

Abstract
Listeria monocytogenes requires listeriolysin O (LLO) and ActA, the products of hly and actA, respectively, to establish a productive intracellular infection. LLO is essential for vacuolar lysis and entry into the cytosol, while ActA is required for bacterial spread to adjacent cells. We have used a transcriptional reporter gene system to compare the expression of actA and hly during intracellular growth to that during growth in broth cultures. The hly and actA genes were transcriptionally fused to Escherichia coli lacZ and Bacillus pumilus cat-86 (cat), and the fusions were integrated in single copies into the L. monocytogenes chromosome. A chloramphenicol resistance assay indicated that the hly fusion but not the actA fusion was significantly activated in Luria-Bertani (LB) broth, and this finding correlated with LLO and ActA levels detectable in broth cultures. Quantitation of promoter activity on the basis of β-galactosidase activity revealed up to 10-fold-higher level of expression of the hly fusion relative to the actA fusion in LB broth. In contrast, both fusions were active in the cytosol of J774 cells, and the activity of the actA fusion was approximately 3-fold higher than that of the hly fusion under these conditions. However, quantitative immunoprecipitation of ActA and LLO from infected J774 cells demonstrated approximately 70-fold more cytosolic ActA than cytosolic LLO. Finally, in comparison to induction in broth cultures, actA was highly induced (226-fold) and hly was moderately induced (20-fold) in J774 cells. Collectively, these results indicate that actA and hly are differentially regulated in response to the growth environment and that both genes are preferentially expressed during intracellular growth. Further, while the lower level of production of ActA than of LLO in broth can be accounted for by transcriptional regulation, the relative abundance of intracellular ActA compared to that of intracellular LLO is a function of additional, possibly host-mediated, factors.
Listeria monocytogenes is a facultative intracellular bacterial pathogen which replicates in the cytosol of mammalian cells. Several bacterial proteins have been shown to play a role in the intracellular growth cycle (reviewed in reference 29). Listeriolysin O (LLO, encoded by hly) is a pore-forming cytolysin which mediates lysis of the phagosomal membrane, allowing bacterial access to the cytosol, where replication begins. Once bacteria are in the cytosol, a surface protein, ActA (encoded by actA), mediates the formation of polarized actin tails that propel the bacteria toward the cytoplasmic membrane. At the membrane, bacteria become enveloped in filopodium-like structures which are recognized and engulfed by adjacent cells. This process results in the formation of double-membrane vacuoles from which the bacteria rapidly free themselves by the cooperative action of two bacterial phospholipases, phosphatidylinositol-specific phospholipase C and broad-range phospholipase C (encoded by plcA and plcB, respectively) (34), and LLO (11). Once in the cytosol of the adjacent cell, L. monocytogenes repeats the cycle, and the infection is propagated. All of these gene products, along with a metalloprotease, Mpl (encoded by mpl), are expressed under the control of a transcriptional regulator, PrfA (encoded by prfA).
L. monocytogenes requires LLO and ActA to establish a productive infection in vivo and in cultured mammalian cells (10, 16, 17, 30). These proteins, which function in vacuolar lysis and actin-based motility, respectively, are clearly associated with growth in the mammalian cell environment and have no known role in the extracellular growth of L. monocytogenes. Nevertheless, it is clear that LLO is produced by wild-type L. monocytogenes 10403S growing in vitro in a variety of media (14, 16, 22). Paradoxically, it has not been possible to demonstrate the presence of LLO in infected-cell lysates by conventional methods of immunodetection. Villaneuva et al. (36) reported the detection of cytosolic LLO, but only under conditions in which the 26S proteasome, the multicatalytic protease complex present in the cytosol of mammalian cells (12), was inhibited. This finding suggests that the amount of LLO present in the cytosol is controlled, at least in part, by host cell-mediated degradation. In support of this idea, an internal LLO peptide, LLO 91-99, has been found in association with major histocompatibility complex class I molecules on the surface of L. monocytogenes-infected cells (25). In direct contrast to the production of LLO, the production of ActA is minimal in bacteriologic media, but ActA is the most abundant surface or secreted bacterial protein detected during intracellular growth (3). These observations suggest that environmental signals specifically associated with the intracellular environment activate the expression of actA.
In this report, the basis for the apparent differential expression of LLO and ActA during L. monocytogenes growth in vitro and in the mammalian cell cytosol is further investigated. Using a reporter gene system, we found that the observed differential expression of LLO and ActA by L. monocytogenes cultured in vitro is controlled at the level of transcription. Further, in comparison to bacterial growth in vitro, we found that the expression of actA and, to a lesser extent, hly is induced during growth in the intracellular environment. Finally, we demonstrated the production of LLO by bacteria actively growing in the cytosol in the absence of proteasome inhibitors and, using quantitative immunoprecipitation, provided evidence that transcriptional regulation alone cannot account for the high levels of ActA relative to LLO present in the cytosol of infected macrophages.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The wild-type L. monocytogenes isolate used for these studies, 10403S, belongs to serotype 1, is resistant to 1 mg of streptomycin per ml, and has a 50% lethal dose for BALB/c mice of 3.3 × 104. The Bacillus subtilis host for plasmid constructions was BD170 (28). The Escherichia coli host for plasmid constructions was HB101. L. monocytogenes was cultured in Luria-Bertani (LB) or brain heart infusion (BHI) broth. E. coli and B. subtilis were cultured in LB broth.
Construction of pLCR.
A transcriptional reporter plasmid containing tandem promoterless lacZ and cat-86 genes was generated by in vivo recombination in B. subtilis as follows. Plasmid pTV53 (37) was used as a source of the promoterless Bacillus pumilus cat-86 gene (1). The BamHI site upstream of cat-86 was eliminated by digestion with BamHI, treatment with the Klenow fragment of DNA polymerase, and religation of the blunt-ended DNA. B. subtilis PY256 harboring plasmid pTV30 (28) was transformed with pTV53 lacking the BamHI site by the method of Cutting and Vander Horn (8). The presence of a weak cryptic promoter upstream of lacZ in pTV30 (28) allowed selection for the desired pTV30–cat-86 recombinants (pTV30cat) on a low level of chloramphenicol (3 μg/ml) at 30°C. The pE194 origin of replication in pTV30cat was replaced with a temperature-sensitive derivative of the origin of replication from the broad-host-range plasmid pWVO1 (18), which replicates in gram-positive and gram-negative bacteria at temperatures of <35°C, as follows. Plasmid pTV1OK (13), which carries the pWVO1ts origin of replication and a kanamycin resistance gene that functions in both gram-positive and gram-negative bacteria (35), was used to transform B. subtilis harboring pTV30cat. Among the products of in vivo recombination was plasmid pTV30catts, which was isolated by selection for kanamycin (10 μg/ml), erythromycin (1 μg/ml), and lincomycin (25 μg/ml) resistance at 28°C. Standard cloning techniques were used to remove approximately 4.5 kb of DNA containing a Tn917 sequence by partial digestion of pTV30catts with XbaI, religation of linear DNA in the 12-kb size range, and transformation of E. coli HB101 to kanamycin resistance (40 μg/ml). The resulting plasmid was further modified by cloning a 400-bp PCR-generated fragment containing the trpA transcription terminator (7) into the NcoI and BamHI sites upstream of lacZ, yielding plasmid pLCR.
Cloning of L. monocytogenes virulence gene promoters into pLCR.
DNA fragments containing the hly (920-bp) and actA (1,191-bp) promoters were amplified from L. monocytogenes genomic DNA with the following primer pairs: for hly, 5′-TTTATGTGGATCCATTAACATTTGT-3′ and 5′-GGGGATCCTTCACTGATTGCGCC-3′, and for actA, 5′-CGGGATCCTGAAGCTTGGGAAGCAG-3′ and 5′-GGGGATCCAAGAAGCATTGGCGTC-3′; primers contained BamHI recognition sequences at the 5′ ends (underlined) to allow cloning of the PCR products into the unique BamHI site upstream of lacZ in pLCR. Clones containing the hly and actA promoters in the forward orientation and a clone containing the actA promoter in the opposite orientation with respect to the transcription of lacZ and cat were constructed.
Transformation of L. monocytogenes with pLCR constructs and integration into the chromosome.
Recombinant plasmids were used to transform L. monocytogenes by electroporation (27), with kanamycin (15 μg/ml) selection at 28°C. pLCR promoter constructs were integrated via homologous recombination between cloned promoter-containing fragments and the corresponding regions on the chromosome. This process was accomplished by growing the strains at 37°C, a nonpermissive temperature for pLCR replication, in the presence of 15 μg of kanamycin per ml. Integration was confirmed by Southern blotting with probes derived from the hly and actA promoter regions.
Intracellular chloramphenicol resistance assay.
Monolayers of J774 cells grown on coverslips as described previously (30) were infected for 1 h with 2.5 × 106 bacteria from stationary-phase BHI cultures. Since cat-86 is an inducible antibiotic resistance gene (1), a subinhibitory concentration of chloramphenicol (0.5 μg/ml) was present in the cell culture medium at the time of infection and for the duration of the assay. After 1 h, monolayers were washed and fresh medium was added. At 1.5 h after infection, 50 μg of gentamicin per ml was added to kill extracellular bacteria. Chloramphenicol (10 μg/ml) was added at various times postinfection. Growth in the presence and absence of chloramphenicol was measured at various times by lysing monolayers and plating the lysates on LB agar to determine the number of CFU as previously described (30).
In vitro chloramphenicol resistance assay.
Bacteria from stationary-phase BHI cultures were washed and subcultured 1:100 in LB medium (pH 7.4) buffered with 50 mM morpholinepropanesulfonic acid (MOPS) and containing 15 μg of kanamycin and 0.5 μg of chloramphenicol per ml. Cultures were grown at 37°C with aeration to an optical density at 600 nm (OD600) of approximately 0.1, at which time 10 μg of chloramphenicol per ml was added. Cultures were grown for an additional 5 h, after which OD600 readings were taken to measure relative levels of bacterial growth in the presence and absence of chloramphenicol.
Extracellular β-galactosidase assay.
Bacteria from stationary-phase BHI cultures were washed and subcultured 1:100 in buffered LB medium (pH 7.4) containing 15 μg of kanamycin per ml. After various periods of growth at 37°C with aeration, an OD600 reading was taken and β-galactosidase activity was measured essentially as described by Youngman (37), but with the following modifications. Bacteria in 0.10 or 0.25 ml of culture were pelleted and resuspended in 50 μl of phosphate-buffered saline (PBS) (pH 8.0)–0.1% Triton X-100. Ten microliters of 4-methylumbelliferyl-β-d-galactopyranoside (MUG) was added, and tubes were incubated for 60 to 100 min at room temperature. The extent of conversion of MUG to the fluorescent product 4-methylumbelliferone by β-galactosidase was measured with a Sequoia-Turner model 450 fluorometer. Units of β-galactosidase activity were calculated as described previously (37) but were normalized to CFU rather than OD600. The number of CFU per milliliter of culture was extrapolated from a standard curve which related CFU per milliliter at hourly time points during bacterial growth in buffered LB medium to OD600.
Intracellular β-galactosidase assay.
J774 cells were seeded into 60-mm dishes and infected as described above for the intracellular chloramphenicol resistance assay, except that chloramphenicol was not present. Monolayers were washed and lysed 5 to 6 h after infection by scraping cells into 200 μl of PBS (pH 8.0)–0.1% Triton X-100. Twenty microliters of MUG was added to 100 μl of lysate, and samples were incubated for 60 to 100 min at room temperature. β-Galactosidase activity was measured as described above for the extracellular assay. A series of 60-mm dishes in which monolayers were seeded onto glass coverslips was set up in parallel. At the time of the assay, monolayers on three coverslips per sample were lysed, and the number of CFU per coverslip was determined by plating the lysates on LB agar. The total number of CFU per dish was extrapolated by multiplication by a factor which corrected for the area of the coverslip relative to that of the 60-mm dish. Units of β-galactosidase activity, normalized to the number of CFU per dish, were calculated as described previously (37).
Production of ActA and LLO in vitro.
Bacteria from stationary-phase BHI cultures were inoculated 1:100 into buffered LB broth and incubated at 37°C with aeration. After 5.5 h, bacteria in 10 ml of culture were pelleted and washed twice with PBS. Bacterial pellets were resuspended in 100 μl of 2× sodium dodecyl sulfate (SDS)–polyacrylamide gel electrophoresis (PAGE) buffer (0.06 M Tris [pH 6.8], 2% SDS, 10% glycerol, 10% 2-mercaptoethanol, 0.01% bromophenol blue) and boiled for 5 min. L. monocytogenes is resistant to lysis under these conditions; therefore, this treatment solubilizes ActA and other membrane proteins (3). Supernatant proteins, including LLO, were precipitated on ice with 10% trichloroacetic acid for 1 h, pelleted, and resuspended in 100 μl of 1× SDS-PAGE buffer containing 0.1 N NaOH. All samples were boiled for 5 min prior to being subjected to SDS-PAGE (8% polyacrylamide). Proteins were visualized by staining of the gel with Coomassie brilliant blue. Western blotting was performed as previously described (20).
Detection of ActA and LLO produced in J774 cells.
Monolayers of J774 cells seeded into 60-mm dishes were infected with 3.3 × 106 L. monocytogenes 10403S organisms. Monolayers were washed after 30 min, and 5 μg of gentamicin per ml was added after an additional 30 min. At 5 h after infection, the medium was replaced with methionine-free Dulbecco minimal essential medium (DMEM) containing 10% dialyzed fetal calf serum, 225 μg of cycloheximide per ml, 30 μg of anisomycin per ml, and 5 μg of gentamicin per ml. After 30 min, monolayers were pulse-labeled for 1 h with 200 μCi of 35S-methionine (Express35S protein labeling mix; NEN Research Products, Boston, Mass.), washed, and lysed with 1 ml of RIPA buffer (150 mM NaCl, 50 mM Tris-HCl [pH 8.0], 1% Nonidet P-40, 0.5% deoxycholate, 0.1% SDS) containing phenylmethylsulfonyl fluoride (1 mM), aprotinin (0.3 μM), leupeptin (1 μM), pepstatin A (1 μM), and EDTA (10 mM). Immunoprecipitation was performed as previously described (3) with monoclonal anti-LLO antibody B3-19 (24), a kind gift from Pascale Cossart, or polyclonal anti-ActA antibody. Immunoprecipitated samples were boiled and loaded on 8% polyacrylamide gels. Following SDS-PAGE, gels were processed for autoradiography and phosphorimaging analysis.
To determine the efficiency of immunoprecipitation of ActA from infected J774 cells, duplicate monolayers were infected and labeled as described above. At 5.5 h postinfection, one monolayer was lysed with 2× SDS-PAGE buffer and all labeled proteins, including ActA, were resolved by SDS-PAGE. The other monolayer was lysed with RIPA buffer, and ActA was immunoprecipitated. Samples containing total and immunoprecipitated proteins were subject in parallel to SDS-PAGE, autoradiography, and phosphorimaging analysis. The efficiency of immunoprecipitation was determined by dividing the value obtained from phosphorimaging analysis of immunoprecipitated ActA by that for the total ActA protein.
Since LLO is not detectable in the population of metabolically labeled bacterial proteins produced in J774 cells (36, 23), the efficiency of immunoprecipitation of LLO was determined with in vitro-labeled LLO. Specifically, exponential-phase bacteria (OD600, 1.0) from 10 ml of BHI culture were pelleted, washed, and resuspended in 1.5 ml of methionine-free DMEM. After 30 min at 37°C in 5% CO2, bacteria were labeled for 1 h with 24 μCi of 35S-methionine, and proteins from 900 μl of supernatant were precipitated for 1.5 h on ice with 25 μg of bovine serum albumin per ml and 10% trichloroacetic acid. Labeled proteins were pelleted and resuspended in PBS. One half of the sample was used to spike a lysate of J774 cells, and LLO was immunoprecipitated. The immunoprecipitated LLO was subjected, along with the other half of the labeled protein sample, to SDS-PAGE and phosphorimaging analysis. The efficiency of immunoprecipitation of LLO was determined as described above for ActA.
RESULTS
Construction of reporter gene vector pLCR.
In order to monitor the activity of L. monocytogenes virulence gene promoters, pLCR, containing tandem promoterless lacZ and cat-86 (hereafter referred to as cat) reporter genes, was constructed (Fig. 1). Features of this vector include (i) a unique BamHI site upstream of lacZ and cat for cloning L. monocytogenes promoter fragments, (ii) a kanamycin resistance gene for selection in both E. coli and L. monocytogenes, and (iii) a temperature-sensitive origin of replication derived from the broad-host-range plasmid pWVO1 (18), which replicates in E. coli and L. monocytogenes at an optimum temperature of 28°C and fails to replicate at temperatures of >35°C. In the presence of kanamycin, L. monocytogenes transformed with pLCR containing cloned promoter fragments can be selected at 28°C, and clones with integrated pLCR can be selected at 37°C.
FIG. 1.
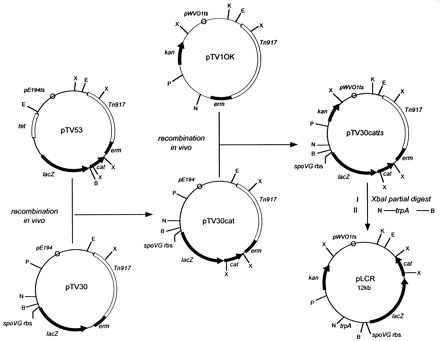
Construction of transcriptional reporter vector pLCR. Plasmids pTV53 and pTV30, which provided the promoterless B. pumilus cat-86 gene and the E. coli lacZ gene with an upstream spoVG ribosome binding site (rbs) and a BamHI site, respectively, were described previously (28, 37). Plasmid pTV1OK (13) provided the kanamycin resistance gene (kan), a type III aminoglycoside phosphotransferase derived from Streptococcus faecalis (35), and the pWVO1ts origin of replication, a temperature-sensitive derivative of the origin of replication from Lactococcus lactis plasmid pWVO1 (18). Plasmid pTV30cat was isolated from B. subtilis by selection for chloramphenicol resistance at 30°C following recombination between plasmids pTV30 and pTV53. Similarly, pTV30catts was isolated by selection for kanamycin resistance at 28°C following recombination between plasmids pTV30cat and pTV1OK. Details of plasmid construction by in vivo recombination in B. subtilis are described in Materials and Methods. pLCR was generated in two conventional cloning steps by partial XbaI digestion and religation of pTV30catts followed by insertion of a PCR-generated fragment containing the trpA terminator (7) into the NcoI and BamHI sites. X, XbaI; E, EcoRI; K, KpnI; P, PstI; N, NcoI; B, BamHI. E, N, and B are unique restriction sites in pLCR.
Construction of L. monocytogenes strains carrying integrated lacZ-cat transcriptional fusions to virulence gene promoters.
DNA fragments (approximately 1 kb) containing L. monocytogenes virulence gene promoters were cloned into the BamHI site upstream of lacZ and cat. Three constructs were made; two constructs contained the hly and actA promoters cloned in the forward orientation with respect to the transcription of lacZ and cat, and a control construct contained the actA promoter cloned in the opposite orientation with respect to the transcription of the reporter genes. Transformation of L. monocytogenes 10403S followed by growth of transformants at 37°C in the presence of 15 μg of kanamycin per ml selected for clones with integrated copies of each construct, as depicted in Fig. 2. Southern blotting with probes derived from the hly and actA regions indicated that the initial strains constructed in this manner contained more than one copy of pLCR integrated in tandem into the chromosome. Further passage of the strains at 28°C (a permissive temperature for plasmid replication) in the absence of kanamycin, followed by screening of individual colonies by Southern blotting, allowed for the isolation of clones containing a single integrated copy of pLCR (data not shown). Stable integration of pLCR in DP-L2986 (phly::lacZ-cat), DP-L2989 (pactA::lacZ-cat), and DP-L2812 (reverse-orientation pactA::lacZ-cat) was demonstrated by repeated passages at 37°C in the presence of kanamycin. Note that in strain DP-L2989, the integration of pLCR places lacZ and cat downstream of the chromosomal mpl promoter as well as the cloned actA promoter. Therefore, transcription of reporter genes in this strain may originate from one or both promoters, which are referred to as the actA/mpl promoters.
FIG. 2.
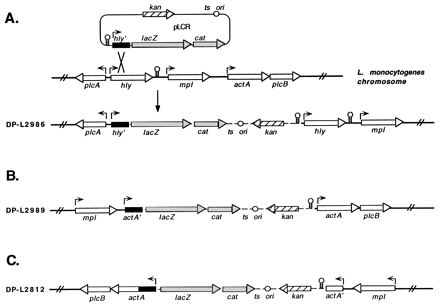
Schematic of pLCR fusion constructs integrated into the L. monocytogenes chromosome. (A) Integration of pLCR by homologous recombination between the cloned hly promoter-containing fragment (hly′) and the corresponding region of the L. monocytogenes chromosome, yielding strain DP-L2986. (B and C) Integration structures which resulted from recombination between the actA promoter-containing fragment (actA′) cloned in the forward (B) and reverse (C) orientations with respect to the transcription of lacZ and cat and the corresponding region of the L. monocytogenes chromosome, yielding strains DP-L2989 and DP-L2812, respectively. Broken lines represent plasmid sequences, and solid lines represent flanking chromosomal regions. Arrows depict promoter positions and the direction of transcription for each of the indicated genes. Stem and loop structures represent transcriptional terminators.
Chloramphenicol resistance in LB broth and in J774 cells.
The activity of the hly and actA/mpl promoter fusions in LB broth was assessed by measuring bacterial growth in the presence and absence of chloramphenicol. As shown in Fig. 3, growth of DP-L2986 was equivalent in the presence and absence of chloramphenicol, indicating that the hly promoter is active during bacterial growth in LB broth. In contrast, DP-L2989 grew normally in the absence of chloramphenicol but failed to reach levels of growth significantly greater than that of the control strain in the presence of chloramphenicol, suggesting that the actA and mpl promoters are relatively inactive during growth in LB broth.
FIG. 3.
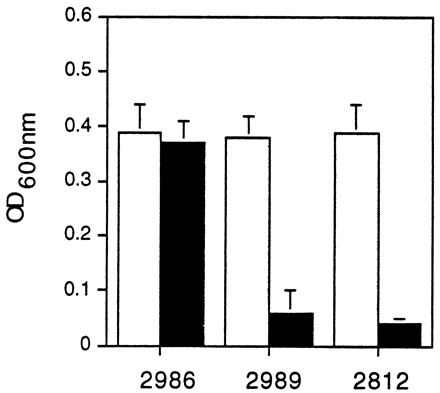
Promoter activity in LB broth as measured by chloramphenicol resistance. L. monocytogenes DP-L2986, DP-L2989, and DP-L2812 carrying the hly, actA, and control reverse-orientation actA promoter fusions, respectively, were cultured in buffered LB broth in the absence (light bars) or presence (dark bars) of 10 μg of chloramphenicol per ml. After 5 h, bacterial growth was assessed by measuring the OD600. The data represent the mean ± standard deviation for three individual experiments. The addition of increasing doses of chloramphenicol at the onset of L. monocytogenes 10403S culturing in LB broth and measurement of the OD600 at various time points demonstrated an MIC of 5 μg/ml (data not shown).
To assay for virulence gene expression during intracellular growth, the ability of strains to replicate in J774 cells in the presence of chloramphenicol was measured (Fig. 4). Both strains grew equally well in the presence and absence of chloramphenicol, indicating that the hly and actA/mpl promoters are active during intracellular growth. The control strain, DP-L2812, failed to grow in the presence of chloramphenicol but grew normally in the absence of chloramphenicol. The doubling times of all three strains in J774 cells were similar to that of wild-type 10403S, indicating that the integration of pLCR constructs in these strains did not cause intracellular growth defects (data not shown).
FIG. 4.
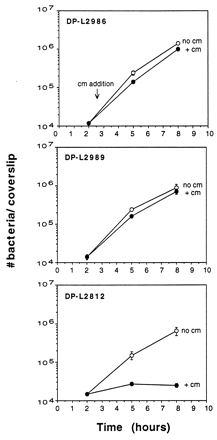
Promoter activity in J774 cells as measured by chloramphenicol resistance. Monolayers of J774 cells grown on glass coverslips were infected with DP-L2986, DP-L2989, or DP-L2812. Chloramphenicol (10 μg/ml) (cm) was added at 2.5 h postinfection. Monolayers were lysed at the indicated time points, and the number of bacteria per coverslip was determined. Results are expressed as the mean number of bacteria ± the standard deviation for three coverslips per time point. One of three experiments with similar results is shown. The MIC of chloramphenicol for L. monocytogenes 10403S grown in J774 cells was determined to be 5 μg/ml by measuring the number of CFU per monolayer in the presence and absence of increasing doses of chloramphenicol at various time points after infection (data not shown).
These results suggest that hly and actA are differentially transcribed in response to the growth environment. hly is expressed during intracellular growth as well as in broth cultures, while actA is significantly activated only in the intracellular environment.
β-Galactosidase activity in LB broth and in J774 cells.
The activity of the hly and actA/mpl promoter fusions during bacterial growth in LB broth was quantitated by assaying for the product of the lacZ gene, β-galactosidase. As shown in Fig. 5, the actA/mpl promoter fusion was not completely inactive in LB broth, as suggested by the chloramphenicol resistance assay, but rather was activated to low levels relative to the hly promoter fusion. Transcription mediated by the hly promoter was 4- to 10-fold higher than transcription mediated by the actA/mpl promoters over 8 h of growth in LB broth.
FIG. 5.
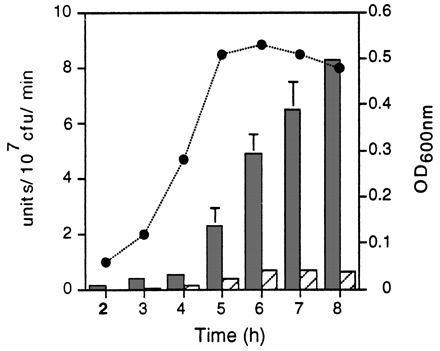
Promoter activity in LB broth as measured by a β-galactosidase assay. L. monocytogenes DP-L2986 (dark bars), DP-L2989 (hatched bars), and DP-L2812 (see below) carrying the hly, actA, and control reverse-orientation actA promoter fusions, respectively, were cultured in buffered LB broth, and β-galactosidase activity was measured at the indicated time points. Results are expressed as units of β-galactosidase activity per 107 CFU per minute. DP-L2812 background activity, which in no case exceeded 0.08 U, was subtracted from each value. The circles indicate OD600 values for DP-L2986 and DP-L2989, which were identical over 8 h of growth. The data represent the mean ± standard deviation for three individual experiments.
We next quantitated promoter activity during intracellular growth. J774 cells were infected with L. monocytogenes, and after 5 to 6 h of growth, bacterial numbers in the cytosol reached 1 × 107 to 3 × 107 CFU per monolayer and bacteria were in log-phase growth (Fig. 4). J774 cells were lysed, and β-galactosidase activity was measured. Since there was some variability associated with this assay, four individual experiments are reported in Table 1. In contrast to the results obtained with LB broth, the activity of the actA/mpl promoter fusion was on average three-fold higher than the activity of the hly promoter fusion in J774 cells.
TABLE 1.
Promoter activity during bacterial growth in J774 cells
| Expt | DP-L2986
|
DP-L2989
|
||
|---|---|---|---|---|
| β-Galactosidase activity (U/107 CFU/min)a | 107 CFU/dishb | β-Galactosidase activity (U/107 CFU/min) | 107 CFU/dish | |
| 1 | 13.6 | 2.4 | 31.1 | 1.5 |
| 2 | 7.1 | 2.6 | 21.2 | 1.6 |
| 3 | 15.4 | 1.8 | 41.8 | 1.0 |
| 4 | 7.1 | 2.6 | 32.9 | 1.4 |
| Mean ± SDc | 10.8 ± 4.3 | 31.7 ± 8.4 | ||
Background activity, derived from DP-L2812-infected monolayers, was subtracted from each value. β-Galactosidase activity derived from DP-L2812-infected monolayers never exceeded the endogenous β-galactosidase activity produced by uninfected J774 cells.
The number of CFU per dish was determined by lysing infected J774 cells grown on coverslips in duplicate dishes and plating a portion of the lysate on LB agar. The total number of CFU per dish was extrapolated by multiplication by a factor which corrected for the area of the coverslip relative to that of the 60-mm dish.
Derived from four individual experiments. The P value, determined by Student’s t test, for a comparison of the values shown was <0.01.
The number of units of β-galactosidase activity produced by DP-L2986 and DP-L2989 in J774 cells after approximately 5 h of growth (Table 1), a time point which corresponds to the log phase (Fig. 4), was divided by the number of units of β-galactosidase activity produced in LB broth at the log phase (4 h) (Fig. 5). The results indicated that the transcription of actA is highly induced in the intracellular environment compared to broth. The level of transcription of the actA/mpl promoter fusion was 226-fold higher in J774 cells than in LB broth (mean ± SD of 31.7 ± 8.4 U in J774 cells and 0.14 ± 0.02 U in LB broth). In comparison, transcription of the hly promoter fusion was also induced in the intracellular environment, but only 20-fold relative to that in LB broth (10.8 ± 4.3 U in J774 cells and 0.53 ± 0.11 U in LB broth).
LLO and ActA production during growth in LB medium.
Reporter assays indicated that hly is transcribed to higher levels than actA in vitro. Consistent with these results, the 58-kDa LLO protein was easily detectable in the secreted protein fraction of L. monocytogenes grown in LB broth, while ActA, a 97-kDa surface protein, was undetectable (data not shown) or was detectable as only a faint band in the SDS-extractable protein fraction (Fig. 6). A strain of L. monocytogenes (SLCC-5764) which produces elevated levels of all virulence proteins, including ActA, in vitro (5), and its isogenic actA deletion mutant, DP-L1955 (20), were included as controls to indicate the position of the ActA protein upon migration through 8% polyacrylamide. Western blotting analysis indicated that the kinetics of LLO and ActA production in LB broth correlated with the β-galactosidase activity time course shown in Fig. 5, providing evidence that the activity of the hly and actA transcriptional fusions accurately reflects the regulation of the natural genes (data not shown).
FIG. 6.
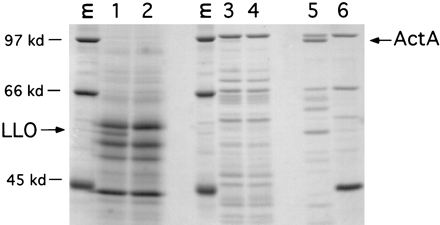
SDS-PAGE of L. monocytogenes secreted and membrane-anchored proteins produced in LB broth. Wild-type L. monocytogenes 10403S was grown in buffered LB broth for 5.5 h, and secreted proteins (lanes 1 and 2) from 10 ml of culture were precipitated with trichloroacetic acid and resuspended in SDS-PAGE sample buffer. Membrane-anchored proteins (lanes 3 to 6) were extracted from bacterial pellets by boiling for 5 min in SDS-PAGE sample buffer. Eighty percent of each sample was subjected to SDS-PAGE, and the gel was stained with Coomassie brilliant blue. Lane 1, 10403S; lane 2, DP-L2161 (10403SΔhly) (14); lane 3, 10403S; lane 4, DP-L1942 (10403SΔactA) (3); lane 5, SLCC-5764 (5); lane 6, DP-L1955 (SLCC-5764ΔactA) (20). Arrows indicate the positions of LLO and ActA. Lanes M show molecular mass standards (kilodaltons [kd]).
LLO and ActA production during growth in J774 cells.
The results above indicate that while hly expression is greater than actA expression in broth cultures, the converse is true in the cytosol of J774 cells. We next wished to evaluate the relative amounts of LLO and ActA produced during bacterial growth in the cytosol of J774 cells. However, with conventional methods of detection, LLO has been difficult to demonstrate in lysates of infected cells. Villanueva et al. (36) provided evidence that LLO secreted by cytosolic bacteria is degraded by the proteasome. In that study, LLO could be immunoprecipitated from infected J774 cell lysates only when an inhibitor of the proteasome was present during infection. Upon optimization of our infection and metabolic labeling protocols (see Materials and Methods for details), we were able to directly compare the steady-state levels of cytosolic LLO and ActA.
Using monoclonal anti-LLO antibody B3-19, and in the absence of proteasome inhibitors, we were able to immunoprecipitate LLO from metabolically labeled 10403S-infected J774 cell lysates after 5.5 h of bacterial growth in the cytosol (Fig. 7). We observed two LLO bands (Fig. 7, lane 1), one comigrating at 58 kDa with in vitro-labeled LLO (lane 3) and a smaller species, which was likely a product of proteolytic degradation. As a control to demonstrate that the bands immunoprecipitated by the anti-LLO antibody were indeed LLO, we used strain DP-L2817, in which chromosomal LLO has been replaced by perfringolysin O (PFO) in a 10403S background. PFO is a related cytolysin which has a slightly lower molecular mass (54 kDa) and which, like LLO, allows L. monocytogenes to escape from vacuoles (15). It is important to note that DP-L2817 produces a mutant PFO molecule which, unlike wild-type PFO, is not toxic for host cells (15); therefore, we were able to achieve an infection level similar to that of 10403S at the time of lysis (data not shown). As illustrated in Fig. 7, lane 2, we did not observe a 58-kDa band in lysates of DP-L2817-infected cells, indicating that the bands detected in 10403S lysates (lane 1) were indeed LLO. However, we did observe a faint band at 54 kDa, indicating that anti-LLO antibody B3-19 weakly cross-reacts with PFO (Fig. 7, lane 2). Similarly, ActA was immunoprecipitated with a polyclonal anti-ActA antibody. As reported previously (3), ActA migrates as three bands, with the two slower-migrating species representing phosphorylated forms (Fig. 7, lane 4). The negative control for this immunoprecipitation was J774 cells infected with DP-L1942 (Fig. 7, lane 5), from which actA has been deleted (3).
FIG. 7.
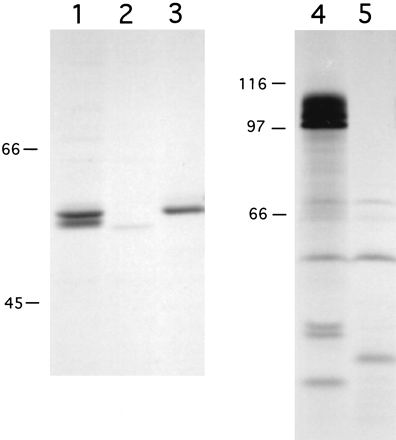
Immunoprecipitation of LLO and ActA from L. monocytogenes-infected J774 cells. Bacterial proteins were metabolically labeled during growth in J774 cells or in vitro and immunoprecipitated with monoclonal anti-LLO antibody B3-19 (lanes 1 to 3) or polyclonal anti-ActA antibody 2553 (lanes 4 and 5). Lanes 1 and 4, 10403S-infected J774 cells; lane 2, DP-L2817 (10403S::pfoH438Y)-infected J774 cells; lane 3, supernatant from 10403S cultured in vitro as described in Materials and Methods; lane 5, DP-L1942 (10403SΔactA)-infected J774 cells. An autoradiograph depicting one of two experiments with similar results is shown. The exposure times for the visualization of ActA and LLO by autoradiography were 18 h and 7 days, respectively. For quantitation of ActA and LLO as described in Results, the gel was scanned with a Molecular Dynamics PhosphorImager, and the resulting images were analyzed with ImageQuant software (Molecular Dynamics). Numbers at left are in kilodaltons.
The relative levels of intracellular LLO and ActA depicted in Fig. 7 by autoradiography were quantitated by phosphorimaging analysis. First, it was determined that both the anti-LLO and the anti-ActA antibodies precipitated approximately 25% of the available protein by our protocol (see Materials and Methods; data not shown). Taking into account that LLO and ActA have four and nine methionines, respectively, the results indicated that approximately 70-fold more ActA than LLO was present in the cytosol of infected J774 cells. As shown in Table 1, transcriptional control accounted for approximately threefold more intracellular actA expression than hly expression. Therefore, our results indicated that additional mechanisms, possibly mediated by host cells, control the relative levels of ActA and LLO produced during intracellular growth.
DISCUSSION
The results of this study demonstrate differential regulation of two essential virulence genes, LLO and ActA, during L. monocytogenes growth in vitro and in the cytosol of mammalian cells. Whereas LLO was produced both in broth cultures and in the cytosol, ActA was efficiently produced only in the cytosol. Further, ActA was present in significantly larger quantities than LLO in the cytosol, and our results indicate that while this finding was due in part to transcriptional control, it is clear that other factors play a significant role in regulating the levels of cytosolic ActA compared to LLO.
hly and actA, like all of the known L. monocytogenes virulence genes, are under the control of PrfA, which activates transcription by binding to 14-bp palindromic DNA sequences in the −35 promoter regions of these genes (6, 9, 21, 32). PrfA interacts with perfect palindromic sequences in the hly and plcA promoter regions, while upstream of actA and mpl, PrfA binds to sequences with single-base-pair substitutions which create imperfect palindromes. It has been postulated that due to a high-affinity interaction with the perfect palindromic sequences of the hly and plcA promoters, the activation of hly and plcA requires less PrfA than the activation of actA and mpl (9, 33). In this scenario, the hly and plcA promoters would be activated more readily under conditions in which PrfA is limiting than the mpl and actA promoters. Support for this heirarchical model of virulence gene expression was provided by use of transcriptional lacZ fusions expressed under the control of isopropyl-β-D-thiogalactopyranoside (IPTG)-inducible PrfA in a heterologous B. subtilis host (33). In that study, hly was activated more efficiently and with faster kinetics than actA in broth cultures. Similar results were obtained in a study in which the L. monocytogenes p60 gene expressed from a multicopy plasmid was used as a transcriptional reporter (4).
We confirmed these results by using single-copy transcriptional fusions expressed under the control of endogenous PrfA in L. monocytogenes. Our results indicated that hly is activated at levels up to 10-fold higher than actA in broth. In addition, we extended the results of Sheehan et al. (33) by examining promoter activity during intracellular growth. In contrast to the in vitro situation, both actA and hly were expressed at significant levels in the cytosol, and we observed an approximately threefold increase in the activity of the actA fusion relative to the hly fusion in the cytosol. This threefold difference could have been a result of actA transcription proceeding from both the actA and mpl promoters. In this regard, it is known that mpl transcription is activated in the cytosol, where its product plays a role in processing the precurser form of PlcB (20). Results from a number of recent studies have led to speculation that PrfA can undergo a conformational change or associate with itself or another molecule in response to its environment (2, 22, 31, 32). By changing conformation or assuming a higher-order structure, PrfA could gain the capacity to interact differentially with virulence gene promoters or to function both as an activator and as a repressor, depending on the environmental conditions. As such, a mechanism for differential transcription of hly and actA would invoke changes in the quantity or quality of PrfA protein produced by bacteria growing in broth cultures and in the cytosol of mammalian cells.
The results of in vitro transcriptional reporter assays are consistent with the relative amounts of LLO and ActA detected in broth cultures. However, the relative amounts of LLO and ActA detected in the cytosol could not be attributed to control at the transcriptional level alone. While transcription appears to account for only threefold more ActA expression than LLO expression, we were able to detect approximately 70-fold more ActA than LLO in the cytosol, indicating an additional level of regulation in the production of these two virulence factors. Our results are consistent with those of Villanueva et al., who demonstrated that cytosolic LLO could be immunoprecipitated from infected cells only in the presence of an inhibitor of the proteasome, suggesting that LLO is made in the cytosol and then rapidly degraded (36). In contrast to LLO, ActA is the most abundant surface or secreted bacterial protein detected in infected cells and has a relatively long half-life, approximately 2.5 h (3, 23). Based on these data and evidence that LLO is rapidly degraded in the cytosol, our results may be explained largely by the relative resistance or susceptibility of the two proteins to degradation by the proteasome or other cytosolic proteases.
Since we have not addressed message stability or translational regulation in this study, we cannot rule out a role for these bacterial control mechanisms in the accumulation of high levels of ActA relative to LLO in the cytosol. However, it is intriguing to speculate that L. monocytogenes is so highly adapted to life in the intracellular environment that it relies primarily on the host cell to regulate the intracellular levels of virulence proteins in such a way as to enhance its intracellular growth and survival. Since LLO is a pore-forming toxin, its rapid degradation in the cytosol may ensure that the host cytoplasmic membrane remains intact during infection, allowing L. monocytogenes to propagate without encountering antimicrobial substances in the extracellular space. This interpretation is supported by the observation that replacement of chromosomal LLO with PFO, a related cytolysin from the extracellular pathogen Clostridium perfringens, is toxic for host cells (14). PFO has a half-life of more than 1 h in the cytosol, and genetic selection for nontoxic PFO molecules led to the isolation of mutants with a shorter intracellular half-life (15). While the degradation of cytosolic LLO may benefit L. monocytogenes, it is also advantageous to the host, as the release of LLO 91-99, a nonameric peptide which associates with major histocompatibility complex class I molecules, leads to the induction of a cellular immune response (26). In fact, the appearance of LLO 91-99 in association with surface class I molecules provided early evidence that LLO, which functions in vacuolar lysis, is also produced in the cytosol. Our results, along with those of Villanueva et al., (36) confirm that LLO continues to be made by L. monocytogenes after it leaves the phagocytic vacuole.
Studies described above in which LLO was replaced with PFO, a cytolysin from a pathogen which has not adapted to the intracellular lifestyle, support the notion that the apparent intracellular instability of LLO is functionally relevant (14, 15). Similarly, evidence that the intracellular stability of ActA is also functionally relevant was recently obtained. L. monocytogenes expressing an ActA mutant molecule which is slightly more susceptible to degradation in the cytosol has a significant defect in its ability to spread to adjacent cells (23). Finally, an example of the importance of regulation at the level of the host cell was provided for the broad-range phospholipase PC-PLC. The inactive precurser form of PC-PLC is processed to the active form in Lamp1+ vacuoles, where it functions in bacterial spread to adjacent cells, but is rapidly degraded in the cytosol (20). Like that of LLO, the production of PC-PLC, a potentially cytotoxic virulence protein, is regulated, at least in part, by the host cell.
Finally, the transcriptional reporter system described in this report may be of use for the isolation of novel L. monocytogenes virulence genes. A similar system based on intracellular chloramphenicol selection was developed to identify novel Salmonella typhimurium genes induced in the intracellular environment (19). Similarly, pLCR may be used to isolate novel L. monocytogenes genes expressed preferentially in the intracellular environment. Because of its relatively rapid intracellular growth rate, we observed powerful selection for L. monocytogenes carrying fusions expressed in the mammalian cytosol over L. monocytogenes carrying an inactive fusion after approximately 5 h of intracellular growth under chloramphenicol selection conditions (Fig. 4). These experiments suggested that L. monocytogenes is particularly amenable to this approach for the isolation of genes which play a role in intracellular growth and virulence.
ACKNOWLEDGMENTS
We thank David Brown for excellent advice and assistance in the construction of plasmids. We also thank Pascale Cossart for the monoclonal anti-LLO antibody.
This work was supported by Public Health Service grants AI-26919 and AI-27655 (to D.A.P.) and American Cancer Society postdoctoral fellowship award PF-3975 (to M.A.M.).
REFERENCES
- 1.Ambulos N P, Jr, Duvall E J, Lovett P S. Analysis of the regulatory sequences needed for induction of the chloramphenicol acetyltransferase gene cat-86 by chloramphenicol and amicetin. J Bacteriol. 1986;167:842–849. doi: 10.1128/jb.167.3.842-849.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bockmann R, Dickneite C, Middendorf B, Goebel W, Sokolovic Z. Specific binding of the Listeria monocytogenes transcriptional regulator PrfA to target sequences requires additional factors and is influenced by iron. Mol Microbiol. 1996;22:643–653. doi: 10.1046/j.1365-2958.1996.d01-1722.x. [DOI] [PubMed] [Google Scholar]
- 3.Brundage R A, Smith G A, Camilli A, Theriot J A, Portnoy D A. Expression and phosphorylation of the Listeria monocytogenes ActA protein in mammalian cells. Proc Natl Acad Sci USA. 1993;90:11890–11894. doi: 10.1073/pnas.90.24.11890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bubert A, Kestler H, Gotz M, Bockmann R, Goebel W. The Listeria monocytogenes iap gene as an indicator gene for the study of PrfA-dependent regulation. Mol Gen Genet. 1997;256:54–62. doi: 10.1007/s004380050545. [DOI] [PubMed] [Google Scholar]
- 5.Camilli A, Tilney L G, Portnoy D A. Dual roles of plcA in Listeria monocytogenes pathogenesis. Mol Microbiol. 1993;8:143–157. doi: 10.1111/j.1365-2958.1993.tb01211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chakraborty T, Leimeister-Wachter M, Domann E, Hartl M, Goebel W, Nichterlein T, Notermans S. Coordinate regulation of virulence genes in Listeria monocytogenes requires the product of the prfA gene. J Bacteriol. 1992;174:568–574. doi: 10.1128/jb.174.2.568-574.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Christie G E, Farnham P J, Platt T. Synthetic sites for transcription termination and a functional comparison with tryptophan operon termination sites in vitro. Proc Natl Acad Sci USA. 1981;78:4180–4184. doi: 10.1073/pnas.78.7.4180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cutting S M, Vander Horn P B. Genetic analysis. In: Harwood C R, Cutting S M, editors. Molecular biological methods for Bacillus. New York, N.Y: John Wiley & Sons, Inc.; 1990. pp. 27–74. [Google Scholar]
- 9.Freitag N E, Rong L, Portnoy D A. Regulation of the prfA transcriptional activator of Listeria monocytogenes: multiple promoter elements contribute to intracellular growth and cell-to-cell spread. Infect Immun. 1993;61:2537–2544. doi: 10.1128/iai.61.6.2537-2544.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gaillard J L, Berche P, Sansonetti P. Transposon mutagenesis as a tool to study the role of hemolysin in the virulence of Listeria monocytogenes. Infect Immun. 1986;52:50–55. doi: 10.1128/iai.52.1.50-55.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gedde, M., and D. A. Portnoy. Unpublished data.
- 12.Goldberg A L. The mechanism and functions of ATP-dependent proteases in bacterial and animal cells. Eur J Biochem. 1992;203:9–23. doi: 10.1111/j.1432-1033.1992.tb19822.x. [DOI] [PubMed] [Google Scholar]
- 13.Gutierrez J A, Crowley P J, Brown D P, Hillman J D, Youngman P, Bleiweis A S. Insertional mutagenesis and recovery of interrupted genes of Streptococcus mutans by using transposon Tn917: preliminary characterization of mutants displaying acid sensitivity and nutritional requirements. J Bacteriol. 1996;178:4166–4175. doi: 10.1128/jb.178.14.4166-4175.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jones S, Portnoy D A. Characterization of Listeria monocytogenes pathogenesis in a strain expressing perfringolysin O in place of listeriolysin O. Infect Immun. 1994;62:5608–5613. doi: 10.1128/iai.62.12.5608-5613.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones S, Preiter K, Portnoy D A. Conversion of an extracellular cytolysin into a phagosome-specific lysin which supports the growth of an intracellular pathogen. Mol Microbiol. 1996;21:1219–1225. doi: 10.1046/j.1365-2958.1996.00074.x. [DOI] [PubMed] [Google Scholar]
- 16.Kathariou S, Metz P, Hof H, Goebel W. Tn916-induced mutations in the hemolysin determinant affecting virulence of Listeria monocytogenes. J Bacteriol. 1987;169:1291–1297. doi: 10.1128/jb.169.3.1291-1297.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kocks C, Gouin E, Tabouret M, Berche P, Ohayon H, Cossart P. L. monocytogenes-induced actin assembly requires the actA gene product, a surface protein. Cell. 1992;68:521–531. doi: 10.1016/0092-8674(92)90188-i. [DOI] [PubMed] [Google Scholar]
- 18.Maguin E, Duwat P, Hege T, Ehrlich D, Gruss A. New thermosensitive plasmid for gram-positive bacteria. J Bacteriol. 1992;174:5633–5638. doi: 10.1128/jb.174.17.5633-5638.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mahan M J, Tobias J W, Slauch J M, Hanna P C, Collier R J, Mekalanos J J. Antibiotic-based selection for bacterial genes that are specifically induced during infection of a host. Proc Natl Acad Sci USA. 1995;92:669–673. doi: 10.1073/pnas.92.3.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marquis H, Goldfine H, Portnoy D A. Proteolytic pathways of activation and degradation of a bacterial phospholipase C during intracellular infection by Listeria monocytogenes. J Cell Biol. 1997;137:1381–1392. doi: 10.1083/jcb.137.6.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mengaud J, Dramsi S, Gouin E, Vazquez-Boland J A, Milon G, Cossart P. Pleiotropic control of Listeria monocytogenes virulence factors by a gene that is autoregulated. Mol Microbiol. 1991;5:2273–2283. doi: 10.1111/j.1365-2958.1991.tb02158.x. [DOI] [PubMed] [Google Scholar]
- 22.Milenbachs A A, Brown D P, Moors M, Youngman P. Carbon-source regulation of virulence gene expression in Listeria monocytogenes. Mol Microbiol. 1997;23:1075–1085. doi: 10.1046/j.1365-2958.1997.2711634.x. [DOI] [PubMed] [Google Scholar]
- 23.Moors, M. A., and D. A. Portnoy. Unpublished data.
- 24.Nato F, Reich K, Lhopital S, Rouyre S, Geoffroy C, Mazie J C, Cossart P. Production and characterization of neutralizing and nonneutralizing monoclonal antibodies against listeriolysin O. Infect Immun. 1991;59:4641–4646. doi: 10.1128/iai.59.12.4641-4646.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pamer E G. Cellular immunity to intracellular bacteria. Curr Opin Immunol. 1993;5:492–496. doi: 10.1016/0952-7915(93)90028-q. [DOI] [PubMed] [Google Scholar]
- 26.Pamer E G, Harty J T, Bevan M J. Precise prediction of a dominant class I MHC-restricted epitope of Listeria monocytogenes. Nature. 1991;353:852–855. doi: 10.1038/353852a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park S F, Stewart G S. High-efficiency transformation of Listeria monocytogenes by electroporation of penicillin-treated cells. Gene. 1990;94:129–132. doi: 10.1016/0378-1119(90)90479-b. [DOI] [PubMed] [Google Scholar]
- 28.Perkins J B, Youngman P J. Construction and properties of Tn917-lac, a transposon derivative that mediates transcriptional gene fusions in Bacillus subtilis. Proc Natl Acad Sci USA. 1986;83:140–144. doi: 10.1073/pnas.83.1.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Portnoy D A, Chakraborty T, Goebel W, Cossart P. Molecular determinants of Listeria monocytogenes pathogenesis. Infect Immun. 1992;60:1263–1267. doi: 10.1128/iai.60.4.1263-1267.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Portnoy D A, Jacks P S, Hinrichs D J. Role of hemolysin for the intracellular growth of Listeria monocytogenes. J Exp Med. 1988;167:1459–1471. doi: 10.1084/jem.167.4.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ripio M T, Dominguez-Bernal G, Lara M, Suarez M, Vazquez-Boland J A. A Gly145Ser substitution in the transcriptional activator PrfA causes constitutive overexpression of virulence factors in Listeria monocytogenes. J Bacteriol. 1997;179:1533–1540. doi: 10.1128/jb.179.5.1533-1540.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sheehan B, Klarsfeld A, Ebright R, Cossart P. A single substitution in the putative helix-turn-helix motif of the pleiotropic activator PrfA attenuates Listeria monocytogenes virulence. Mol Microbiol. 1996;20:785–797. doi: 10.1111/j.1365-2958.1996.tb02517.x. [DOI] [PubMed] [Google Scholar]
- 33.Sheehan B, Klarsfeld A, Msadek T, Cossart P. Differential activation of virulence gene expression by PrfA, the Listeria monocytogenes virulence regulator. J Bacteriol. 1995;177:6469–6476. doi: 10.1128/jb.177.22.6469-6476.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith G A, Marquis H, Jones S, Johnston N C, Portnoy D A, Goldfine H. The two distinct phospholipases C of Listeria monocytogenes have overlapping roles in escape from a vacuole and cell-to-cell spread. Infect Immun. 1995;63:4231–4237. doi: 10.1128/iai.63.11.4231-4237.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trieu-Cuot P, Courvalin P. Nucleotide sequence of the Streptococcus faecalis plasmid gene encoding the 3′5"-aminoglycoside phosphotransferase type III. Gene. 1983;23:331–341. doi: 10.1016/0378-1119(83)90022-7. [DOI] [PubMed] [Google Scholar]
- 36.Villanueva M S, Sijts A J, Pamer E G. Listeriolysin is processed efficiently into an MHC class I-associated epitope in Listeria monocytogenes-infected cells. J Immunol. 1995;155:5227–5233. [PubMed] [Google Scholar]
- 37.Youngman P. Use of transposons and integrational vectors for mutagenesis and construction of gene fusions in Bacillus species. In: Harwood C R, Cutting S M, editors. Molecular biological methods for Bacillus. New York, N.Y: John Wiley & Sons, Inc.; 1990. pp. 221–266. [Google Scholar]