

Abstract
Viruses are important pathogens in tropical areas; most of them, especially the tropical hemorrhagic fevers, produce mucocutaneous manifestations. More than any other kind of pathogen, viruses have the possibility for being widespread, since they have a greater probability of mutation than do bacteria, can cross species barriers easily, and infect both human beings and animals in habitats with a great biodiversity. Tropical habitats also have been subject to major ecologic changes in the last few decades, exposing humans to direct contact with these viruses and allowing hemorrhagic fevers due to new emergent viruses such as flaviviruses, filoviruses, arenaviruses, and hantaviruses to become major threats to public health. The collapse of eradication programs in many countries, as well as population increases and ecologic modifications, have led to the spread of dengue and yellow fever to large portions of the world owing to the dissemination of vectors, especially mosquitoes, with broad ecologic ranges. Viruses previously restricted to some geographic areas, such as Rift Valley fever, Crimean-Congo hemorrhagic fever, West Nile fever, and monkeypox are now affecting new countries and populations. Other viruses such as herpes B infection often affect travelers and animal handlers in most parts of the world. Dermatologic lesions occur in all these diseases and can facilitate a rapid diagnosis, leading to control of the virus and helping prevent possible outbreaks.
Tropical viral infections with dermatologic manifestations are common in many tropical countries of the Old World and the New (Table I). Most of these viruses used to be restricted to specific geographic areas where the viruses, their hosts, and vectors coevolved for long periods. There are, however, some situations in which the tropical virus disseminates to areas previously free from the pathogen. A common situation involves an accidental contamination of a traveler, tourist, or worker who has contact with a tropical virus and spreads the disease.1, 2 A contaminated animal can also act as a vector of the disease to new areas or countries, as was recently reported with monkeypox in several midwestern states of the United States.2 Both apes and monkeys, because of their close genetic relationship to humans, are efficient vectors of this kind of dissemination, as the study of monkeypox infection and Herpesvirus simiae can easily show.
Table I.
%Tropical viruses with dermatologic findings
| Virus family | Characteristics | Virus |
|---|---|---|
| RNA viruses | ||
| Flaviviridae | Single-stranded RNA, positive-sense, enveloped | Dengue virus |
| Yellow fever virus | ||
| West Nile virus | ||
| Filoviridae | Single-stranded RNA, negative-sense, enveloped | Ebola virus |
| Marburg virus | ||
| Bunyaviridae | Single-stranded RNA, negative-sense, enveloped | Hantavirus complex, Rift Valley fever virus, and Crimean-Congo hemorrhagic fever virus |
| Arenaviridae | Single-stranded RNA, negative-sense, enveloped | Lassa virus, Tacaribe complex virus (Junin, Sabia, Guanarito, and Machupo viruses) |
| Retroviridae | Single-stranded RNA, positive-sense | Human T-cell leukemia virus type I (HTLV-I) |
| DNA viruses | ||
| Herpesviridae | Double-stranded DNA, enveloped | Herpesvirus simiae (B virus) |
| Poxviridae | Double-stranded DNA, enveloped | Monkeypox virus |
Another situation that allows dissemination of tropical viruses involves a major modification in the environment, with deep ecologic consequences, such as the uncontrolled urbanization and increasing poverty in much of the developing countries.3, 4 The resurgence of dengue and yellow fever in Central and South America, as well as the West Nile virus in the United States and Canada in the last decade, is due, in large part, to the erosion of eradication programs against the Aedes aegypti mosquito and human population growth.5
Other common themes in the story of these tropical viruses are that humans enter new areas where viruses are circulating or rodents that harbor the viruses can enter ecologically disturbed areas to carry infection to humans, and viruses may then spread to involve larger geographic areas.3, 4, 5 These are the main patterns involved in filovirus, hantavirus, and arenavirus infections, some of the most deadly causes of hemorrhagic fevers.
Arenaviruses
The Arenaviridae are a family of viruses whose members are generally associated with benign infections in restricted rodent hosts but cause a severe, often lethal, disseminated disease in humans.6 Arenaviruses are pleomorphic enveloped RNA viruses, 110-130 nm in diameter, with cellular ribosomes incorporated into the virion.7, 8
The genome consists of 2 distinct single-stranded viral RNA species, called L and S.6 The arenaviruses have ambisense genomes: the 3′ half is antisense, whereas the 5′ half is positive-sense.7 The envelope that surrounds the virion contains 2 major glycoprotein components (GP1, GP2) that appear as spikelike or clublike projections with variable spacing along the virus envelope.6, 7, 8 During morphogenesis, sandy-appearing granules are found within the unstructured interior of new virions. These particles give arenaviruses their name (arena is the Latin word for sand).6, 9 Suitable conditions for transmission occur in areas where humans come in contact with rodent urine that contains virus.10 These viruses are transmitted from mother rodents to their offspring during pregnancy and thus remain in the rodent population generation after generation.11, 12 Persistent viremia and viruria in rodents result from a slow or insufficient immune response when immunologically immature rodent fetuses or neonates are infected. The disease in humans is acute. Although the cellular receptor has not been identified, it must be highly conserved and widely distributed because arenaviruses replicate in a wide variety of cell types.6, 11
In every case in which a human arenavirus disease has been studied, an interface between humans and rodents has been described; the one common characteristic of these zoonotic infection patterns is human contact with rodent excreta.10, 11, 12 The most common situations are the exposure of agricultural workers to rodents during crop harvesting or the incidental infection of hunters who have contact with rodents in forests.11, 12, 13 The fact that only a specific species of rodent is infected in each geographic area and the severe disease that arenaviruses cause in all other species, including other rodents, suggest that the benign infection in restricted rodent hosts is an important measure developed by these animals to survive.6, 8 They probably adopted this symbiotic relationship with these viruses to eliminate competition for food and shelter in the local ecology. The infection of human beings is incidental to the natural cycle of the viruses.10, 12
Although aerosol and respiratory spread, as well as cuts and abrasions in the skin, are suspected, the portal of entry of arenaviruses is still unknown.7, 8 Most infections are asymptomatic or so mild that they cannot be distinguished from common respiratory or gastrointestinal viruses, but about 30% of infected persons may present a more severe clinical course.10 The infection presents such a wide spectrum of manifestations that a typical case is difficult to characterize. Nevertheless, headache, photophobia, apathy, and confusion are common. This infection can be temporarily debilitating but is rarely fatal, and complete recovery is the rule.6, 11
In 1934, the prototypic arenavirus, lymphocytic choriomeningitis (LCM) virus, was first isolated during serial monkey passage of human material that was obtained from a fatal infection in the first documented epidemic of St. Louis encephalitis, a totally unrelated virus.8 LCM virus was the first recognized cause of aseptic meningitis in humans.6, 8, 9 Arenaviruses have been divided into 2 groups on the basis of whether the virus is found in the Old World (Eastern Hemisphere) or the New World (Western Hemisphere).8, 9 Of the 15 arenaviruses known to infect animals, all live in tropical regions.10 LCM virus is the only arenavirus to exist in both areas but is classified as an Old World virus.8, 9 The following are the major viruses and the other recognized Arenaviridae listed in relationship to their rodent reservoirs (Table II). 13
Table II.
%Arenaviruses according to their ecologic characteristics and rodent reservoirs
| Arenavirus | Rodent | Location | Habitat | Human contact |
|---|---|---|---|---|
| Old World arenaviruses | ||||
| LCM virus | Mus musculus, Mus domesticus (house mouse), Mesocricetus auratus (Syrian hamster) | Europe, Asia, and the Americas | Peridomestic, grasslands | Primarily within households |
| Lassa virus | Mastomys natalensis | West Africa | Savanna, forest clearing | Primarily within houses |
| Classic New World arenaviruses | ||||
| Junin virus | Calomys masculinus (corn mouse), Akodon azarae (grass field mouse), Bolomys obscurus (dark field mouse) | Argentina (pampas) | Grasslands, cultivated fields, and hedgerows | Occupational in fields |
| Machupo virus | Calomys callosus (vesper mouse) | Bolivia | Peridomestic, grasslands | Primarily within houses |
| Guanarito virus | Sigmodon alstoni (cane mouse) | Venezuela | Grasslands, brush | Within houses |
| Sabia virus | Unknown | Brazil | Unknown | Associated with several human cases, including a laboratory worker in Connecticut |
| Selected less common New World arenaviruses | ||||
| Tamiami virus | Sigmodon hispidus (cotton rat) | Florida | Grasslands, marsh | Unclear |
| Whitewater Arroyo virus | Neotoma albigula (white-throated wood rat) | California, New Mexico | Grasslands | Unclear |
Of the 6 common arenaviruses pathogenic to humans, 5 cause severe hemorrhagic fever associated with a mortality of about 15% among hospitalized patients.14 They affect circumscribed areas in West Africa (Lassa fever),15, 16 the Argentine pampas (Junin virus),17, 18, 19 the savannas of the Beni province of Bolivia (Machupo virus),20 Venezuela (Guanarito virus),21, 22, 23 and the southeast province of Brazil (Sabia virus).24, 25 The sixth, LCM virus, is the only one more widely distributed but causes only milder neurologic infection.6, 9 LCM and Lassa viruses are associated with Old World rats and mice (family Muridae, subfamily Murinae),13 while the Latin American viruses, also known as the Tacaribe complex viruses,10, 11 are associated with New World rodents (family Muridae, subfamily Sigmodontinae).13
The restricted areas affected by most of these viruses, West Africa and Latin America, may reflect the geographic distribution of their natural hosts.13 Most of these regions in South America and Africa are near forests or still have a low population density.6, 8 The progressive occupation of previous wild areas will expose humans to new emergent viruses.26 In recent years, a significant number of LCM infections has been attributed to silently infected pet hamsters and field mice (Mus musculus) in biomedical laboratory colonies, which explains the wide distribution of LCM virus.6 Other arenaviruses have been associated, sporadically, with laboratory contamination, such as the Sabia virus at Yale University in 199625 and Machupo virus during necropsies.20
In the United States, the California Department of Health Services identified, between June 1999 and May 2000, evidence of infection with an arenavirus in 3 female patients hospitalized with similar fatal illnesses.27 All 3 patients had acute respiratory distress syndrome, and 2 developed liver failure and hemorrhagic manifestations.27, 28 Arenavirus-specific RNA was detected in 1 or more materials from each patient by means of polymerase chain reaction.27 The assay confirmed the presence of the Whitewater Arroyo virus prototype strain,27, 28, 29 an arenavirus recovered from a Neotoma albigula (white-throated wood rat) from New Mexico in early 1996 (Table II).30 Whitewater Arroyo virus is found in North America among wood rats (Neotoma spp) and has not been known previously to cause disease in humans.28 Of 20 Neotoma species, 9 occur in the United States.28, 29 The geographic range of these species incorporates most of the United States. At least 5 of the 9 U.S. species may harbor the virus; however, a complete description of its distribution requires further study. The abundance and habits of wood rats suggest a great potential contact between Neotoma species and humans.28 Other arenaviruses, such as the Tamiami virus, are present in cotton rats, rodents with a large distribution in the United States (Table II).31
Knowledge of the multiplication of arenaviruses is fragmentary. Most of what is known comes from studies of the LCM virus.6 LCM virus replicates in a wide variety of cell types.6, 8 Although the virus receptor has not been identified, it must be highly conserved and widely distributed. Transcription of the genome and replication is confined to the cytoplasm. The small RNA in the virion encodes in the negative sense a nucleoprotein and in the positive, or message, sense a precursor glycoprotein, which is cleaved into 2 virion glycoproteins (GP1 and GP2).6, 7, 8, 9, 14 The large RNA in the virion encodes in the negative sense an RNA-dependent RNA polymerase and in the positive sense a zinc-binding protein that binds to the ribonucleoprotein complex.9 The virus buds from the plasma membrane, incorporating host lipids into the virus membrane.9, 14
The onset of the hemorrhagic fevers caused by Lassa, Junin, Machupo, Sabia, and Guanarito viruses may be insidious, with the disease presentation within 7 to 14 days after infection simply as pyrexia, headache, sore throat, and myalgia.17, 18, 19, 20, 21, 25 Virus can be recovered from the blood and serum for up to 3 weeks after onset of the infection, and Lassa virus can be recovered from the urine for up to 5 weeks.15 Hemorrhagic phenomena, heralded by unremitting high fever, can begin after day 5 of illness and are followed by dehydration and hemoconcentration, shock syndrome, hemorrhagic manifestations, and cardiovascular collapse.15, 17, 18, 19, 20, 32 The pantropic nature of these viruses is revealed by their presence in various dysfunctional organs.32
Compared with the dramatic clinical course and mortality, the gross pathology is unimpressive and of little help in constructing a pathogenetic scheme.32 Complete autopsies have not been performed on patients with Lassa15, 16 and Bolivian hemorrhagic fever10, 11, 20; however, autopsies performed on patients with Argentine hemorrhagic fever show a lack of deposited immunoglobulin and complement component C3 in the kidneys and small blood vessels.17, 18, 32 Mediators released from infected cells have a potential role in the pathogenesis of dysfunction of some target organs.32 Although LCM virus can produce severe human disease, characterized by prominent neurologic manifestations, pathologic lesions have not been studied extensively.16 However, in the mouse model, the immune response against LCM virus (specifically in the T-cell compartment) is central to the development of fatal neurologic disease.32 Furthermore, mice infected with a lethal dose of this virus can invariably be saved by means of treatment with antibody to interferon-α and -β, which raises the possibility that endogenous interferon-α and -β enhance the immunopathology.6, 14, 32
Antibodies develop after overt human infection with arenaviruses and are detectable with enzyme-linked immunosorbent assay (ELISA), complement fixation, neutralization, and fluorescent antibody techniques.33 The humoral response is exceptionally slow, but ultimately, a long-lasting and vigorous production of antibodies occurs.7, 33 Antibodies demonstrable by means of immunofluorescence usually are the first to appear and are followed by complement-fixing antibodies. The complement-fixing antibodies are short-lived, with titers diminishing rapidly 5 to 12 months after onset. In contrast, neutralizing antibodies remain detectable for many years.33, 34 Cell-mediated immunity is important in arenavirus infections of experimental animals; it is sometimes harmful but is probably beneficial in human infections, at least for Lassa fever.14 In Lassa fever, passive transfer of early-convalescent-phase human antibodies does not protect monkeys or guinea pigs, whereas late antibodies neutralize virus and are protective.14, 15, 16 Induction of interferon-α has been shown in patients with Argentine hemorrhagic fever. In general, arenaviruses are relatively resistant to the antiviral action of interferon-α and -β.6 Interferon titers are significantly higher in those who die than in survivors (perhaps owing to higher levels of virus in the former).6, 9, 34 All evidence suggests that viral clearance in humans is complete and that chronic infection is not established. Reinfection with Lassa virus is possible but appears to be uncommon.15
The clinical presentation of all these South American hemorrhagic fever diseases and Lassa fever is similar in several ways.10, 11, 14, 15 The incubation period is around 2 weeks.14 Disease onset usually begins with insidious progression of general malaise and fever over a 5-day period. In clinical illness, the onset is gradual, with fever, malaise, headache, sore throat, cough, nausea, vomiting, diarrhea, myalgia, and chest and abdominal pain.8, 14 The fever may be either constant or intermittent with spikes. Inflammation of the throat and eyes is common.10
Progression beyond this stage is the norm for all but Lassa fever.10, 11, 15 In severe cases, hypotension or shock, pleural effusion, hemorrhage, seizures, encephalopathy, and swelling of the face and neck are frequent. Approximately 15% of hospitalized patients die. The disease is more severe in pregnant patients, and fetal loss occurs in more than 80% of cases.10, 11
Hemorrhaging, neurologic signs and symptoms, and leukopenia and thrombocytopenia are common.11 Dehydration and unremitting high fever are followed by hemoconcentration, shock syndrome, hemorrhagic manifestations on skin and mucous membranes (Fig 1), and cardiovascular collapse.18, 20, 32 Purpura, minor gingival bleeding, hemorrhagic bullae on mucous membranes, and disseminated nonpalpable petechiae on the skin are frequently seen.20, 21
Fig 1.
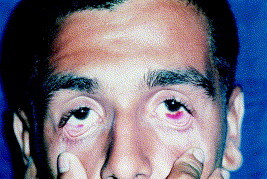
Brazilian hemorrhagic fever with conjunctival purpura. (Courtesy of A. Pereira, MD, PhD.)
Hair loss and loss of coordination may occur during convalescence. In addition, deafness occurs in 25% of patients, with only half recovering some function after 1 to 3 months.8, 10, 14 Immunity to reinfection occurs after infection, but the length of this period of protection is unknown.34
Other hemorrhagic fevers from viral origin, such as Ebola infection and hemorrhagic dengue fever, should be considered in the differential diagnosis. Meningococcemia and other diseases leading to sepsis, with disseminated intravascular coagulation and shock, can be confused with the Latin American hemorrhagic fever diseases.18
Junin18 and Machupo20 viruses are isolated by intracerebral inoculation of newborn hamsters. Lassa virus can be isolated by inoculation of Vero cells.15 All arenaviruses share some antigenic determinants detected with ELISA,33 and all appear to share antigenic determinants in the ribonucleoproteins, as well as antigenically distinct determinants in their outer glycoproteins.6 Positive immunofluorescent staining of acetone-fixed infected cells is definitive for more than just family identification, since with limiting dilutions of antibody, Old World arenaviruses can be readily distinguished from New World viruses (Table II).6, 9, 10 Arenavirus species may be identified from their unique surface glycoproteins and infectivity neutralization.6 Most of the cases, however, are diagnosed from the epidemiologic and clinical data. The arenaviruses must be suspected if they are prevalent in geographic areas where infections have occurred and in regions known to harbor reservoir rodent species.
Although several classes of antiviral compounds have been found with specific in vitro activity against arenaviruses, only ribavirin has been proven to be effective against Lassa fever15, 16 and Machupo viral infection in humans.35 The efficacy of this drug against the other members of the family has not been established yet.36, 37 It may be used at any point in the illness, as well as for postexposure prophylaxis.14 Junin and Guanarito viruses have been found to be highly sensitive to ribavirin in vitro, but efficacy in humans has not been established yet.17, 21, 38 The adult dose for Lassa fever (with hepatitis or hemorrhagic manifestations, or both) is 2 g (30 mg/kg) intravenously initially; 1 g (15 mg/kg) intravenously every 6 hours for 4 days, and then 500 mg (7.5 mg/kg) intravenously every 8 hours for 6 days.14, 15, 16 The suggested prophylactic dose is 600 mg by mouth 4 times daily for 10 days. Ribavirin is contraindicated in pregnant women. Systemic ribavirin use causes dose-related anemia and hyperbilirubinemia related to extravascular hemolysis, and at higher doses, a bone marrow suppression of the erythroid elements may occur.35, 38
The only measure that already has been proven to be safe and effective is a successful rodent control program in areas affected by these viruses,13 as described for Bolivian hemorrhagic fever.13, 20 Although elimination of rodents that shed virus has protected humans, it is not a reasonable long-term approach for other arenaviruses, because of the rodent ecology.13
Vaccines are under development.11, 18 A live attenuated Junin virus vaccine has been tested in about 100 volunteers, with a resultant humoral and cell-mediated response frequency of more than 95 percent.39, 40 A Lassa virus glycoprotein gene has been cloned and expressed in vaccinia virus. This vaccine has offered a high degree of protection against disease and death in monkeys challenged with the intact Lassa virus.14 Plasma from convalescent patients has become the single specific therapeutic adjunct for patients severely ill with Bolivian and Argentine hemorrhagic fevers.20, 40 Physicians attending patients are convinced that such plasma is valuable if given during the first 8 days of disease, but more controlled trials are needed.38 Use of plasma is not indicated yet for patients with Lassa fever.14 At least 7 serologically distinct strains of Lassa virus have been isolated; animal studies suggest that effective therapy should involve geographic matching of immune plasma and virus strain. In view of the frequency of Lassa virus transmission from person to person in a hospital setting, strict measures must be taken to isolate patients who have or are suspected to have the disease.14, 15, 16 Isolation of patients with the other pathogenic arenaviruses is desirable.
Filoviruses
Filoviruses are filamentous, enveloped particles with a negative-sense, single-stranded RNA genome, approximately 19 kilobase pairs long. Genes are defined by conserved transcriptional start and termination signals and are arranged linearly.41 A single glycoprotein forms the spikes on the virion surface. The nucleocapsid contains the RNA and four viral structural proteins, including the virus-encoded polymerase.41, 42 Within the Filoviridae there is a single genus, Filovirus, and a separation into 2 genotypes, Marburg and Ebola. Ebola is subdivided into 3 subtypes: Zaire, Sudan, and Reston.42, 43 They are similar in morphology, density, and electrophoresis profile, with a close serologic relationship among them. At electron microscopy, Filoviridae virions are pleomorphic, usually b-shaped, containing a nucleocapsid (20 nm in diameter) surrounded by a helical capsid (50 nm in diameter).41, 42 Transcription of the virions takes place in the cytoplasm of the infected cells, but the mechanism of virus entry is still unknown.41
Filovirus transcription and replication are mediated by a single virus-encoded polymerase in the cytoplasm of the infected cell. The negative-sense RNA genome is transcribed into monocystronic, polyadenylated subgenomic RNA species, which are translated into 7 structural proteins.41, 42 In the case of Ebola viruses, a single glycosylated nonstructural protein is expressed by means of RNA editing or frameshifting at a specific place in the glycoprotein open reading frame.41, 42 Replication works through a full-length, positive-sense antigenome that serves as a template for negative-sense progeny genomes. Particles mature at the plasma membrane.42, 44
This family is indigenous to Africa. Marburg and Ebola both cause severe hemorrhagic fevers. Marburg virus was first recognized in laboratory workers exposed to tissues and blood from African green monkeys (Cercopithecus aethiops) in Marburg, Germany, in 1967.45 Since then, sporadic, virologically confirmed Marburg disease cases have occurred in Zimbabwe, South Africa, and Kenya.46 Ebola virus first emerged in 2 major disease outbreaks, with mortalities of 88%, in Zaire47 and Sudan48 in 1976. Sporadic cases and minor outbreaks occurred again in the same locations previously affected and in Gabon,49 the Ivory Coast, and Uganda after 1977.50, 51
The Ebola-Reston virus was first discovered in 1989 in monkeys imported from the Philippines, which had died in a holding facility in Reston, Virginia, just outside Washington, DC.52 While monkeys suffer a severe disease often leading to death, the limited information available indicates that humans may not become clinically ill. However, this information is based only on the isolation of Reston virus from one asymptomatically infected animal handler identified during the original outbreak and a few seroconversions that were not associated with clinical disease.52, 53 A formal quarantine procedure for imported monkeys was developed after the original Reston episode, and it is with this system that the current cases apparently were identified.52 The Ebola-Reston virus also has been isolated from 2 nonhuman primates (cynomolgus) held at a quarantine facility in Texas.53 The monkeys had also been imported from the Philippines.
Serologic studies suggest filoviruses are endemic in many countries of the central African region.54, 55 Although serologic data based on ELISA are of only limited reliability, they at least suggest the possible occurrence of subclinical infections caused by known or unknown filoviruses.54, 55 The mode of primary infection in any natural setting is unknown for Marburg and Ebola viruses.55 All secondary cases, however, were due to intimate contact with infected patients.56, 57 After hospitalization of an infected person, the disease spreads rapidly via contaminated needles and contact with blood, which seems to be the most important route of contamination.56 In each Ebola outbreak in Africa, the initial patient spreads the disease to close family members through intimate contact.47, 48, 49, 50 The natural reservoir of these 2 viruses remains unknown. However, on the basis of available evidence and the nature of similar viruses, they must be zoonotic and normally maintained in an animal host that is native to the African continent. The infected monkeys were not considered to be natural hosts, since they also died with hemorrhagic symptoms.56, 57
Filoviridae viruses are usually recovered from acute-phase sera and have been found in throat washes, urine, soft tissue effusates, semen, and anterior eye fluid.46 They have also been regularly isolated from autopsy material, such as spleen, lymph node, liver, and kidney tissues.51 Clinical and biochemical findings support anatomic observations of extensive liver involvement, renal damage, changes in vascular permeability, and activation of the clotting cascade.32, 57 Visceral organ necrosis is the consequence of viral replication in parenchymal cells. However, no single organ is sufficiently damaged to cause death. Fluid distribution problems and platelet abnormalities indicate dysfunction of endothelial cells and platelets.32, 44, 46 The shock syndrome in severe and fatal cases seems to be mediated by virus-induced release of humoral factors such as cytokines.46, 56, 57 Filoviral glycoproteins carry a presumably immunosuppressive domain, and immunosuppression has been observed in infected monkeys.46
Filoviruses cause a severe hemorrhagic fever in both human and nonhuman primates.32 After an incubation period of 4-15 days, onset is sudden, marked by high fever, fatigue, headache, erythematous transient rashes, and myalgia.50, 51 Abdominal pain, sore throat, nausea, vomiting, cough, arthralgia, diarrhea, and pharyngeal and conjunctival vasodilatation58 may follow these symptoms. Patients are dehydrated, apathetic, and disoriented. They may develop a characteristic, nonpruritic, maculopapular centripetal rash associated with varying degrees of erythema, which desquamates by day 5 or 7 of the illness.58
Hemorrhagic manifestations develop at the peak of the illness and are of prognostic value. Bleeding into the gastrointestinal tract is the most prominent, besides petechiae and hemorrhages from puncture wounds and mucous membranes.58 Most of the patients develop severe hemorrhagic manifestations in the next few days, with bleeding from multiple sites such as the gastrointestinal tract, oropharynx, and lungs.50, 58 The skin and mucous membranes also have ecchymoses, disseminated nonpalpable petechiae, and massive gingival bleeding that usually herald a fatal outcome.58
Laboratory parameters are less characteristic, but the following are associated with the disease: leukopenia (as low as 1,000 per microliter); left shift with atypical lymphocytes; thrombocytopenia (50,000-100,000 per microliter); markedly elevated serum transaminase levels (typically aspartate aminotransferase exceeding alanine aminotransferase); hyperproteinemia; and proteinuria.58, 59 Prothrombin and partial thromboplastin times are prolonged, and fibrin split products are detectable. In a later stage, secondary bacterial infection may lead to elevated white blood counts. There is fever in patients, who eventually recover after about 5 to 9 days.58, 59 In cases ending in death, clinical signs occur at an early stage and the patient dies between day 6 and 16, from hemorrhage and hypovolemic shock. Mortality is between 30% and 90%, depending on the virus, and the highest rate has been reported for Ebola Zaire.47, 54, 55 Ebola Reston seems to possess a low pathogenicity in humans and may even be nonpathogenic.54 Convalescence is prolonged and sometimes is associated with myelitis, recurrent hepatitis, psychosis, or uveitis. An increased risk of abortion exists for pregnant women, and clinical observations indicate a high death rate among children of infected mothers.54, 55, 58
In tropical settings, identification of filoviral hemorrhagic fever may be difficult, since the most common causes of severe, acute, febrile disease are malaria and typhoid fever. The differential diagnosis should also include other viral hemorrhagic fevers, such as yellow fever, dengue infection, and arenaviral hemorrhagic fevers, as well as meningococcemia, leptospirosis, and idiopathic thrombocytopenic purpura.51 Travel, treatment in local hospitals, and contact with sick persons or wild and domestic monkeys are useful historical features in returning travelers, especially in those from Africa.3, 4 Diagnosis of single cases is extremely difficult, but occurrence of clusters of cases with prodromal fever followed by cases of hemorrhagic diatheses and person-to-person transmission are suggestive of viral hemorrhagic fever, and containment procedures must be initiated. In filoviral hemorrhagic fever, prostration, lethargy, wasting, and diarrhea are usually more severe than those symptoms observed in patients with other viral hemorrhagic fever. The rash is characteristic and extremely useful in the differential diagnosis.3, 4
Laboratory diagnosis can be achieved in 2 different ways: by measurement of the host-specific immunologic response to the infection and by detection of viral antigen and genomic RNA in the infected host. The most commonly used assays to detect antibodies to filoviruses are the indirect immunofluorescence assay, immunoblot, and ELISA (direct IgG and IgM ELISA, and IgM capture assay).44, 57, 60 Direct detection of viral particles, viral antigen, and genomic RNA can be achieved with electron microscopy (negative contrast, thin-section), immunohistochemistry, immunofluorescence on impression smears of tissues, antigen detection ELISA, and reverse transcriptase polymerase chain reaction.53, 54
Attempts to isolate the virus from serum or other clinical material should be performed with Vero or MA-104 cells (monkey kidney cells).53, 60 However, most filoviruses do not cause extensive cytopathogenic effects at primary isolation. The most useful animal, besides nonhuman primates, is the guinea pig, which develops fever within 10 days upon primary infection.60
There is no standard treatment for Ebola and Marburg infection, and virus-specific treatment does not exist.61 Supportive therapy should be directed toward the maintenance of effective blood volume and electrolyte balance.58 Shock, cerebral edema, renal failure, coagulation disorders, and secondary bacterial infection must be managed. Heparin treatment should be considered only when there is clear evidence of disseminated intravascular coagulopathy.61 Filoviruses are resistant to the antiviral effects of interferon, and administration of interferon to monkeys has failed to increase survival rate or to reduce virus titer.42, 61 Ribavirin does not affect filoviruses in vitro and thus is probably not of clinical value, in contrast to its efficacy against other viral hemorrhagic fevers.61 Isolation of patients is recommended, and protection of medical and nursing staff is required. Monkeys caught in the wild are an important source of the introduction of filoviruses. Quarantine of imported nonhuman primates and professional handling of animals will help prevent introduction into humans.42, 52
Even though filoviral hemorrhagic fever outbreaks have been rare and were mainly restricted to a small number of cases, vaccines would be of value for both medical personnel in Africa and for laboratory personnel.57 Cross protection among different Ebola subtypes in experimental animal systems has been reported, a finding that suggests the general value of vaccines.42 Inactivated vaccines have been developed by means of treatment with formalin or heating of cell culture-propagated Marburg and Ebola, subtypes Sudan and Zaire.57, 58, 61 Protection, however, has been achieved only by carefully balancing the challenge dose and virulence. Because of the biohazardous nature of the agents, recombinant vaccines would be an attractive approach in the future.61
Dengue and yellow fever
The Flavivirus genus is composed of more than 68 arthropod-transmitted viruses, of which 30 are known to cause human disease.62, 63 The flaviviral infections include dengue and yellow fever, as well as Japanese encephalitis and tick-borne encephalitis.63 It is important to consider this group of viruses in the clinical differential of central nervous system infection, hemorrhagic fever, and acute febrile illnesses with arthropathy.64 Two diseases, however— yellow fever and dengue infection—have caused large epidemics in Africa and the Americas for more than 400 years. Dengue is also found in large portions of southeast Asia, and the prevalence is increasing worldwide. Both diseases are transmitted by the bite of mosquitoes.63, 64, 65
The pathophysiology of both infections was largely inferred from vaccine studies in rhesus monkeys with the attenuated vaccines.62 After inoculation in rhesus monkeys, initial replication of the virus in local lymph nodes was followed by blood-borne spread and subsequent replication mostly in regional lymph tissue, spleen tissue, and bone marrow, and later in the liver, lung, and adrenal glands.62
Dengue, also known as breakbone fever, occurs as an epidemic disease in tropical and subtropical regions of Asia and Africa, but transmission has been increasing geographically during the past few decades.66 Successive introduction of new serotypes into the Caribbean and Central and South America has occurred since 1977.64 Dengue hemorrhagic fever (DHF) was first reported in the Caribbean in 1981, and since 1982, epidemics have occurred in 14 Latin American countries.67 One hundred million cases of dengue fever are reported yearly, making it one of the most important viral diseases in the world.64 Some 2.5 billion people—two fifths of the world's population—are now at risk of dengue.68
In 2001 alone, there were more than 609,000 reported cases of dengue in the Americas, of which 15,000 cases were DHF.68, 69 This figure is more than double the number of dengue cases recorded in the same region in 1995. Not only is the number of cases increasing as the disease spreads to new areas, but also explosive outbreaks are occurring.68 In 2001, Brazil reported more than 390,000 cases, including more than 670 cases of DHF.70
Yellow fever is constantly present with low levels of infection in some tropical areas of Africa and the Americas. The viral presence can amplify into regular epidemics under the right demographic and environmental conditions (hot and humid climates). The name of the disease is due to the jaundice that affects some patients.71, 72, 73, 74
A biting mosquito transmits both viruses (horizontal transmission) and can also pass the virus via infected eggs to its offspring (vertical transmission).63, 75, 76, 77 Therefore, the mosquito is the true reservoir and the vector for both diseases. Several different species of mosquitoes of the genera Aedes and Haemogogus transmit the yellow fever virus.77 Control programs successfully eradicated mosquito habitats in the past, especially in South America.76 However, these programs have been reduced over the last 30 years and the mosquito population has increased. The main vector of dengue, Aedes aegypti, flourishing in mankind's urban and suburban environments, has spread the disease to many parts of the world.63 Aedes aegypti, which can also transmit yellow fever, presents a broad ecologic range and spread throughout the world mostly via ships.
It is surmised that slave-trading vessels, infested with Aedes aegypti, introduced the yellow fever virus from West Africa, with similar outbreaks occurring in port cities in the New World and in Europe.72 Sanitation measures, such as piped water, greatly diminished the transmission of the disease. The viral cause of yellow fever was not discovered until after 1928, which led to Theiler's discovery of the attenuated 17D vaccine strain in the 1930s that earned him a Nobel Prize.78 The last epidemic of yellow fever in North America occurred in New Orleans in 1905, during which more than 3,000 cases were associated with 452 deaths.71, 72 Because Aedes aegypti now has reinfested the southeastern United States, autochthonous transmission in the United States is possible.69
There are 4 serotypes of the dengue virus, an RNA-containing flavivirus, which explain the major features of the disease: classic dengue, DHF, and mild dengue.64, 66 Some of the findings about this disease are, however, still an enigma. Children are generally unaffected by classic dengue but are subject to as high as 50% mortality with DHF.73 Persons recently arriving in endemic regions usually contract the classic form, while long-time residents in the same area generally get DHF.74 Yellow fever presents a similar pattern, with 2 distinct genetic types (called topotypes) distributed in East and West Africa and South America.63, 75 While dengue is mostly an urban disease,63 yellow fever has 2 different transmission cycles: one sylvatic, in which infected mosquitoes bite humans entering the forest, and the other characterized by the urban cycle in which migrants introduce the virus into areas with high human population density.75, 76 Yellow fever has a more restricted geographic distribution than dengue, occurring from 15° north to 10° south of the equator, an area that includes most of the north and central areas of Africa, nine South American countries, and several Caribbean islands. Residents of Brazil, Bolivia, Colombia, Ecuador, and Peru are considered at greatest risk.75, 76
Classic dengue fever is a febrile viral syndrome of sudden onset, characterized by fever for 2-5 days, severe headache, intense myalgia, arthralgia, retro-orbital pain, and sometimes a diffuse morbilliform rash that may be pruritic and heals with desquamation.79 DHF is more likely to develop if an person previously infected with one serotype is later inoculated with a different viral strain.73, 74 It is recognized primarily in children younger than 15 years and is associated with a more severe course, characterized by vomiting, facial flushing, circumoral cyanosis, and weakness, as well as cool and clammy extremities.73 Minor bleeding phenomena such as epistaxis, petechiae, and gingival bleeding may occur at any time, but major bleeding phenomena such as menorrhagia and gastrointestinal hemorrhage are poor prognostic indicators (Fig 2).80
Fig 2.
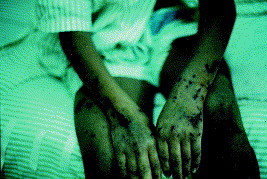
Dengue hemorrhagic fever with petechiae and ecchymoses.
Diffuse capillary leakage of plasma is responsible for the hemoconcentration. With hemoconcentration and thrombocytopenia, the patient is considered to have DHF and is classified according to the World Health Organization classification (Table III).81, 82 DHF is a potentially deadly complication that is characterized by high fever, hemorrhagic phenomena—often with enlargement of the liver—and in severe cases, circulatory failure.81, 82 The illness commonly begins with a sudden rise in temperature accompanied by facial flush and other nonspecific constitutional symptoms of dengue fever. The fever usually continues for 2 to 7 days and can be as high as 40°C or 41°C, possibly with febrile convulsions and hemorrhagic phenomena.81, 82 In moderate DHF cases, all signs and symptoms abate after the fever subsides (grades I and II). In severe cases, the patient's condition may suddenly deteriorate after a few days of fever; a drop in temperature is followed by signs of circulatory failure, and the patient may go rapidly into a critical state of shock and die within 12 to 24 hours (dengue shock syndrome) or quickly recover after appropriate volume replacement therapy.67, 73, 74 The case-fatality of DHF with dengue shock syndrome is 10% or higher if untreated. With supportive treatment, fewer than 1% of such cases succumb.
Table III.
%Dengue hemorrhagic fever classification
| Grade | Signs and symptoms |
|---|---|
| I | Thrombocytopenia and hemoconcentration; absence of spontaneous bleeding |
| II | Thrombocytopenia and hemoconcentration; presence of spontaneous bleeding |
| III | Thrombocytopenia and hemoconcentration; hemodynamic instability: filiform pulse, narrowing of the pulse pressure (<20 mmHg), cold extremities, and mental confusion |
| IV | Thrombocytopenia and hemoconcentration; declared shock, patient pulseless and with arterial blood pressure of 0 mmHg (dengue shock syndrome) |
Yellow fever is characterized by an initial acute phase that includes fever, muscle pain, headache, and vomiting. The high fever often is associated paradoxically with a slow pulse. After 4 days, the condition of most patients improves and symptoms disappear.83 However, 15% enter a “toxic phase,” during which jaundice and severe abdominal pain with vomiting develop. Bleeding can occur from the mouth, nose, and eyes. Hemorrhage and erosion of the gastric mucosa lead to hematemesis, popularly known as “black vomit.”83, 84 Hepatocellular damage is characterized by lobular necrosis with the subsequent formation of Councilman bodies.84 Findings in the central nervous system can be attributed to cerebral edema and hemorrhages compounded on metabolic disturbances. The bleeding diathesis of this disease can be attributed to reduced hepatic synthesis of clotting factors, thrombocytopenia, and platelet dysfunction.83, 84 Kidney function deteriorates, and 50% of the patients in this phase will die within 10 to 14 days, with complete kidney failure.76, 84
Yellow fever and dengue can be confused with malaria, typhoid, hemorrhagic viral fevers, viral hepatitis, and poisoning.85 Serologic assays can help detect antibodies in both diseases, and virus-specific nucleic acid sequences can be detected with polymerase chain reaction.66 Laboratory diagnosis of yellow fever in travelers is dependent principally on serologic testing of serum immunoglobulins. IgM testing by means of ELISA is the preferred method.86 This assay is 95% sensitive when serum specimens are collected 7 to 10 days after the onset of illness. A positive tourniquet test is usually present in dengue patients, even with mild clinical symptoms.80
Treatment of both diseases is nonspecific and supportive. For patients with severe bleeding and shock syndrome, measures to correct hypovolemia, hypoxia, and shock can reduce complications and death. The use of high doses of corticosteroids has not been shown to alter mortalities in this situation.63, 84 No specific antiviral therapy is available for yellow fever. Specific chemotherapies under investigation include interferon and ribavirin.86 When interferon is administered to monkeys within 8 hours of infection, mortality is reduced; however, interferon is ineffective when given 24 hours after infection.86 The use of interferon in combination with other immune-enhancing drugs still is undergoing research. Ribavirin, although effective in vitro, has not been shown to be effective in vivo because of an inability to achieve sufficient concentrations in the blood.86, 87
Vaccination is the single most important measure for preventing yellow fever, since it is safe and highly effective.76 A single dose provides protection for 10 years, and the side effects are rare. Vaccination is highly recommended for travelers to high-risk areas in South America and Africa, and several of these countries have agreed to incorporate yellow fever vaccine into their routine national vaccination programs.88, 89 An effective vaccine against dengue is a far more difficult task, since different strains of the dengue virus are present in various countries and a future infection with another serotype can predispose to DHF.90
Prevention and control of the arthropod vector is reliant on insecticides, barrier measures, protective clothing, bed netting, and insect repellents.76 In Asia and the Americas, Aedes aegypti breeds primarily in man-made containers like earthenware jars, metal drums, and concrete cisterns used for domestic water storage, as well as discarded plastic food containers, used automobile tires, and other items that collect rainwater. In Africa it also breeds extensively in natural habitats such as tree holes and leaf axils. In recent years, Aedes albopictus, a secondary dengue vector in Asia, has become established in the United States, several Latin American and Caribbean countries, and parts of Europe.91 The rapid geographic spread of this species has been attributed largely to the international trade in used tires.67, 70
Vector control is implemented by means of environmental management and chemical methods. Proper solid waste disposal and improved water storage practices, including the covering of containers to prevent access by egg-laying female mosquitoes, are among methods encouraged through community-based programs.70 The application of appropriate insecticides to larval habitats, particularly those considered useful by householders (eg, water storage vessels), prevent mosquito breeding for several weeks but must be reapplied periodically.67, 70 Biocontrol efforts include the use of predatory fish to reduce larvae populations and plankton management, which is sometimes effective.75 During outbreaks, emergency control measures may also include the application of insecticides as space sprays to kill adult mosquitoes using portable or truck-mounted machines or even aircraft.67, 70
West nile virus
West Nile virus (WNV), another member of the family Flaviviridae, is responsible for West Nile encephalitis, which in most cases presents in subtle or asymptomatic forms.92, 93 The infection may affect human beings, horses, dogs, and some domestic poultry.92 The main reservoir for the virus is birds of the crow family: crows, ravens, magpies, and blue and gray jays. The WNV is spread from bird to bird and accidentally to other animals by means of mosquitoes. Humans and other mammals are a dead-end host for WNV.92, 93
The disease is endemic in East Africa,90 first appeared in eastern North America in 1999, and has spread steadily across the country over the past few years.93, 94, 95 The virus was detected in birds as far west as Manitoba, and mosquito pools in Winnipeg tested positive in 2002.96 Previous studies show that Culex salinarius transmits WNV efficiently by bite; furthermore, WNV has been isolated from Cx. salinarius after the 2000 outbreak in New York.97 Culex salinarius is also known to carry similar viruses like St. Louis encephalitis. Because it has an opportunistic feeding habit and can transmit the virus by bite, Cx. salinarius may be an ideal bridge vector between the bird-mosquito cycle of WNV and humans.96, 98 Because Cx. tarsalis feeds mainly on birds and its close relative is a known threat to spread WNV, this species also must be considered a major threat in the United States.95, 96
Approximately 80% of persons infected with WNV have no signs or symptoms. Only a small number (<1%) develop severe neurologic disease. The incubation period is approximately 3 to 14 days. The West Nile fever is a febrile illness of sudden onset often accompanied by malaise, anorexia, nausea, eye pain, headache, and myalgia.94, 98 The clinical manifestations of the severe cases include fever and neurologic disease, with ataxia and extrapyramidal signs, myelitis, seizures, and optic neuritis. Severe muscle weakness and flaccid paralysis are common findings, together with maculopapular or morbilliform rash involving the neck, trunk, arms, or legs in about 20% of patients.94, 98
The most efficient diagnostic method is detection of IgM antibody to WNV in serum or cerebrospinal fluid collected within 8 days of illness onset using the IgM antibody capture ELISA.98, 99
Treatment is supportive, often involving hospitalization, intravenous fluids, respiratory support, and prevention of secondary infections in patients with severe disease. Ribavirin in high doses and interferon-α-2b were found to have some activity against WNV in vitro, but there have been no reports of controlled studies of the use of these or other medications, including steroids, antiseizure drugs, or osmotic agents, in the management of WNV encephalitis.99
Bunyaviruses
The Bunyaviridae family encompasses about 300 different viruses, 2 of them associated with severe hemorrhagic fevers and mucocutaneous manifestations in humans: Rift Valley fever (RVF) and Crimean-Congo hemorrhagic fever (CCHF).100, 101
Although primarily a zoonosis, sporadic cases and outbreaks of CCHF do affect humans.101 The disease is endemic in many countries in Africa, Europe, and Asia, and during 2001, cases or outbreaks were recorded in Kosovo, Albania, Iran, Pakistan, and South Africa.102 CCHF was first described in the Crimea (central Asia) in 1944 and was given the name Crimean hemorrhagic fever.100, 101 In 1969 it was recognized that the pathogen causing Crimean hemorrhagic fever was the same as that responsible for an illness identified in 1956 in the Congo, and linkage of the 2 place names resulted in the current name for the disease and the virus.100, 101 CCHF is a severe disease in humans, with a high mortality.101 Fortunately, human illness occurs infrequently, although animal infection may be more common. The CCHF virus may infect a wide range of domestic and wild animals. Many birds are resistant to infection, but ostriches are susceptible and may show a high prevalence of infection in endemic areas.102, 103 Animals become infected with CCHF from the bite of infected ticks. The most efficient and common vectors for CCHF appear to be members of the Hyalomma genus.104, 105 Humans who become infected with CCHF acquire the virus from direct contact with blood or other infected tissues from livestock, or they may become infected from a tick bite. The majority of cases have occurred in people involved with the livestock industry, such as agricultural workers, slaughterhouse workers, and veterinarians.105
RVF virus was first isolated in 1930 near Lake Naivasha in Kenya in 1931.100, 101 Since then, the virus has been shown to be widespread in sub-Saharan Africa and in Egypt.36, 37 Major epidemics and epizootics occurred in Egypt in 197737 (200,000 human infections with 600 deaths) and 1993, Mauritania in 1987 (200 human deaths),106 Madagascar in 1991, and eastern Africa (89,000 infections and more than 500 deaths) with the recent outbreak in 1997-98 in Kenya, Tanzania, and Somalia.36 In 2000, the ministries of health of Yemen and Saudi Arabia received reports of unexplained hemorrhagic fever in humans and associated animal deaths during the first confirmed occurrence of RVF outside Africa.107 More than 315 persons with suspected severe RVF were reported from primary health-care centers and hospitals, and at least 66 patients (21%) died with hemorrhagic manifestations.107 The epidemiology of RVF consists of both epizootic and interepizootic cycles. Epizootics of RVF in Africa often occur after unusually heavy rainfall. During an epizootic, virus circulates among infected arthropod vectors and mammalian hosts, particularly cattle and sheep, which represent the most significant livestock amplifiers of RVF virus.106, 108 The interepizootic survival of RVF virus is believed to depend on transovarian transmission of virus in floodwater Aedes mosquitoes.100, 101 Virus can persist in mosquito eggs until the next period of heavy rainfall, when they hatch and yield mosquitoes infected with the RVF virus.100, 101
Although RVF has a more circumscribed distribution than CCHF, the disease presents a greater potential of dissemination because of the more widespread vector. Selected North American mosquito species were evaluated as potential vectors of RVF. Field populations of Aedes canadensis, Ae cantator, Ae excrucians, Ae sollicitans, Ae taeniorhynchus, Ae triseriatus, Anopheles bradleyi-crucians, Culex salinarius, Cx tarsalis, and Cx territans perorally exposed to RVF virus readily became infected.109, 110 Infection rates ranged from 51% (65 of 127) for Cx salinarius to 96% (64 of 67) for Ae Canadensis. Disseminated infection rates were generally greater at 14 days than at 7 days after the infectious blood meal, and with the exception of An bradleyi-crucians, they were not significantly different from the pooled rate of 59% for each species tested. For most of the species, about half of the mosquitoes with a disseminated infection transmitted an infectious dose of virus to hamsters.109 While all species, with the exception of An bradleyi-crucians, transmitted virus, Ae canadensis, Ae taeniorhynchus, and Cx tarsalis had the highest vector potential of the species tested. After inoculation of virus, 100% of the mosquitoes of each species became infected.110 For most species, transmission rates were similar for inoculated mosquitoes and those that developed a disseminated infection after peroral infection. Viral titers of transmitting and nontransmitting-disseminated mosquitoes were similar for all species tested. These data suggest that, if RVF virus were introduced into North America, several mosquito species would be capable of transmitting it.109, 110
The clinical presentation of both diseases is similar. The incubation period is about 6 days, with a documented maximum of 13 days.103 Onset of symptoms is sudden, with fever, myalgia, dizziness, neck pain and stiffness, backache, headache, sore eyes, and photophobia. There may be nausea, vomiting, and sore throat early after infection, which may be accompanied by diarrhea and generalized abdominal pain. Over the next few days, the patient may experience sharp mood swings and may become confused and aggressive.36, 103, 111 After 2 to 4 days, the agitation may be replaced by sleepiness, depression, and lassitude, and the abdominal pain may localize to the right upper quadrant, with detectable hepatomegaly. Other clinical signs that emerge include tachycardia, lymphadenopathy, and a petechial rash both on internal mucosal surfaces, such as the mouth and throat, and on the skin.36, 103, 111 The petechiae may give way to ecchymoses and other hemorrhagic phenomena such as melena, hematuria, epistaxis, and bleeding from the gums. There is usually evidence of hepatitis. The severely ill may develop hepatorenal and pulmonary failure after the fifth day of illness.36, 103, 111
Mortality from CCHF is approximately 30%, with death occurring in the second week of illness. In those patients who recover, improvement generally begins on the ninth or tenth day after the onset of illness. RVF presents a more benign course, with severe eye disease (2%), meningoencephalitis, and hemorrhagic fever syndrome in 1% of the patients.36, 111
Diagnosis of suspected CCHF and RVF is performed in specially equipped, high-biosafety-level laboratories.102, 105, 111 IgG and IgM antibodies may be detected in serum by means of ELISA from about day 6 of illness.102, 112 IgM remains detectable for up to 4 months, and IgG levels decline but remain detectable for up to 5 years.105 General supportive therapy is the mainstay of patient management in both diseases. Intensive monitoring to guide volume and blood component replacement is required. Ribavirin has been used in the treatment of established CCHF infection with apparent benefit.112, 113 Both oral and intravenous formulations seem to be effective.113 The value of immune plasma from recovered patients for therapeutic purposes has not been demonstrated, although such plasma has been employed on several occasions.
There is no safe and effective vaccine widely available for human use against either virus.101 Persons living in endemic areas should use personal protective measures that include avoidance of areas containing tick vectors and mosquitoes. When patients with CCHF are admitted to a hospital, there is a risk of nosocomial spread of infection. In the past, serious outbreaks have occurred in this way, and it is imperative that adequate infection control measures be observed to prevent this outcome.113 Patients with suspected or confirmed CCHF should be isolated and cared for with barrier-nursing technique.112, 113 Specimens of blood or tissues taken for diagnostic purposes should be collected and handled with the use of universal precautions. Sharps (needles and other penetrating surgical instruments) and body wastes should be safely disposed of by means of appropriate decontamination procedures.112
Hantaviruses
Hantaviruses are single-stranded, negative-sense RNA viruses that encompass 25 antigenically distinguishable viral species.114 They are enveloped virus particles, measuring 80-115 nm in diameter, and belong to the family Bunyaviridae.100, 101, 115 Hantavirus has become a serious concern in the United States recently, but the disease was first identified during the Korean War in the early 1950s when about 3,000 U.S. and United Nations forces were infected.114, 115 This elusive virus was named Hantaan, in recognition of the Hantaan River, which flows through Korea, and the major clinical manifestations were hemorrhagic fever with renal syndrome (HFRS).114 Hantaviruses have caused outbreaks of hemorrhagic fevers in Russia (1913), Manchuria and Scandinavia (1932-35), and Finland (1945), but the Hantaan virus was isolated only in 1976, in Korea, from the rodent Apodemus agrarius. 114, 115, 116
A similar, though more virulent, virus gripped America in 1993 when several young, basically healthy, adult Navajo Indians died mysteriously in the Four Corners region of the United States.117 Hantavirus pulmonary syndrome (HPS), first recognized in 1993, is caused by several related viruses in the genus Hantavirus.117 The primary reservoir of one of these, Sin Nombre virus, is the deer mouse, Peromyscus maniculatus. 117, 118 Deer mice adapt to a variety of habitats and are found across most of the United States; their presence in and around homes has been implicated as a risk factor for HPS. Case-control studies of the original outbreak in the Four Corners region of the southwestern United States showed that the prevalence of Hantavirus infection in deer mice in and around urban and rural homes was 27.5% to 32.5%, with no significant difference in prevalence between homes of case and control patients.117 The first cases were identified in the southwestern United States: New Mexico, Arizona, and California.118 Now HPS has been identified in 31 states and Canada, with 280 cases reported since 1993.118
Hantaviruses are transmitted by aerosols of rodent excreta, saliva, and urine. The most common mode of transmission is inhalation of dust or dried particles that carry dried saliva or waste products of an infected rodent.119 Hantaviruses are found worldwide and appear to be host-specific to rodent genera and possibly rodent species (Table IV). 120, 121, 122, 123, 124, 125, 126, 127, 128, 129
Table IV.
%Worldwide distribution of hantaviruses
| Virus | Natural Reservoir | Distribution |
|---|---|---|
| US hantavirus | ||
| Sin Nombre Virus | Deer mouse (Peromyscus maniculatus) | Southwest United States |
| Black Creek Canal virus | Cotton rat (Sigmondon hispidus) | Southeast United States |
| Bayou virus | Rice rat (Oryzomys palustris) | Southeast United States |
| New York virus | White-footed mouse (Peromyscus leucopus) | Northeast United States |
| Other countries | ||
| Hantaan virus | Striped field mouse (Apodemus agrarius) | Asia, mainly Korea |
| Seoul virus | Domestic rat (Rattus norvegicus and Rattus rattus) | Asia and seaports worldwide |
| Puumala virus | Bank vole (Clethrionomys glareolus) | Scandinavia and western Russia |
| Dobrava virus | Yellow-necked mouse (Apodemus flavicollis) | Eastern Europe, mainly Greece |
| Khabarovsk virus | Microtus fortis | Far-eastern Russia |
Once infected, a rodent experiences a brief viremia that lasts 5 to 10 days. After this stage, the viral antigens remain present in many major organs for weeks to months.130 In spite of the presence of antibody in the rodent's serum, infectious virus is shed in the rodent's saliva, urine, and feces possibly throughout the rest of its life. Mice appear to be at their most infectious 40 days after infection with the virus.114, 115, 130 No arthropod vector has been established for hantaviruses.
In general, there are 2 seasonal peaks for almost all outbreaks of Hantavirus diseases: a small one in spring and a large one in fall.115, 130 It is suspected that this phenomenon corresponds with seasonal increases in the infection rate of the rodents and with farming cycles, by which farmers are exposed to rodents in the fields during planting and harvest periods.115, 130 Unusually great rainfall in dry parts of the country result in increased food sources for rodents and, subsequently, increased rodent populations. Fall and winter outbreaks, such as those in Greece,123 correspond to the movement of rodents from the fields into man-made structures.
Symptoms of HFRS usually occur 1 to 6 weeks after exposure to the virus. Initial onset is marked by nonspecific flulike symptoms: fever, myalgia, headache, abdominal pain, nausea, and vomiting.114, 130 There is a characteristic facial flushing and usually a petechial rash (often limited to the axilla).114, 115 Sudden and extreme albuminuria occurs about day 4; this finding is characteristic of severe HFRS.130 The hemorrhagic manifestations also include skin hemorrhages with petechiae, purpura, ecchymoses, gingival and nasal bleeding, and hematuria. Gastrointestinal bleeding is also common with hematemesis, melena, and hematochezia, with increased menstrual flow.114, 115, 116, 130 Additional symptoms include hypotension, shock, respiratory distress or failure, and renal impairment or failure.130
HPS, caused mainly by Sin Nombre virus, is primarily a lung infection; the kidneys and the skin are largely unaffected.117, 118, 119 HPS is characterized by flulike symptoms: fever, myalgia, headache, and cough. Other symptoms can include chills, abdominal pain, diarrhea, and malaise. Subsequent symptoms include coughing and shortness of breath, tachypnea, tachycardia, dizziness, arthralgia, sweating, and back or chest pain.118 Uncommon symptoms include inflammation of the eardrum, sinuses, and throat; conjunctivitis; and rhinorrhea.119 The disease progresses rapidly; further symptoms can include thrombocytopenia, hypoxemia, and interstitial pulmonary edema. Eventually, the patient experiences hypotension, shock, and respiratory distress.130 In severe cases, respiratory distress is followed by respiratory failure.130, 131
At examination, peripheral blood smears of patients with both HPS and HFRS are found to have thrombocytopenia, increased immature granulocytes, and large immunoblastoid lymphocytes, accompanied by an elevated white blood cell count.132, 133 While coagulopathy is not common, increased partial thromboplastin and prothrombin times are observed. In addition, several enzymes are elevated, including serum lactate, lactate dehydrogenase, aspartate aminotransferase, and alanine aminotransferase. Chest radiographs of HPS patients show diffuse interstitial pulmonary infiltrates, advancing to alveolar edema with severe bilateral involvement. Severe infections may have pulmonary secretions with a total protein ratio of edema fluid/serum greater than 80%. Pleural effusions may be seen in the radiograph, along with peribronchial cuffing.132, 133
Serologic examination will help detect the presence of IgG or IgM to Hantavirus strains Seoul, Hantaan, Puumala, Dobrava, and Sin Nombre, which are the most clinically important strains.133, 134 Individual strains cannot be distinguished with this assay because of antigenic similarities between the Hantavirus strains; however, geographic distribution of rodent hosts, as well as clinical manifestations of the infection, can aid in further identification.133, 134 The CDC reports that to date, primary isolation of any Sin Nombre–like viruses has never been achieved.118
The illness progresses rapidly to severe respiratory failure and shock. The crude mortality is approximately 40%-50%. The patients usually need intensive cardiopulmonary support with invasive hemodynamic monitoring. Ribavirin given intravenously has been shown to be effective during the early phase of the HFRS illness but has not shown any effectiveness for HPS to date.114, 130
Monkeypox
Monkeypox is an orthopoxvirus with enzootic circulation in rain forests of central and western Africa; the virus can be transmitted to humans and can cause a syndrome clinically similar to smallpox, with a pustular rash, fever, respiratory symptoms, and in some cases, death.135 Smallpox no longer occurs since its worldwide eradication in 1977, whereas monkeypox is still seen as a sporadic disease in parts of Africa.136 Most cases occur in remote villages of central and western Africa close to tropical rain forests where there is frequent contact with infected animals. Monkeypox is usually transmitted to humans from squirrels and primates through contact with the animal's blood or through a bite.135
African arboreal squirrels of the Funisciurus and Heliosciurus species (sun squirrels) have been implicated previously as probable reservoir hosts for monkeypox virus in Zaire on the basis of antibody data and a single viral isolate from a Funisciurus anerythrus squirrel.135, 137 To assess the potential role of squirrels as a reservoir for monkeypox virus and to estimate the seroprevalence in wild-caught species, animals were hunted by local villagers and trapped by the study team. Over 4 days, 84 animals representing 16 species were captured; all animals were examined for lesions, and serum specimens were collected from 64. Except for 1 squirrel from which skin biopsy specimens were collected, lesions suspected to be associated with monkeypox were not present on any other animal.137
After reports of ongoing cases of human monkeypox in the Democratic Republic of the Congo that represented a new pattern of the disease, the CDC organized 2 investigations in February and October 1997.138, 139 In the past, an outbreak of monkeypox would have been limited to the village and would not last long because it did not spread extensively after the first patients recovered.135, 140 However, new outbreaks in Africa indicate that monkeypox disease is changing its pattern of infection in humans.138, 139, 141 The outbreak had a much higher rate of person-to-person transmission than seen previously and spread through many generations of transmission, thus maintaining the outbreak for more than a year.139, 141 Previous studies over a 20-year period had shown that the rate of transmission of monkeypox within households was low, suggesting that the disease had a low potential for transmission from person to person.135 Outbreaks were generally self-limiting after 1 or 2 sequential transmissions.135
The ending of vaccination programs against smallpox in the late 1970s has probably led to an increase in susceptibility to monkeypox and could explain the larger size of the most recent outbreak, the higher proportion of patients aged 15 and older, and the spread through many generations of transmission.142 Since smallpox vaccination is not a routine in any country of the world, it is possible to consider that monkeypox can also disseminate to other countries and continents if the chance occurs.142
In Zaire between 1981 and 1986, 977 persons with skin eruptions not clinically diagnosed as human monkeypox underwent laboratory testing.143 About 3% of human monkeypox cases were found among 730 patients with diagnoses of chickenpox, 7.3% among cases diagnosed as “atypical chickenpox,” and 6.1% among cases of skin rash for which clinical diagnosis could not be established.143 The diagnostic difficulties were mainly based on clinical features characteristic of chickenpox: regional pleomorphism (in 46% of misdiagnosed cases), indefinite body distribution of skin eruptions (49%), and centripetal distribution of skin lesions (17%). Lymph-node enlargement was observed in 76% of patients with misdiagnoses. In the absence of smallpox, the main clinical diagnostic problem is the differentiation of human monkeypox from chickenpox. Lymphadenopathy, preeruptive fever, and slower maturation of skin lesions were the most important clinical signs to support correct diagnosis of monkeypox.142, 143
From February through August 1996, another outbreak occurred in 13 villages in the Katako-Kombe health zone in the Congo.140 A total of 92 possible monkeypox cases were identified (attack rate, 2%); 7 cases had typical active vesiculopustular skin lesions.140 In Akungula, the village with the highest attack rate (11 per 100 population), the 45 reported cases were clustered in 8 of the 44 housing compounds. Three (3%) of the 92 patients died; all were aged less than 3 years and died within 3 weeks of disease onset.140 Seventy-one clinical cases of monkeypox were submitted to a preliminary DNA phylogenetic study of this strain of virus.136 The evaluation indicated only minor genetic variation compared with other strains of monkeypox virus from previous outbreaks during the 1970s.
Vaccination against smallpox also gave protection against monkeypox.144 but during the Congo's outbreak of 1996, 18% of the infected patients had a vaccination scar on the upper left arm suggesting receipt of vaccinia vaccine.140 It is possible to speculate that ecologic modifications in Africa and the possible emergence of new serotypes of monkeypox virus are modifying the pattern of the disease, changing not only the clinical and epidemiologic characteristics of the disease but also the immunologic response to the infection, with unpredictable consequences.
In June 2003, the first cases of monkeypox in the Western Hemisphere were reported in Wisconsin, Illinois, and Indiana. All cases had direct or close contact with ill prairie dogs, which were kept as pets. These prairie dogs had been purchased from pet stores where they had been caged adjacent to Gambian rats, which had been imported from West Africa and were thought to be the source of the virus. While the most prevalent clinical feature of monkeypox in these patients was the pustular eruption (Fig 3), other signs and symptoms, in decreasing order of frequency, were fever, respiratory difficulties, lymphadenopathy, sweating, sore throat, chills, headache, and nausea or vomiting. No deaths were reported in this outbreak, which was probably related to better nutrition than in areas of Africa where the virus is endemic. Other possible reasons for the lack of mortality were the rapid diagnosis and treatment of these patients. Recommendations for therapy included smallpox vaccination within 14 days of exposure, as well as the use of cidofovir.
Fig 3.
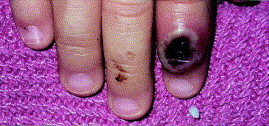
Monkeypox: primary inoculation site on right index finger, 14 days after prairie dog bites, 11 days after start of fever. Photograph courtesy of John Melski, MD, Marshfield Clinic, Marshfield, WI.
Herpes b virus
Herpesvirus simiae (B virus) is a member of the herpes group of viruses that is enzootic in rhesus (Macaca mulatta), cynomolgus (M. fascicularis), and other Old World monkeys of the genus Macaca, commonly used in biomedical research.145, 146 The seroprevalence of neutralizing antibodies to B virus in captive adult macaque populations is 73% to 100%.146, 147 Like herpesvirus simplex viral infection in human beings, B virus infection in monkeys is characterized by lifelong infection with intermittent reactivation and shedding of the virus in saliva or genital secretions, particularly during periods of stress or immunosuppression.148 B virus infection is transmitted among free-ranging or group-housed animals, primarily through sexual activity and bites. In captivity, as well as in the wild, mature macaques are more likely than immature animals to have been infected with and to shed the virus. Antibody titer to B virus indicates infection but can neither help confirm nor eliminate actual viral shedding at the time of the bite.148
B virus disease in humans usually results from macaque bites or scratches. Incubation periods may be as short as 2 days but usually are 2 to 5 weeks.148, 149 The report of a case of encephalitis caused by B virus in a monkey handler in 1932 indicated that B virus could be highly pathogenic in humans.150 Seventeen additional cases of B virus infection in humans were described through 1973,151 and 4 cases, including the first known case of person-to-person transmission of the virus, occurred in Pensacola, Florida, in 1987.152
Most documented infections, however, have occurred among biomedical research employees who had occupational exposure to macaques, although transmission also has been documented among laboratory workers handling infected central nervous system and kidney tissues.148, 153 From 1990 to 1998, 48 patients reported nonoccupational macaque bites in the United States.154 Of the six macaques for which herpes B serologic results were available, four (67%) were positive.154 Most free-ranging monkey populations are thought to be part of the exotic fauna of distant tourist destinations and wild animal parks; however, macaque species have established free-ranging feral populations in Texas and Florida.154 In such settings, contact between humans and macaques cannot be safely controlled, and workers and visitors are at risk.
Symptomatic human infection with B virus is rare; fewer than 40 cases were reported from 1933 to 1994.155, 156, 157 However, the consequences of symptomatic infection may be severe. Viral infection rapidly progresses to central loci in the spinal cord and, eventually, the brain. Of 24 known symptomatic patients whose cases were reviewed in 1992, 19 (79%) died. B virus–related disease is characterized by a variety of symptoms, which generally occur within 1 month of exposure.148 These symptoms include vesicular skin lesions at or near the site of inoculation, localized neurologic symptoms, and ultimately, encephalitis.148
In the past, most surviving human patients had moderate to severe neurologic impairment, sometimes requiring lifelong institutionalization. Acyclovir has, however, prevented progression of the disease in a limited number of patients.148 Rapid diagnosis and initiation of therapy are of paramount importance in preventing death or permanent disability in surviving patients.145, 148 The extremely high prevalence of B virus, along with the behavioral characteristics of the macaque species, makes the animals unsuitable as pets.
Human t-cell leukemia/lymphoma virus
The first pathogenic human retrovirus was discovered in 1980. At that time, Poiesz et al isolated a retrovirus from lymphocytes of a patient with cutaneous T-cell lymphoma.158 Yoshida et al also isolated the same retrovirus from a Japanese leukemia patient.159 Both viruses were shown to be indistinguishable at nucleic acid comparison, and the name human T-cell leukemia/lymphoma virus (HTLV), type 1, was proposed. In 1982, a second human retrovirus, related to but distinct from HTLV-1, was isolated from a cell line derived from a patient with hairy cell leukemia and was named HTLV-2.160 Although isolated from a leukemic patient, a clear association to a distinct disease has not been shown for HTLV-2.
HTLV-1 and -2 are related but distinct human retroviruses from the Retroviridae family.158, 159, 160 Infection of human, monkey, and rabbit T lymphocytes by HTLV-I in vitro leads to their continuous growth in tissue culture and the development of cell lines with growth characteristics of transformed cells.161 In infected patients, HTLV-1 is mainly present in CD4+ T cells but has also been found in blood dendritic cells and cells from the synovial lining of arthritic joints.161
The mode of transmission of HTLV-1 appears to be analogous to that of HIV-1.162 However, HTLV-1, which is mainly infectious by cell-to-cell contact, is less infectious than HIV-1, which can be transmitted cell-free. It has become well established that both sexual and parenteral transmission of HTLV-I are effective.162 An estimated 10 million to 20 million persons are infected with HTLV-I worldwide.163 Seroprevalence ranges from 1% up to 36%, in the Caribbean islands and in countries of equatorial Africa such as Gabon, Zaire, and the Ivory Coast.163 Small clusters of HTLV-1– and HTLV-2–seropositive populations have been reported from restricted areas in the northeast province of Brazil,164 Argentina,165 and the Middle East.163 Except for the endemic cluster in Japan, which may have been introduced by Portuguese sailors and African slaves during the 15th century, the incidence of HTLV-1 is much greater in tropical regions than in Europe or the United States.163
Pruritus is the most common cutaneous symptom of HTLV-1 infection.166 Xerosis is the most frequent dermatologic sign, and acquired ichthyosis the clinical course most observed.166, 167 However, other chronic dermatologic conditions are also associated with the virus, such as adult T-cell leukemia/lymphoma (ATL)168 and infective dermatitis.169 Uchiyama and colleagues first described ATL in 1977 as a distinct malignancy of mature T cells occurring primarily in patients born in southwestern Japan.170 At the beginning of the 1980s, HTLV-1 was linked to ATL by means of virus isolation from leukemic cells, demonstration of integrated HTLV-1 provirus in leukemic cells, and extensive seroepidemiologic studies.168, 171 Although HTLV-1 is a prerequisite for ATL development, infection with this virus does not necessarily lead to development of leukemia.168 More than 90% of infected persons remain asymptomatic carriers.162 The latent period from infection to outbreak of leukemia is 20 years or more.
Acute, prototypic ATL is a fatal malignancy of adult onset with a similar clinical presentation in all endemic areas and is the most common manifestation of ATL.171 It is characterized by the variable combination of multiorgan involvement, including hepatosplenomegaly, generalized lymphadenopathy, hepatic involvement, and the presence of multilobar leukemic cells in the peripheral blood.168 Skin lesions are prominent and polymorphous and may appear as uncharacteristic erythematous plaques, as nodular tumors, and as erythroderma.166 Both clinical and histopathologic patterns are similar to those of mycosis fungoides and Sézary syndrome. However, while most of these T-cell markers can be found in other T-cell malignancies, the strong expression of CD25 can be regarded as a distinguishing marker for ATL.168 Erythematous and edematous plaques that can progress anytime into an acute stage characterize chronic ATL.172
HTLV-1 also has been associated with chronic progressive myelopathy, known as tropical spastic paraparesis (TSP).173 Patients with TSP are generally younger than ATL patients, and the latency from infection to development of clinical neurologic symptoms has been described to be short in individual cases. In contrast to ATL, which is associated with HTLV-1 infection early in life, TSP patients frequently become infected only in adolescence.173, 174 In particular, the acquisition of HTLV-1 by means of blood transfusion has been reported to play an important role in the development of TSP.173
HTLV-1–associated infective dermatitis is a distinctive pattern of dermatitis first described in Jamaican children in 1966 by Sweet.175 It was found to be associated with HTLV-1 infection in 1990.176 Since then, infective dermatitis also has been reported from other HTLV-1 endemic areas.177, 178 It is characterized by severe chronic relapsing eczema associated with infection with Staphylococcus aureus and β-hemolytic Streptococcus. 176 Bacterial infection in infective dermatitis is resistant to treatment, and although it responds to antibiotic treatment, prolonged therapy is necessary and relapses are common after discontinuation. The condition is characterized by a perioral, perinasal, and retroauricular papular rash that progresses to eczematous plaques or nodules.176, 177, 178 The characteristic clinical picture, the recalcitrant course, and HTLV-1 seropositivity set infective dermatitis apart from other forms of recurrent eczema, including atopic eczema and seborrheic dermatitis.179 HTLV-1 genome has been detected in cultured skin biopsy material from patients with infective dermatitis, and genetic factors may predispose to the development of this cutaneous manifestation.179
LaGrenade and colleagues evaluated infective dermatitis in 50 children and observed that the mean age was 6.9 years.179 The disease was also characterized by dermopathic lymphadenitis in 67% of the cases and with a particular immunologic profile. T-cell subsets among patients with infective dermatitis revealed T-cell activation with a high percentage of HLA-DR antigen positivity, elevated CD4 and CD8 cell counts, and an increased CD4/CD8 ratio.179 Some patients with infective dermatitis also developed TSP.154 Goncalves and co-workers suggested that HTLV-1–associated infective dermatitis might be an indolent HTLV-1–associated lymphoma and recommend that tissue immunohistochemistry analyses be performed in any patient with this cutaneous manifestation.177
Conclusion
Recent reports of the first outbreak of monkeypox in the Western Hemisphere emphasize that patients with viral infections endemic to the tropics occasionally present to dermatologists in North America, Europe, and other places with temperate climates. As more immigrants move from tropical to temperate areas and as more persons from temperate climates travel or work in the tropics, it is expected that such infections will become increasingly common outside their usual range. Therefore it is important that physicians be familiar with the mucocutaneous manifestations of such infections and that they be prepared to diagnose and treat the conditions of these patients.
Footnotes
Funding source: Conselho Nacional de Desenvolvimento Cientifico e Tecnologico–Brazil.
Conflict of interest: None identified.
References
- 1.Feldmann H., Czub M., Jones S., Dick D., Garbutt M., Grolla A., et al. Emerging and re-emerging infectious diseases. Med Microbiol Immunol (Berl) 2002;191:63–74. doi: 10.1007/s00430-002-0122-5. [DOI] [PubMed] [Google Scholar]
- 2.Joyce M.P. Skin diseases of travelers. Prim Care. 2002;29:971–981. doi: 10.1016/s0095-4543(02)00039-8. [DOI] [PubMed] [Google Scholar]
- 3.Isaacson M. Viral hemorrhagic fever hazards for travelers in Africa. Clin Infect Dis. 2002;33:1707–1712. doi: 10.1086/322620. [DOI] [PubMed] [Google Scholar]
- 4.Rweyemamu M., Paskin R., Benkirane A., Martin V., Roeder P., Wojciechowski K. Emerging diseases of Africa and the Middle East. Ann N Y Acad Sci. 2000;916:61–70. doi: 10.1111/j.1749-6632.2000.tb05275.x. [DOI] [PubMed] [Google Scholar]
- 5.Colebunders R., Van Esbroeck M., Moreau M., Borchert M. Imported viral haemorrhagic fever with a potential for person-to-person transmission: review and recommendations for initial management of a suspected case in Belgium. Acta Clin Belg. 2002;57:233–240. doi: 10.1179/acb.2002.047. [DOI] [PubMed] [Google Scholar]
- 6.Albarino C.G., Posik D.M., Ghiringhelli P.D., Lozano M.E., Romanowski V. Arenavirus phylogeny: a new insight. Virus Genes. 1998;16:39–46. doi: 10.1023/a:1007993525052. [DOI] [PubMed] [Google Scholar]
- 7.Peters C.J. Human infection with arenaviruses in the Americas. Curr Top Microbiol Immunol. 2002;262:65–74. doi: 10.1007/978-3-642-56029-3_3. [DOI] [PubMed] [Google Scholar]
- 8.Charrel R.N., Lamballerie X. Arenaviruses other than Lassa virus. Antiviral Res. 2003;57:89–100. doi: 10.1016/s0166-3542(02)00202-4. [DOI] [PubMed] [Google Scholar]
- 9.Charrel R.N., Feldmann H., Fulhorst C.F., Khelifa R., de Chesse R., de Lamballerie X. Phylogeny of New World arenaviruses based on the complete coding sequences of the small genomic segment identified an evolutionary lineage produced by intrasegmental recombination. Biochem Biophys Res Commun. 2002;296:1118–1124. doi: 10.1016/s0006-291x(02)02053-3. [DOI] [PubMed] [Google Scholar]
- 10.Vainrub B., Salas R. Latin American hemorrhagic fever. Infect Dis Clin North Am. 1994;8:47–59. [PubMed] [Google Scholar]
- 11.Doyle T.J., Bryan R.T., Peters C.J. Viral hemorrhagic fevers and hantavirus infections in the Americas. Infect Dis Clin North Am. 1998;12:95–110. doi: 10.1016/s0891-5520(05)70411-6. [DOI] [PubMed] [Google Scholar]
- 12.Fritz C.L., Fulhorst C.F., Enge B., Winthrop K.L., Glaser C.A., Vugia D.J. Exposure to rodents and rodent-borne viruses among persons with elevated occupational risk. J Occup Environ Med. 2002;44:962–967. doi: 10.1097/00043764-200210000-00016. [DOI] [PubMed] [Google Scholar]
- 13.Salazar-Bravo J., Ruedas L.A., Yates T.L. Mammalian reservoirs of arenaviruses. Curr Top Microbiol Immunol. 2002;262:25–63. doi: 10.1007/978-3-642-56029-3_2. [DOI] [PubMed] [Google Scholar]
- 14.McCormick J.B. In: Encyclopedia of virology. Webster R.G., Granoff A., editors. Academic Press; San Diego: 1994. Lassa, Junin, Machupo and guanarito viruses; pp. 776–837. [Google Scholar]
- 15.Demby A.H., Chamberlain J., Brown D.W.G., Clegg C.S. Early diagnosis of Lassa fever by reverse transcription-PCR. J Clin Invest. 1994;32:2898–2903. doi: 10.1128/jcm.32.12.2898-2903.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bowen M.D., Rolin P.E., Ksiazek T.G., Hustad H.L., Baush D.G., Amby A.H., et al. Genetic diversity among Lassa virus strains. J Virol. 2000;74:6992–7004. doi: 10.1128/jvi.74.15.6992-7004.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maiztegui J.I. Clinical and epidemiological patterns of Argentine hemorrhagic fever. Bull World Health Organ. 1975;52:567–575. [PMC free article] [PubMed] [Google Scholar]
- 18.Weissenbacher M.C. Advances in Argentinean hemorrhagic fever. Medicina (B Aires) 1979;39:278–281. [PubMed] [Google Scholar]
- 19.Carballal G., Videla C.M., Merani M.S. Epidemiology of Argentine hemorrhagic fever. Eur J Epidemiol. 1988;4:259–274. doi: 10.1007/BF00144764. [DOI] [PubMed] [Google Scholar]
- 20.CDC Bolivian hemorrhagic fever—El Beni Department, Bolivia, 1994. JAMA. 1995;273:194–196. [PubMed] [Google Scholar]
- 21.Salas R., Manzione N., Tesh R.B., Rico-Hesse R., Shope R.E., Betancourt A., et al. Venezuelan hemorrhagic fever. Lancet. 1991;338:1033–1036. doi: 10.1016/0140-6736(91)91899-6. [DOI] [PubMed] [Google Scholar]
- 22.Weaver S.C., Salas R.A., Manzione N., Fulhorst C.F., Duno G., Utrera A., et al. Guanarito virus (Arenaviridae) isolates from endemic and outlying localities in Venezuela: sequence comparison among and within strains isolated from Venezuelan hemorrhagic fever patients and rodents. Virology. 2000;266:189–195. doi: 10.1006/viro.1999.0067. [DOI] [PubMed] [Google Scholar]
- 23.Fulhorst C.F., Ksiazek T.G., Peters C.J., Tesh R.B. Experimental infection of the cane mouse Zygodontomys brevicauda (family Muridae) with Guanarito virus (Arenaviridae), the etiologic agent of Venezuelan hemorrhagic fever. J Infect Dis. 1999;180:966–969. doi: 10.1086/315029. [DOI] [PubMed] [Google Scholar]
- 24.Gonzalez J.P., Bowen M.D., Nichol S.T., Rico-Hesse R. Genetic characterization and phylogeny of Sabia virus, an emergent pathogen in Brazil. Virology. 1996;221:318–324. doi: 10.1006/viro.1996.0381. [DOI] [PubMed] [Google Scholar]
- 25.Gandsman E.J., Aaslestad H.G., Ouimet T.C., Rupp W.D. Sabia virus incident at Yale University. Am Ind Hyg Assoc J. 1997;58:51–53. doi: 10.1080/15428119791013080. [DOI] [PubMed] [Google Scholar]
- 26.Garcia J.B., Morzunov S.P., Levis S., Lowe J., Calderon G., Enria D., et al. Genetic diversity of the Junin virus in Argentina: geographic and temporal patterns. Virology. 2000;272:127–136. doi: 10.1006/viro.2000.0345. [DOI] [PubMed] [Google Scholar]
- 27.Fulhorst C.F., Milazzo M.L., Carroll D.S., Charrel R.N., Bradley R.D. Natural host relationships and genetic diversity of Whitewater Arroyo virus in southern Texas. Am J Trop Med Hyg. 2002;67:114–118. doi: 10.4269/ajtmh.2002.67.114. [DOI] [PubMed] [Google Scholar]
- 28.Fulhorst C.F., Charrel R.N., Weaver S.C., Ksiazek T.G., Bradley R.D., Milazzo M.L., et al. Geographic distribution and genetic diversity of Whitewater Arroyo virus in the southwestern United States. Emerg Infect Dis. 2001;7:403–407. doi: 10.3201/eid0703.010306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Charrel R.N., de Lamballerie X., Fulhorst C.F. The Whitewater Arroyo virus: natural evidence for genetic recombination among Tacaribe serocomplex viruses (family Arenaviridae) Virology. 2001;283:161–166. doi: 10.1006/viro.2001.0874. [DOI] [PubMed] [Google Scholar]
- 30.Fulhorst C.F., Bowen M.D., Ksiazek T.G., Rollin P.E., Nichol S.T., Kosoy M.Y., et al. Isolation and characterization of Whitewater Arroyo virus, a novel North American Arenavirus. Virology. 1996;224:114–120. doi: 10.1006/viro.1996.0512. [DOI] [PubMed] [Google Scholar]
- 31.Winn W.C., Jr, Murphy F.A. Tamiami virus infection in mice and cotton rats. Bull World Health Organ. 1975;52:501–506. [PMC free article] [PubMed] [Google Scholar]
- 32.Peters C.J., Zaki S.R. Role of the endothelium in viral hemorrhagic fevers. Crit Care Med. 2002;30(5 Suppl):S268–273. doi: 10.1097/00003246-200205001-00016. [DOI] [PubMed] [Google Scholar]
- 33.Oldstone M.B., Lewicki H., Homman D., Nguyen C., Julien S., Gairin J.E. Common antiviral cytotoxic T-lymphocyte epitope for diverse Arenaviruses. J Virol. 2001;75:6273–6278. doi: 10.1128/JVI.75.14.6273-6278.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Damonte E.B., Coto C.E. Treatment of arenavirus infections: from basic studies to the challenge of antiviral therapy. Adv Virus Res. 2002;58:125–155. doi: 10.1016/s0065-3527(02)58004-0. [DOI] [PubMed] [Google Scholar]
- 35.Kilgore P.E., Ksiazek T.G., Rollin P.E., Mills J.N., Villagra M.R., Montenegro M.J., et al. Treatment of Bolivian hemorrhagic fever with intravenous ribavirin. Clin Infect Dis. 1997;24:718–722. doi: 10.1093/clind/24.4.718. [DOI] [PubMed] [Google Scholar]
- 36.Gerdes G.H. Rift valley fever. Vet Clin North Am Food Anim Pract. 2002;18:549–555. doi: 10.1016/s0749-0720(02)00029-4. [DOI] [PubMed] [Google Scholar]
- 37.Abd el-Rahim I.H., Abd el-Hakim U., Hussein M. An epizootic of Rift Valley fever in Egypt in 1997. Rev Sci Tech. 1999;18:741–748. doi: 10.20506/rst.18.3.1195. [DOI] [PubMed] [Google Scholar]
- 38.Armstrong L.R., Dembry L.M., Rainey P.M., Russi M.B., Khan A.S., Fischer S.H., et al. Management of a Sabia virus-infected patient in a US hospital. Infect Control Hosp Epidemiol. 1999;20:176–182. doi: 10.1086/501607. [DOI] [PubMed] [Google Scholar]
- 39.Enria D.A., Barrera Oro J.G. Junin virus vaccines. Curr Top Microbiol Immunol. 2002;263:239–261. doi: 10.1007/978-3-642-56055-2_12. [DOI] [PubMed] [Google Scholar]
- 40.del Carmen Saavedra M., Sottosanti J.M., Riera L., Ambrosio A.M. IgG subclasses in human immune response to wild and attenuated (vaccine) Junin virus infection. J Med Virol. 2003;69:447–450. doi: 10.1002/jmv.10308. [DOI] [PubMed] [Google Scholar]
- 41.Ikegami T., Calaor A.B., Miranda M.E., Niikura M., Saijo M., Kurane I., et al. Genome structure of Ebola virus subtype Reston: differences among Ebola subtypes. Arch Virol. 2001;146:2021–2027. doi: 10.1007/s007050170049. [DOI] [PubMed] [Google Scholar]
- 42.Casillas A.M., Nyamathi A.M., Sosa A., Wilder C.L., Sands H. A current review of Ebola virus: pathogenesis, clinical presentation, and diagnostic assessment. Biol Res Nurs. 2003;4:268–275. doi: 10.1177/1099800403252603. [DOI] [PubMed] [Google Scholar]
- 43.Groseth A., Stroher U., Theriault S., Feldmann H. Molecular characterization of an isolate from the 1989/90 epizootic of Ebola virus Reston among macaques imported into the United States. Virus Res. 2002;87:155–163. doi: 10.1016/s0168-1702(02)00087-4. [DOI] [PubMed] [Google Scholar]
- 44.Cohen J. Virology. New findings heats up the hot zone. Science. 2001;293:191. doi: 10.1126/science.293.5528.191a. [DOI] [PubMed] [Google Scholar]
- 45.Sleuczka W.G. The Marburg virus outbreak of 1967 and subsequent episodes. Curr Trop Microbiol Immunol. 1999;235:49–75. doi: 10.1007/978-3-642-59949-1_4. [DOI] [PubMed] [Google Scholar]
- 46.Takada A., Kawaoka Y. The pathogenesis of Ebola hemorrhagic fever. Trends Microbiol. 2001;9:506–511. doi: 10.1016/s0966-842x(01)02201-6. [DOI] [PubMed] [Google Scholar]
- 47.World Health Organization Ebola haemorrhagic fever in Zaire, 1976. Report of an international commission. Bull WHO. 1978;56:271–293. [PMC free article] [PubMed] [Google Scholar]
- 48.World Health Organization Ebola hemorrhagic fever in Sudan, 1976. Report of a World Health Organization international study team. Bull WHO. 1978;56:247–270. [PMC free article] [PubMed] [Google Scholar]
- 49.Georges A.J., Leroy E.M., Renault A.A. Ebola hemorrhagic fever outbreaks in Gabon, 1994-7: epidemiologic and health control issues. J Infect Dis. 1999;179(Suppl):S65–S75. doi: 10.1086/514290. [DOI] [PubMed] [Google Scholar]
- 50.Jezek Z., Szczeniowski M.Y., Muyembe T., McCormick J.B., Heymann D.L. Ebola between outbreaks: intensified Ebola hemorrhagic fever surveillance in the Democratic Republic of the Congo, 1981-1985. J Infect Dis. 1999;179(Suppl):S60–S64. doi: 10.1086/514295. [DOI] [PubMed] [Google Scholar]
- 51.Bwaka M.A., Bonnet M.J., Calain P. Ebola hemorrhagic fever in Kikwit, Democratic Republic of the Congo: clinical observation in 103 patients. J Infect Dis. 1999;17 (Suppl)9:S1–S7. doi: 10.1086/514308. [DOI] [PubMed] [Google Scholar]
- 52.Ikegami T., Miranda M.E., Calaor A.B., Manalo D.L., Miranda N.J., Niikura M., et al. Histopathology of natural Ebola virus subtype Reston infection in cynomolgus macaques during the Philippine outbreak in 1996. Exp Anim. 2002;51:447–455. doi: 10.1538/expanim.51.447. [DOI] [PubMed] [Google Scholar]
- 53.Schou S., Hansen A.K. Marburg and Ebola virus infections in laboratory non-human primates: a literature review. Comp Med. 2000;50:108–123. [PubMed] [Google Scholar]
- 54.Freed E.O. Virology. Rafting with Ebola. Science. 2002;296:279. doi: 10.1126/science.1070868. [DOI] [PubMed] [Google Scholar]
- 55.Portela Camara F. Epidemiology of the Ebola virus: facts and hypotheses. Braz J Infect Dis. 1998;2:265–268. [PubMed] [Google Scholar]
- 56.Mwanatambwe M., Yamada N., Arai S., Shimizu-Suganuma M., Shichinohe K., Asano G. Ebola hemorrhagic fever: mechanism of transmission and pathogenicity. J Nippon Med Sch. 2001;68:370–375. doi: 10.1272/jnms.68.370. [DOI] [PubMed] [Google Scholar]
- 57.Leroy E.M., Baize S., Debre P., Lansoud J., Mavoungou E. Early immune responses accompanying human asymptomatic Ebola. Clin Exp Immunol. 2001;124:453–460. doi: 10.1046/j.1365-2249.2001.01517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rowe A.K., Bertolli J., Khan A.S., Mukunu R. Clinical, virologic, and immunologic follow-up of convalescent Ebola hemorrhagic fever patients and their household contacts, Kikwit, Democratic Republic of the Congo. J Infect Dis. 1999;179(Suppl):S28–S35. doi: 10.1086/514318. [DOI] [PubMed] [Google Scholar]
- 59.Jerek Z. Ebola fever: an emerging disease. Epidemiol Mikrobiol Imunol. 2001;50:54–66. [PubMed] [Google Scholar]
- 60.Morikawa S. Ebola virus and Marburg virus. Nippon Rinsho. 2003;61(Suppl 3):544–549. [PubMed] [Google Scholar]
- 61.Bray M., Paragas J. Experimental therapy of filovirus infections. Antiviral Res. 2002;54:1–17. doi: 10.1016/s0166-3542(02)00005-0. [DOI] [PubMed] [Google Scholar]
- 62.Chambers T.J., Liang Y., Droll D.A., Schlesinger J.J., Davidson A.D., Wright P.J., et al. Yellow fever virus/dengue-2 virus and yellow fever virus/dengue-4 virus chimeras: biological characterization, immunogenicity, and protection against dengue encephalitis in the mouse model. J Virol. 2003;77:3655–3668. doi: 10.1128/JVI.77.6.3655-3668.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Solomon T., Mallewa M. Dengue and other emerging flaviviruses. J Infect. 2001;42:104–115. doi: 10.1053/jinf.2001.0802. [DOI] [PubMed] [Google Scholar]
- 64.Rosen L. Pathogenesis of hemorrhagic dengue: critical discussion of current hypothesis. Bull Soc Pathol Exot Filiales. 1986;79:342–349. [PubMed] [Google Scholar]
- 65.Massad E., Coutinho F.A., Burattini N.M., Lopez L.F. The risk of yellow fever in a dengue-infested area. Trans R Soc Trop Med Hyg. 2001;95:370–374. doi: 10.1016/s0035-9203(01)90184-1. [DOI] [PubMed] [Google Scholar]
- 66.Guzman M.G., Kouri G. Advances in dengue diagnosis. Clin Diagn Lab Immunol. 1996;3:621–627. doi: 10.1128/cdli.3.6.621-627.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Isturiz R.E., Gubler D.J., Brea del Castillo J. Dengue and dengue hemorrhagic fever in Latin America and Caribbean. Infect Dis Clin North Am. 2000;14:121–140. doi: 10.1016/s0891-5520(05)70221-x. [DOI] [PubMed] [Google Scholar]
- 68.Derouich M., Boutayeb A., Twizell E. A model of dengue fever. Biomed Eng Online. 2003;2:4. doi: 10.1186/1475-925X-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Halstead S.B. Dengue. Curr Opin Infect Dis. 2002;15:471–476. doi: 10.1097/00001432-200210000-00003. [DOI] [PubMed] [Google Scholar]
- 70.Penna M.L. Dengue control: a challenge for the public health system in Brazil. Cad Saude Publica. 2003;19:305–309. doi: 10.1590/s0102-311x2003000100034. [DOI] [PubMed] [Google Scholar]
- 71.Reed W., Carroll J. The etiology of yellow fever: a supplemental note. Mil Med. 2001;166:62–66. [PubMed] [Google Scholar]
- 72.W, Carroll J, Agramonte A, Lazear JW. The etiology of yellow fever: a preliminary note. Mil Med 2001;166:29–36 [PubMed]
- 73.Lindback H., Lindback J., Tegnell A., Janzon R., Vene S., Ekdahl K. Dengue fever in travelers to the tropics, 1998 and 1999. Emerg Infect Dis. 2003;9:438–442. doi: 10.3201/eid0904.020267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gubler D.J. Dengue and dengue hemorrhagic fever. Clin Microbiol Rev. 1998;11:480–496. doi: 10.1128/cmr.11.3.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vasconcelos P.F., Rosa A.P., Rodrigues S.G., Rosa E.S., Monteiro H.A., Cruz A.C., et al. Yellow fever in Para State, Amazon region of Brazil, 1998-1999: entomologic and epidemiologic findings. Emerg Infect Dis. 2001;7:565–569. doi: 10.3201/eid0707.010738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vasconcelos P.F., Costa Z.G., Travassos Da Rosa E.S., Luna E., Rodrigues S.G., Barros V.L., et al. Epidemic of jungle yellow fever in Brazil, 2000: implications of climatic alterations in disease spread. J Med Virol. 2001;65:598–604. [PubMed] [Google Scholar]
- 77.Kozoka V., Ahmed A., Wimmer E.A., Raikhel A.S. Efficient transformation of the yellow fever mosquito Aedes aegypti using the piggyBac transposable element vector pBac[3xP3-EGFP afm] Insect Biochem Mol Biol. 2001;31:1137–1143. doi: 10.1016/s0965-1748(01)00120-5. [DOI] [PubMed] [Google Scholar]
- 78.Theiler M., Smith H.H. The use of yellow fever virus modified by in vitro cultivation for human immunization J. Exp. Med. 65, 787-800 (1937) Rev Med Virol. 2000;10(1):6–16. [PubMed] [Google Scholar]
- 79.Monath T.P. Early indicators in acute dengue infection. Lancet. 1997;350:1719–1720. doi: 10.1016/S0140-6736(05)63567-2. [DOI] [PubMed] [Google Scholar]
- 80.Mahe A., Lamaury I., Strobel M. Mucocutaneous manifestations of dengue. Presse Med. 1998;27:1909–1913. [PubMed] [Google Scholar]
- 81.John T.J. Dengue fever and dengue hemorrhagic fever. Lancet. 2003;361:181–182. doi: 10.1016/S0140-6736(03)12220-9. [DOI] [PubMed] [Google Scholar]
- 82.Morita K. Dengue/dengue hemorrhagic fever. Nippon Rinsho. 2003;61(Suppl 2):302–305. [PubMed] [Google Scholar]
- 83.Gear J.H. Clinical aspects of African viral hemorrhagic fevers. Rev Infect Dis. 1989;11(Suppl 4):S777–782. doi: 10.1093/clinids/11.supplement_4.s777. [DOI] [PubMed] [Google Scholar]
- 84.Ishak K.G., Walker D.H., Coetzer J.A., Gardner J.J., Gorelkin L. Viral hemorrhagic fevers with hepatic involvement: pathologic aspects with clinical correlations. Prog Liver Dis. 1982;7:495–515. [PubMed] [Google Scholar]
- 85.Lacy M.D., Smego R.A. Viral hemorrhagic fevers. Adv Pediatr Infect Dis. 1996;12:21–53. [PubMed] [Google Scholar]
- 86.Balmaseda A., Guzman M.G., Hammond S., Robleto G., Flores C., Tellez Y., et al. Diagnosis of dengue virus infection by detection of specific immunoglobulin M (IgM) and IgA antibodies in serum and saliva. Clin Diagn Lab Immunol. 2003;10:317–322. doi: 10.1128/CDLI.10.2.317-322.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Crance J.M., Scaramozzino N., Jouan A., Garin D. Interferon, ribavirin, 6-azauridine and glycyrrhizin: antiviral compounds active against pathogenic flaviviruses. Antiviral Res. 2003;58:73–79. doi: 10.1016/s0166-3542(02)00185-7. [DOI] [PubMed] [Google Scholar]
- 88.LeGuenno B. Viral hemorrhagic fevers: what is the risk for travelers? Med Trop (Mars) 1997;57:511–513. [PubMed] [Google Scholar]
- 89.David K.P., O'Hare M.R., Skinhoj P. Dengue fever among 44 Danish travelers investigated in the department of epidemics at the Rigshospitalet during 1988-1998. Ugeskr Laeger. 2000;162:5074–5077. [PubMed] [Google Scholar]
- 90.Jacobs M., Young P. Dengue vaccines: preparing to roll back dengue. Curr Opin Invest Drugs. 2003;4:168–171. [PubMed] [Google Scholar]
- 91.Castleberry J.S., Mahon C.R. Dengue fever in the Western Hemisphere. Clin Lab Sci. 2003;16:34–38. [PubMed] [Google Scholar]
- 92.Ruef C. West Nile virus—an emerging pathogen. Infection. 2003;31:1–2. doi: 10.1007/s15010-003-7103-5. [DOI] [PubMed] [Google Scholar]
- 93.Nedry M., Mahon C.R. West Nile virus: an emerging virus in North America. Clin Lab Sci. 2003;16:43–49. [PubMed] [Google Scholar]
- 94.Asnis D.S., Conetta R., Waldman G., Teixeira A.A. The West Nile virus encephalitis outbreak in the United States (1999-2000): from Flushing, New York, to beyond its borders. Ann N Y Acad Sci. 2001;951:161–171. doi: 10.1111/j.1749-6632.2001.tb02694.x. [DOI] [PubMed] [Google Scholar]
- 95.Jones T.F., Gottfried K.L., McCauley T.A., Swinger G.L. West Nile virus in Tennessee. Tenn Med. 2003;96:37–39. [PubMed] [Google Scholar]
- 96.Rappole J.H., Hubalek Z. Migratory birds and West Nile virus. J Appl Microbiol. 2003;94(Suppl 1):47–58. doi: 10.1046/j.1365-2672.94.s1.6.x. [DOI] [PubMed] [Google Scholar]
- 97.Komar N., Burns J., Dean C., Panella N.A., Dusza S., Cherry B. Serologic evidence for West Nile virus infection in birds in Staten Island, New York, after an outbreak in 2000. Vector Borne Zoonotic Dis. 2001;1:191–196. doi: 10.1089/153036601753552558. [DOI] [PubMed] [Google Scholar]
- 98.Cupples S. West Nile virus infection. Prog Transplant. 2003;13:7271. doi: 10.1177/152692480301300115. [DOI] [PubMed] [Google Scholar]
- 99.Bren L. West Nile virus: reducing the risk. FDA Consum. 2003;37:20–27. [PubMed] [Google Scholar]
- 100.Sidwell R.W., Smee D.F. Viruses of the Bunya and Togaviridae families: potential as bioterrorism agents and means of control. Antiviral Res. 2003;57:101–111. doi: 10.1016/s0166-3542(02)00203-6. [DOI] [PubMed] [Google Scholar]
- 101.Zeller H., Bouloy M. Infections by viruses of the families Bunyaviridae and Filoviridae. Rev Sci Tech. 2000;19:79–91. doi: 10.20506/rst.19.1.1208. [DOI] [PubMed] [Google Scholar]
- 102.Burt F.J., Leman P.A., Abbott J.C., Swanepoel R. Serodiagnosis of Crimean-Congo haemorrhagic fever. Epidemiol Infect. 1994;113:551–562. doi: 10.1017/s0950268800068576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Drosten C., Minnak D., Emmerich P., Schmitz H., Reinicke T. Crimean-Congo hemorrhagic fever in Kosovo. J Clin Microbiol. 2002;40:1122–1123. doi: 10.1128/JCM.40.3.1122-1123.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yashina L., Vyshemirskii O., Seregin S., Petrova I., Samokhvalov E., Lvov D., et al. Genetic analysis of Crimean-Congo hemorrhagic fever virus in Russia. J Clin Microbiol. 2003;41:860–862. doi: 10.1128/JCM.41.2.860-862.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Burt F.J., Swanepoel R., Braack L.E. Enzyme-linked immunosorbent assays for the detection of antibody to Crimean-Congo hemorrhagic fever virus in the sera of livestock and wild vertebrates. Epidemiol Infect. 1993;111:547–557. doi: 10.1017/s0950268800057277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nabeth P., Kane Y., Abdalahi M.O., Diallo M., Ndiaye K., Ba K., et al. Rift Valley fever outbreak, Mauritania, 1998: seroepidemiologic, virologic, entomologic, and zoologic investigations. Emerg Infect Dis. 2001;7:1052–1054. doi: 10.3201/eid0706.010627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Balkhy H.H., Memish Z.A. Rift Valley fever: an uninvited zoonosis in the Arabian peninsula. Int J Antimicrob Agents. 2003;21:153–157. doi: 10.1016/s0924-8579(02)00295-9. [DOI] [PubMed] [Google Scholar]
- 108.Digoutte J.P. Present status of an arbovirus infection: yellow fever, its natural history of hemorrhagic fever, Rift Valley fever. Bull Soc Pathol Exot. 1999;92:343–348. [PubMed] [Google Scholar]
- 109.Gargan T.P., Clark G.G., Dohm D.J., Turell M.J., Bailey C.L. Vector potential of selected North American mosquito species for Rift Valley fever virus. Am J Trop Med Hyg. 1988;38:440–446. doi: 10.4269/ajtmh.1988.38.440. [DOI] [PubMed] [Google Scholar]
- 110.Turell M.J., Bailey C.L., Beaman J.R. Vector competence of a Houston, Texas, strain of Aedes albopictus for Rift Valley fever virus. J Am Mosq Control Assoc. 1988;4:94–96. [PubMed] [Google Scholar]
- 111.Shawky S. Rift Valley fever. Saudi Med J. 2000;21:1109–1115. [PubMed] [Google Scholar]
- 112.Drosten C., Gottig S., Schilling S., Asper M., Panning M., Schmitz H., et al. Rapid detection and quantification of RNA of Ebola and Marburg viruses, Lassa virus, Crimean-Congo hemorrhagic fever virus, Rift Valley fever virus, dengue virus, and yellow fever virus by real-time reverse transcription-PCR. J Clin Microbiol. 2002;40:2323–2330. doi: 10.1128/JCM.40.7.2323-2330.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bangash S.A., Khan E.A. Treatment and prophylaxis with ribavirin for Crimean-Congo hemorrhagic fever—is it effective? J Pak Med Assoc. 2003;53:39–41. [PubMed] [Google Scholar]
- 114.Clement J.P. Hantavirus. Antiviral Res. 2003;57:121–127. doi: 10.1016/s0166-3542(02)00205-x. [DOI] [PubMed] [Google Scholar]
- 115.Hawes S., Seabolt J.P. Hantavirus. Clin Lab Sci. 2003;16:39–42. [PubMed] [Google Scholar]
- 116.Schmaljohn C., Hjelle B. Hantaviruses: a global disease problem. Emerg Infect Dis. 1997;3:95–104. doi: 10.3201/eid0302.970202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Botten J., Mirowsky K., Kusewitt D., Ye C., Gottlieb K., Prescott J., et al. Persistent Sin Nombre virus infection in the deer mouse (Peromyscus maniculatus) model: sites of replication and strand-specific expression. J Virol. 2003;77:1540–1550. doi: 10.1128/JVI.77.2.1540-1550.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Boone J.D., McGwire K.C., Otteson E.W., DeBaca R.S., Kuhn E.A., St. Jeor S.C. Infection dynamics of Sin Nombre virus after a widespread decline in host populations. Am J Trop Med Hyg. 2002;67:310–318. doi: 10.4269/ajtmh.2002.67.310. [DOI] [PubMed] [Google Scholar]
- 119.Calisher C.H., Mills J.N., Root J.J., Beaty B.J. Hantaviruses: etiologic agents of rare but potentially life-threatening zoonotic diseases. J Am Vet Med Assoc. 2003;222:163–166. doi: 10.2460/javma.2003.222.163. [DOI] [PubMed] [Google Scholar]
- 120.Avsic-Zupanc T., Nemirov K., Petrovec M., Trilar T., Poljak M., Vaheri A., et al. Genetic analysis of wild-type Dobrava hantavirus in Slovenia: co-existence of two distinct genetic lineages within the same natural focus. J Gen Virol. 2000;81:1747–1755. doi: 10.1099/0022-1317-81-7-1747. [DOI] [PubMed] [Google Scholar]
- 121.Kosoy M., Slonova R., Mills J., Mandel E., Childs J. Community structure and prevalence of hantavirus infection in rodents: a geographic division of the enzootic area in Far Eastern Russia. J Vect Ecol. 1997;22:52–63. [PubMed] [Google Scholar]
- 122.Yashina L., Patrushev N., Ivanov L., Slonova R., Mishin V., Kompanez G., et al. Genetic diversity of hantaviruses associated with hemorrhagic fever with renal syndrome in the far east of Russia. Virus Res. 2000;70:31–44. doi: 10.1016/s0168-1702(00)00203-3. [DOI] [PubMed] [Google Scholar]
- 123.Nemirov K., Vapalahti O., Papa A., Plyusnina A., Lundkvist A., Antoniadis A., et al. Genetic characterization of new Dobrava hantavirus isolate from Greece. J Med Virol. 2003;69:408–416. doi: 10.1002/jmv.10304. [DOI] [PubMed] [Google Scholar]
- 124.Hutchinson K.L., Rollin P.E., Shieh W.J., Zaki S., Greer P.W., Peters C.J. Transmission of Black Creek Canal virus between cotton rats. J Med Virol. 2000;60:70–76. doi: 10.1002/(sici)1096-9071(200001)60:1<70::aid-jmv12>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 125.Hutchinson K.L., Rollin P.E., Peters C.J. Pathogenesis of a North American hantavirus, Black Creek Canal virus, in experimentally infected Sigmodon hispidus. Am J Trop Med Hyg. 1998;59:58–65. doi: 10.4269/ajtmh.1998.59.58. [DOI] [PubMed] [Google Scholar]
- 126.Torrez-Martinez N., Bharadwaj M., Goade D., Delury J., Moran P., Hicks B., et al. Bayou virus–associated hantavirus pulmonary syndrome in eastern Texas: identification of the rice rat, Oryzomys palustris, as reservoir host. Emerg Infect Dis. 1998;4:105–111. doi: 10.3201/eid0401.980115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gavrilovskaya I., LaMonica R., Fay M.E., Hjelle B., Schmaljohn C., Shaw R., et al. New York 1 and Sin Nombre viruses are serotypically distinct viruses associated with hantavirus pulmonary syndrome. J Clin Microbiol. 1999;37:122–126. doi: 10.1128/jcm.37.1.122-126.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Rose A.M., Vapalahti O., Lyytikainen O., Nuorti P. Patterns of Puumala virus infection in Finland. Euro Surveill. 2003;8:9–13. doi: 10.2807/esm.08.01.00394-en. [DOI] [PubMed] [Google Scholar]
- 129.Wasser W.G., Rossi C.A., Glass G.E. Hantavirus antibodies in New York. Ann Intern Med. 1997;127:166–167. doi: 10.7326/0003-4819-127-2-199707150-00028. [DOI] [PubMed] [Google Scholar]
- 130.Lednicky J.A. Hantaviruses. A short review. Arch Pathol Lab Med. 2003;127:30–35. doi: 10.5858/2003-127-30-. [DOI] [PubMed] [Google Scholar]
- 131.Bausch D.G., Ksiazek T.G. Viral hemorrhagic fevers including hantavirus pulmonary syndrome in the Americas. Clin Lab Med. 2002;22:981–1020. doi: 10.1016/s0272-2712(02)00019-7. [DOI] [PubMed] [Google Scholar]
- 132.Khaiboullina S.F., St. Jeor S.C. Hantavirus immunology. Viral Immunol. 2002;15:609–625. doi: 10.1089/088282402320914548. [DOI] [PubMed] [Google Scholar]
- 133.Hujakka H., Koistinen V., Kuronen I., Eerikainen P., Parviainen M., Lundkvist A., et al. Diagnostic rapid tests for acute hantavirus infections: specific tests for Hantaan, Dobrava and Puumala viruses versus a hantavirus combination test. J Virol Methods. 2003;108:117–122. doi: 10.1016/s0166-0934(02)00282-3. [DOI] [PubMed] [Google Scholar]
- 134.Araki K., Yoshimatsu K., Ogino M., Ebihara H., Lundkvist A., Kariwa H., et al. Truncated hantavirus nucleocapsid proteins for serotyping Hantaan, Seoul, and Dobrava hantavirus infections. J Clin Microbiol. 2001;39:2397–2404. doi: 10.1128/JCM.39.7.2397-2404.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Pattyn S.R. Monkeypoxvirus infections. Rev Sci Tech. 2000;19:92–97. doi: 10.20506/rst.19.1.1207. [DOI] [PubMed] [Google Scholar]
- 136.Shchelkunov S.N., Totmenin A.V., Babkin I.V., Safronov P.F., Ryazankina O.I., Petrov N.A., et al. Human monkeypox and smallpox viruses: genomic comparison. FEBS Lett. 2001;509:66–70. doi: 10.1016/S0014-5793(01)03144-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Merouze F., Matton T., Bertherat E., Dalco O. Present status of monkeypox. Med Trop (Mars) 1999;59(2 Suppl):53–54. [PubMed] [Google Scholar]
- 138.Centers for Disease Control and Prevention Human monkeypox—Kasai Oriental, Democratic Republic of Congo, February 1996-October 1997. JAMA. 1998;279:189–190. [PubMed] [Google Scholar]
- 139.Hutin Y.J., Williams R.J., Malfait P., Pebody R., Loparev V.N., Ropp S.L., et al. Outbreak of human monkeypox, Democratic Republic of Congo, 1996 to 1997. Emerg Infect Dis. 2001;7:434–438. doi: 10.3201/eid0703.010311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Heymann D.L., Szczeniowski M., Esteves K. Re-emergence of monkeypox in Africa: a review of the past six years. Br Med Bull. 1998;54:693–702. doi: 10.1093/oxfordjournals.bmb.a011720. [DOI] [PubMed] [Google Scholar]
- 141.Meyer H., Perrichot M., Stemmler M., Emmerich P., Schmitz H., Varaine F., et al. Outbreaks of disease suspected of being due to human monkeypox virus infection in the Democratic Republic of Congo in 2001. J Clin Microbiol. 2002;40:2919–2921. doi: 10.1128/JCM.40.8.2919-2921.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Breman J.G., Henderson D.A. Poxvirus dilemmas—monkeypox, smallpox, and biologic terrorism. N Engl J Med. 1998;339:556–559. doi: 10.1056/NEJM199808203390811. [DOI] [PubMed] [Google Scholar]
- 143.Jezek Z., Szczeniowski M., Paluku K.M., Mutombo M., Grab B. Human monkeypox: confusion with chickenpox. Acta Trop. 1988;45:297–307. [PubMed] [Google Scholar]
- 144.Baker R.O., Bray M., Huggins J.W. Potential antiviral therapeutics for smallpox, monkeypox and other orthopoxvirus infections. Antiviral Res. 2003;57:13–23. doi: 10.1016/S0166-3542(02)00196-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Greenwood A. Herpesvirus B in macaque monkeys. Vet Rec. 2002;151:516. [PubMed] [Google Scholar]
- 146.Mahalingam R., Traina-Dorge V., Wellish M., Smith J., Gilden D.H. Naturally acquired simian varicella virus infection in African green monkeys. J Virol. 2002;76:8548–8550. doi: 10.1128/JVI.76.17.8548-8550.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Orcutt R.P., Pucak G.J., Foster H.L., Kilcourse J.Y. Multiple testing for the detection of B virus antibody in specially handled rhesus monkeys after capture from virgin trapping grounds. Lab Anim Sci. 1976;26:70–74. [PubMed] [Google Scholar]
- 148.Lupi O., Lamy F. In: Herpes: clinica, diagnostico e tratamento. Lupi O., Silva A.G., Pereira A.C. Jr, editors. Medsi; Rio de Janeiro: 2000. Herpes símio; pp. 235–247. [Google Scholar]
- 149.Holmes G.P., Hilliard J.K., Klontz K.C., Rupert A.H., Schindler C.M., Parrish E. B-virus (Herpesvirus simiae) infection in humans: epidemiologic investigations of a cluster. Ann Intern Med. 1990;112:833–839. doi: 10.7326/0003-4819-112-11-833. [DOI] [PubMed] [Google Scholar]
- 150.Sabin A.B., Wright A.M. Acute ascending myelitis following a monkey bite, with the isolation of a virus capable of reproducing the disease. J Exp Med. 1934;59:115–136. doi: 10.1084/jem.59.2.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Palmer A.E. B virus, Herpesvirus simiae: historical perspective. J Med Primatol. 1987;16:99–130. [PubMed] [Google Scholar]
- 152.CDC B-virus infection in humans—Pensacola, Florida. MMWR. 1987;36:289–290. 95-6. [PubMed] [Google Scholar]
- 153.Centers for Disease Control Guidelines for prevention of Herpesvirus simiae (B-virus) infection in monkey handlers. MMWR Morb Mortal Wkly Rep. 1987;36:681–682. 687-9. [PubMed] [Google Scholar]
- 154.Ostrowski S.R., Leslie M.J., Parrott T., Abelt S., Piercy P.E. B-virus from pet macaque monkeys: an emerging threat in the United States? Emerg Infect Dis. 1998;4:117–121. doi: 10.3201/eid0401.980117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Jainkittivong A., Langlais R.P. Herpes B virus infection. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1998;85:399–403. doi: 10.1016/s1079-2104(98)90064-6. [DOI] [PubMed] [Google Scholar]
- 156.Centers for Disease Control B-virus infection in humans—Pensacola, Florida. MMWR Morb Mortal Wkly Rep. 1987;36:289–290. 295-6. [PubMed] [Google Scholar]
- 157.Centers for Disease Control B-virus infection in humans—Michigan. MMWR Morb Mortal Wkly Rep. 1989;38:453–454. [PubMed] [Google Scholar]
- 158.Poiesz B.J., Ruscetti F.W., Gadzar A.F., Bunn P.A., Minna J.D., Gallo R.C. Detection and isolation of type C retrovirus particles from fresh cultured lymphocytes of a patient with cutaneous T cell lymphoma. Proc Natl Acad Sci USA. 1980;77:7415–7419. doi: 10.1073/pnas.77.12.7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Yoshida M., Miyoshi J., Hinuma Y. Isolation and characterization of retrovirus from cell lines of human T-cell leukemia and its complication in the disease. Proc Natl Acad Sci USA. 1982;79:2031–2035. doi: 10.1073/pnas.79.6.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kaltanaraman V.S., Sarngadharan M.G., Robert-Guroff M. A new subtype of human T-cell leukemia virus (HTLV-II) associated with a T-cell variant of hairy cell leukemia. Science. 1982;218:571–573. doi: 10.1126/science.6981847. [DOI] [PubMed] [Google Scholar]
- 161.Franchini G. Molecular mechanisms of human T-cell/leukemia/lymphotropic virus type I infection. Blood. 1995;86:3619–3622. [PubMed] [Google Scholar]
- 162.Khabbaz R.F., Darrow W.W., Harthey T.M. Seroprevalence and risk factors for HTLV-I/II infection among female prostitutes in the United States. JAMA. 1990;263:60–64. [PubMed] [Google Scholar]
- 163.Asher D.M., Goudsmit J., Pomeroy K.L. Antibodies to HTLV-I in populations of the southwestern Pacific. J Med Virol. 1988;26:339–351. doi: 10.1002/jmv.1890260402. [DOI] [PubMed] [Google Scholar]
- 164.Andrade-Serpa M.J., Dobbin J.A., Gomes P. Incidence of retrovirus in some Brazilian groups. Immunol Lett. 1988;18:15–18. doi: 10.1016/0165-2478(88)90063-6. [DOI] [PubMed] [Google Scholar]
- 165.Bouzas M.B., Picchio G., Muchinik G., Gallo D. HTLV-1 in Argentina. J Acquir Immune Defic Syndr. 1992;5:743–747. [PubMed] [Google Scholar]
- 166.Lenzi M.E.R., Araujo A.Q., Maya T.C., Serapiao M.J., Leite A.C.C., Schor D., et al. Dermatite infectiva associada ao HTLV-1: relato de caso. An Bras Dermatol. 1996;71:115–118. [Google Scholar]
- 167.Simpson R.M., Leno M., Hubbard B.S., Kindt T.J. Cutaneous manifestations of HTLV-1 infection in an experimental model. J Infect Dis. 1996;173:722–726. doi: 10.1093/infdis/173.3.722. [DOI] [PubMed] [Google Scholar]
- 168.Dosaka N., Tanaka T., Miyachi Y., Immamura S., Kakizuka A. Examination of HTLV-I integration in the skin lesions of various types of adult T-cell leukemia (ATL): independence of cutaneous-type ATL confirmed by Southern blot analysis. J Invest Dermatol. 1991;96:196–200. doi: 10.1111/1523-1747.ep12461033. [DOI] [PubMed] [Google Scholar]
- 169.LaGrenade L. HTLV-1-associated infective dermatitis: past, present, and future. J Acquir Immune Defic Syndr Hum Retrovirol. 1996;13(Suppl 1):S46–49. doi: 10.1097/00042560-199600001-00009. [DOI] [PubMed] [Google Scholar]
- 170.Uchiyama T., Yodoi J., Sagawa K., Takatsuki K., Uchino H. Adult T-cell leukemia: clinical and hematologic features of 16 cases. Blood. 1977;50:481–492. [PubMed] [Google Scholar]
- 171.Catovsky J.W., Greaves M.F., Rose M. Adult T-cell lymphoma/leukaemia in blacks from the West Indies. Lancet. 1982;1:639–643. doi: 10.1016/s0140-6736(82)92200-0. [DOI] [PubMed] [Google Scholar]
- 172.Giudice P., Marie D.S., Gerard Y. Is crusted (Norwegian) scabies a marker of adult T cell leukemia/lymphoma in human T lymphotropic virus type I–seropositive patients? J Infect Dis. 1997;176:1090–1092. doi: 10.1086/516518. [DOI] [PubMed] [Google Scholar]
- 173.Araujo A.Q., Afonso C.R., Leite A.C. Clinical and demographic features of HTLV-I associated myelopathy/tropical spastic paraparesis (HAM\TSP) in Rio de Janeiro, Brazil. Acta Neurol Scand. 1993;88:59–62. doi: 10.1111/j.1600-0404.1993.tb04188.x. [DOI] [PubMed] [Google Scholar]
- 174.Hashiguchi T., Osame M., Arimura K. In: HTLV-I and nervous system. Roman C.G., Vemant J.C., Osame M., editors. Alan R. Liss; New York: 1989. Skin manifestations in HTLV-associated myelopathy (HAM): xerosis and erythema; pp. 443–448. [Google Scholar]
- 175.Sweet R.D. A pattern of eczema in Jamaica. Br J Dermatol. 1966;78:93–100. doi: 10.1111/j.1365-2133.1966.tb12181.x. [DOI] [PubMed] [Google Scholar]
- 176.LaGrenade L., Hanchard B., Fletcher V. Infective dermatitis of Jamaican children: a marker for HTLV-I infection. Lancet. 1990;336:1345–1347. doi: 10.1016/0140-6736(90)92896-p. [DOI] [PubMed] [Google Scholar]
- 177.Goncalves D.U., Guedes A.C., Carneiro-Proietti A.B., Lambertucci J.R. HTLV-1 associated infective dermatitis may be an indolent HTLV-1 associated lymphoma. Braz J Infect Dis. 2000;4:100–102. [PubMed] [Google Scholar]
- 178.Mahe A., Chollet-Martin S., Gessain A. HTLV-1-associated infective dermatitis. Lancet. 1999;16:354. doi: 10.1016/S0140-6736(05)76239-5. :1386. [DOI] [PubMed] [Google Scholar]
- 179.LaGrenade L., Manns A., Fletcher V., Derm D., Carberry C., Hanchard B., et al. Clinical, pathologic, and immunologic features of human T-lymphotropic virus type I–associated infective dermatitis in children. Arch Dermatol. 1998;134:439–444. doi: 10.1001/archderm.134.4.439. [DOI] [PubMed] [Google Scholar]