

Abstract
In the 21st century we are faced with the potential use of natural or recombinant VARV and MPXV as biological weapons, and the emergence of human MPXV. Such an occurrences would require therapeutic and prophylactic intervention with antivirals. Cidofovir, an antiviral approved for the treatment of cytomegalovirus retinitis in AIDS patients, has activity against poxviruses, but must be administered intravenously and is associated with nephrotoxicity. An ether–lipid analogue of CDV, CMX001 (HDP-CDV), has potent antiviral activity against a range of DNA viruses including poxviruses, excellent oral bioavailability and minimal nephrotoxicity. CMX001 and CDV are equally efficacious at protecting mice from mortality following high ectromelia virus doses (10,000 × LD50) introduced by the intra-nasal route or small particle aerosol. Using CMX001 at a 10 mg/kg dose followed by 2.5 mg/kg doses every other-day for 14 days provided solid protection against mortality and weight loss following an intra-nasal challenge of (100–200) × LD50 of ectromelia virus. Furthermore, complete protection against mortality was achieved when administration was delayed until as late as 5 days post-infection, which is 3–4 days prior to the death of the untreated controls. This therapeutic window would be equivalent to intervening during the rash stage of ordinary smallpox.
Keywords: Antiviral, Bioterrorism, Ectromelia, Monkeypox, Oral drug, Smallpox
1. Introduction
Many vertebrates, including humans, are susceptible to orthopoxvirus infections. Humans are most at risk from smallpox (variola, VARV) and human monkeypox (MPXV). Although smallpox was eliminated in the late 1970s, it is still considered a threat because official, and most likely clandestine, stocks of the virus still exist. Furthermore, human infections with MPXV have been reported with increasing frequency throughout central and western Africa (reviewed by Parker et al., 2007). To counter these threats, prophylactic vaccines have been stockpiled by many governments. Although vaccines can provide good protection against VARV and MPXV, the difficulty of vaccinating en masse populations before the virus has become widely disseminated may limit their utility in a public health emergency. Also, a large section of the population cannot be vaccinated due to a growing list of contraindications (Baker et al., 2003, Bray, 2003, Rosenthal et al., 2001). Moreover, techniques to drastically increase the virulence of the mousepox virus, ectromelia (ECTV), could be applied to other orthopoxviruses making vaccinated individuals susceptible to severe disease (Jackson et al., 2001). For these reasons, effective, highly efficacious antivirals are crucial.
Cidofovir (CDV), a wide-spectrum antiviral approved for the treatment of cytomegalovirus retinitis in AIDS patients, has been investigated for the treatment of smallpox and for the treatment of smallpox vaccination complications. However, CDV must be administered intravenously due to its poor oral bioavailability (Safrin et al., 1997). This drawback, coupled with its inherent nephrotoxicity, makes CDV an unrealistic therapeutic following a wide-scale natural or bioterrorist VARV attack or MPXV outbreak. However, esterification of CDV with alkoxyalkanols decreases poxviral EC50 values by 24–910-fold compared to CDV (Buller et al., 2004, Keith et al., 2004, Kern et al., 2002). Crucially, esterification allows the drug to be delivered orally without diminishing its efficacy and prevents accumulation of CDV in the kidneys (Ciesla et al., 2003, Hostetler et al., 2007, Quenelle et al., 2004). The hexadecyloxypropyl ester of CDV (CMX001) has been demonstrated to have a good balance between high efficacy and low toxicity.
Using the aerosol mousepox challenge model, we previously showed that treatment of A/Ncr mice with five consecutive, daily, 10 mg/kg doses of CMX001 commencing 4 h post-infection provided full protection against lethal disease (Buller et al., 2004). The mousepox model is arguably the best small animal model for the evaluation of smallpox therapeutics due, in part, to the low doses of virus required for lethal infections producing a disease course that accurately reflects the progress of natural infection. The LD50 for ectromelia in A/Ncr mice infected via the intra-nasal route and by aerosol are 0.3 and 0.36 PFU/mouse, respectively (Buller et al., 2004). The intra-nasal route mimics an upper respiratory tract infection which is thought to be a natural route of infection by VARV. A small particle aerosol delivers virus to the lower respiratory tract with the goal of modelling a biowarfare attack.
In this study, using aerosol and intra-nasal mousepox challenge models, we show orally administered CMX001 to be equal or superior to intraperitoneally administered CDV for protection from lethal disease and control of virus replication in key tissues. We show that CMX001 protects over a broad range of virus challenge doses, and using a low-dose challenge model we optimise the treatment regimen to provide complete protection against an ECTV challenge, yet minimize exposure of the animal to drug and consequently to potential toxicity. Finally, we show therapeutic intervention with the optimised treatment regimen provides complete protection against mortality when administered as late as 5 days following challenge, which is 3–4 days prior to the death of the untreated controls.
2. Materials and methods
2.1. Cells and virus
BSC-1 cells (ATCC CCL 26) were grown in Eagle's minimum essential medium (MEM) containing 10% fetal clone III (Hyclone, Logan, UT), 2 mM l-glutamine (GIBCO, Grand Island, NY), 100 U/ml penicillin (GIBCO, Grand Island, NY), and 100 μg/ml streptomycin (GIBCO, Grand Island, NY). A plaque-purified isolate of the MOS strain of ECTV (ATCC VR-1374) designated MOS-3-P2 was propagated in an African green monkey kidney cell line, BSC-1 (Chen et al., 1992). Virus was purified through a sucrose cushion as described elsewhere (Moss and Earl, 1998). Virus infectivity was estimated as described previously (Wallace and Buller, 1985). Briefly, virus suspensions were serially diluted in PBS + 1% sera, absorbed to monolayers for 1 h at 37 °C, and overlaid with a suspension of 1% carboxyl methyl cellulose in DMEM + 5% fetal clone III. After 4 days at 37 °C, virus plaques were visualised and virus inactivated by the addition to each well of 0.5 ml of a 0.3% crystal violet/10% formalin solution.
2.2. Animals
Four- to 6-week-old female A/Ncr mice were obtained from the National Cancer Institute, Frederick, MD, housed in filter-top microisolator cages and fed commercial mouse chow and water, ad libitum. The mice were housed in an animal biosafety level 3 containment area. Animal husbandry and experimental procedures were in accordance with PHS policy, and approved by the Institutional Animal Care and Use Committee.
2.3. Antiviral compounds
Cidofovir ([S]-1-[3-hydroxy-2-phosphonylmethoxypropyl]cytosine, HPMPC, Vistide®) was a gift from Gilead Sciences, Inc. (Foster City, CA). The CMX001 analogue of CDV was a gift from Chimerix Inc. (Durham, NC). Solutions of CDV and CMX001 were prepared fresh prior to each experiment by dissolving the compounds in sterile, distilled water, and stored at 4 °C over the course of the experiment.
2.4. Aerosol challenge
Mice were exposed to aerosolized ECTV suspended in MEM using a nose-only inhalation exposure system (NOIES; CH Technologies) equipped with a 1-jet BioAerosol Nebulizing Generator, and operated within a class 2 biological safety cabinet. The NOIES was operated with a primary air pressure of 20 psi giving 1.5 l/min flow rate to the aerosol chamber (without secondary air), a virus suspension flow rate of 0.5 ml/min, and a system operating pressure of ∼−0.5 in vacuum relative to the outside atmospheric pressure. The quantity of virus delivered to the mice over the course of exposure was estimated by multiplying the concentration of virus in the aerosol (C A expressed in PFUs) by the total volume (V M) of air respired by a mouse of given body weight over the exposure time using Guyton's formula for minute volumes administered to rodents (Guyton, 1947). This presented virus dose is likely an upper limit as it assumes that the entire virus challenge was optimally aerosolized and completely taken up on inhalation.
2.5. Intra-nasal challenge
Mice were anesthetised with 0.1 ml/10 g body weight of ketamine HCl (9 mg/ml) and xylazine (1 mg/ml) by intraperitoneal injections. Anesthetised mice were laid on their dorsal side with their bodies angled so that the anterior end was raised 45° from the surface, a plastic mouse holder was used to ensure conformity. ECTV was diluted in PBS to the required concentration and slowly loaded into each nare (5 μl/nare). Mice were subsequently left in situ for 2–3 min before being returned to their cages.
At indicated times following exposure to ECTV, groups of mice were treated by gavage with 0.1 ml sterile, distilled water (placebo) or water containing the desired concentration of CMX001. CDV was delivered by an intraperitoneal injection at the desired dose. This treatment was repeated as described throughout the results. To determine infectious viral titres, mice were sacrificed at 4, 6, and 8 days post-challenge, and lung, spleen, and liver tissues and nasal wash were isolated. Tissue was ground in PBS (10%, w/v), frozen and thawed three times, and sonicated for 20 s. Virus infectivity (PFU/ml) in tissue homogenates was estimated by titration on BSC-1 monolayers (see Section 2.1). Arithmetic means were calculated for PFU/ml values above the limit of detection (102 PFU/ml). Remaining mice were observed for clinical signs of disease (morbidity) and mortality. Moribund mice were euthanized.
2.6. Statistics
An unpaired t-test was used to compare the means of two groups of mice. P values below 0.05 were considered statistically significant. Mortality rates were measured using Fisher's exact test.
2.7. Pharmacokinetics
Pooled CD-1 mouse, cynomolgus monkey and human liver S9 fractions were obtained from a commercial source and stored at −70 °C prior to use. Mouse, cynomolgus monkey and human S9 fractions were incubated with 1 or 10 μM CMX001 in triplicate in 0.1 M potassium phosphate buffer (pH 7.4) containing 2 mM magnesium chloride, 0.2 mM 3′-phosphoadenosine, 5′-phosphosulfate, and 2 mM uridine 5′-diphosphogucuronic acid. The appropriate positive and negative controls were performed in parallel to ensure the validity of the assay. The reaction was initiated by the addition of 2 mM NADPH (final concentration). At the end of each incubation time period, an aliquot of 50 μl was obtained from each incubation mixture and transferred to a clean tube containing 50 μl acetonitrile and 1 μM internal standard. CMX001 remaining in the sample was quantified using an LC/MS/MS method.
Pooled, mixed-sex, primary, human, CD-1 mouse, and cynomolgus monkey hepatocytes were obtained from commercial sources as cryopreserved suspensions. The cells were thawed according to the supplier's instructions and maintained in supplemented hepatocyte maintenance media from Clonetics (San Diego, CA). 14C-CMX001 (1 or 10 μM) was added to hepatocyte suspensions (1 × 106 viable cells/ml) and incubated (in duplicate) at 37 °C and 5% CO2. The appropriate positive and negative controls were performed in parallel to ensure the validity of the assay. The incubation was terminated by the addition of one volume of ice-cold methanol and a brief sonication. Cell debris was removed by centrifugation and the supernatant was stored at −20 °C prior to analysis. 14C-CMX001 remaining in the sample was quantified by HPLC using radiochemical detection.
3. Results
3.1. Efficacy of CDV and CMX001 in high-dose ectromelia infections
3.1.1. High-dose aerosol challenge
The efficacy of CDV and CMX001 at approximately equivalent molar doses was determined following a high-dose (5 × 104 PFU, 1.6 × 105 × LD50) ECTV aerosol challenge in A/Ncr mice. Challenged mice were treated with either an intraperitoneal CDV injection at 5 mg/kg on day 0 and at 1.25 mg/kg on day 3; or, an orally administered CMX001 gavage at 10 mg/kg on day 0 and 2.5 mg/kg on day 3. Mice gavaged with a sterile water placebo had 100% mortality by day 12 (Fig. 1A), uninfected mice had 0% mortality and a steady increase in body weight (Fig. 1B). Mice treated with CDV had an initial decrease in mass followed by a steady mass increase in survivors from day 21. A mortality of 50% was observed by day 20 in mice treated with CDV (P = 0.0325). In comparison, mice treated with CMX001 have a smaller initial reduction in body mass compared to the CDV-treated mice and have a mortality of 25% by day 20 (P = 0.0014) (Fig. 1A and B). Titres from spleen, liver, lung and nasal washes were calculated from days 4, 6 and 8 (Fig. 1C–F). Spleen and liver titres in mice treated with CMX001 or CDV were between 2 and 5 logs lower than those treated with placebo. Mice treated with CMX001 consistently had lower average splenic (P = 0.35, 0.27 and 0.1 for days 4, 6 and 8, respectively) and liver (P = 1.0, 0.24 and 0.19 for days 4, 6 and 8, respectively) titres compared to CDV-treated animals. Nasal wash titres were similar between CDV- and CMX001-treated mice. Titres in the lungs were 2 logs lower in CDV-treated animals on day 4 compared to those treated with CMX001 (P values from a comparison of CDV and CMX001 titres in the lung = 0.02, 0.37 and 0.07 for days 4, 6 and 8, respectively). Neither drug had a significant effect on lung titres compared to placebo mice (CDV, P = 0.35, 0.22 and 0.11 on days 4, 6 and 8, respectively; CMX001, P = 0.25, 0.22 and 0.10 on days 4, 6 and 8, respectively) (Fig. 1E).
Fig. 1.
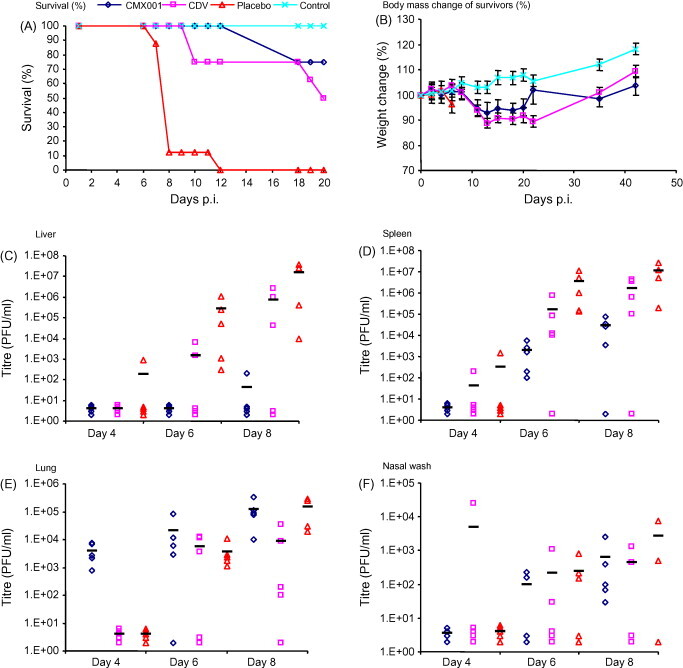
The efficacy of CMX001 following a high-dose aerosol challenge. Groups of five A/Ncr mice were infected with an aerosol challenge of 5 × 104 PFU of ECTV and treated with CDV (5 mg/kg on day 0 and 1.25 mg/kg on day 3), CMX001 (10 mg/kg on day 0 and 2.5 mg/kg on day 3) or placebo (sterile water). Mice were observed for mortality for 20 days (A) and the body mass of surviving mice was recorded (B). Mice were sacrificed on days 4, 6 and 8 and liver (C), spleen (D) and lung (E) samples were isolated and measured for virus infectivity. A nasal wash sample (F) was also isolated and tested for virus infectivity.
3.1.2. High-dose intra-nasal challenge
The efficacy of CDV and CMX001 in mice infected with a high-dose intra-nasal challenge (3.3 × 103 PFU, 9.1 × 103 × LD50) was also tested. The efficacies were evaluated according to the method used for the aerosol challenge (outlined in Section 3.1.1). Both CDV and CMX001 protected 100% of the animals from lethality and CMX001-treated mice lost less mass overall (Fig. 2A and B). All mice treated with placebo died on, or before, day 7. In the spleen (Fig. 2D), CMX001 consistently outperformed CDV by 1–3 logs (P = 0.35, 0.31 and 0.25 for days 4, 6 and 8, respectively). CDV and CMX001 have similar protective abilities in the lung, liver and nasal wash samples (Fig. 2C, E and F). These results reveal that CMX001-treated mice have similar or lower tissue viral titres as compared with CDV following high-dose intra-nasal or high-dose aerosol challenges.
Fig. 2.
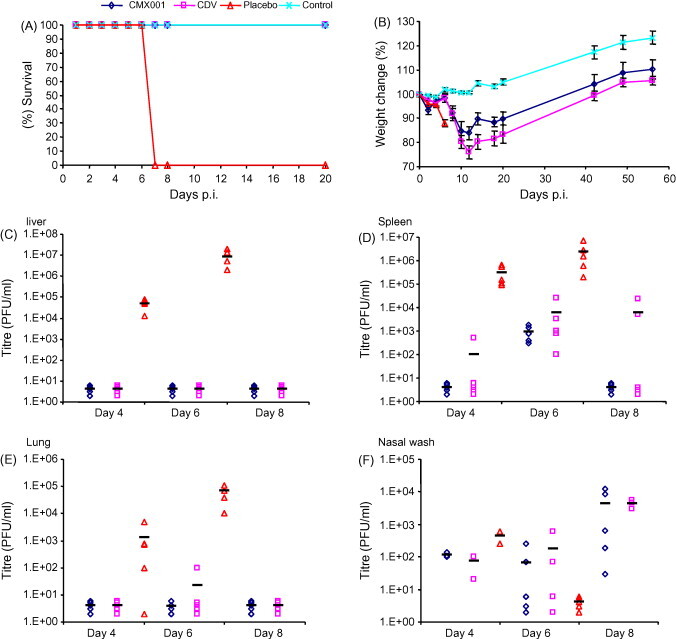
The efficacy of CMX001 following a high-dose intra-nasal challenge. Groups of five A/Ncr mice were infected with an intra-nasal challenge of 9.1 × 103 PFU of ECTV and treated with CDV (5 mg/kg on day 0 and 1.25 mg/kg on day 3), CMX001 (10 mg/kg on day 0 and 2.5 mg/kg on day 3) or placebo (sterile water). Mice were observed for mortality for 20 days (A) and the body mass of surviving mice was recorded (B). Mice were sacrificed on days 4, 6 and 8 and liver (C), spleen (D) and lung (E) samples were isolated and measured for virus infectivity. A nasal wash sample (F) was also isolated and tested for virus infectivity.
3.2. CMX001 protection against an escalating intra-nasal mousepox dose
The intra-nasal route of infection was employed for all subsequent experimental infections because it is believed to best mimic the natural transmission of smallpox and monkeypox. Groups of mice were infected with 5000, 500, 50, 5, 1.2, 0.12 or 0.012 PFU of ECTV and treated with 1, 2, 4 or 8 mg/kg of CMX001 4 h p.i. and then everyday for 5 days. At 8 mg/kg all groups of mice survived (not shown). At high doses, such as 5000–500 PFU, protection was reduced to 62.5 and 75%, respectively, when the dose of CMX001 was reduced to 4 mg/kg (Fig. 3C). At 50 PFU, protection was reduced to 87.5% at the 2 mg/kg dose. In mice infected with up to 5 PFU of ECTV, a dose of 2 mg/kg of CMX001 provided complete protection (Fig. 3B). All surviving mice continued to loose body weight until approximately day 14, after which body weight increases were observed (Fig. 4A–D). These data indicate that a minimum dose of 2 mg/kg of CMX001 everyday for 5 days is required to protect A/Ncr mice from low-dose (<5 PFU) intra-nasal ECTV infections.
Fig. 3.
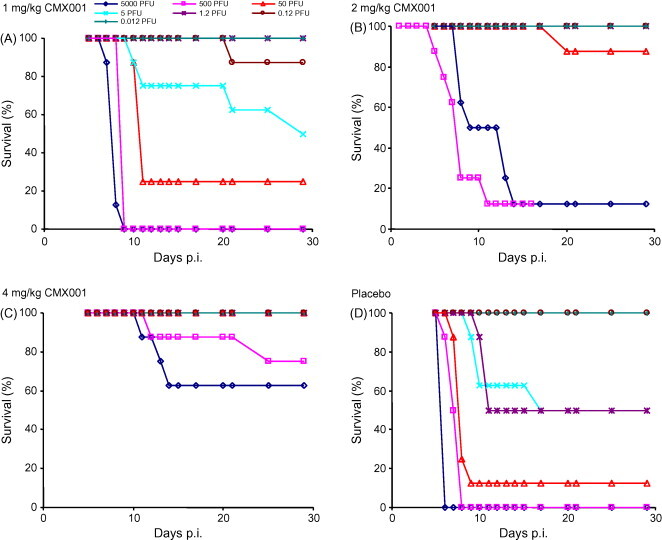
Determining the dose of CMX001 required to protect against an escalating challenge. Groups of four A/Ncr mice were intra-nasally infected with 5000, 500, 50, 5, 1.2, 0.12 and 0.012 PFU of ECTV (data from two experiments were combined). All groups of mice were treated with CMX001 at 1 mg/kg (A), 2 mg/kg (B) and 4 mg/kg (C) daily for 5 days, or received placebo (D).
Fig. 4.
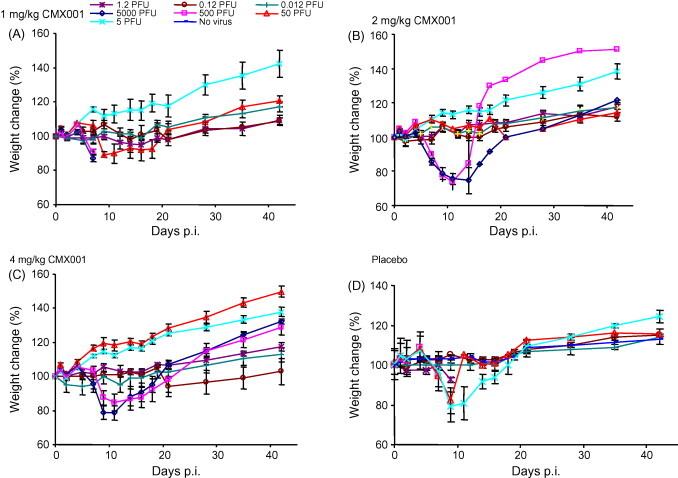
Survivors’ mass changes following escalating challenges. Groups of four A/Ncr mice were intra-nasally infected with 5000, 500, 50, 5, 1.2, 0.12 and 0.012 PFU of ECTV. All groups of mice were analysed for mass change and treated with CMX001 at 1 mg/kg (A), 2 mg/kg (B) and 4 mg/kg (C) daily for 5 days. The placebo control is shown in (D).
3.3. Optimisation of the treatment regimen to protect against a low-dose challenge
As outlined in Section 3.2, a daily, 5-day treatment with 2 mg/kg of CMX001 does not fully protect mice infected with 50 PFU of ECTV, a dose that is likely to approach the upper limit of the natural infectious dose for variola (see Section 4) (Fig. 3B). In an attempt to determine the effective dose at a 50 PFU intra-nasal challenge, mice were treated daily, for 14 days, with CMX001 at 5, 2.5, 1.25, 0.6 and 0.3 mg/(kg day) beginning 4 h p.i. Daily treatment with ≥1.25 mg/(kg day) of CMX001 significantly protected mice from lethal infections (P < 0.0001) (Fig. 5A). Remarkably, even doses as low as 0.3 mg/(kg day) delay the day of death. Daily treatment with 2.5 mg/(kg day) of CMX001 provides 100% protection and significantly less weight loss than does daily treatment with 1.25 mg/(kg day) (P = 0.0045 and 0.0051 on days 13 and 15, respectively) (Fig. 5A and B).
Fig. 5.
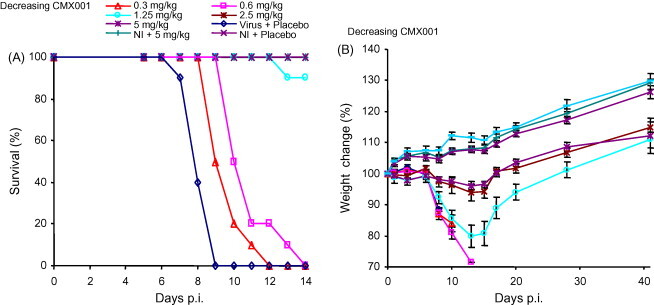
Determining the minimum dose of CMX001 to protect mice. Groups of five A/Ncr mice were intra-nasally challenged with 50 PFU of ECTV and treated with a decreasing dose of CMX001. Groups were treated with 5, 2.5, 1.25, 0.6 and 0.3 mg/kg of CMX001 daily for 14 days and survival (A) and weight loss (B) are presented. NI: not infected.
To further optimise the treatment regimen, groups of mice were infected with 100 PFU of an ECTV intra-nasal challenge and treated with 1.25 or 2.5 mg/kg of CMX001 on days 0 and then every day (regimen B), every second day (regimen C), every third day (regimen D) or every fourth day (regimen E) to investigate the minimum CMX001 exposure that provides 100% protection (regimen A received daily placebo). Regimen E, at 1.25 mg/kg of CMX001, provided mice with the lowest exposure to drug but failed to provide any protection from morbidity. Indeed, none of the 1.25 mg/kg dose regimens provided 100% protection (Fig. 6A). At the 2.5 mg/kg dose, protection could be increased to 50, 60 and 100% using regimens E, D and C, respectively. All mice treated with placebo died by day 9. These data reveal that protection from a viral challenge that mimics natural infection requires, minimally, treatment every 2 days with 2.5 mg/kg of CMX001 commencing 4 h p.i. (P < 0.0001). Interestingly, mice receiving a daily treatment of 1.25 mg/(kg day) of CMX001 (regimen B) do not have 100% (P = 0.0325) protection despite receiving the same cumulative dose of drug as mice treated every 2 days with 2.5 mg/kg of CMX001 (Fig. 6A).
Fig. 6.

Optimising the treatment regimen to protect mice. Groups of five A/Ncr mice were intra-nasally infected with 100 PFU of ECTV. Groups were treated with 1.25 or 2.5 mg/kg of CMX001 on day 0 and then every day (regimen B), every second day (regimen C), every third day (regimen D) or every fourth day (regimen E). Survival is shown in (A) and weight change in (B). NI: not infected.
3.4. Delayed treatment with CMX001
The treatment regimen was therapeutically evaluated using mice infected intra-nasally with 5 PFU of ECTV. This model extends the disease course, but remains 100% lethal in untreated mice. Groups of mice were administered CMX001 on days 0 (4 h p.i.), 1, 2, 3, 4, 5 or 6 p.i. Mice were treated with an initial dose of 10 mg/kg followed by 2.5 mg/kg doses of CMX001 on every second day. We chose to employ an initial dose of 10 mg/kg because we previously found that it helps to ensure a lower morbidity rate in infected mice compared to infected mice not receiving the initial high-dose treatment (not shown). Control mice were treated with CDV on days 0 and 3 p.i. We found that CDV treatment delayed until 3 days p.i. provided complete protection, but CDV treatment initiated at day 0 did not (P < 0.0001) (Fig. 7A). In the case of CMX001, delaying treatment for 1–5 days affords 100% protection from lethality and mortality (Fig. 7C). Initiating treatment 1 day later, i.e. 6 days p.i., yields 100% morbidity (P < 0.0001) (Fig. 7C and D). These data reveal that CMX001 can be administered several days following a lethal infectious dose of ECTV and still provide complete protection from mortality.
Fig. 7.
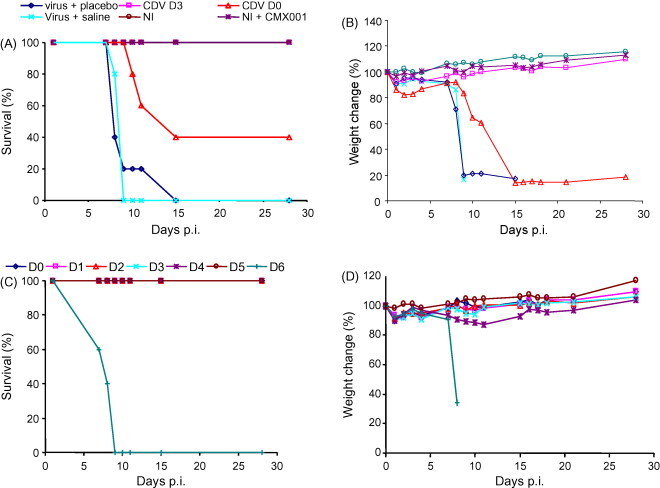
Delayed treatment with CMX001 following a low-dose infection. Groups of five A/Ncr mice were intra-nasally infected with 5 PFU of ECTV. Treatment began with a 10 mg/kg loading dose of CMX001 followed by 2.5 mg/kg doses of CMX001 on every second day. Initiation of treatment commenced on day 0, 1, 2, 3, 4, 5 or 6 (indicated by D0, D1, D2, D3, D4, D5 or D6) post-infection (C and D). Infected control mice were treated with 100 mg/kg of CDV on day 0 or 3 (CDV D0 and CDV D3), virus + placebo or virus + saline (A and B). Uninfected mice received no treatment (NI) or a treatment with CMX001 (A and B).
4. Discussion
The success of the 20th century smallpox eradication program was based on the lack of an animal reservoir for VARV, the availability of inexpensive stable vaccines, a low mobility population, a long incubation period that allowed for tracing of post-exposure contacts, and a relatively high level of herd immunity. In the 21st century, we are faced with the potential use of natural or recombinant VARV and MPXV as biological weapons, and the emergence of MPXV as a more important zoonotic disease. Currently, the human population lacks solid herd immunity to orthopoxviruses due to the cessation of smallpox vaccination in the 1970s. Also, the traditional smallpox vaccine is not suitable for a growing percentage of the world's population due to several contraindications (Marris, 2007, Wiser et al., 2007). The threat posed by the intentional release of VARV, MPXV or an epizoonosis will require a capacity to rapidly diagnose the disease and to intervene therapeutically and prophylactically with antivirals. Intervention is likely to take place during the diagnosis of the primary (incident) cases after onset of disease 7–14 days p.i. (Fenner et al., 1988). Vaccine pre-immunisation of ‘at risk’ cohorts with vaccines is not practical, and therapeutic use of vaccines is likely ineffective beyond day 4 of infection based on less than optimal clinical data acquired during the smallpox eradication program (Mortimer, 2003). Intramuscular administration of vaccinia immune globulin (VIG), a product derived from the pooled plasma of vaccinated individuals, is indicated as a suitable treatment of generalised vaccinia, progressive vaccinia (vaccinia necrosum), eczema vaccinatum, and certain auto-inoculations (Bray, 2003). However, the efficacy of VIG has not been demonstrated in controlled clinical trials and its robustness is doubtful. One large study did suggest that vaccination and VIG treatment of individuals that were in contact with smallpox patients appeared more efficacious than vaccination alone, but a VIG only arm was not evaluated (Rosenthal et al., 2001). There is a need for an antiviral that is efficacious when administered during frank diseases; however, none are currently licensed or approved for emergency use.
CDV is a nucleotide analogue that selectively inhibits viral DNA polymerase and has been shown to be highly efficacious against most, and possibly all, double-stranded DNA viruses that cause human morbidity and mortality (De Clercq, 1996, Lalezari et al., 1995). It has been approved for the treatment of cytomegalovirus retinitis in HIV/AIDS patients (Safrin et al., 1997), and has potent antiviral activities against poxviral infections in mice and monkeys (Bray et al., 2002, De Clercq, 2002, Quenelle et al., 2003). CDV is currently the only drug available for the treatment of smallpox infections under an Investigational New Drug Application; however, its use in a large smallpox or monkeypox outbreak would be limited due to its route of administration and its potential to cause nephrotoxicity—both of which require rigorous clinical management. There is a need for an orally bioavailable drug with strong and rapid antiviral activity against VARV and MPXV. To address this need, several ether–lipid conjugates of CDV have been synthesized, with CMX001 demonstrating the best therapeutic index (Quenelle et al., 2004, Buller et al., 2004, Ciesla et al., 2003). CMX001 was designed to mimic the structure of lysophosphatidylcholine, and utilise its uptake pathway (Hostetler et al., 1997). CMX001 is efficiently absorbed by the mouse small intestine and approximately 88% of the orally administered drug can be accounted for in plasma over a 72 h period, but because CMX001 is not concentrated in the kidney as efficiently as CDV, it has not been shown to cause nephrotoxicity in studies employing CMX001 for up to 14 days (Ciesla et al., 2003). The potential stability of CMX001 in blood was studied in vitro using assays derived from S9 liver fractions (Table 1 ) and primary cryopreserved hepatocyctes (Table 2 ). A similar stability of CMX001 was detected in cultures of mouse and human primary hepatocytes and S9 liver fractions supporting the relevance of mouse models to generate efficacy data for licensure under the Animal Efficacy Rule (Table 1). Interestingly, CMX001 was metabolised much faster in similar assays based on hepatocytes and extracts from livers of cynomolgus monkeys, suggesting that non-human primates are not an appropriate animal model for predicting the efficacy of CMX001 in humans.
Table 1.
Metabolism of CMX001 in S9 liver fractions
| Species | Concentration (μM) | Percentage of CMX001 remaining at incubation time point |
|||||
|---|---|---|---|---|---|---|---|
| 0 min | 15 min | 30 min | 60 min | 90 min | Half-life | ||
| CD-1 mouse | 1 | 100 | 83 | 113 | 99 | 95 | >100 |
| 10 | 100 | 75 | 104 | 66 | 40 | 79 | |
| Cynomolgus monkey | 1 | 100 | 13 | 5 | 1 | 1 | 8 |
| 10 | 100 | 17 | 7 | 2 | 0 | 9 | |
| Human | 1 | 100 | 82 | 107 | 90 | 55 | 97 |
| 10 | 100 | 63 | 54 | 35 | 26 | 37 | |
Table 2.
Metabolism of CMX001 in primary cryopreserved hepatocytes
| Species | Concentration (μM) | Percentage of CMX001 remaining at incubation time point |
|||||
|---|---|---|---|---|---|---|---|
| 0 min | 60 min | 120 min | 180 min | 240 min | Half-life | ||
| CD-1 mouse | 1 | 96 | 77 | 59 | 55 | 46 | 213 |
| 10 | 99 | 88 | 72 | 67 | 61 | >260 | |
| Cynomolgus monkey | 1 | 96 | 49 | 20 | 8 | 4 | 58 |
| 10 | 99 | 86 | 74 | 66 | 61 | >260 | |
| Human | 1 | 96 | 85 | 67 | 57 | 48 | 228 |
| 10 | 100 | 97 | 88 | 85 | 77 | >260 | |
In this study we have shown CMX001 to be as efficacious as CDV when treatment was initiated at the time of aerosol or intra-nasal infections with ECTV. Following high-dose aerosol and intra-nasal infections, both CDV and CMX001 significantly reduced titres in the liver and spleen, but not lungs (Fig. 1, Fig. 2). This suggests that following an aerosol infection as a result of a bioterrorist attack, CMX001 will likely reduce mortality and morbidity in the human population, but may not reduce transmissibility of the virus to first-generation contacts. However, CMX001 radically reduced lung titres following an intra-nasal infection, a route thought to mimic natural transmission. Presumably this resulted from the drug treatment reducing the efficiency of the virus replication cycles as the virus spread from the initial site of replication in the upper respiratory tract via the lymphatics (primary viremia) to internal organs (e.g. liver, spleen) and back to the mucosal epithelium of the respiratory tract (see review by Parker et al., 2007). The transmission cycle will be broken without efficient seeding of the respiratory tract. Hence, the risk of second-generation infections from an initial aerosol infection would be expected to be greatly diminished in CMX001 contacts.
In a series of experiments to explore the relationship between the level of virus infectivity present in the challenge inoculum and the mg/kg of CMX001 exposure required to protect from lethal disease, we demonstrated that higher infectivity challenges required higher doses of CMX001 to provide complete protection. We found that following a low-dose challenge we could protect 100% of mice when treatment initiation was delayed until 5 days p.i., over halfway through a disease course that ends in 100% uniform mortality 8–9 days following infection. This therapeutic window would be equivalent to intervening during the rash period of ordinary smallpox (>12 days p.i.) (Fenner et al., 1988).
The 5–100 PFU dose range utilised in these studies is reflective of a dose that results in uniform infection of other hosts by related and distinct viruses, such as influenza virus (Alford et al., 1966). The LD50 of ECTV by the intra-nasal route for the A/Ncr mouse is 0.3 PFU (Buller et al., 2004). If the LD50 is calculated using virus particle concentrations as measured by electron microscopy with latex spheres as standards instead of PFUs, the value for the A/Ncr mouse by the footpad route is approximately 1.6 particles (Chen et al., 1992). Thus, using a particle to PFU ratio of 1:20, which is typical for orthopoxviruses, a 5–100 PFU dose of ECTV is actually 100–2000 particles, the vast majority of which are potentially infectious. It is reasonable to assume that the natural infectious dose of variola virus does not exceed the 5–100 PFU dose utilized in these studies.
Based on several lines of evidence we hypothesise that the 5–100 PFU dose used in these studies actually exceeds the natural infectious dose of VARV. Primarily, at peak levels of infectiousness VARV titers in oropharyngeal secretions of smallpox patients were rarely above 7 × 104 plaque forming units per milliliter (Sarkar et al., 1973), for comparison, human influenza A virus reaches titers of 107 TCID50 units per milliliter of oropharyngeal secretion (Murphy et al., 1975). Thus, even at the peak level of infectiousness, only small amounts of virus would be expelled in the respiratory gases and be available for infection, which makes the 58% secondary attack rate all the more impressive (Fenner et al., 1988).
CMX001 is currently in phase 1 clinical trials and continues to show promise for use as the first oral antiviral to treat poxvirus infections. Another orally administered antiviral is in phase II clinical trials. ST-246 has been shown to be highly efficacious against orthopoxviruses in vivo and in vitro (Yang et al., 2005). In animal studies it has been reported to be effective against lethal viral doses and can be administered several days p.i. (Quenelle et al., 2007, Yang et al., 2005). ST-246 has also been shown to be effective against CDV resistant poxviruses in accord with its targeting a different stage of the replication cycle than CMX001 (Yang et al., 2005). The distinct mechanism of action of ST-246 and CMX001 suggests that combination therapy could be a viable therapeutic option. Such therapy could retard the generation of resistance to each drug, reduce the inherent efficacy variation in the out-bred human population, and provide a more potent therapy for late stage disease. Studies to investigate the use of both drugs in combination to treat late stage poxvirus disease are eagerly anticipated.
Acknowledgements
This work was supported by NIAID NOI-AI-15436 and through a subcontract with Chimerix Inc. We thank Monica Allen for administrative assistance, Ed Hembrador for assaying virus infectivity, and Elsa Taricone and Jill Schriewer for technical assistance.
References
- Alford R.H., Kasel J.A., Gerone P.J., Knight V. Human influenza resulting from aerosol inhalation. Proc. Soc. Exp. Biol. Med. 1966;122:800–804. doi: 10.3181/00379727-122-31255. [DOI] [PubMed] [Google Scholar]
- Baker R.O., Bray M., Huggins J.W. Potential antiviral therapeutics for smallpox, monkeypox and other orthopoxvirus infections. Antiviral Res. 2003;57:13–23. doi: 10.1016/S0166-3542(02)00196-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray M. Pathogenesis and potential antiviral therapy of complications of smallpox vaccination. Antiviral Res. 2003;58:101–114. doi: 10.1016/s0166-3542(03)00008-1. [DOI] [PubMed] [Google Scholar]
- Bray M., Martinez M., Kefauver D., West M., Roy C. Treatment of aerosolized cowpox virus infection in mice with aerosolized cidofovir. Antiviral Res. 2002;54:129–142. doi: 10.1016/S0166-3542(01)00220-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buller R.M., Owens G., Schriewer J., Melman L., Beadle J.R., Hostetler K.Y. Efficacy of oral active ether lipid analogs of cidofovir in a lethal mousepox model. Virology. 2004;318:474–481. doi: 10.1016/j.virol.2003.11.015. [DOI] [PubMed] [Google Scholar]
- Chen W., Drillien R., Spehner D., Buller R.M. Restricted replication of ectromelia virus in cell culture correlates with mutations in virus-encoded host range gene. Virology. 1992;187:433–442. doi: 10.1016/0042-6822(92)90445-u. [DOI] [PubMed] [Google Scholar]
- Ciesla S.L., Trahan J., Wan W.B., Beadle J.R., Aldern K.A., Painter G.R., Hostetler K.Y. Esterification of cidofovir with alkoxyalkanols increases oral bioavailability and diminishes drug accumulation in kidney. Antiviral Res. 2003;59:163–171. doi: 10.1016/s0166-3542(03)00110-4. [DOI] [PubMed] [Google Scholar]
- De Clercq E. Therapeutic potential of cidofovir (HPMPC, Vistide) for the treatment of DNA virus (i.e. herpes-, papova-, pox- and adenovirus) infections. Verh. K. Acad. Geneeskd. Belg. 1996;58:19–47. [PubMed] [Google Scholar]
- De Clercq E. Cidofovir in the treatment of poxvirus infections. Antiviral Res. 2002;55:1–13. doi: 10.1016/S0166-3542(02)00008-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenner F., Henderson D.A., Arita I., Jezek Z., Ladnyi I.D. World Health Organisation; Geneva: 1988. Smallpox and its Eradication. [Google Scholar]
- Guyton A.C. Measurement of the respiratory volumes of laboratory animals. Am. J. Physiol. 1947;150:70–77. doi: 10.1152/ajplegacy.1947.150.1.70. [DOI] [PubMed] [Google Scholar]
- Hostetler K.Y., Beadle J.R., Kini G.D., Gardner M.F., Wright K.N., Wu T.H., Korba B.A. Enhanced oral absorption and antiviral activity of 1-O-octadecyl-sn-glycero-3-phospho-acyclovir and related compounds in hepatitis B virus infection, in vitro. Biochem. Pharmacol. 1997;53:1815–1822. doi: 10.1016/s0006-2952(97)82446-x. [DOI] [PubMed] [Google Scholar]
- Hostetler K.Y., Beadle J.R., Trahan J., Aldern K.A., Owens G., Schriewer J., Melman L., Buller R.M. Oral 1-O-octadecyl-2-O-benzyl-sn-glycero-3-cidofovir targets the lung and is effective against a lethal respiratory challenge with ectromelia virus in mice. Antiviral Res. 2007;73:212–218. doi: 10.1016/j.antiviral.2006.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson R.J., Ramsay A.J., Christensen C.D., Beaton S., Hall D.F., Ramshaw I.A. Expression of mouse interleukin-4 by a recombinant ectromelia virus suppresses cytolytic lymphocyte responses and overcomes genetic resistance to mousepox. J. Virol. 2001;75:1205–1210. doi: 10.1128/JVI.75.3.1205-1210.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith K.A., Wan W.B., Ciesla S.L., Beadle J.R., Hostetler K.Y., Kern E.R. Inhibitory activity of alkoxyalkyl and alkyl esters of cidofovir and cyclic cidofovir against orthopoxvirus replication in vitro. Antimicrob. Agents Chemother. 2004;48:1869–1871. doi: 10.1128/AAC.48.5.1869-1871.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern E.R., Hartline C., Harden E., Keith K., Rodriguez N., Beadle J.R., Hostetler K.Y. Enhanced inhibition of orthopoxvirus replication in vitro by alkoxyalkyl esters of cidofovir and cyclic cidofovir. Antimicrob. Agents Chemother. 2002;46:991–995. doi: 10.1128/AAC.46.4.991-995.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalezari J.P., Drew W.L., Glutzer E., James C., Miner D., Flaherty J., Fisher P.E., Cundy K., Hannigan J., Martin J.C. (S)-1-[3-Hydroxy-2-(phosphonylmethoxy)propyl]cytosine (cidofovir): results of a phase I/II study of a novel antiviral nucleotide analogue. J. Infect. Dis. 1995;171:788–796. doi: 10.1093/infdis/171.4.788. [DOI] [PubMed] [Google Scholar]
- Marris E. Dramatic rescue relieves rare case of smallpox infection. Nat. Med. 2007;13:517. doi: 10.1038/nm0507-517. [DOI] [PubMed] [Google Scholar]
- Mortimer P.P. Can postexposure vaccination against smallpox succeed? Clin. Infect. Dis. 2003;36:622–629. doi: 10.1086/374054. [DOI] [PubMed] [Google Scholar]
- Moss B., Earl P.L. Current Protocols in Molecular Biology. Wiley; 1998. Expression of proteins in mammalian cells using vaccinia cirus vectors. Overview of the vaccinia virus expression system. pp. 16.15.1–16.15.5. [Google Scholar]
- Murphy B.R., Richman D.D., Chalhub E.G., Uhlendorf C.P., Baron S., Chanock R.M. Failure of attenuated temperature-sensitive influenza A (H3N2) virus to induce heterologous interference in humans to parainfluenza type 1 virus. Infect. Immun. 1975;12:62–68. doi: 10.1128/iai.12.1.62-68.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker S., Nuara A., Buller R.M., Schultz D.A. Human monkeypox: an emerging zoonotic disease. Future Microbiol. 2007;2:17–34. doi: 10.2217/17460913.2.1.17. [DOI] [PubMed] [Google Scholar]
- Quenelle D.C., Buller R.M., Parker S., Keith K.A., Hruby D.E., Jordan R., Kern E.R. Efficacy of delayed treatment with ST-246 given orally against systemic orthopoxvirus infections in mice. Antimicrob. Agents Chemother. 2007;51:689–695. doi: 10.1128/AAC.00879-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quenelle D.C., Collins D.J., Kern E.R. Efficacy of multiple- or single-dose cidofovir against vaccinia and cowpox virus infections in mice. Antimicrob. Agents Chemother. 2003;47:3275–3280. doi: 10.1128/AAC.47.10.3275-3280.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quenelle D.C., Collins D.J., Wan W.B., Beadle J.R., Hostetler K.Y., Kern E.R. Oral treatment of cowpox and vaccinia virus infections in mice with ether lipid esters of cidofovir. Antimicrob. Agents Chemother. 2004;48:404–412. doi: 10.1128/AAC.48.2.404-412.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal S.R., Merchlinsky M., Kleppinger C., Goldenthal K.L. Developing new smallpox vaccines. Emerg. Infect. Dis. 2001;7:920–926. doi: 10.3201/eid0706.010602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safrin S., Cherrington J., Jaffe H.S. Clinical uses of cidofovir. Rev. Med. Virol. 1997;7:145–156. doi: 10.1002/(sici)1099-1654(199709)7:3<145::aid-rmv196>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Sarkar J.K., Mitra A.C., Mukherjee M.K., De S.K., Mazumdar D. Bulletin of the World Health Organization. 1973. Virus excretion in smallpox 1. pp. 517–522. [PMC free article] [PubMed] [Google Scholar]
- Wallace G.D., Buller R.M. Kinetics of ectromelia virus (mousepox) transmission and clinical response in C57BL/6j, BALB/cByj and AKR/J inbred mice. Lab. Anim. Sci. 1985;35:41–46. [PubMed] [Google Scholar]
- Wiser I., Balicer R.D., Cohen D. An update on smallpox vaccine candidates and their role in bioterrorism related vaccination strategies. Vaccine. 2007;25:976–984. doi: 10.1016/j.vaccine.2006.09.046. [DOI] [PubMed] [Google Scholar]
- Yang G., Pevear D.C., Davies M.H., Collett M.S., Bailey T., Rippen S., Barone L., Burns C., Rhodes G., Tohan S., Huggins J.W., Baker R.O., Buller R.L., Touchette E., Waller K., Schriewer J., Neyts J., DeClercq E., Jones K., Hruby D., Jordan R. An orally bioavailable antipoxvirus compound (ST-246) inhibits extracellular virus formation and protects mice from lethal orthopoxvirus challenge. J. Virol. 2005;79:13139–13149. doi: 10.1128/JVI.79.20.13139-13149.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]