

ABSTRACT
The mechanisms by which the ATG16L1T300A polymorphism affects cell function and causes an increased risk for the development of Crohn disease remain incompletely understood. Here we report that healthy individuals and mice bearing this polymorphism, even as heterozygotes, manifest enhanced TLR, and NLR cytokine and chemokine responses due to increased activation of NFKB. We elucidated the mechanism of the NFKB abnormality and found that in the ATG16L1T300A cell, there is enhanced polyubiquitination of TRAF6 or RIPK2 resulting from the accumulation of SQSTM1/p62. Indeed, knockout of Sqstm1 in autophagy-deficient cells almost completely normalized TRAF6 or RIPK2 polyubiquitination and NFKB activation in these cells. Thus, by identifying that autophagy is a pathway-intrinsic homeostatic mechanism that restricts excessive TLR- or NLR-mediated inflammatory signaling, our findings shed new light on how the ATG16L1T300A polymorphism sets the stage for the occurrence of Crohn disease.
Abbreviations: 3-MA: 3-methyladenine; ATG16L1: autophagy related 16 like 1; ATG7: autophagy related 7; BMDM: bone marrow-derived macrophage; CD: Crohn disease; CXCL: C-X-C motif chemokine ligand; IBD: inflammatory bowel disease; iBMDM: immortalized mouse BMDM; IL1B/IL-1β: interleukin 1 beta; IL6: interleukin 6; KI: knockin; KO: knockout; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; LPS: lipopolysaccharide; MDP: muramyl dipeptide; MEF: mouse embryonic fibroblast; NFKB/NF-κB: nuclear factor kappa B; NFKBIA/IKBA: NFKB inhibitor alpha; NLR: NOD-like receptor; NOD: nucleotide-binding oligomerization domain containing; RIPK2: receptor interacting serine/threonine kinase 2; SNP: single nucleotide polymorphism; SQSTM1/p62: sequestosome 1; TLR: toll like receptor; TNF/TNF-α: tumor necrosis factor; TRAF6: TNF receptor associated factor 6; Ub: ubiquitin; WT: wild type
KEYWORDS: ATG16L1T300A, autophagy, NFKB, NLR, TLR4
Introduction
The ATG16L1 gene encodes a protein essential to the function of macroautophagy/autophagy, an evolutionarily conserved phagosome-like process that facilitates the disposal of discarded intracellular proteins and participates in some aspects of host defense. Over a decade ago, it was shown that a single nucleotide polymorphism (SNP) in the ATG16L1 gene (ATG16L1T300A) confers increased risk for the development of Crohn disease (CD) [1,2]. However, despite considerable study, the mechanisms by which the variant ATG16L1 gene contributes to Crohn disease pathogenesis are still not fully clarified.
The risk polymorphism associated with CD consists of an A > G SNP in the ATG16L1 gene that results in a Thr-to-Ala mutation at position 300 (thus designated: T300A) [1,2]. Recently, it was shown that the T300A variant of the ATG16L1 gene sensitizes the latter to CASP3 (caspase 3)-mediated cleavage [3,4]. This important finding implies that the pro-inflammatory effect of this polymorphism may require the presence of a more primary abnormality resulting in the release of cytokines (such as TNF/TNF-α) capable of inducing CASP3 activation [3,5].
One hypothesis that satisfies this requirement is that the ATG16L1 polymorphism, through its impact on autophagy, affects pro-inflammatory hematopoietic cell function. Support for this idea comes first from the well-established fact that the presence of the polymorphism is associated with increased NLRP3 inflammasome activity [6]; however, there is no as yet convincing evidence indicating that this inflammasome plays an important role in most cases of CD [7]. There are also studies showing that the ATG16L1 polymorphism causes decreased clearance of pathogenic organisms and increased proinflammatory cytokine production [8–11]. This suggests that the ability of this polymorphism to increase the risk for CD occurrence may consist in its association of impaired autophagy with TLR- and NLR-mediated effects on NFKB/NF-κB activation. However, the direct link between impaired autophagy and TLR or NLR mediated signaling has never been fully established.
In this study, we utilize human macrophages bearing the Crohn disease-associated ATG16L1T300A polymorphism and Atg16l1T300A/T300A KI mouse cells to directly address the influence of ATG16L1T300A on the regulation of TLR- and NLR-mediated inflammatory signaling. The data obtained provided strong evidence that the disturbance in autophagy by the ATG16L1T300A polymorphism causes an increased risk of CD by inducing a fundamental defect in NFKB-mediated inflammation.
Results
Human macrophages and mouse cells bearing the Crohn disease-associated ATG16L1T300A polymorphism exhibit enhanced pro-inflammatory responses
The Crohn disease risk-associated ATG16L1T300A polymorphism is present in a large fraction of normal individuals as well as in individuals with Crohn disease and can be functionally evaluated in the former group in the relative absence of confounding genetic or environmental factors that cause CD. Accordingly, we assembled a group of healthy volunteers who were negative or positive for the (ATG16L1T300A) on one or both alleles by appropriate screening and then utilized peripheral monocyte-derived macrophages obtained from these individuals to determine responses to TLR and NLR ligand stimulation. We found that when we stimulated macrophages from these individuals with TLR4 ligand (LPS) or NOD2 ligand (MDP) for the indicated time and then evaluated cell lysates by RT-PCR, lysates from donors that carry the risk-associated polymorphism on one or both alleles exhibited increased expression of chemokine (CXCL1) and cytokine (IL1B and IL6) mRNA compared to cells from WT donors that are negative for the risk-associated polymorphism on both alleles (Figure 1 A and B).
Figure 1.
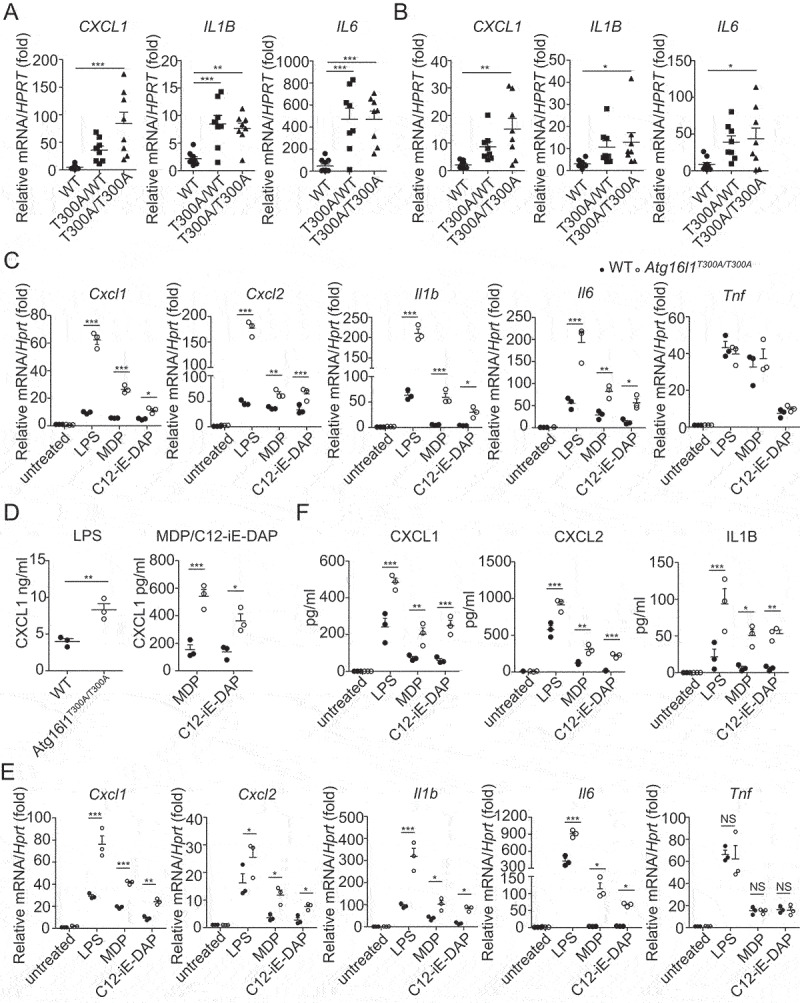
Human macrophages and mouse cells bearing the Crohn disease–associated ATG16L1T300A variant exhibit enhanced pro-inflammatory responses. (A-B) Expression of CXCL1, IL1B, and IL6 in the human monocyte-derived macrophages obtained from WT and ATG16L1T300A donors (WT: n = 10; ATG16L1T300A/WT: n = 8; ATG16L1T300A/T300A: n = 8) treated for 3 h in vitro with LPS (A) or MDP (B). Each symbol represents an individual donor; (C-D) Expression of the chemokines and cytokines in the MEF cells obtained from WT and Atg16l1T300A/T300A KI mice treated with LPS, MDP, C12-iE-DAP for indicated times were detected by real-time PCR (C) and ELISA (D); (E-F) Expression of the chemokines and cytokines in the BMDM cells obtained from WT and Atg16l1T300A/T300A KI mice treated as in (C) and (D) were assessed by real-time PCR (E) and ELISA (F). p-values were determined using one-way ANOVA followed by Tukey’s multiple-comparisons test (A-F). *p < 0.05. **p < 0.01. ***p < 0.001.
In parallel studies of murine cells, we investigated TLR and NLR responses of both mouse embryonic fibroblasts (MEFs) and bone marrow-derived macrophages (BMDMs) derived from Atg16l1T300A/T300A knockin (KI) mice. In initial studies, Atg16l1T300A/T300A KI MEFs were cultured with TLR4 ligand (LPS), NOD1 ligand (C12-iE-DAP), or NOD2 ligand (MDP) for the indicated time and the cell lysates and culture supernatants were then assayed by quantitative RT-PCR or ELISA respectively to determine levels of various chemokines and cytokines. We found that MEFs from Atg16l1T300A/T300A KI mice treated with LPS, MDP, or C12-iE-DAP exhibited significantly elevated Cxcl1, Cxcl2, Cxcl15 as well as Il1b and Il6 production compared with that of WT MEFs (Figure 1 C and D), whereas Tnf production resulting from the various types of stimulation was not significantly different. In subsequent studies, we evaluated BMDMs from WT or Atg16l1T300A/T300A KI mice in a similar manner and found that BMDMs from Atg16l1T300A/T300A KI mice also exhibited significantly increased Cxcl1, Cxcl2, Il1b, and Il6 but not Tnf production as compared to WT BMDMs (Figure 1 E and F).
Taken together, these observations of human macrophages and mouse MEFs or BMDMs established that the presence of the ATG16L1T300A polymorphism results in cells displaying a hyper-inflammatory response to TLR4 and NOD ligand stimulation and suggested that ATG16L1 negatively regulates TLR4- or NLR-mediated chemokine and cytokine production in stimulated cells.
Enhanced TLR and NLR responses in human macrophages and mouse cells bearing the Crohn disease-associated ATG16L1T300A polymorphism are mediated by NFKB activation
Having observed that the presence of ATG16L1T300A in both human and mouse cells led to upregulated chemokine and pro-inflammatory cytokine production, we sought to determine at which step of the TLR4, NOD1, and NOD2 signaling pathways this polymorphism exerts its regulatory role. Because CXCL1 and CXCL2 transcription is solely regulated by NFKB [12–14], we first determined whether TLR- or NLR- stimulated NFKB activation was altered in human macrophages bearing the ATG16L1T300A polymorphism or mouse ATG16L1T300A cells. In these studies, human monocyte-derived macrophages from ATG16L1T300A polymorphism-positive and -negative human donors were stimulated with LPS or MDP for the indicated time and whole-cell lysates of the cells were subjected to immunoblotting to determine NFKB activation by assessment of p-NFKBIA/IKBA levels. In line with the chemokine and cytokine findings, we found that NFKB activation was increased in macrophages from polymorphism-positive donors upon LPS (Figure 2A) or MDP (Figure 2B) stimulation as compared with that of macrophages from WT donors (Figure 2 A and B). Thus, the presence of the ATG16L1T300A polymorphism led to enhanced NFKB activation upon TLR or NLR stimulation in human macrophages.
Figure 2.
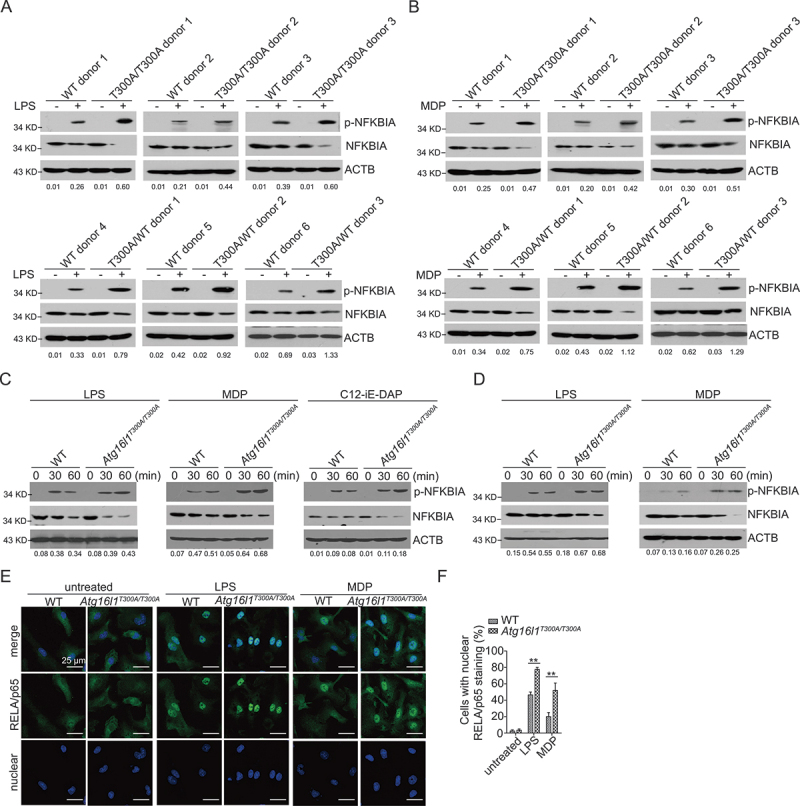
Enhanced TLR and NLR responses in human and mouse ATG16L1T300A/T300A KI cells are mediated by NFKB activation. (A-B) Human monocyte-derived macrophages from WT and ATG16L1T300A donors were treated with LPS (A) or MDP (B) for 1 h after which whole cell extracts were obtained and subjected to immunoblot analysis with anti-p-NFKBIA, anti-NFKBIA, and anti-ACTB/β-actin. (C-D) WT and Atg16l1T300A/T300A KI MEFs (C) or BMDMs (D) were treated with LPS, MDP, or C12-iE-DAP after which whole cell extracts were obtained and subjected to immunoblot analysis with anti-p-NFKBIA, anti-NFKBIA, and anti-ACTB. (E) WT and Atg16l1T300A/T300A KI BMDMs were treated with LPS or MDP and then subjected to immunofluorescent staining to detect RELA/p65 translocation from the cytosol to the nuclear (RELA/p65, green; DAPI, blue). Scale bar: 25 µm. (F) Quantitative assessment of the percentage of cells with RELA/p65 translocation in (E); at least 100 cells in 3 random fields were assessed. Numbers underneath the blot (A-D) represent the fold change of p-NFKBIA intensity compared with ACTB. p-values were determined using one-way ANOVA followed by Tukey’s multiple-comparisons test (F). *p < 0.05. **p < 0.01.
Studies of MEFs and BMDMs derived from Atg16l1T300A/T300A KI mice corroborated the above findings from studies of human cells. Here we first assessed NFKB activation in LPS-, MDP- or C12-iE-DAP-stimulated MEFs obtained from Atg16l1T300A/T300A KI or WT mice for indicated times and then subjected whole-cell lysates to immunoblotting to determine NFKB activation by assessment of p-NFKBIA levels. We found that Atg16l1T300A/T300A KI MEF cell lysates contained greatly increased amounts of p-NFKBIA as compared with lysates of WT cells (Figure 2C).
In parallel studies of WT and Atg16l1T300A/T300A KI BMDMs, we again found increased p-NFKBIA expression in LPS and MDP-stimulated BMDMs from mice bearing the polymorphism (Figure 2D). In addition, in confocal microscopy-based studies assessing NFKB activation by translocation of the RELA/p65 subunit of NFKB into the nucleus [15], we found that while after stimulation with either LPS or MDP, both BMDMs from Atg16l1T300A/T300A KI and WT mice exhibited increased nuclear RELA/p65, this increase was far greater in BMDMs from Atg16l1T300A/T300A KI mice than BMDMs from WT mice (Figure 2 E and F). These findings suggested that ATG16L1T300A regulates TLR4, NOD1, and NOD2 signaling upstream or at the level of NFKB activation.
Finally, we found that as in studies of macrophages from heterozygous humans described above and in line with a previous study in which we showed that heterozygous (Atg16l1T300A/+) mice are similar to homozygous mice with respect to autophagy induction and bacterial clearance [9], BMDMs from both Atg16l1T300A homozygous and heterozygous mice display equivalent levels of TLR and NLR-induced enhancement of NFKB activation (FigureS 1 A and B).
Taken together, these data established that the increased pro-inflammatory cytokine production in human or mouse ATG16L1T300A cells is due to increased upregulation of TLR- or NLR-mediated NFKB activation.
Upregulated NFKB activation and pro-Inflammatory cytokine production in ATG16L1T300A cells are autophagy dependent
To determine whether the upregulated TLR- and NLR-induced NFKB signaling in Atg16l1T300A cells is autophagy-dependent as implied by the studies above, i.e., due to a diminished capacity to mount a TLR- and NLR-induced autophagic response and not due to an effect of the polymorphism on a function unrelated to autophagy, we compared the ability of TLR and NLR stimuli to induce autophagy in Atg16l1T300A cells with that in atg16l1 KO (Atg16l1f/f-Lyz2/LysM-Cre) cells and WT cells. In initial phenotypic studies, we found that Atg16l1T300A cells do not differ from WT cells (or, from atg16l1 KO cells) with regard to the level of expression of TLR4 so it is not likely that differences in levels of autophagy are due to differences in initial TLR ligand signaling (FigureS2 A and B). In subsequent functional studies, BMDMs from WT, Atg16l1T300A/T300A KI, and atg16l1 KO BMDMs were stimulated with LPS, MDP, and C12-iE-DAP for the indicated times, and the cultured cells were subjected to immunofluorescence staining to detect LC3-II puncta. We found that stimulation of the WT BMDMs induced a sharp increase in the number of cells displaying LC3-II puncta as well as increased numbers of puncta per cell, whereas stimulated Atg16l1T300A/T300A KI BMDMs exhibited a much lower number of cells displaying LC3-II puncta and puncta per cell that was comparable to that in atg16l1 KO BMDMs (FigureS2 C and D). These findings relating to LC3-II puncta were supported by studies of LC3-II generation in which cell lysates of WT, Atg16l1T300A/T300A KI, and atg16l1 KO BMDMs stimulated with LPS, MDP, or C12-iE-DAP were subjected to immunoblotting to detect the conversion of endogenous LC3-I to LC3-II. We found that in agreement with the puncta results LC3-II generation was more robust in WT BMDMs as compared to that in Atg16l1T300A/T300A KI and atg16l1 KO BMDMs (FigureS2 E). In addition, lysates of the Atg16l1T300A/T300A and atg16l1 KO BMDMs contained increased amounts of SQSTM1/p62 (sequestosome 1) as compared to WT BMDMs (FigureS2 E), in line with a previous report showing that this protein is normally constitutively degraded by autophagy and accumulates when autophagy is inhibited [16]. Inasmuch as TLR and NLR stimulation of ATG16L1T300A/T300A BMDM resulted in a level of autophagy similar to that observed in cells with total deletion of ATG16L1, these studies provide evidence that the ATG16L1T300A/T300A polymorphism confers a diminished capacity to undergo TLR- and NLR-induced autophagy.
We next compared BMDMs from Atg16l1T300A/T300A KI mice with BMDMs from atg16l1 KO mice and WT mice with respect to their abilities to activate NFKB upon TLR and NLR stimulation. Here, we treated BMDMs from WT, Atg16l1T300A/T300A KI, atg16l1 KO mice with LPS, MDP, or C12-iE-DAP for the indicated times and then assessed phosphorylation of NFKBIA in cell lysates by immunoblotting. We found that the level of p-NFKBIA was greatly increased in Atg16l1T300A/T300A KI cells as well as in atg16l1 KO cells compared with that of WT cells (Figure 3A). In addition, immunofluorescence studies disclosed that the RELA/p65 subunit of NFKB exhibited greater nuclear localization in stimulated Atg16l1T300A/T300A KI and atg16l1 KO BMDMs than in the stimulated WT BMDMs (Figure 3 B and C). Thus, these studies show that TLR and NLR induction of NFKB activation in Atg16l1T300A/T300A KI cells is similar to that in atg16l1 KO BMDMs and therefore is likely due to loss of autophagic function.
Figure 3.
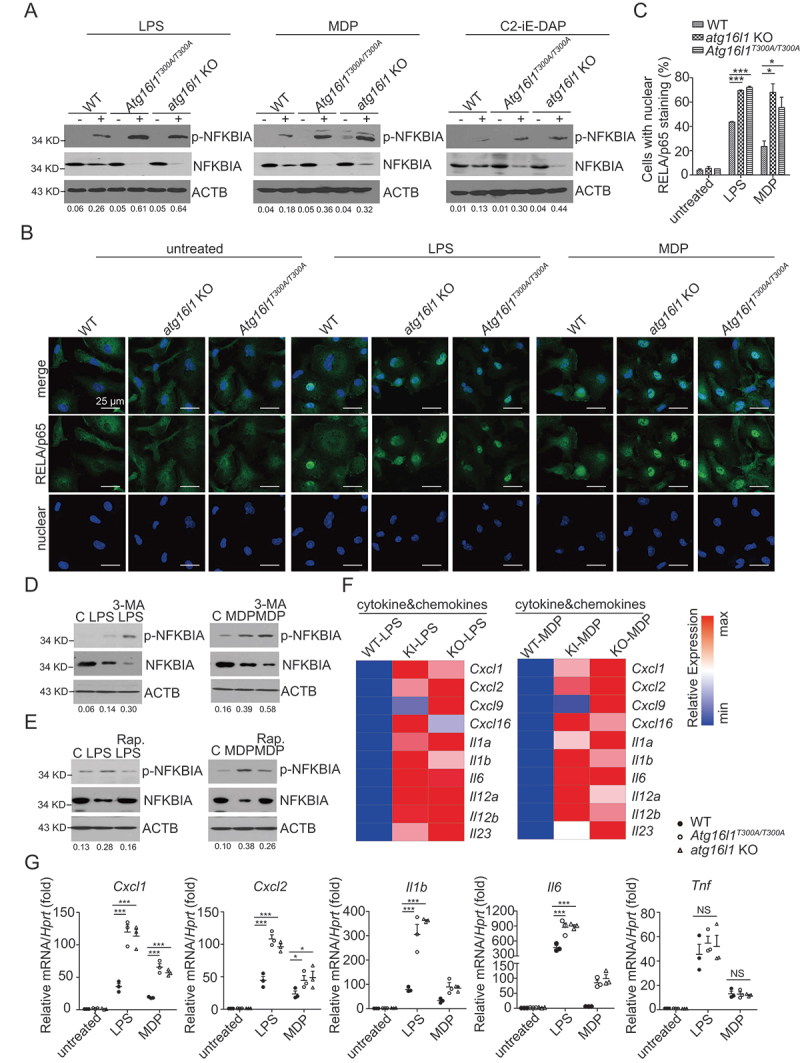
Upregulated NFKB activation and pro-inflammatory cytokine production in ATG16L1T300A/T300A KI cells are autophagy dependent. (A) BMDMs from WT, Atg16l1T300A/T300A KI, atg16l1 KO mice were treated with LPS, MDP, or C12-iE-DAP for 1 h after which whole cell extracts of the treated cells were obtained and subjected to immunoblot analysis with anti-p-NFKBIA, anti-NFKBIA, and anti-ACTB. Numbers underneath the blot represent the fold change of p-NFKBIA intensity compared with ACTB. (B) BMDMs from WT, atg16l1 KO, Atg16l1T300A/T300A KI mice were treated as in A and then subjected to immunofluorescent staining to detect RELA/p65 translocation (RELA/p65, green; DAPI, blue). Scale bar: 25 µm. (C) Quantitative assessment of the percentage of the cells with RELA/p65 translocation in (B); at least 100 cells in 3 random fields were assessed. (D-E) MEFs from WT mice were pretreated with 3-MA (D) or rapamycin (E) for 16 h and then treated with LPS (left) or MDP (right) after which the cells were subjected to immunoblot analysis to assess p-NFKBIA, NFKBIA; ACTB as a loading control. Numbers underneath the blot represent the fold change of p-NFKBIA intensity compared with ACTB. (F) Heat map of Atg16l1 KI-LPS vs. KO-LPS or Atg16l1 KI-MDP vs. KO-MDP regulated genes. (G) Real-time qPCR of selected pro-inflammatory genes identified in RNA-seq. p-values were determined using two-way ANOVA followed by Bonferroni post hoc test (C and G). *p < 0.05. **p < 0.01. ***p < 0.001.
In further studies designed to show that cells bearing the ATG16L1T300A/T300A polymorphism manifest enhanced NFKB activation because of impaired autophagy, we determined if alterations in autophagy in WT cells induced by a pharmacologic agent also influence the level of NFKB activation. In these studies, we treated TLR- or NLR-stimulated MEFs with one of two pharmacological reagents: 3-methyladenine (3-MA), a class III phosphatidylinositol 3-kinase inhibitor that suppresses autophagy [17] or rapamycin, an inhibitor of MTOR kinase that induces autophagy [18]. We reasoned that if NFKB activation is regulated by TLR or NLR stimulation via an effect on autophagy, 3-MA treatment should enhance TLR- or NOD-mediated NFKB activation, whereas rapamycin treatment should inhibit NFKB activation; we therefore evaluated the effect of 3-MA or rapamycin on stimulated MEFs with respect to their ability to generate p-NFKBIA. We found that lysates of 3-MA-treated cells subjected to immunoblotting exhibited increased LPS- and MDP-induced p-NFKBIA compared to untreated WT cells (Figure 3D, compare lane LPS or MDP only with lane LPS+ or MDP+3-MA). In contrast, we found that lysates of rapamycin-treated cells displayed decreased LPS- and MDP-induced p-NFKBIA compared to untreated WT MEFs (Figure 3E, compare lanes LPS or MDP only with lane LPS+ or MDP+rapamycin). These studies indicate that induced increases or decreases in the level of autophagy in WT cells affect the level of NFKB activation, and thus further support the idea that the autophagy changes in ATG16L1T300A/T300A cells explain the enhanced TLR- and NLR-induced NFKB activation displayed by these cells.
In an final set of studies addressing the relation of enhanced NFKB activity to autophagy, we constructed becn1 and atg7 KO BMDMs using CRISPR-Cas9 methodology and, after verification that the mutated cells had autophagy defects (FigureS3 A), we cultured these cells with LPS or MDP and subjected lysates of the cultured cells to immunoblotting for detection of p-NFKBIA; alternatively, we examined the cultured cells with confocal microscopy for detection of RELA/p65 nuclear translocation. We found that as in the above studies of atg16l1 KO cells, the becn1 and atg7 KO cells exhibited enhanced p-NFKBIA expression and RELA/p65 translocation, indicative of enhanced NFKB activation (FigureS3 B-D). These data indicate that ATG16L1T300A/T300A cells are similar to cells with a variety of mutations causing loss of autophagy with respect to their enhanced TLR- and NLR-induced activation of NFKB.
The findings above, in aggregate, provide strong evidence that autophagy suppresses TLR and NLR induction of NFKB activation, and that cells with defective autophagy such as those bearing the ATG16L1T300A polymorphism, manifest increased TLR- and NLR-induced NFKB activation; thus, in these cells, the increased TLR- and NLR-induced NFKB activation is in fact autophagy-dependent. It should be noted that since this enhancing effect is a strictly intracellular event, it is likely mediated by canonical autophagy rather than non-canonical autophagy, the latter a mechanistically different form of autophagy concerned with endocytosis or exocytosis. To verify this probability, we conducted additional experiments to determine if the Atg16l1T300A/T300A cells used in the above studies (MEFs and BMDMs) exhibit decreased autophagy when investigated under conditions known to elicit canonical autophagy, i.e., Atg16l1T300A/T300A MEFs or BMDMs cultured with bafilomycin A1 alone or cultured with rapamycin -/+ bafilomycin A1. We found that Atg16l1T300A/T300A MEFs and BMDMs cultured under these conditions, consistent with our previous findings [9], exhibited impaired autophagy as indicated by reduced LC3II conversion or LC3 puncta expression and increased SQSTM1/p62 expression as compared to WT cells (FigureS3 E-G). Thus, the Atg16l1T300A/T300A MEFs or BMDMs used in these studies to examine effects of impaired autophagy on TLR- and NLR-induced NFKB activation exhibit impaired canonical autophagy. In further studies discussed below we present evidence that these cells also support canonical autophagy induced by TLR and NLR stimulation.
Global pro-inflammatory gene expression upregulation in Atg16l1T300A/T300A KI and atg16l1 KO cells are similar
Given the data above showing that TLR- and NLR-induced NFKB activation is enhanced in both Atg16l1T300A/T300A KI and atg16l1 KO macrophages, we performed RNA-based next-generation sequencing (RNA-Seq) analysis of LPS- or MDP-treated Atg16l1T300A/T300A KI, atg16l1f/f-Lyz2/LysM-Cre (atg16l1 KO) and WT macrophages to assess whether the enhanced NFKB activation associated with these autophagy defects resulted in similar downstream effects on pro-inflammatory cytokine and chemokine production. These RNA-seq studies showed that most of the pro-inflammatory cytokine and chemokine genes up-regulated in the cells from Atg16l1T300A/T300A KI mice were also up-regulated in cells from atg16l1 KO mice (such as Cxcl1, Cxcl2, Il6, and Il1b, etc) (Figure 3F left (LPS), right (MDP). We subsequently confirmed these findings by selective RT-PCR studies that showed, consistent with the RNA-seq analysis, that a significantly greater abundance of transcripts of Cxcl1, Cxcl2, Il6, and Il1b was noted in BMDMs from atg16l1 KO mice relative to their abundance in BMDMs from WT mice (Figure 3G) and that this increase was not different from that of BMDMs from Atg16l1T300A/T300A KI mice. These observations thus provide evidence that the effect of ATG16L1T300A polymorphism is similar to complete ATG16L1 deletion with respect to its regulation of TLR and NLR pro-inflammatory signaling in macrophages.
Accumulation of ubiquitinated TRAF6 and RIPK2 in ATG16L1T300A/T300A KI cell upon LPS and MDP stimulation
We next turned our attention to the mechanism by which autophagy regulates TLR- and NLR-mediated NFKB activation. Recognizing that TLR and NLR activation of NFKB requires the polyubiquitination (hereafter referred to more simply as ubiquitination) of several signaling intermediates including TRAF6 and RIPK2 [19–21], we first determined whether autophagy regulates the ubiquitination of TRAF6 following TLR stimulation and RIPK2 following NLR stimulation. Accordingly, we treated LPS- or MDP-stimulated Raw cells with the autophagy regulators mentioned above, 3-MA or rapamycin, and then assessed ubiquitination of TRAF6 or RIPK2 in cell lysates of LPS- and MDP-stimulated cells respectively by immunoprecipitation with anti-TRAF6 or anti-RIPK2 antibody followed by immunoblotting with anti-ubiquitin. We found that 3-MA-treated cells exhibited increased ubiquitination of TRAF6 and RIPK2, whereas rapamycin-treated cells exhibited decreased ubiquitination of TRAF6 and RIPK2 as compared with that of untreated cells (Figure 4 A-D). On the basis of these results, we next assessed ubiquitination of TRAF6 or RIPK2 in TLR- or NLR-stimulated Atg16l1T300A/T300A KI and atg16l1 KO cells. Here, we stimulated BMDMs from WT, atg16l1 KO, and Atg16l1T300A/T300A KI mice with LPS or MDP and then subjected lysates of the stimulated cells to immunoprecipitation with anti-TRAF6 or anti-RIPK2 antibody followed by immunoblotting with anti-ubiquitin as above. We found that in LPS-stimulated cells, ubiquitination of endogenous TRAF6 was increased in both Atg16l1T300A/T300A KI and atg16l1 KO BMDMs as compared with that of WT BMDMs (Figure 4E). Similarly, in MDP-stimulated cells, ubiquitination of endogenous RIPK2 was increased in both Atg16l1T300A/T300A KI and atg16l1 KO BMDMs compared with that of WT BMDMs (Figure 4F). Similar observations were obtained with LPS- or MDP-stimulated becn1 KO Raw cells (FigureS4 A and B).
Figure 4.
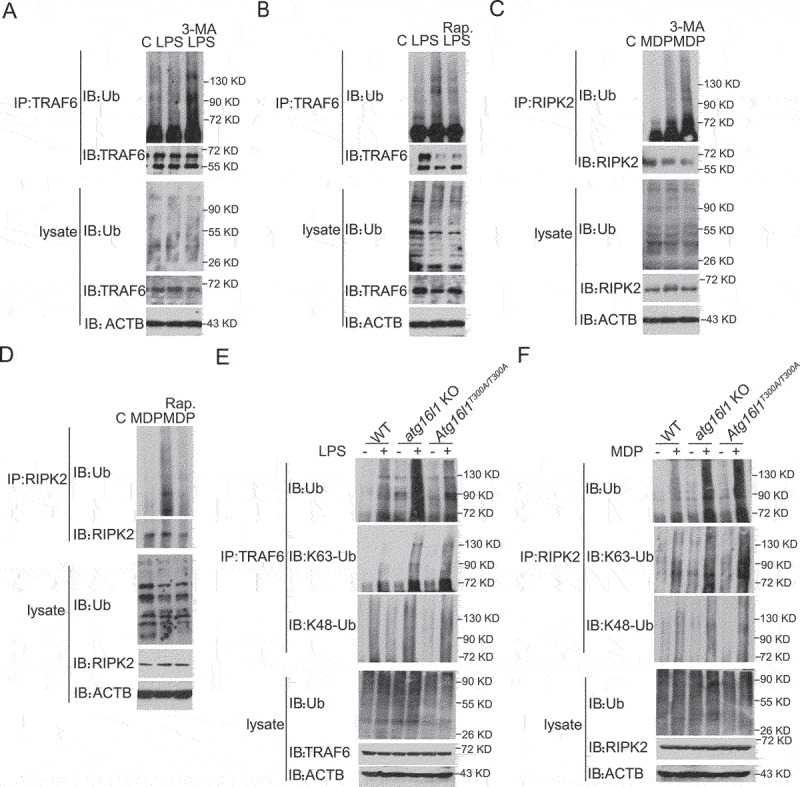
Accumulation of ubiquitinated TRAF6 and RIPK2 in Atg16l1T300A/T300A KI cell upon TLR4 and MDP stimulation. (A-B) Raw cells were pretreated with 3-MA (A) or rapamycin (B) for 16 h and then cultured with LPS for 1 h; extracts of the cells were then subjected to immunoprecipitation with anti-TRAF6 followed by immunoblotting with anti-TRAF6, anti-Ub, and anti-ACTB. (C-D) Raw cells were pretreated with 3-MA (C) or rapamycin (D) for 16 h and then cultured with MDP for 1 h; extracts of the cells were then subjected to immunoprecipitation with anti-RIPK2 followed by immunoblotting with anti-RIPK2, anti-Ub, and anti-ACTB. (E-F) BMDMs from WT, atg16l1 KO, and Atg16l1T300A/T300A KI mice were treated with LPS (E) or MDP (F) for 1 h; extracts of the cells were then subjected to immunoprecipitation with anti-TRAF6 (E) or anti-RIPK2 (F) followed by immunoblotting with anti-TRAF6 (E) or anti-RIPK2 (F), anti-Ub, and anti-ACTB.
It has been shown that K63-linked ubiquitination of TRAF6 or RIPK2 rather than K48-linked ubiquitination is associated with downstream activation of NFKB [22]; we therefore determined whether the increased ubiquitination of TRAF6 and RIPK2 in Atg16l1T300A/T300A KI or atg16l1 KO cells is K63- or K48-linked. In immunoblot studies of stimulated BMDMs from Atg16l1T300A/T300A KI and atg16l1 KO mice similar to those described above, but in this case, immunoblotting with anti-K63-specific or anti-K48-specific ubiquitin, we found that both TRAF6 and RIPK2 exhibited increased K63-linked ubiquitination compared to cells from WT mice, whereas K48-linked ubiquitination was comparable among cells from WT, Atg16l1T300A/T300A KI and atg16l1 KO mice (Figure 4 E and F)
In further studies relating the above findings to canonical autophagy, we took advantage of recent findings that have shown that canonical autophagy is independent of the WD40 repeat domain of ATG16L1 whereas non-canonical autophagy is dependent on this domain [23]. On this basis, we determined if the effect of autophagy on NFKB activation is dependent or independent of the ATG16L1 WD40 repeat domain. In these studies, we stably transduced both full-length ATG16L1 and ATG16L1 lacking the WD40-repeat domain into atg16l1-KO BMDM cells, stimulated the cells with TLR or NLR ligands, and then determined the level of NFKB activation and TRAF6 or RIPK2 ubiquitination. We found that transduction of atg16l1 KO cells with both full-length ATG16L1 and ATG16L1 lacking the WD40-repeat domain exhibited equally decreased NFKB activation and TRAF6 or RIPK2 ubiquitination compared to non-transduced cells, suggesting that the WD40-repeat domain of ATG16L1 is not required for autophagic effects on NFKB activation and TRAF6 or RIPK2 ubiquitination. (FigureS4 C and D).
Together, these studies suggest that the enhanced TLR- and NLR-induced NFKB activation in Atg16L1T300A/T300A or in atg16l1 KO cells is due to increased K63-ubiquitination of TRAF6 or RIPK2 occurring in cells with impaired autophagy; in addition, since the effects of loss of autophagy on K63-linked ubiquitination and NFKB activation is independent of the ATG16L1 WD40-repeat domain, these effects are due to loss of canonical autophagy.
The autophagy receptor SQSTM1/p62 interacts with TRAF6 or RIPK2 and augments TRAF6 or RIPK2 ubiquitination and NFKB activation
The observation that TRAF6 and RIPK2 ubiquitination is enhanced in TLR- and NLR-stimulated autophagy-deficient cells prompted us to search for a candidate molecule that is involved in this enhancement. One possibility that required consideration in this context was SQSTM1/p62, a sequestosome-like receptor (SLR) cargo-binding agent that has an LC3-interacting region and thus targets proteins for autophagosomal degradation [24]. This molecule was also of interest because it was reported to facilitate K63-polyubiquitination of TRAF6 and thereby promote nerve growth factor-induced NFKB activation [19] and to bind to the adapter protein RIPK1 and thereby regulate the activation of NFKB induced by TNF/TNF-α [25]. In our study, we observed that SQSTM1/p62 was increased in ATG16L1T300A/T300A KI cells, consistent with previous studies showing that SLRs are themselves autophagic cargo and exhibit accumulation in a cell-type- and stimulus-dependent manner when autophagy is perturbed [26]. We therefore investigated whether SQSTM1/p62 enhances TRAF6 and RIPK2-mediated NFKB activity and, if so, whether it interacts with TRAF6 or RIPK2 and contributes to the enhanced accumulation of ubiquitinated TRAF6 and RIPK2 proteins found in autophagy-deficient cells.
In initial studies addressing the NFKB question, we co-transfected HEK293T cells with constructs expressing SQSTM1/p62, TRAF6 (or RIPK2) and NFKB luciferase reporter plasmids and assayed luciferase activity in the transfected cells. We found that whereas SQSTM1/p62 had no capacity to induce NFKB activity alone, it enhanced both TRAF6-induced and RIPK2-induced NFKB activity (Figure 5 A and B). Next, we examined whether SQSTM1/p62 binds to TRAF6 or RIPK2 and regulates ubiquitination of these signaling molecules. In this study we co-transfected SQSTM1/p62-, TRAF6- (or RIPK2-) and ubiquitin plasmids into HEK293T cells and after short-term culture, subjected cell-lysates to immunoprecipitation or immunoblotting for detection of SQSTM1/p62 binding to TRAF6 or RIPK2 as well as ubiquitination of these molecules. We found that SQSTM1/p62 and TRAF6 or SQSTM1/p62 and RIPK2 bind to one another (Figure 5C lane 3, Figure 5D, lane 3); in addition, we found that whereas transfected TRAF6 or RIPK2 displayed low basal ubiquitination when transfected alone into HEK293T cells, both TRAF6 and RIPK2 displayed dramatically enhanced ubiquitination when co-transfected with SQSTM1/p62 (Figure 5 C and D).
Figure 5.
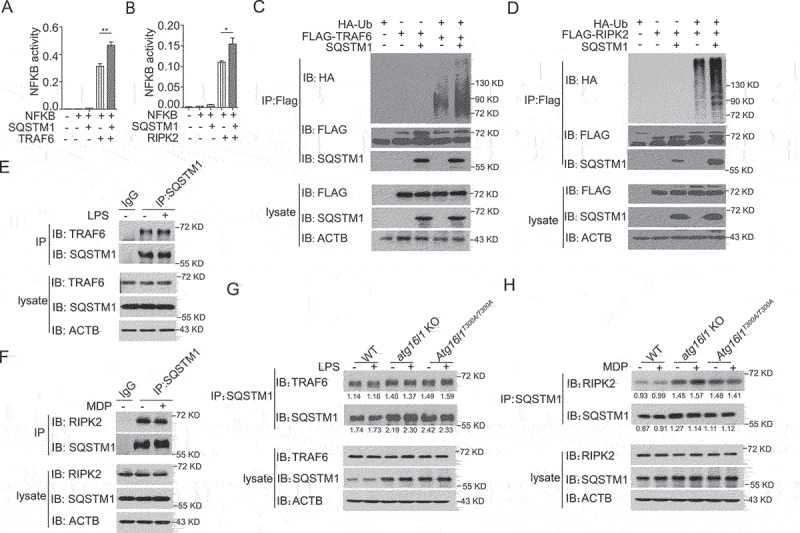
The autophagy receptor SQSTM1/p62 interacts with RIPK2 or TRAF6 and augments RIPK2 or TRAF6 polyubiquitination and NFKB activation. (A-B) HEK293T cells were co-transfected with SQSTM1/p62, TRAF6 (A) or RIPK2 (B) plasmids as well as an NFKB luciferase reporter plasmid and cultured for 24 h; the cells were then assessed for NFKB activity by luciferase assay. (C-D) HEK293T cells were co-transfected with HA-Ub, SQSTM1/p62, and FLAG-TRAF6 or FLAG-RIPK2 plasmids and cultured for 24 h; cell extracts were then obtained and subjected to immunoprecipitation with anti-FLAG and immunoblotting with anti-Ub, anti- SQSTM1/p62, and anti-FLAG. (E-F) WT BMDMs were stimulated with LPS (E) or MDP (F) for 1 h following which cell extracts were obtained and subjected to immunoprecipitation with anti-TRAF6 (E) or anti-RIPK2 (F) or IgG and immunoblotting with anti-TRAF6 (E) or anti-RIPK2 (F), anti-SQSTM1/p62 and anti-ACTB. (G-H) BMDMs from WT, atg16l1 KO, and Atg16l1T300A/T300A KI mice were stimulated with LPS (G) or MDP (H) for 1 h following which cell extracts were obtained and subjected to immunoprecipitation with anti-SQSTM1/p62 and immunoblotting with anti-TRAF6 (G) or anti-RIPK2 (H), anti-SQSTM1/p62 and anti-ACTB. Numbers underneath the blot represent the fold change of indicated band intensity compared with ACTB. Values are depicted as mean ± SD. P values were determined using one-way ANOVA followed by Tukey’s multiple-comparisons test (A). *p < 0.05. **p < 0.01.
Finally, to assess endogenous physical interaction between SQSTM1/p62 and TRAF6 or between SQSTM1/p62 and RIPK2, BMDMs from WT mice were left untreated or stimulated with LPS or MDP respectively, and the cell lysates subsequently obtained were subjected to immunoprecipitation/immunoblotting for detection of TRAF6 or RIPK2 binding to SQSTM1/p62 as described above. We found that endogenous TRAF6 or RIPK2 interacts with SQSTM1/p62 under basal and under conditions in which cells are stimulated with TLR4 or NOD2 ligand (Figure 5 E and F). In contrast, BMDMs from Atg16l1T300A/T300A KI or atg16l1 KO cells displayed increased TRAF6-SQSTM1 and RIPK2-SQSTM1 interaction with or without LPS or MDP stimulation respectively (Figure 5 G and H) as compared with that of WT cells. These findings suggest that the decreased SQSTM1/p62 degradation and increased SQSTM1/p62 accumulation that accompanies impaired autophagy leads to enhanced TRAF6 or RIPK2 interaction with SQSTM1/p62 and thereby to enhanced ubiquitination of these components.
Sqstm1/p62 deletion in autophagy-deficient cells normalizes ubiquitination of TRAF6 and RIPK2 and the NFKB activation
Based on the above conclusion, we reasoned that the increased NFKB activation characteristic of cells with impaired autophagy is dependent on SQSTM1/p62 accumulation and should disappear in the absence of this autophagy-related component. To test this hypothesis, we deleted sqstm1/p62 in atg16l1 KO (Raw) cells by CRISPR-Cas9 methodology (FigureS5 A and B) to obtain atg16l1 sqstm1 double knockout (DKO) Raw cells. We then stimulated WT, atg16l1 KO, and atg16l1 sqstm1 DKO cells with LPS or MDP and subjected lysates of the cultured cells to immunoprecipitation with anti-TRAF6 or anti-RIPK2 antibody followed by immunoblotting with anti-ubiquitin as above. We found that the ubiquitination of TRAF6 and RIPK2 in atg16l1 sqstm1 DKO cells was decreased compared to that of atg16l1 KO cells, but comparable to that of WT cells (Figure 6 A and B). Moreover, atg16l1 KO cells exhibited increased phosphorylation of NFKBIA compared to that of WT cells, whereas the phosphorylation of NFKBIA in atg16l1 sqstm1 DKO cells was similar to that of WT cells (Figure 6C). Finally, atg16l1 KO cells exhibited greater nuclear localization of the RELA/p65 subunit of NFKB than that in WT cells whereas atg16l1 sqstm1 DKO cells exhibited RELA/p65 localization comparable to that of WT cells (Figure 6 D and E).
Figure 6.
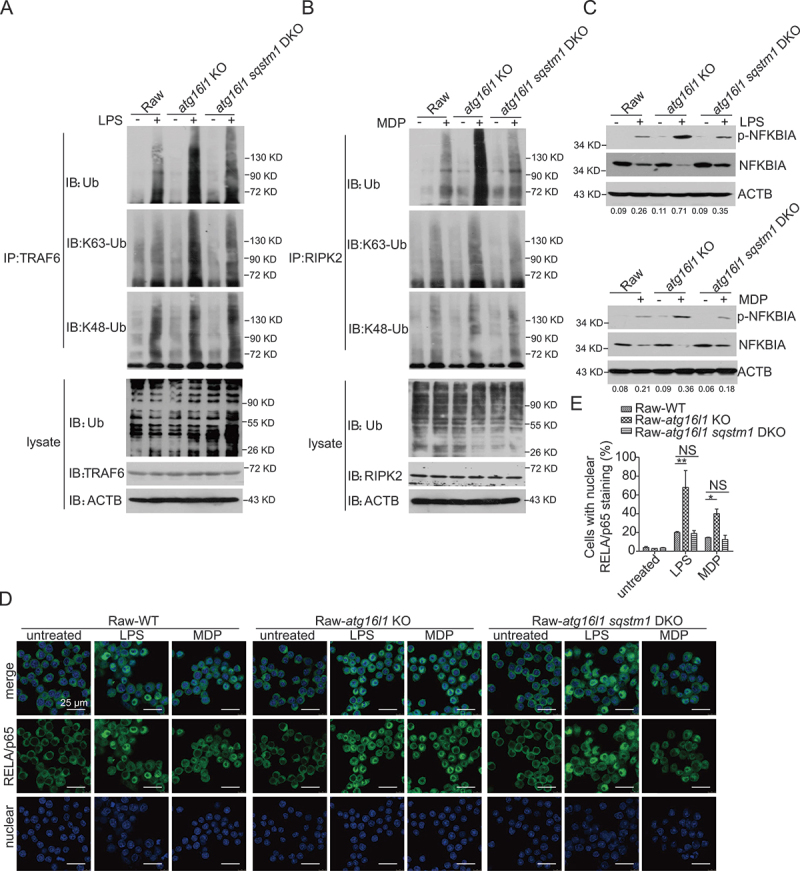
Sqstm1/p62 deletion in autophagy-deficient cells normalizes ubiquitination of TRAF6, RIPK2, and NFKB activation. (A-B) WT, atg16l1 KO, atg16l1 sqstm1 DKO Raw cells were treated with LPS (A) or MDP (B) for 1 h; cell extracts were then obtained and subjected to immunoprecipitation with anti-TRAF6 (A) or anti-RIPK2 (B) and immunoblotting with anti-TRAF6 (A) or anti-RIPK2 (B), anti-Ub and anti-ACTB. (C) WT, atg16l1 KO, atg16l1 sqstm1 DKO Raw cells were treated with LPS or MDP for 1 h; cell extracts were then obtained and subjected to immunoblotting with anti-p-NFKBIA, anti-NFKBIA, anti-ACTB. Numbers underneath the blot represent the fold change of p-NFKBIA intensity compared with ACTB. (D) WT, atg16l1 KO, Atg16l1 sqstm1 DKO Raw cells were treated as in (C) and then subjected to immunofluorescent staining and confocal microscopy to detect RELA/p65 translocation (RELA/p65, green; DAPI, blue). Scale bar: 25 µm. (E) Quantitative assessment of the percentage of the cells with RELA/p65 translocation in (D); at least 100 cells in 3 random fields were assessed. P values were determined using two-way ANOVA followed by Bonferroni post hoc test (E). *p < 0.05. **p < 0.01.
In further studies to show that the effect of sqstm1/p62 deletion noted above was a general property of impaired autophagy, not just that caused by knockout of Atg16l1, we conducted studies of atg7 KO or becn1 KO cells as well as studies of atg7 sqstm1 DKO and becn1 sqstm1 DKO cells again by constructing double KO cells using CRISPR-Cas9 methodology (FigureS5 A and B). In studies of LPS- and MDP-stimulated WT, atg7 KO and atg7 sqstm1 DKO cells processed as above, we found that the ubiquitination of TRAF6 and RIPK2 in atg7 sqstm1 DKO cells was decreased compared to that of atg7 KO cells but comparable to that of WT cells (FigureS5 C and D). Similarly, the phosphorylation of the NFKBIA in atg7 sqstm1 DKO or becn1 sqstm1 DKO cells was decreased compared to atg7 KO and becn1 KO cells and comparable to that of WT cells (FigureS5 E and F).
These results indicate that SQSTM1/p62 plays a structural role in the formation of some if not all ubiquitinated protein aggregates, and are thus consistent with previous observations showing that the formation of such aggregates in autophagy-null mice is abrogated when Sqstm1/p62 is knocked out [27,28]. Moreover, these results show that the accumulation of ubiquitinated TRAF6 and RIPK2 substrates after inhibition of autophagy is dependent on SQSTM1/p62 and that induction of autophagy thus plays an essential role in the maintenance of NFKB signal homeostasis.
Discussion
In the present study, we marshal compelling evidence showing the effect of the ATG16L1T300A polymorphism on TLR- or NLR-mediated signaling in macrophages (and perhaps in dendritic cells as well), is likely to be a major contributor to Crohn disease inflammation. The key findings reported here supporting this view are first that intact autophagy in macrophages regulates NFKB activation, and that defective autophagy in macrophages caused by the polymorphism or indeed caused by other molecular abnormalities resulting in loss of autophagy is accompanied by increased TLR- or NLR-induced NFKB activation and a concomitant increase in pro-inflammatory cytokines and chemokines. Second, we show that defective autophagy due to the ATG16L1T300A polymorphism leads to increased ubiquitination of NFKB activating factors (TRAF6 and RIPK2) as a result of cellular accumulation of SQSTM1/p62, a sequestosome receptor molecule that has the capacity to bind to and ubiquitinate TRAF6 and RIPK2. Finally, we show that deletion of SQSTM1/p62 in autophagy-defective cells results in substantial normalization of the enhanced NFKB activation otherwise displayed by these cells and thus proves that the autophagy effect on NFKB activation is mainly related to SQSTM1/p62 accumulation. The conclusion that emerges from these findings is that defective autophagy in TLR- or NLR-stimulated macrophages cause increased NFKB-mediated pro-inflammatory responses which itself can be a cause of the hyper-responsiveness to commensal organisms in the GI tract that underlies Crohn disease.
The fact that the ATG16L1T300A polymorphism in particular or defective autophagy, in general, might result in enhanced NFKB-mediated inflammation has not been clearly demonstrated until now, and, consequently, its mechanism has not been explored. In one previous study touching on this issue, it was shown that NOD2-induced NFKB activation is diminished in cells subjected to atg16l1 knockdown or autophagy inhibition; however, monocytes from humans bearing the ATG16L1T300A polymorphism displayed no abnormalities of NOD2-induced TNF/TNF-α secretion, and while an epithelial cell expressing ATG16L1T300A exhibited decreased Salmonella killing, the latter function was shown to be independent of NFKB activation [29]. In two other studies, it was found that intact ATG16L1 interferes with the function of RIPK2, most likely by binding to RIPK2 and inhibiting its ubiquitination. In one of these studies, this led to diminished NOD1-induced activation of NFKB and in the other to diminished TLR2-induced, but not NOD2-induced, activation of NFKB. These inhibitory effects of ATG16L1, however, were shown in accompanying studies to be independent of its role in autophagy [10,30]. Thus, these studies did not provide a clear conclusion regarding the role of autophagy in TLR- or NLR-mediated signaling. Here, by using macrophages from both mice and humans bearing the ATG16L1T300A polymorphism, we provided strong evidence substantiating this relationship as well studies clarifying its underlying mechanism.
The central player in the mechanism linking NFKB activation with autophagy described above, SQSTM1/p62, has previously been shown to participate in a number of important signaling pathways [24,31]. This multi-functionality is at least in part attributable to the fact that the SQSTM1/p62 molecule contains several binding motifs that allows it to interact with a diverse set of signaling components. These include a zinc finger domain allowing interaction with RIP (Receptor-Interacting Protein) kinase, a TB domain allowing interaction with TRAF6, a UBA domain facilitating ubiquitination, and an LIR domain allowing binding to LC3 present on the elongating autophagosome. The latter binding interaction empowers SQSTM1/p62 to serve as a cargo carrier that delivers intra-cellular molecules (or aggregates) to the autophagosome for eventual elimination. In addition, SQSTM1/p62 is itself an autophagic cargo (as are other SLRs) and exhibits accumulation in a cell-type- and stimulus-dependent manner when autophagy is perturbed [26]. These SQSTM1/p62 characteristics account for the fact that autophagy-deficient mice in which SQSTM1/p62 has accumulated exhibit intracellular speckles composed of ubiquitinated aggregates that have not been cleared by autophagosomes [27,28]. With respect to the role of SQSTM1/p62 in the augmentation of NFKB activation, it has been shown that SQSTM1/p62 participates in the TRAF6-mediated polyubiquitination of IKKγ (NEMO) [19,32], probably, as shown in this study, by first binding to TRAF6 and enhancing its level of polyubiquitination. In addition, SQSTM1/p62 has been shown to interact with RIPK1 (receptor interacting serine/threonine kinase 1), thus facilitating TNF activation of NFKB [25] and in the present study we showed that SQSTM1/p62 interacts with and enhances the ubiquitination of RIPK2, a kinase important to NOD1 and NOD2 activation of NFKB as well as to NOD1 and NOD2-independent NFKB activation [33,34]. These findings are parallel to those obtained in a previous study showing that SQSTM1/p62 also binds to BCL10, suggesting that T cell receptor activation of NFKB is also influenced by the absence of autophagy. Taken together, these observations support the conclusion that whereas SQSTM1/p62 enhances the activity of pro-inflammatory adaptor molecules that drive NFKB activation, such enhancement is limited by autophagy which serves to degrade SQSTM1/p62-bound molecules; on the other hand, in the absence of autophagy, SQSTM1/p62-bound molecules persist and SQSTM1/p62-enhancement of NFKB activation is unrestrained.
The studies described here emphasize that regulation of NFKB-induced inflammation by autophagy is greatly dependent on the latter’s effect on SQSTM1/p62 levels and function. It is possible, however, that autophagy regulation of inflammation can also be exerted via other sequestosome receptor molecules. Evidence for this possibility comes from studies showing that following TLR4 stimulation autophagy controls TICAM1/TRIF (toll like receptor adaptor molecule 1) levels and production of β-interferon by its regulation of both SQSTM1/p62 and another sequestosome receptor molecule termed TAX1BP1 (Tax1 binding protein 1) [35]. This latter finding indicates that autophagic regulation is multifaceted and wide-ranging.
As briefly mentioned in the introduction, autophagy plays a role in host defense. With respect to the GI tract, this includes the provision of a mechanism for limiting the proliferation of pathogens that penetrate the GI epithelial barrier and thus the prevention of systemic spread to other organs. Previous studies showing that mice bearing the ATG16L1T300A polymorphism exhibit more severe Salmonella infection provides verification of this fact [8–10]. This raises the question of why the proportion of the human population bearing a polymorphism that impairs autophagy is so high (greater than 50%). The finding in the present study showing that macrophages with impaired autophagy mount an increased NFKB-dependent cytokine response suggests a possible answer to this question: the impairment of autophagy in macrophages at the onset of infection ultimately results in a more robust cytokine response that provides better protection from systemic infections. Thus, loss of autophagy consequent to the ATG16L1T300A polymorphism may eventually be shown to confer a selective advantage for survival in human populations.
Materials and methods
Mice
Atg16l1T300A/T300A KI mice and Atg16l1f/f mice were generated on a C57BL/6 J background as described previously [9]. To obtain atg16l1 KO cells, the Atg16l1f/f mice were crossed with Lyz2/LysM-Cre mice or ERT2-Cre mice. Studies were carried out under animal care guidelines of the Institute of Microbiology, Chinese Academy of Science as well as the National Institute of Allergy and infectious Diseases, NIH.
Mouse bone marrow–derived macrophage cell culture
Bone marrow-derived macrophages (BMDMs) were obtained as previously described [36]. Briefly, bone marrow cells flushed out of femurs were plated on sterile petri dishes and cultured for one week in RPMI 1640 medium (Gibco, C11875500BT) containing 10% fetal bovine serum (Gibco, 16,140,071) and 20% L929 cell conditioned medium for 7 days. L-929 cells (ATCC, CCL-1) were grown in DMEM high glucose (Gibco, C11995500BT) supplemented with 10% FBS for 1 week to secrete CSF2/M-CSF (colony stimulating factor 2), then the medium was filtered using a 0.22-µm filter and store at −20°C. The cells obtained were washed and resuspended in RPMI 1640 medium supplemented with 10% fetal bovine serum.
Mouse embryonic fibroblast (MEF) culture
For the preparation of MEFs, embryos were dissected from pregnant females at day 13 after mating. MEFs isolated by standard procedures were cultured in DMEM supplemented with 10% (vol:vol) FBS and containing penicillin (100 U/mL) and streptomycin (100 µg/mL) [37].
Human samples collection and macrophage culture
Healthy volunteers of both sexes who were within the age range of 20–30 years inclusive and had body mass index values within the range 18.5–27 participated in the study. Institutional ethics committee approval was sought before the study was started. Informed consent was obtained from all individual participants in the study. All volunteers were healthy and without previous history of chronic disease.
Monocytes were isolated and differentiated into macrophages as described [38], and were cultured in RPMI 1640 medium plus 10% FBS plus 100 ng/mL rhM-CSF (R&D Systems, 216-MC-025) for 7 days. Adherent macrophages were washed and re-suspended in RPMI 1640 medium supplemented with 10% FBS.
Knock out cell line construction by CRISPR/Cas9
The atg16l1, becn1, atg7 and sqstm1 single or DKO iBMDM (a gift from Feng Shao, National institute of Biological Sciences, Beijing, China) and Raw (ATCC, TIB-71) cells were constructed using CRISPR-Cas9 system as described [39]. All cell types were maintained in humidified atmosphere at 37°C with 5% CO2.
RNA extraction and RT- PCR
RNA isolation and cDNA generation was performed in accordance with the manufacturer’s instructions. Cytokines and chemokines qPCR were performed with Power SYBR Green (Applied Biosystems, 4,367,659). Gene expression values were calculated by using 2−ΔΔCt normalized to Hprt.
RNA-seq
Total RNA was extracted from BMDMs from WT, atg16l1 KO, and Atg16l1T300A/T300A KI mice. RNA sequencing (RNA-seq) and bioinformatics analyses were conducted by Novogene. Differential expression analysis of two conditions was performed using the DEGSeq R package (1.20.0). The p values were adjusted using the Benjamini–Hochberg method. p = 0.05 and log2 (fold change) = 1 were set as the threshold for significantly differential expression.
Immunoprecipitation and immunoblot analysis
Immunoprecipitation and immunoblot was performed as previous described [40]. Briefly, after treated with LPS (Invivogen, tlrl-eblps) or MDP (Invivogen, tlrl-mdp) for indicated time, Cell extracts were prepared and immunoprecipitations were performed with indicated antibody: anti-Traf6 (Medical & Biological Laboratories co., LTD.,597); anti-RIPK2 (abcam, ab75257); anti-SQSTM1/p62 (Medical & Biological Laboratories co., LTD., PM045); anti-FLAG (Sigma-Aldrich, F7425). Immunoprecipitates were electrophoresed on SDS-PAGE and transferred to nitrocellulose membranes and subjected to immunodetection by standard procedures. Blots were probed with the following primary antibodies: anti-p-NFKBIA/IKBA (Cell Signaling Technology, 2859S), anti-NFKBIA/IKBA (Cell Signaling Technology, 9242S), anti-ATG16L1 (Medical & Biological Laboratories co., LTD., M-150–3), anti-ATG7 (Sigma-Aldrich, A2856), anti-BECN1 (BD biosciences, 612,113), anti-LC3 (Sigma-Aldrich, L7543), anti-HA (Santa Cruz Biotechnology, sc-7392), anti-Ub (Cell Signaling Technology, 3933S), anti-K63-Ub (Cell Signaling Technology, 5621S), anti-K48-Ub (Cell Signaling Technology,), anti-ACTB (Santa Cruz Biotechnology, sc-47,778).
Generation of stable cell line by lentivirus transduction
For lentivirus packaging, FUIPW plasmid (a gift from Feng Shao, National institute of Biological Sciences, Beijing, China) harboring the full length or ΔWD domain of ATG16L1, psPAX2 (Addgene, 12,260; deposited by Didier Trono), and pMD2.G (Addgene, 12,259; deposited by Didier Trono) plasmids (4:3:1) were co-transfected into 293 T cells (ATCC, CRL-1573). 24 h post transfection, the medium was changed into 2 mL fresh DMEM supplemented with 10% FBS. Forty-eight h later, supernatants containing lentivirus particles were collected and used to infect iBMDM-atg16l1 KO cells. Forty-eight h post infection, stable cells were selected with puromycin for 2–5 days.
Immunofluorescence microscopy
Immunofluorescence microscopy assay was performed as previous described [41]. Briefly, treated BMDMs or iBMDMs on glass coverslips were fixed and permeabilized. The cells were then blocked with PBS (Sigma-Aldrich, D5652)-BSA (Amresco, 0332) and incubated with primary antibodies after which they were washed with PBS for 3 times. Finally, the cells were incubated with secondary antibody Alexa Fluor 488 goat anti-rabbit IgG (Invitrogen, A11008) and again washed with PBS for 3 times. The coverslips were mounted with Permeafluor Aqueous mounting medium (Leagene Biotech Co Ltd., IH-0252) and examined by confocal microscopy Leica SP8.
Flow cytometry
To detect TLR4 expression levels in BMDMs, the cells were stimulated with LPS for indicated times and then detached from the plastic surface by incubation in trypsin for 5–10 min. The cells obtained were centrifuged, dispersed, and were incubated with anti-TLR4-PE (eBioscience, 12–9041–80) at a 1:50 dilution in FACS buffer (eBioscience, 00–4222) for 30 min at 4°C. After washed with PBS, the cells were analyzed on a FACS Canto II (BD Biosciences).
Luciferase reporter assay
HEK293T cells were seeded in 48-well plates. Cells were cotransfected with plasmids encoding NFKB-regulated firefly luciferase and thymidine kinase–controlled Renilla luciferase and designated amounts of expression constructs encoding TRAF6 or RIPK2 and SQSTM1/p62 for 24 h. Cell extracts were collected in Reporter Lysis Buffer (Promega Corporation, E1960), and luciferase activity was measured in accordance with the manufacturer’s instructions (Promega Corporation, GloMax® 20/20 Luminometer).
ELISA
Primary BMDMs and MEFs were seeded in 12-well plates and were allowed to grow for 24 h. Cells were then stimulated with indicated ligand for 16 h. Cell supernatant were collected, CXCL1 (R&D Systems, DY453–05), CXCL2 (R&D Systems, DY452–05), IL1B (BD biosciences, 559,603) were analyzed by ELISA in accordance with the manufacturer’s instructions.
Rs2241880 (ATG16L1T300A) genotype analysis
Genomic DNA from human donors was extracted and genotyping for the ATG16L1 variation was performed by TaqMan allelic discrimination (Invitrogen, 4,351,379). Experiments were conducted on a subgroup of 10 WT, 16 ATG16L1T300A carriers matched for age and gender.
Statistical analysis
GraphPad Prism v7 was used for statistics analysis. A one or two-way ANOVA analysis of variance was initially done to determine whether an overall statistically significant change existed before analysis with a Tukey/Bonferroni’s post hoc test for multiple comparisons. P values of less than 0.05 were considered statistically significant.
Supplementary Material
Acknowledgments
We thank Feng Shao (National institute of Biological Sciences, Beijing, China) for providing cells and plasmids. We also thank Xiaolan Zhang and Yihui Xu for expert technical support with confocal microscopy.
Funding Statement
The work was supported by National Key Research and Development Project Grant (2021YFC2300502 and 2016YFC1200302) and National Natural Science Foundation of China Grants (31300719 and 31470861).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here
References
- [1].Hampe J, Franke A, Rosenstiel P, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39(2):207–211. [DOI] [PubMed] [Google Scholar]
- [2].Rioux JD, Xavier RJ, Taylor KD, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39(5):596–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Murthy A, Li Y, Peng I, et al. A Crohn’s disease variant in Atg16l1 enhances its degradation by caspase 3. Nature. 2014;506(7489):456–462. [DOI] [PubMed] [Google Scholar]
- [4].Lassen KG, Kuballa P, Conway KL, et al. Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc Natl Acad Sci U S A. 2014;111(21):7741–7746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Eriguchi Y, Nakamura K, Yokoi Y, et al. Essential role of IFN-gamma in T cell-associated intestinal inflammation. JCI Insight. 2018;3(18). DOI: 10.1172/jci.insight.121886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Saitoh T, Fujita N, Jang MH, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456(7219):264–268. [DOI] [PubMed] [Google Scholar]
- [7].Mao L, Kitani A, Strober W, et al. The Role of NLRP3 and IL-1beta in the Pathogenesis of Inflammatory Bowel Disease. Front Immunol. 2018;9:2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Conway KL, Kuballa P, Song JH, et al. Atg16l1 is required for autophagy in intestinal epithelial cells and protection of mice from Salmonella infection. Gastroenterology. 2013;145(6):1347–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gao P, Liu H, Huang H, et al. The Inflammatory Bowel Disease-Associated Autophagy Gene Atg16L1T300A Acts as a Dominant Negative Variant in Mice. J Immunol. 2017;198(6):2457–2467. [DOI] [PubMed] [Google Scholar]
- [10].Sorbara MT, Ellison LK, Ramjeet M, et al. The protein ATG16L1 suppresses inflammatory cytokines induced by the intracellular sensors Nod1 and Nod2 in an autophagy-independent manner. Immunity. 2013;39(5):858–873. [DOI] [PubMed] [Google Scholar]
- [11].Takahama M, Akira S, Saitoh T.. Autophagy limits activation of the inflammasomes. Immunol Rev. 2018;281(1):62–73. [DOI] [PubMed] [Google Scholar]
- [12].Devalaraja MN, Wang DZ, Ballard DW, et al. Elevated constitutive IkappaB kinase activity and IkappaB-alpha phosphorylation in Hs294T melanoma cells lead to increased basal MGSA/GRO-alpha transcription. Cancer Res. 1999;59(6):1372–1377. [PubMed] [Google Scholar]
- [13].Lee YH, Kim SH, Kim Y, et al. Inhibitory effect of the antidepressant imipramine on NF-kappaB-dependent CXCL1 expression in TNFalpha-exposed astrocytes. Int Immunopharmacol. 2012;12(4):547–555. [DOI] [PubMed] [Google Scholar]
- [14].Burke SJ, Lu D, Sparer TE, et al. NF-kappaB and STAT1 control CXCL1 and CXCL2 gene transcription. Am J Physiol Endocrinol Metab. 2014;306(2):E131–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Moynagh PN. The NF-kappaB pathway. J Cell Sci. 2005;118(Pt 20):4589–4592. [DOI] [PubMed] [Google Scholar]
- [16].Lee J, Kim HR, Quinley C, et al. Autophagy suppresses interleukin-1beta (IL-1beta) signaling by activation of p62 degradation via lysosomal and proteasomal pathways. J Biol Chem. 2012;287(6):4033–4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Seglen PO, Gordon PB. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci U S A. 1982;79(6):1889–1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Noda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem. 1998;273(7):3963–3966. [DOI] [PubMed] [Google Scholar]
- [19].Wooten MW, Geetha T, Seibenhener ML, et al. The p62 scaffold regulates nerve growth factor-induced NF-kappaB activation by influencing TRAF6 polyubiquitination. J Biol Chem. 2005;280(42):35625–35629. [DOI] [PubMed] [Google Scholar]
- [20].Sanz L, Diaz-Meco MT, Nakano H, et al. The atypical PKC-interacting protein p62 channels NF-kappaB activation by the IL-1-TRAF6 pathway. EMBO J. 2000;19(7):1576–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hasegawa M, Fujimoto Y, Lucas PC, et al. A critical role of RICK/RIP2 polyubiquitination in Nod-induced NF-kappaB activation. EMBO J. 2008;27(2):373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Abbott DW, Yang Y, Hutti JE, et al. Coordinated regulation of Toll-like receptor and NOD2 signaling by K63-linked polyubiquitin chains. Mol Cell Biol. 2007;27(17):6012–6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Fletcher K, Ulferts R, Jacquin E, et al. The WD40 domain of ATG16L1 is required for its non-canonical role in lipidation of LC3 at single membranes. EMBO J. 2018;37(4). DOI: 10.15252/embj.201797840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Moscat J, Diaz-Meco, M.t MT. Diaz-Meco, p62 at the crossroads of autophagy, apoptosis, and cancer. Cell. 2009;137(6):1001–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Sanz L, Sanchez P, Lallena MJ, et al. The interaction of p62 with RIP links the atypical PKCs to NF-kappaB activation. EMBO J. 1999;18(11):3044–3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Rogov V, Dotsch V, Johansen T, et al. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol Cell. 2014;53(2):167–178. [DOI] [PubMed] [Google Scholar]
- [27].Komatsu M, Waguri S, Koike M, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131(6):1149–1163. [DOI] [PubMed] [Google Scholar]
- [28].Bjorkoy G, Lamark T, Brech A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171(4):603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Homer CR, Richmond AL, Rebert NA, et al. ATG16L1 and NOD2 interact in an autophagy-dependent antibacterial pathway implicated in Crohn’s disease pathogenesis. Gastroenterology. 2010;139(5):1630–41, 1641 e1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Honjo H, Watanabe T, Arai Y, et al. ATG16L1 negatively regulates RICK/RIP2-mediated innate immune responses. Int Immunol. 2020. DOI: 10.1093/intimm/dxaa062. [DOI] [PubMed] [Google Scholar]
- [31].Sanchez-Martin P, Saito T, Komatsu M. p62/SQSTM1: ‘Jack of all trades’ in health and cancer. FEBS J. 2019;286(1):8–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zotti T, Scudiero I, Settembre P, et al. TRAF6-mediated ubiquitination of NEMO requires p62/sequestosome-1. Mol Immunol. 2014;58(1):27–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Park JH, Kim YG, McDonald C, et al. RICK/RIP2 mediates innate immune responses induced through Nod1 and Nod2 but not TLRs. J Immunol. 2007;178(4):2380–2386. [DOI] [PubMed] [Google Scholar]
- [34].Watanabe T, Minaga K, Kamata K, et al. RICK/RIP2 is a NOD2-independent nodal point of gut inflammation. Int Immunol. 2019;31(10):669–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Samie M, Lim J, Verschueren E, et al. Selective autophagy of the adaptor TRIF regulates innate inflammatory signaling. Nat Immunol. 2018;19(3):246–254. [DOI] [PubMed] [Google Scholar]
- [36].Sutterwala FS, Ogura Y, Szczepanik M, et al. Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity. 2006;24(3):317–327. [DOI] [PubMed] [Google Scholar]
- [37].Xu J. Preparation, culture, and immortalization of mouse embryonic fibroblasts. Chapter 28: p. Unit. 2005;:28 1. Curr Protoc Mol Biol [DOI] [PubMed] [Google Scholar]
- [38].Li J, Moran T, Swanson E, et al. Regulation of IL-8 and IL-1beta expression in Crohn’s disease associated NOD2/CARD15 mutations. Hum Mol Genet. 2004;13(16):1715–1725. [DOI] [PubMed] [Google Scholar]
- [39].Ran FA, Hsu PD, Wright J, et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8(11):2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Yang S, Wang B, Humphries F, et al. Pellino3 ubiquitinates RIP2 and mediates Nod2-induced signaling and protective effects in colitis. Nat Immunol. 2013;14(9):927–936. [DOI] [PubMed] [Google Scholar]
- [41].Gao P, Bauvy C, Souquere S, et al. The Bcl-2 homology domain 3 mimetic gossypol induces both Beclin 1-dependent and Beclin 1-independent cytoprotective autophagy in cancer cells. J Biol Chem. 2010;285(33):25570–25581. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.