

Abstract
Duchenne muscular dystrophy (DMD) is a severe, progressive X-linked recessive disorder, caused by the absence of the dystrophin protein. A resolutive therapy for DMD is not yet available. The first approved drug for DMD patients with nonsense mutations is ataluren, approved for the treatment of children aged ≥ 2 yrs, that seems effective in slowing the disease progression. An earlier introduction of ataluren seems to give better results.
We report the case of a 14-year-old DMD patient with a nonsense mutation in exon 70, still ambulant, who started taking ataluren at 12 years and remained stable for the following two years. The patient was on steroid since the age of 6, with beneficial effects. At two-years follow-up, an optimal disease evolution was observed, associated with a constant decrease of creatine kinase blood levels. Despite the late start of the treatment, ataluren seems to have significantly contributed to the stabilization of the functional status in this patient though it cannot be excluded that the result may have been influenced by the previous favorable course of the disease. However, further studies should be planned in patients with similar age treated with ataluren to better evaluate the treatment’s results compared to the natural course of the disease.
Key words: Duchenne muscular dystrophy, target therapy, nonsense mutation, ataluren, time function tests
Introduction
Duchenne muscular dystrophy (DMD) is a disabling, life-shortening X-linked neuromuscular disease 1. Causative mutations (deletions, duplications, point mutations) involve the DMD gene, which codes for dystrophin, a large protein responsible for muscle membrane stabilization and signaling mediation 1. About 13% of DMD patients have a nonsense mutation that converts an amino acid into a premature stop codon in the dystrophin mRNA, generating a nonfunctional protein 2. Typical disease trajectory involves delayed walking, worsening motor skills, loss of ambulation, and premature death due to cardiorespiratory failure. In the last decade, an improved standard of care focused on the preservation of the ambulatory function 2 has positively modified the natural history of DMD. Corticosteroids are still the gold standard treatment to increase muscle strength 1 nevertheless, they cannot target the molecular cause of the disease. For DMD patients with nonsense mutations, a novel treatment approach has been essayed with ataluren (PTC124), able to restore the function of dystrophin by introducing an amino acid at the stop codon site to continue the mRNA translation. Ataluren is an orally bioavailable drug evaluated in two randomized, double-blind, placebo-controlled trials in Phase IIb and Phase III 3-5.
In DMD patients, motor function changes with age, anthropometric and myometric variables, genetic predisposition, training and steroid use 1. Therefore, baseline disease trajectories and reliable measures of outcome are crucial to interpret new drugs’ efficacy.
We report the case of a DMD patient with a nonsense mutation, who started ataluren at 12 years and 9 months, and maintained ambulation and an optimal functional status at two-years follow-up.
Case report
The patient, currently aged 14 years, reached independent ambulation at 15 months showing speech delay. He received a DMD diagnosis at one year of age, after an incidental registration of elevated creatin kinase (CK) values (13.369 U/L). Muscle biopsy showed a severe dystrophic muscle picture. The immunohistochemical staining for dystrophin was absent with COOH and rod domains monoclonal antibodies, while a faint signal was detected with NH2 domain antibody. The WB analysis showed the absence of the dystrophin band, while traces of an immunoreactive band were detected with the rod-domain antibody. Genetic analysis showed the nonsense mutation c.10141C > T; R3381X in exon 70 of the DMD gene. The stop codon generated by the nonsense mutation is TGA and the 3’ adjacent nucleotide is A (TGAA tetranucleotide).
At the age of 6 years, daily deflazacort was started (0.9 mg/kg/day) without major side effects. At 10 years, though ejection fraction and heart volumes were within the normal limits, angiotensin-converting-enzyme inhibitor therapy was introduced to delay the left ventricle dysfunction. Motor skills and performance remained stable, showing a positive profile as demonstrated by 6MWT and time function tests values (TFTs) (Fig. 1). The patient practiced physical rehabilitation since the time of diagnosis and regularly utilized ankle- and leg-foot orthoses. A cognitive evaluation with Weschler III test showed an Intelligence Quotient (IQ) of 75. At 12 years and 3 months, a muscle MRI was performed that showed hypertrophy of triceps surae and a mild to moderate fat substitution of adductor magnus, vastus lateralis and intermedius (Fig. 2).
Figure 1.
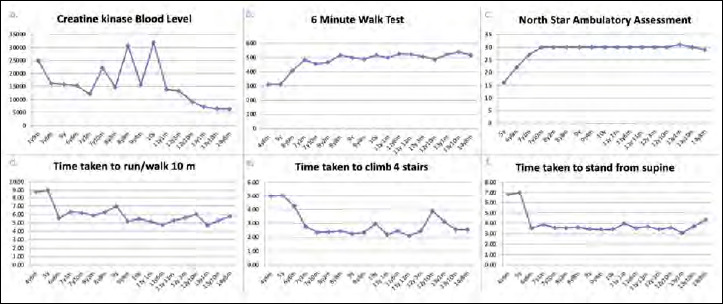
Time function tests and CK levels during follow-up. A) creatine kinase value (U/L) at different follow-up visits; B) 6 minute walk test results (meters) at different follow-up visits; C) North Star Ambulatory Assessment (score) at different follow-up visits; D) time taken to run/walk 10 meters (seconds) at different follow-up visits; E) time taken to climb 4 stairs (seconds) at different follow-up visits; F) time taken to stand from supine (seconds) at different follow-up visits.
Figure 2.
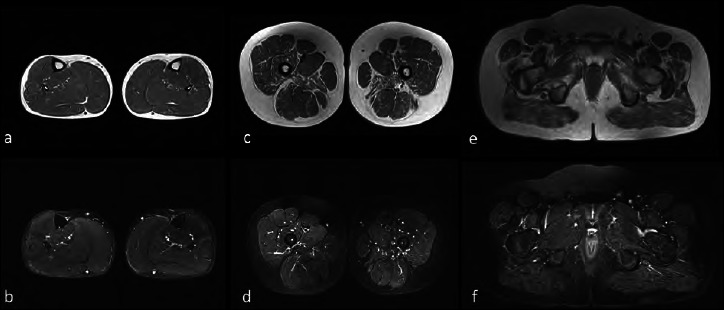
Muscle MRI. Muscle MRI findings performed at the age of 12 years and 3 months. (A,C,E) Axial turbo spin echo T2 weighted (w), and (B,D,F) short tau inversion recovery (STIR) axial images at the level of the lower leg (A,B), thigh (C,D) and pelvic girdle (E,F) are shown. No significant fat substitution or edema is evident at the level of the leg muscles, in which, by contrast, a mild triceps surae hypertrophy can be detected (A,B). At the level of the thigh (C,D) the adductor magnus, vastus lateralis and intermedius show signs of fat substitution bilaterally, without associated muscle edema. At the level of the pelvic girdle the involvement of the glutei is evident with fat substitution (E) and no corresponding edema (F).
The treatment with ataluren was started at 12 years and 9 months, at the dosage of 40 mg/kg/day. At that time, he was still ambulant with a waddling gait, lumbar lordosis and showed a positive Gowers’ sign. He was in the pre-pubertal period with short stature (129 cm, less than 3rd percentile) and a body mass index (BMI) of 26; mild cushingoid features, tightness of the Achilles tendons, and proximal muscle weakness were present. Echocardiography revealed normal kinetic and dimensional parameters. Spirometric examination showed normal vital capacity and expiratory volumes. CK values were about 12.000/13.000 U/L in several determinations.
Soon after the ataluren introduction, the patient reported a prompt subjective improvement in muscle strength. The 6MWT values registered in the two years after the start of the drug were superimposable to those achieved in the previous years, with the best performance obtained at one year follow-up (covered distance: 540 m). Also the North Star Ambulatory Assessment (NSAA) score and the other TFTs remained stable over the last 2 years. Interestingly, the CK blood levels decreased to 6000-7000 U/L, at last follow-up visits (Fig. 1).
Discussion
The only approved drugs for DMD concern deletions amenable for exon 53 skipping (Viltolarsen, in the USA and Japan), and deletions amenable for exon 51 skipping (Eteplirsen). Ataluren was recently approved for the treatment of nonsense mutations. Several randomized clinical trials 3-5 showed that ataluren is well-tolerated and able to improve dystrophin expression, reducing the rate of muscle degeneration as measured using 6MWT. In Italy, ataluren is approved by the Italian Drug Agency (AIFA) for DMD boys since two years of age. Recent studies show that ataluren can be of benefit in non-ambulatory as well as in ambulatory patients 6. However, for a better interpretation of the data in clinical practice, different outcome measures are necessary according to the different stages of the disease.
Furthermore, a positive relation between earlier drug introduction and better results has also been described 7.
Our patient has shown a good response to the steroid treatment and physiotherapy, compared to DMD pairs. According to the model of Mercuri et al. 8, he relies on the stable/improvement disease class. Moreover, according to the most recent studies 7, our patient was not among those most likely to benefit from treatment with ataluren, as 6MWD was 439 m at baseline. Nonetheless, the response to ataluren contributed to the stabilization of the previously shown functional profile and even to the improvement of some motor performances. Compared to the population of nonsense and deletion DMD patients described by Hamuro et al. 9, our patient shows a disease course that can be considered exceptional (6MWT > 500 m at > 14 years of age). Furthermore, in our patient, both values of 6MWT and of 10 m walking test - which measure the walking function - are significantly related each other, a result usually more evident in boys with preserved functional activity. Finally, the improvement in motor performance seems to have a corresponding biochemical trend in a decrease of CK levels (more than half), observed in the last two years of treatment compared to the values recorded at the start of treatment. This pattern may reflect a treatment effect as described in the study by Finkel et al. 3, in which the majority of subjects treated with ataluren showed a decrease in CK values without any correlation with the magnitude of the response.
Given the increasing availability of trials aimed at establishing new drugs’s efficacy in DMD, reliable tools are needed to measure the outcomes and more predictable trajectories of natural diseases. DMD patients having a nonsense mutation do not appear to show a significantly different disease course than patients with other mutations. In the study of Pane et al. 10, boys with DMD gene duplications generally performed better than those with other mutations, and those skipping exon 44 had better baseline results and less drastic changes than those eligible for skipping of exon 45 or 53 10.
Our patient has a point mutation involving exon 70, for which predictable responses to active therapeutic management have not yet been reported. Considering the mild natural disease evolution, the results obtained could be explained not only by the steroid treatment or the ataluren association, but by the set of therapeutic interventions implemented. The significant results observed in particular in the 6MWT after the ataluren introduction, are likely due to the greater reliability of this test in measuring muscle endurance compared to the MRC scale.
The good response to ataluren could also be explained by predisposing genetic factors not yet fully explored. For instance, the stop+4 model may help in predicting functional changes, as shown by the recent papers of Wangen et al. 11. In this report while no difference in 6MWT were observed in patients with point mutations according to the type of stop codon, frame status of exons involved and protein domain affected, a significant difference (p < 0.05) was observed when considering the stop codon together with the 3’ adjacent nucleotide (“stop+4 model”), as patients with stop codon TGA and 3’ adjacent nucleotide G (TGAG) have a more rapid decline 12.
Limitations of the study
We are aware that the results obtained in a single case report cannot be extended to the population of nonsense DMD patients. Furthermore, the lack of a second muscle biopsy (refused by the patient’s parents) which might show the state of dystrophin after the ataluren introduction, and the relative long-term follow-up required to better describe the evolution pattern in the patient, can be considered as limitations of the study. However, while there is now supporting evidence for an early introduction of ataluren in patients with DMD, indications regarding ataluren introduction at older ages are lacking.
We are convinced that a deep clinical and genetic characterization of patients with exceptional course of disease and possibly good response to ataluren may be of interest for both clinical and research purposes, as well as that further studies on larger cohorts, exploring modifying genes and other genetic determinants are necessary to clarify the stratification criteria for DMD, and to identify patients who can most benefit from the ataluren treatment.
Acknowledgement
We thank the patient and his family for the cooperation. We are grateful to Professor Anna Pichecchio for the imaging analysis.
Conflict of interest statement
The Authors declare no conflict of interest.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author contributions
AB, LP: conceptualization; AB, LP: methodology; AB, LP, AG, DV, MP: investigation; AB, LP, AG, DV, MP: data curation; AB, LP: writing-original draft preparation; AB, LP, AG, DV, MP: writing-review and editing. All Authors have read and agreed to the published version of the manuscript.
Ethical consideration
The informed consent to the treatment was collected from the patient’s parents.
Figures and tables
References
- 1.Bushby K, Finkel R, Birnkrant DJ, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol 2010;9:77-93. https://doi.org/10.1016/S1474-4422(09)70271-6 10.1016/S1474-4422(09)70271-6 [DOI] [PubMed] [Google Scholar]
- 2.Birnkfrant DJ, Bushby K, Bann C, et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol 2018;17:347-361. https://doi.org/10.1016/S1474-4422(18)30025-5 10.1016/S1474-4422(18)30025-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Finkel RS, Flanigan KM, Wong B, et al. Phase 2a study of ataluren-mediated dystrophin production in patients with nonsense mutation Duchenne muscular dystrophy. PLoS One 2013;8:e81302. https://doi.org/10.1371/journal.pone.0081302 10.1371/journal.pone.0081302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bushby K, Finkel R, Wong B, et al. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve 2014:50:477-487. https://doi.org/10.1002/mus.24332 10.1002/mus.24332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McDonald CM, Campbell C, Torricelli RE, et al. Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017;390:1489-1498. https://doi.org/10.1016/S0140-6736(17)31611-2 10.1016/S0140-6736(17)31611-2 [DOI] [PubMed] [Google Scholar]
- 6.Ebrahimi-Fakhari D, Dillmann U, Flotats-Bastardas M, et al. Off-label use of ataluren in four non-ambulatory patients with nonsense mutation Duchenne muscular dystrophy: effects on cardiac and pulmonary function and muscle strength. Front Pediatr 2018;6:316. https://doi.org/10.3389/fped.2018.00316 10.3389/fped.2018.00316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McDonald CM, Muntoni F, Penematsa V, et al. Ataluren delays loss of ambulation and respiratory decline in nonsense mutation Duchenne muscular dystrophy patients. J Comp Eff Res 2022;11:139-155. https://doi.org/10.2217/cer-2021-0196 10.2217/cer-2021-0196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mercuri E, Signorovitch JE, Swallow E, et al. Categorizing natural history trajectories of ambulatory function measured by the 6-minute walk distance in patients with Duchenne muscular dystrophy. Neuromuscul Disord 2016;26:576-583. https://doi.org/10.1016/j.nmd.2016.05.016 10.1016/j.nmd.2016.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hamuro L, Chan P, Tirucherai G, et al. Developing a natural history progression model for Duchenne muscular dystrophy using the six-minute walk test. CPT Pharmacometrics Syst Pharmacol 2017;6:596-603. https://doi.org/10.1002/psp4.12220 10.1002/psp4.12220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pane M, Mazzone ES, Sormani MP, et al. 6 minute walk test in Duchenne MD patients with different mutations: 12 month changes. PLoS One 2014;9:e83400. https://doi.org/10.1371/journal.pone.0083400 10.1371/journal.pone.0083400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wangen JR, Green R. Stop codon context influences genome-wide stimulation of termination codon readthrough by aminoglycosides. Elife 2020;23;9:e52611. https://doi.org/10.7554/eLife.52611 10.7554/eLife.52611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brogna C, Coratti G, Rossi R, et al. The nonsense mutation stop+4 model correlates with motor changes in Duchenne muscular dystrophy. Neuromuscul Disord 2021;31:479-488. https://doi.org/10.1016/j.nmd.2021.02.015 10.1016/j.nmd.2021.02.015 [DOI] [PubMed] [Google Scholar]