is absent in MProWT. The Glu166 movement attracts the Leu141 side chain that rotates closer to Glu166, making hydrophobic interactions, leading to a somewhat distorted oxyanion loop geometry and apparent closure of the S1 subsite. A similar conformational change of Glu166 side chain was observed in the recently described structure of MPro1-199 which lacks the helical domain.30 But, in MPro1-199, the oxyanion loop unwinding and reshaping into a 310-helix resulted in Glu166 hydrogen bonding with the main chain amide of Gly143. Due to the formation of the Asp166…His163 hydrogen bond in (-6*)MProH41A we can speculate that His163 becomes positively charged (i.e., doubly protonated) to neutralize the negative charge on the Glu166 carboxylate, whereas this histidine was found neutral (i.e., singly protonated) in MProWT.34 The considerable conformational plasticity of the MPro helical domain was also previously noted in the crystal structures of the fully monomeric enzyme containing single mutations G11A, S139A, and R298A.32, 33, 38 However, in these structures the substantial geometry change of the helical domain may be partially or solely due to the monomeric nature of the mutant enzymes crystallographic studies, which were carried out under cryogenic conditions.
Figure 6.

A close-up view of the (-6*)MProH41A homodimer active sites. Superposition of (-6*)MProH41A (colored like in Figure 3, panel A) and MProWT (light grey) showing the close-up view of the active sites in protomer A (panel A) and protomer B (panel B). Changes in the positions of the oxyanion loop residues and of Glu166 are shown by the curved black arrows. A new hydrogen bond formed between Glu166 and His163 in protomer B of (-6*)MProH41A is indicated by a blue dotted line with a measured distance in panel B.
To determine whether inhibitor binding to the miniprecursor enzyme restores its quaternary structure to the native state we obtained a room-temperature X-ray crystal structure of (-6*)MProH41A in complex with covalent inhibitor GC373 at 1.80 Å resolution. (-6*)MProH41A-GC373 crystallizes with the isomorphous unit cell parameters relative to the inhibitor-free (-6*)MProH41A, indicating that their quaternary structures are similar. Indeed, superimpositions of the corresponding protomers in the inhibitor-free and GC373-bound structures result in RMSDs of 0.23 Å for protomers A and 0.27 Å for protomers B. GC373 is found in the active sites of both protomers in (-6*)MProH41A-GC373 (Figure 7 (A)). GC373 binding, however, has no effect on the protomer B tertiary structure that maintains the semi-open conformation of its helical domain observed in the inhibitor-free (-6*)MProH41A structure. Conversely, the presence of GC373 in the protomer B active site fully restores the geometry of the oxyanion loop and the Glu166 orientation. Thus, covalent inhibitor binding reverts the oxyanion loop and S1 subsite geometries to their active configurations. GC373 binding to (-6*)MProH41A is nearly identical to its binding to MProWT; the inhibitor makes similar hydrogen bonding interactions with the active sites even when H41A is present (Figure 7(B), (C), and S4). Hence, the structural analysis demonstrates that covalent binding of GC373 leads to conformational changes in the oxyanion loop and the S1 subsite to their mature-like geometries but has no impact on the overall tertiary and quaternary structures of the enzyme dimer that maintains the semi-open conformation of the helical domains found in the inhibitor-free precursor (-6*)MProH41A.
Figure 7.

Binding of GC373 to (-6*)MProH41A. (A) Electron density of GC373 covalently bonded to Cys145 is shown at 1.5σ level (protomer A). (B, C) Hydrogen bonds formed between GC373 and the active site residues in protomer A (panel B) and protomer B (panel C) of (-6*)MProH41A. Distances are in Å.
Discussion
In this study, we partially succeeded in our attempt to characterize a miniprecursor MPro mimetic prior to the cleavage at its N-terminus. The miniprecursor consisted of 6 amino acids (-6) of the flanking nsp4 sequence appended to the N-terminus of MPro. An effective approach to retain the N-terminal flanking sequence for biochemical and structural analyses requires inactivating the enzyme by mutating either one of the catalytic dyad residues, H41 or C145. As the thermal denaturation profile is different for MProC145A, contrary to MProH41A, which closely resembles the wild type, H41A mutation was the logical choice for creating the miniprecursor mimetic (-6)MProH41A. However, as H41A mutation did not completely abolish catalytic activity, which resulted in N-terminal autoprocessing during prolonged periods at high concentrations of (-6)MProH41A, a second mutation was introduced. The second miniprecursor construct, (-6*)MProH41A, bearing the substitution mutation P1-Q to E of the residue flanking the N-terminus of MProH41A and is devoid of cleavage, was systematically analyzed. Clearly, the lower stability of the miniprecursor also points to the importance of the N-terminal cleavage and the requirement of the free N-terminal residues, which form the interface for stable dimer formation. The Tm changes from 49.3-49.7 °C for the miniprecursors to 53.9 °C for MProH41A. This Tm difference (ΔTm) of 4-5 °C correlates with the total interface forming upon dimerization. Of note, the enhanced thermal stability observed for MProC145A raises the question if this mutant is indeed an ideal surrogate for biochemical studies. Further experiments are needed to explain if the C145A mutation exerts an effect on global stability by perturbing the oxyanion loop equilibrium which is coupled to dimer formation.
The low Kd of MProWT with GC373 of 0.15 µM is mainly a contribution of the hemithioacetal formation between the inhibitor’s aldehyde carbon and the sulfur of C145. Even though there are no contacts < 3.5 Å between the H41 residue and GC373 in MProWT-GC373 complex (PDB ID 7UKK),30 H41A mutation leads to a ∼9-fold increase in Kd for GC373 binding to MProH41A probably due to the difference in the chemical reactivity between thiolate anion and thiol. This trend in Kd is reflected by increases in Tm for MProWT-GC373 (∼20 °C) and MProH41A-GC373 (∼17 °C) complexes. This difference is even more when comparing mature MProH41A-GC373 and miniprecursor (-6*)MProH41A-GC373 complexes such that a 12-fold weaker binding of GC373 to the miniprecursor accounts for a ΔTm of ∼21 °C. In accordance with the lack of thermal response by ITC, indicative of very weak binding, no increase in Tm was observed for MProC145A in the presence of 20-fold molar excess GC373, relative to inhibitor-free MProC145A (Figure 2(A)), again indicating the importance of the reactive group in the inhibitor, which forms a covalent bond with the active site C145 leading to enhanced affinity.
Catalytic activity is governed by an equilibrium in which the oxyanion loop transitions from an unwound state (E), as observed in the monomer, to a wound mature-like state (E*), which is accompanied with interface conformational rearrangements leading to dimer stabilization. The conformational stability assessed by DSF and SEC-MALS indicates that both the N-terminal flanking sequence and the P1-Q to E mutation exert an additive effect by decreasing the Tm by 4–5 °C and increasing the Kdimer by ≫ 85-fold. Notably, just the P1-Q to E mutation contributes to a further increase in Kdimer to > 340 µM and Kd by 12-fold as compared to (-6)MProH41A suggesting that prior to N-terminal processing, P1-Q influences the M-D equilibrium by reshaping (induced fit) the oxyanion loop to an E* state. However, addition of the inhibitor GC373 restores dimerization clearly indicating that E to E*I transition is coupled to dimerization, as shown recently for a full-length MProM 10 and a dissected catalytic domain of MPro, MPro1-199.30 Notably, the large increase in Tm is observed only for the dimeric protein in which the oxyanion loop conformation is mainly in the E* state and readily available for binding. Thus, an inhibitor that is designed to bind to the unwound E conformation selectively and prevent its switching to the E* state may restrict autoprocessing itself. In this context, knowing that inhibitors promote dimer formation and enhance catalytic activity through an E-E* transition in vitro, warrants an understanding of how such inhibitors may alter MPro autoprocessing and the virus maturation itself in space and time in vivo.
Even though the Kdimer of (-6*)MProH41A is > 340 µM in the absence of GC373, under the conditions of crystal growth at ∼9 mg/ml (∼260 µM), selective crystallization of only the dimer population was observed even when the majority of the protein is monomeric. Therefore, failing to attain a monomeric structure of (-6*)MProH41A, only the dimers of inhibitor-free (-6*)MProH41A and GC373 bound (-6*)MProH41A complex could be compared. As seen in both structures, disorder of the terminal residues may account for the lower conformational stability, which again emphasizes the importance of the N-terminal cleavage for stable dimer formation. Unlike MProWT, (-6*)MProH41A dimer is asymmetrical, one major difference arising from a significant movement of the helical domain of protomer B away from the corresponding helical domain of protomer A, thus, defining a semi-open quaternary state. Also, of note is a unique downward flip of the E166 residue in protomer B which plays a critical role in substrate binding.39 This movement and associated changes may relate to the transitioning of the oxyanion loop from the E to E* state and could partly be attributed to the much weaker binding of GC373 to (-6*)MProH41A, relative to its mature counterpart. Accordingly, (-6*)MProH41A-GC373 complex shows a completely restored oxyanion loop conformation, symmetric relative to each other, except for the helical domain, which stays in a semi-open state pointing to the requirement of the N-terminal cleavage to restore the symmetry for this region through interactions of S1 and G2 residues with subsite S1 of the opposite protomer as seen for mature MProWT. In (-6*)MProH41A and its complex with GC373, the N-terminal flanking residues S1 and G2 are disordered and not visible in the electron density maps. A closer examination reveals lack of one of the two hydrogen bonds between R4 of one protomer and E290 of the other, as seen in the mature wild type MPro,34 which may partly account for the increased Kdimer and semi-open conformation in the precursor form of MPro prior to its N-terminal maturation.
In conclusion, being a large multidomain dimeric protein, the appearance of catalytic activity of MPro is governed collectively by several equilibria and conformational changes during its conversion from a polyprotein to a mature fully active dimer as illustrated in Figure 8 . Our results suggest that inhibitors designed to the mature active site oxyanion loop (E*) will bind weakly to monomeric MPro and its miniprecursor, thus, providing an insight that the E state of monomeric MPro could be a strategic target for inhibitor design to effectively restrict MPro maturation from its polyprotein precursor.
Figure 8.

Conformational equilibria for MPro maturation and inhibition. The three domains of MPro are designated with roman numerals and distinct colors. Black wavy line represents the 6 nsp4 residues flanking the N-terminus of MPro. The inactive oxyanion loop (E, unwound yellow) existing in equilibrium with active E* state (wound black) is shown as ribbons. Open and closed red boxes also denote the E and E* states, respectively. Blue and red arrows signify the change in Kd and Tm, respectively (Table 1, Table 2).
Materials and Methods
Construction and designation of MPro constructs
The expression and purification of MProWT (GenBank ID: MN908947.3) were carried out as described.10, 13 Except for MProWT, the rest of the constructs were expressed without a fusion partner flanking the N-terminus of MPro. All constructs were synthesized and cloned into pJ414 vector (ATUM, Newark, CA). (-6)MProH41A consists of 6 amino acids of the flanking nsp4 sequence appended to the active site mutant, MProH41A. To restrict residual autoprocessing for prolonged periods, as required for crystallization, another variant of the (-6)MProH41A was engineered, which contains a substitution mutation of residue Q in the P1 position (denoted with an asterisk) of the nsp4/nsp5 cleavage site to E and termed (-6*)MProH41A. All constructs contain a GP-6His tag at the C-terminus of MPro to facilitate initial purification followed by removal of GP-6His via human rhinovirus 3C (HRV-3C) protease cleavage. Amino acid sequence and designations of all MPro constructs used in this study are listed in Figure S1.
Expression and purification
Plasmids were transformed into BL21-DE3 cells (Agilent) and induced for expression at 0.7–0.8 optical density with 1 mM isopropyl β-d-1-thiogalactopyranoside typically for 3 hrs. Proteins were purified from the cell lysate by nickel-affinity chromatography (NAC, step 1). The bound fraction was subjected to isocratic fractionation on Superose-12 column (step 2, Cytiva Life Sciences) and HRV-3C protease cleavage (step 3, purchased from Sigma-Aldrich) overnight at 4 °C followed by repeating NAC and step 2 in a final buffer of 25 mM Tris-HCl, pH 7 or 7.6, 150 mM NaCl and 1 mM TCEP (buffer A). The full-length wild type (MProWT) was expressed and purified similar in strategy to that described previously6 except for substituting the fusion partner GST with maltose binding protein (MBP) followed by a 36 amino acid spacer sequence corresponding to the immunoglobulin binding domain B1 of protein G (ΔGB1).10, 13, 21 Peak fractions were pooled and concentrated to the desired concentration using Amicon Ultra – 15 or 0.5 ml centrifugal filters (Merck Millipore ltd.) and stored in aliquots at −30 °C and for long term storage at −80 °C. Purity was verified both by SDS-PAGE on 4–20 % gradient mini-protean TGX precast gel (Bio-Rad) and reverse-phase liquid chromatography with in-line electrospray ionization mass spectrometry.30 Protein concentrations were measured before storage and prior to the experiment at least in duplicate based on the extinction coefficient (Accelrys Gene v2.0) at 280 nm on a Perkin Elmer Lambda 35 or 40 UV/Vis spectrometer.
Size exclusion chromatography with multi-angle light scattering (SEC-MALS)
Molecular mass was estimated by analytical SEC with in-line MALS (DAWN Heleos-II, Wyatt Technology Inc., Santa Barbara, CA), refractive index (Optilab T-rEX, Wyatt Technology Inc.) and UV (Waters 2487, Waters Corporation, Milford, MA) detectors. Sample (125 µl) was applied onto a pre-equilibrated Superose-12 column (1.0 × 30 cm, Cytiva) and eluted at a flow rate of 0.5 mL/min in buffer A at 25 °C. Molecular mass was calculated using the Astra software provided with the instrument.
Sedimentation velocity analytical ultracentrifugation (SV-AUC)
Various constructs were subjected to SV-AUC either in buffer B (25 mM Tris-HCl, pH 7, 50 mM NaCl and 1 mM TCEP) or buffer C (25 mM Tris-HCl, pH 7.2, 20 mM NaCl and 1 mM TCEP), respectively. Samples containing the inhibitor were prepared using a 10 mM stock solution of GC373 in buffer C to achieve the desired protein and inhibitor ratios and incubated for a period of 1–2 hours prior to filling the cells.
Sedimentation velocity experiments were conducted at 50,000 rpm and 25 °C on a Beckman Coulter ProteomeLab XL-I or Beckman Optima XL-A analytical ultracentrifuge following standard protocols.40 Samples were loaded in 2-channel centerpiece cells and scans were collected using both the absorbance (280 nm) and Rayleigh interference (655 nm, when available) optical detection systems. Sedimentation data were time-corrected and analyzed in SEDFIT 16.1C41 in terms of a continuous c(s) distribution of Lamm equation solutions. Solution densities ρ, solution viscosities η, and protein partial specific volumes were calculated in SEDNTERP.42 To estimate the dimer dissociation constant for (-6)MProH41A and (-6*)MProH41A with GC373, absorbance sedimentation velocity data collected at various concentrations were analyzed globally using Lamm equation modeling in SEDPHAT 15.2b.43 A monomer–dimer self-association model was used and the presence of both monomer and dimer species was confirmed in the analysis. Absorbance extinction coefficients were calculated in SEDNTERP. Data were plotted in GUSSI.44
Differential Scanning Fluorimetry (DSF)
Samples in duplicate were prepared with SYPRO orange dye (5000x, Millipore Sigma product number S5692) to yield a final concentration of 10 µM protein and 5x dye in 25 µl of buffer B (25 mM Tris-HCl, pH 7, 50 mM NaCl and 1 mM TCEP) and 0.1 % DMSO. The FRET signal was monitored as a function of temperature in a Bio-Rad C1000 Touch Thermal Cycler, and data was processed with the provided software and plotted using Sigmaplot (Systat Software Inc.). Experiments were repeated at least twice.
Isothermal titration calorimetry (ITC)
Purified proteins were diluted from a stock solution to slightly above the desired concentration and dialyzed extensively against buffer C (25 mM Tris-HCl, pH 7.2, 20 mM NaCl and 1 mM TCEP). Concentrations were estimated after dialysis based on their 280 nm absorbance at least twice. Stock solutions of inhibitors in buffer C were diluted in the same buffer to the desired concentration. Titrations were performed with proteins (30 to 150 µM) kept in the cell and inhibitors at 10-times the concentration of the protein in the syringe at 28 °C on iTC200 microcalorimeter (Malvern Instruments Inc., Westborough, MA). Data were processed using the Origin software provided with the instrument. For competitive inhibitors that bind at only one site, the dissociation constant (Kd = 1/Ka) is equivalent to the inhibition constant measured by enzyme kinetics (Ki).
Circular dichroism
CD spectra were recorded using 10 µM protein in buffer B (25 mM Tris-HCl, pH 7, 50 mM NaCl and 1 mM TCEP) at 25 °C on a JASCO J-810 spectropolarimeter using Spectra Manager software version 2 (Jasco Analytical Instruments, Easton, MD) and a 0.1 cm pathlength cell. Spectra were processed using the same software. The bandwidth was set to 1.0 nm with an integration time of 0.5 s and 100 nm/min scanning speed. Ten scans were averaged and subtracted from a buffer scan acquired the same way. For thermal denaturation measurements, the change in the CD signal at 222 nm was measured as a function of temperature from 25-60 °C for MProWT and MProH41A and 25–75 °C for MProC145A at 1 °C intervals. The spectra were normalized to the mean residue ellipticity.
Protein crystallization and room-temperature X-ray crystallography
(-6*)MProH41A protein sample was concentrated to 9 mg/ml. GC376 stock was prepared at 10 mM concentration in buffer C (25 mM Tris-HCl, pH 7.2, 20 mM NaCl and 1 mM TCEP) for crystallization purposes and stored at −30 °C. For co-crystallization, (-6*)MProH41A was mixed with GC373 at 1:5 molar ratio and incubated at room temperature for at least 30 minutes before setting up crystal trays. Crystals of inhibitor-free and GC373-bound (-6*)MProH41A were grown by sitting drop vapor diffusion methodology with 18–21 % PEG3350, 0.1 M Bis-Tris pH 6.5 or pH 7.0 (1 mL) as the precipitant solution. Crystallization drops of 20 µL at 1:1 ratio were seed struck using the crystals of the native MPro in complex with a covalent ligand NBH2 as described.13, 21 Crystals appeared after several days and grew to the final size in about 1 month at 14 °C. The crystals suitable for X-ray diffraction measurements were mounted in MiTeGen (Ithaca, NY) room-temperature capillary setups for data collection.
All room temperature X-ray crystallographic data were collected on a Rigaku HighFlux HomeLab instrument equipped with a MicroMax-007 HF X-ray generator, Osmic VariMax optics, and a DECTRIS Eiger R 4 M hybrid photon counting detector. X-ray diffraction data were integrated using the CrysAlis Pro software suite (Rigaku Inc., The Woodlands, TX) then reduced and scaled using Aimless45 from the CCP4 suite.46 Structures were solved by molecular replacement using Phaser.47 MProWT structure (PDB code 6WQF,9 was used as a search model to solve the structures of inhibitor-free (-6*)MProH41A and (-6*)MProH41A-GC373 complex. Each model was iteratively refined with phenix.refine from the PHENIX suite48 and COOT.49 Geometry validation was aided by Molprobity.50 GC373 restraints were generated with eLBOW51 using geometry optimized by quantum mechanical calculations in Gaussian16 at B3LYP/6–31 g(d,p) level of theory.52 Final data collection and refinement statistics can be found in Table S1.
Accession numbers
PDB ID 8E4J, inhibitor-free (-6*)MProH41A; PDB ID 8E4R, (-6*)MProH41A-GC373 complex.
Author contributions
A.Y.K and J.M.L. Conceptualization, Methodology, Investigation, Writing. L.C., D.K., R.G., A.A., N.T.N. Methodology, Investigation, Editing.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This research used resources at the Spallation Neutron Source, and the High Flux Isotope Reactor, which are DOE Office of Science User Facilities operated by the Oak Ridge National Laboratory. The Office of Biological and Environmental Research supported research at ORNL's Center for Structural Molecular Biology (CSMB), a DOE Office of Science User Facility. ORNL is managed by UT-Battelle LLC for DOE’s Office of Science, the single largest supporter of basic research in the physical sciences in the United States. We thank John Lloyd and the NIDDK mass spectrometry core facility. This work was supported by the Intramural Research Program of National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), NIH.
Edited by Eric O. Freed
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jmb.2022.167876.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Wu F., Zhao S., Yu B., Chen Y.M., Wang W., Song Z.G. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.V’kovski P., Kratzel A., Steiner S., Stalder H., Thiel V. Coronavirus biology and replication: implications for SARS-CoV-2. Nat. Rev. Microbiol. 2020;19:155–170. doi: 10.1038/s41579-020-00468-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mariano G., Farthing R.J., Lale-Farjat S.L.M., Bergeron J.R.C. Structural Characterization of SARS-CoV-2: Where We Are, and Where We Need to Be. Front. Mol. Biosci. 2020;7:605236. doi: 10.3389/fmolb.2020.605236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang C., Liu Z., Chen Z., Huang X., Xu M., He T. The establishment of reference sequence for SARS-CoV-2 and variation analysis. J. Med. Virol. 2020;92:667–674. doi: 10.1002/jmv.25762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoshimoto F.K. The Proteins of Severe Acute Respiratory Syndrome Coronavirus-2 (SARS CoV-2 or n-COV19), the Cause of COVID-19. Protein J. 2020;39:198–216. doi: 10.1007/s10930-020-09901-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Groneberg D.A., Hilgenfeld R., Zabel P. Molecular mechanisms of severe acute respiratory syndrome (SARS) Respir. Res. 2005;6:8. doi: 10.1186/1465-9921-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen S., Jonas F., Shen C., Hilgenfeld R. Liberation of SARS-CoV main protease from the viral polyprotein: N-terminal autocleavage does not depend on the mature dimerization mode. Protein Cell. 2010;1:59–74. doi: 10.1007/s13238-010-0011-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kneller D.W., Phillips G., O'Neill H.M., Jedrzejczak R., Stols L., Langan P. Structural plasticity of SARS-CoV-2 3CL M(pro) active site cavity revealed by room temperature X-ray crystallography. Nat. Commun. 2020;11:3202. doi: 10.1038/s41467-020-16954-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nashed N.T., Aniana A., Ghirlando R., Chiliveri S.C., Louis J.M. Modulation of the monomer-dimer equilibrium and catalytic activity of SARS-CoV-2 main protease by a transition-state analog inhibitor. Commun. Biol. 2022;5:160. doi: 10.1038/s42003-022-03084-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ghahremanpour M.M., Tirado-Rives J., Deshmukh M., Ippolito J.A., Zhang C.H., Cabeza de Vaca I. Identification of 14 Known Drugs as Inhibitors of the Main Protease of SARS-CoV-2. ACS Med. Chem. Lett. 2020;11:2526–2533. doi: 10.1021/acsmedchemlett.0c00521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baker J.D., Uhrich R.L., Kraemer G.C., Love J.E., Kraemer B.C. A drug repurposing screen identifies hepatitis C antivirals as inhibitors of the SARS-CoV2 main protease. PLoS ONE. 2021;16:1–13. doi: 10.1371/journal.pone.0245962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kneller D.W., Li H., Galanie S., Phillips G., Labbe A., Weiss K.L. Structural, Electronic, and Electrostatic Determinants for Inhibitor Binding to Subsites S1 and S2 in SARS-CoV-2 Main Protease. J. Med. Chem. 2021 doi: 10.1021/acs.jmedchem.1c01475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Owen D.R., Allerton C.M.N., Anderson A.S., Aschenbrenner L., Avery M., Berritt S. An oral SARS-CoV-2 M(pro) inhibitor clinical candidate for the treatment of COVID-19. Science. 2021:eabl4784. doi: 10.1126/science.abl4784. [DOI] [PubMed] [Google Scholar]
- 15.Yang K.S., Ma X.R., Ma Y., Alugubelli Y.R., Scott D.A., Vatansever E.C. A Quick Route to Multiple Highly Potent SARS-CoV-2 Main Protease Inhibitors*. ChemMedChem. 2021;16:942–948. doi: 10.1002/cmdc.202000924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Konno S., Kobayashi K., Senda M., Funai Y., Seki Y., Tamai I. 3CL Protease Inhibitors with an Electrophilic Arylketone Moiety as Anti-SARS-CoV-2 Agents. J. Med. Chem. 2022;65:2926–2939. doi: 10.1021/acs.jmedchem.1c00665. [DOI] [PubMed] [Google Scholar]
- 17.Qiao J., Li Y.S., Zeng R., Liu F.L., Luo R.H., Huang C. SARS-CoV-2 M(pro) inhibitors with antiviral activity in a transgenic mouse model. Science. 2021;371:1374–1378. doi: 10.1126/science.abf1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deshmukh M.G., Ippolito J.A., Zhang C.H., Stone E.A., Reilly R.A., Miller S.J. Structure-guided design of a perampanel-derived pharmacophore targeting the SARS-CoV-2 main protease. Structure. 2021;29(823–33):e5. doi: 10.1016/j.str.2021.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kitamura N., Sacco M.D., Ma C., Hu Y., Townsend J.A., Meng X. Expedited Approach toward the Rational Design of Noncovalent SARS-CoV-2 Main Protease Inhibitors. J. Med. Chem. 2022;65:2848–2865. doi: 10.1021/acs.jmedchem.1c00509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pillaiyar T., Flury P., Kruger N., Su H., Schakel L., Barbosa Da Silva E. Small-molecule thioesters as SARS-CoV-2 main protease inhibitors: Enzyme inhibition, structure-activity relationships, antiviral activity, and X-ray structure determination. J. Med. Chem. 2022;65:9376–9395. doi: 10.1021/acs.jmedchem.2c00636. [DOI] [PubMed] [Google Scholar]
- 21.Kneller D.W., Li H., Phillips G., Weiss K.L., Zhang Q., Arnould M.A. Covalent narlaprevir- and boceprevir-derived hybrid inhibitors of SARS-CoV-2 main protease. Nat. Commun. 2022;13:2268. doi: 10.1038/s41467-022-29915-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goyal B., Goyal D. Targeting the dimerization of the main protease of coronaviruses: A potential broad-spectrum therapeutic strategy. ACS Comb. Sci. 2020;22:297–305. doi: 10.1021/acscombsci.0c00058. [DOI] [PubMed] [Google Scholar]
- 23.Xia B., Kang X. Activation and maturation of SARS-CoV main protease. Protein Cell. 2011;2:282–290. doi: 10.1007/s13238-011-1034-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsu M.F., Kuo C.J., Chang K.T., Chang H.C., Chou C.C., Ko T.P. Mechanism of the maturation process of SARS-CoV 3CL protease. J. Biol. Chem. 2005;280:31257–31266. doi: 10.1074/jbc.M502577200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muramatsu T., Kim Y.T., Nishii W., Terada T., Shirouzu M., Yokoyama S. Autoprocessing mechanism of severe acute respiratory syndrome coronavirus 3C-like protease (SARS-CoV 3CLpro) from its polyproteins. FEBS J. 2013;280:2002–2013. doi: 10.1111/febs.12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li C., Qi Y., Teng X., Yang Z., Wei P., Zhang C. Maturation mechanism of severe acute respiratory syndrome (SARS) coronavirus 3C-like proteinase. J. Biol. Chem. 2010;285:28134–28140. doi: 10.1074/jbc.M109.095851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Noske G.D., Nakamura A.M., Gawriljuk V.O., Fernandes R.S., Lima G.M.A., Rosa H.V.D. A Crystallographic Snapshot of SARS-CoV-2 Main Protease Maturation Process. J. Mol. Biol. 2021;433:167118. doi: 10.1016/j.jmb.2021.167118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muramatsu T., Takemoto C., Kim Y.T., Wang H., Nishii W., Terada T. SARS-CoV 3CL protease cleaves its C-terminal autoprocessing site by novel subsite cooperativity. Proc. Natl. Acad. Sci. U S A. 2016;113:12997–13002. doi: 10.1073/pnas.1601327113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee J., Worrall L.J., Vuckovic M., Rosell F.I., Gentile F., Ton A.T. Crystallographic structure of wild-type SARS-CoV-2 main protease acyl-enzyme intermediate with physiological C-terminal autoprocessing site. Nat. Commun. 2020;11:5877. doi: 10.1038/s41467-020-19662-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nashed N.T., Kneller D., Coates L., Ghirlando R., Aniana A., Kovalevsky A. Autoprocessing and oxyanion loop reorganization upon GC373 and nirmatrelvir binding of monomeric SARS-CoV-2 main protease catalytic domain. Commun. Biol. 2022;5:976. doi: 10.1038/s42003-022-03910-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bacha U., Barrila J., Velazquez-Campoy A., Leavitt S.A., Freire E. Identification of novel inhibitors of the SARS coronavirus main protease 3CLpro. Biochemistry. 2004;43:4906–4912. doi: 10.1021/bi0361766. [DOI] [PubMed] [Google Scholar]
- 32.Shi J., Sivaraman J., Song J. Mechanism for controlling the dimer-monomer switch and coupling dimerization to catalysis of the severe acute respiratory syndrome coronavirus 3C-like protease. J. Virol. 2008;82:4620–4629. doi: 10.1128/JVI.02680-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen S., Hu T., Zhang J., Chen J., Chen K., Ding J. Mutation of Gly-11 on the dimer interface results in the complete crystallographic dimer dissociation of severe acute respiratory syndrome coronavirus 3C-like protease: crystal structure with molecular dynamics simulations. J. Biol. Chem. 2008;283:554–564. doi: 10.1074/jbc.M705240200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kneller D.W., Phillips G., Weiss K.L., Pant S., Zhang Q., O'Neill H.M. Unusual zwitterionic catalytic site of SARS-CoV-2 main protease revealed by neutron crystallography. J. Biol. Chem. 2020;295:17365–17373. doi: 10.1074/jbc.AC120.016154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fan K., Ma L., Han X., Liang H., Wei P., Liu Y. The substrate specificity of SARS coronavirus 3C-like proteinase. Biochem. Biophys. Res. Commun. 2005;329:934–940. doi: 10.1016/j.bbrc.2005.02.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sayer J.M., Liu F., Ishima R., Weber I.T., Louis J.M. Effect of the active site D25N mutation on the structure, stability, and ligand binding of the mature HIV-1 protease. J. Biol. Chem. 2008;283:13459–13470. doi: 10.1074/jbc.M708506200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Louis J.M., Aniana A., Weber I.T., Sayer J.M. Inhibition of autoprocessing of natural variants and multidrug resistant mutant precursors of HIV-1 protease by clinical inhibitors. Proc. Natl. Acad. Sci. U S A. 2011;108:9072–9077. doi: 10.1073/pnas.1102278108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu T., Zhang Y., Li L., Wang K., Chen S., Chen J. Two adjacent mutations on the dimer interface of SARS coronavirus 3C-like protease cause different conformational changes in crystal structure. Virology. 2009;388:324–334. doi: 10.1016/j.virol.2009.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cheng S.C., Chang G.G., Chou C.Y. Mutation of Glu-166 blocks the substrate-induced dimerization of SARS coronavirus main protease. Biophys. J. 2010;98:1327–1336. doi: 10.1016/j.bpj.2009.12.4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao H., Brautigam C.A., Ghirlando R., Schuck P. Overview of current methods in sedimentation velocity and sedimentation equilibrium analytical ultracentrifugation. Curr. Protoc. Protein Sci. 2013;12 doi: 10.1002/0471140864.ps2012s71. Chapter 20:Unit20 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cole J.L., Lary J.W., Moody T.P., Laue T.M. Analytical ultracentrifugation: sedimentation velocity and sedimentation equilibrium. Methods Cell Biol. 2008;84:143–179. doi: 10.1016/S0091-679X(07)84006-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schuck P. On the analysis of protein self-association by sedimentation velocity analytical ultracentrifugation. Anal. Biochem. 2003;320:104–124. doi: 10.1016/s0003-2697(03)00289-6. [DOI] [PubMed] [Google Scholar]
- 44.Brautigam C.A. Calculations and publication-quality illustrations for analytical ultracentrifugation data. Methods Enzymol. 2015;562:109–133. doi: 10.1016/bs.mie.2015.05.001. [DOI] [PubMed] [Google Scholar]
- 45.Evans P.R., Murshudov G.N. How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 2013;69:1204–1214. doi: 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liebschner D., Afonine P.V., Baker M.L., Bunkoczi G., Chen V.B., Croll T.I. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D Struct. Biol. 2019;75:861–877. doi: 10.1107/S2059798319011471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Casanal A., Lohkamp B., Emsley P. Current developments in coot for macromolecular model building of electron cryo-microscopy and crystallographic data. Protein Sci. 2020;29:1069–1078. doi: 10.1002/pro.3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen V.B., Arendall W.B., 3rd, Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moriarty N.W., Grosse-Kunstleve R.W., Adams P.D. electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr. D Biol. Crystallogr. 2009;65:1074–1080. doi: 10.1107/S0907444909029436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R. Gaussian Inc.; Wallingford, CT: 2016. Gaussian 16, Revision B.01. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.