

ABSTRACT
Cerebral infarction induces angiogenesis in the thalamus and influences functional recovery. The mechanisms underlying angiogenesis remain unclear. This study aimed to investigate the role of RTN4/Nogo-A in mediating macroautophagy/autophagy and angiogenesis in the thalamus following middle cerebral artery occlusion (MCAO). We assessed secondary neuronal damage, angiogenesis, vascular autophagy, RTN4 and S1PR2 signaling in the thalamus. The effects of RTN4-S1PR2 on vascular autophagy and angiogenesis were evaluated using lentiviral and pharmacological approaches. The results showed that RTN4 and S1PR2 signaling molecules were upregulated in parallel with angiogenesis in the ipsilateral thalamus after MCAO. Knockdown of Rtn4 by siRNA markedly reduced MAP1LC3B-II conversion and levels of BECN1 and SQSTM1 in vessels, coinciding with enhanced angiogenesis in the ipsilateral thalamus. This effect coincided with rescued neuronal loss of the thalamus and improved cognitive function. Conversely, activating S1PR2 augmented vascular autophagy, along with suppressed angiogenesis and aggravated neuronal damage of the thalamus. Further inhibition of autophagic initiation with 3-methyladenine or spautin-1 enhanced angiogenesis while blockade of lysosomal degradation by bafilomycin A1 suppressed angiogenesis in the ipsilateral thalamus. The control of autophagic flux by RTN4-S1PR2 was verified in vitro. Additionally, ROCK1-BECN1 interaction along with phosphorylation of BECN1 (Thr119) was identified in the thalamic vessels after MCAO. Knockdown of Rtn4 markedly reduced BECN1 phosphorylation whereas activating S1PR2 increased its phosphorylation in vessels. These results suggest that blockade of RTN4-S1PR2 interaction promotes angiogenesis and secondary neural repair in the thalamus by suppressing autophagic activation and alleviating dysfunction of lysosomal degradation in vessels after cerebral infarction.
Abbreviations: 3-MA: 3-methyladenine; ACTA2/ɑ-SMA: actin alpha 2, smooth muscle, aorta; AIF1/Iba1: allograft inflammatory factor 1; BafA1: bafilomycin A1; BMVECs: brain microvascular endothelial cells; BrdU: 5-bromo-2’-deoxyuridine; CLDN11/OSP: claudin 11; GFAP: glial fibrillary acidic protein; HUVECs: human umbilical vein endothelial cells; LAMA1: laminin, alpha 1; MAP2: microtubule-associated protein 2; MBP2: myelin basic protein 2; MCAO: middle cerebral artery occlusion; PDGFRB/PDGFRβ: platelet derived growth factor receptor, beta polypeptide; RECA-1: rat endothelial cell antigen-1; RHOA: ras homolog family member A; RHRSP: stroke-prone renovascular hypertensive rats; ROCK1: Rho-associated coiled-coil containing protein kinase 1; RTN4/Nogo-A: reticulon 4; RTN4R/NgR1: reticulon 4 receptor; S1PR2: sphingosine-1-phosphate receptor 2; SQSTM1: sequestosome 1.
KEYWORDS: Angiogenesis, autophagy, cerebral infarction, RTN4, secondary neurodegeneration, thalamus
Introduction
Focal cerebral infarction leads not only to direct neuropathological damage in the primary ischemic site but also to delayed neuronal injury in remote nonischemic regions such as the ipsilateral thalamus [1]. The secondary neuronal damage in the ipsilateral thalamus hinders functional recovery of somatosensory and cognitive impairment [2,3]. Therefore, understanding the mechanisms underlying such secondary thalamic neurodegeneration would be of great importance in improving stroke outcomes.
Accumulating evidence has demonstrated that remote neurodegeneration of the thalamus is associated with progressive retrograde or anterograde axon degeneration, inflammatory responses, or direct neuronal injury resulting from excessive apoptosis and autophagy after cerebral infarction [4–7]. In contrast, we have demonstrated that endogenous repair processes such as angiogenesis occur within the ipsilateral thalamus [8], and activating EFNB2 (ephrin B2) reversal signaling enhanced endogenous angiogenesis in concert with ameliorating thalamic neuron loss after experimental cortical cerebral infarction [9]. These findings suggest that modulation of angiogenesis could be a potential avenue to rescue the remote neuronal damage of the thalamus and neurological function recovery after stroke. However, the precise mechanisms underlying angiogenesis in the thalamus after cerebral infarction remain unclear.
Macroautophagy/autophagy is a constitutive and controlled lysosomal catabolic process by which cells maintain cellular homeostasis by degrading damaged components or abnormal protein aggregates in response to stress [10]. Although vascular autophagy has been previously demonstrated [11], its role in vascular biology remains debatable. Previous studies have reported that autophagy is involved in the angiogenic function of endothelial cells. Inhibiting autophagy by 3-methyladenine (3-MA) or Atg5 gene knockdown reduces angiogenic functions, such as tube formation or cell migration, whereas inducing autophagy enhances these functions [12]. Similarly, gene knockout of Ubr4 (ubiquitin protein ligase E3 component n-recognin 4) destroys autophagic flux and results in abnormal vascular network development and thereby embryonic death [13], suggests that autophagy is essential for vascular development. In contrast, other studies have showed that deficiency of the autophagy-related protein BECN1/beclin1 promotes angiogenesis [14], and upregulation of BECN1 inhibits angiogenesis by attenuating tubule formation in tumor issue, implicating its negative effect on angiogenesis [15]. Collectively, these findings indicate that autophagy has diverse effects on angiogenesis, depending on the conditions. Whether autophagy is involved in thalamic angiogenesis secondary to ischemic stroke and its underlying mechanisms remains to be elucidated.
RTN4/Nogo-A inhibits axonal regeneration in response to spinal cord injury [16], and ischemic stroke [6]. RTN4 exerts distinct effects on the extracellular RTN4R/NgR1 (reticulon 4 receptor) or its Δ20-domain specific S1PR2 (sphingosine-1-phosphate receptor 2) [17]. Previously, we have revealed that oligodendrocyte RTN4 is involved in axonal degeneration and neuronal loss of the ipsilateral thalamus in an RTN4R-dependent manner following cerebral ischemia [6]. Additionally, blockade of RTN4-RTN4R interaction with RTN4 extracellular peptide 1–40 has been shown to inhibit autophagic activation in neurons and oligodendrocytes to reduce neuronal damage in the ipsilateral thalamus after cerebral infarction [18]. These findings indicate the potential correlates of RTN4-RTN4R signaling with autophagy and secondary neuronal damage of the thalamus after ischemic stroke. Recently, RTN4 has been shown to negatively regulate angiogenesis and three-dimensional vascular network development in postnatal brain [19,20]. Additionally, genetic deletion of Rtn4 or neutralization of RTN4 with its Δ20-specific antibody improves vascular repair and vessel network formation in the vicinity of ischemic regions after stroke [21], suggesting a possible role for RTN4-S1PR2 in repairing ischemic brain tissues. However, it remains unclear whether RTN4-S1PR2 interaction regulates endogenous vascular biology through autophagy in the thalamus secondary to distal cerebral infarction.
The present study was designed to determine the profiles of the autophagic flux in blood vessels and their correlation with angiogenesis in the ipsilateral thalamus after cerebral infarction. Further, we sought to examine the spatial expression patterns of RTN4 and S1PR2 in the ipsilateral thalamus and explore the potential involvement of RTN4-S1PR2 signaling in vascular autophagy and angiogenesis in the ipsilateral thalamus secondary to cerebral infarction. Additionally, the casual effects of RTN4-S1PR2 on autophagic flux were confirmed in vitro.
Results
Angiogenesis and autophagic activation in the ipsilateral thalamus after cerebral cortical infarction
Immunofluorescence results showed that 5-bromo-2’-deoxyuridine (BrdU)+_LAMA1 (laminin, alpha 1)+ cells markedly increased in the ipsilateral thalamus at 7 days after MCAO in contrast to those in the sham-operated group (Figure 1A). The number of BrdU+_LAMA1+ cells and the density of LAMA1-labeled vessels in the ipsilateral thalamus after MCAO were significantly higher than those in the sham-operated animals (both P < 0.01, (Figure 1B-C)), suggesting the occurrence of angiogenesis in the thalamus after distal cerebral infarction. In parallel, there was an evident increase in double staining of MAP1LC3B/LC3B, an autophagic marker, with RECA-1 (rat endothelial cell antigen-1; the marker for endothelial cells), ACTA2/ɑ-SMA (actin alpha 2, smooth muscle, aorta; the marker for smooth muscle cells), or PDGFRB/PDGFRβ (platelet derived growth factor receptor, beta polypeptide; the marker for pericytes) in the ipsilateral thalamus at 7 days post-MCAO compared to the sham-operated group (Figure 1D). The colocalization of the indicated cell types was further confirmed using a confocal microscopy (Figure S1A-D). Immunoblotting analysis showed that the conversions of MAP1LC3B-II were significantly elevated in vessels of the ipsilateral thalamus at 7 days after MCAO relative to the sham-operated group (P < 0.05, (Figure 1E-F)). These results indicate the potential associations between excessive autophagic activation and angiogenesis in the ipsilateral thalamus after cerebral infarction.
Figure 1.
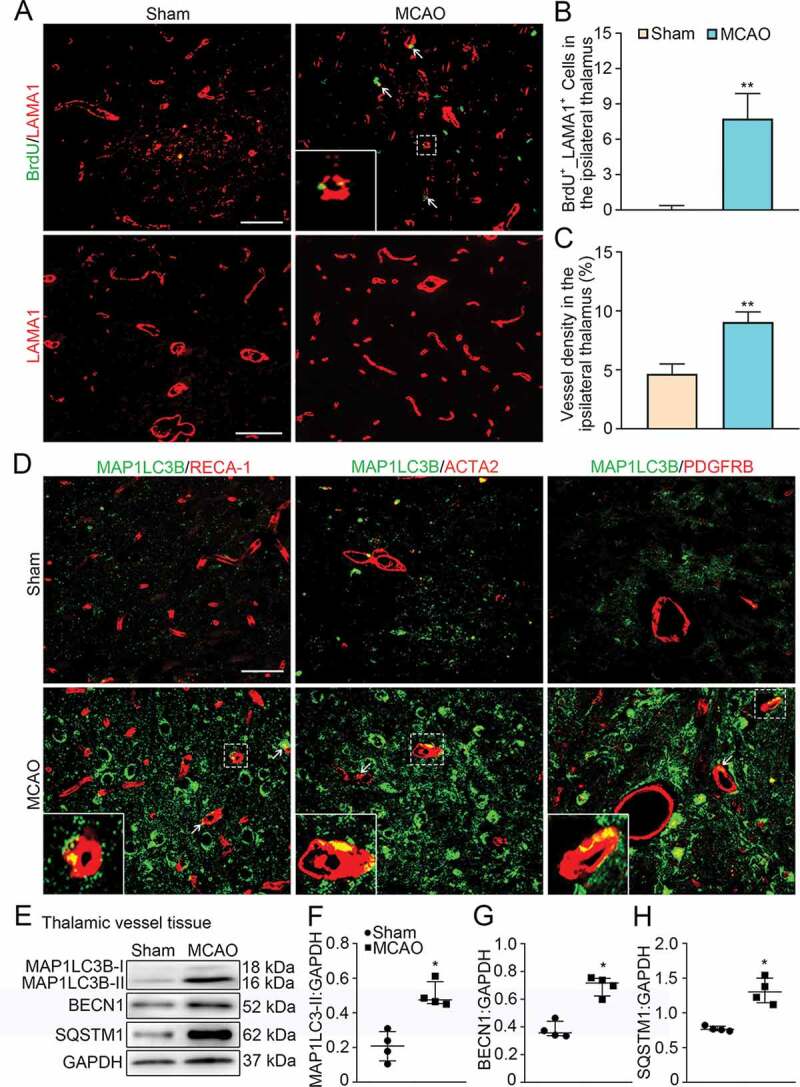
Angiogenesis and vascular autophagic activation in the ipsilateral thalamus after cerebral infarction. (A) Immunostaining of BrdU (green) and LAMA1-labeled vessels (red) within the ipsilateral thalamus in the sham-operated and MCAO groups at 7 days after MCAO (arrows). Insets show colocalization of MAP1LC3B with LAMA1+ cells. Scale bar: 50 μm, 100 μm. (B and C) Quantitative analysis of BrdU+_LAMA1+ cells and vessel density. n = 6, data are expressed as median ± interquartile range. **P < 0.01, compared with the sham-operated group. (D) Co-staining of MAP1LC3B (green) with RECA-1, ACTA2, and PDGFRB (red) in the ipsilateral thalamus of the sham-operated and MCAO groups at 7 days after MCAO (arrows). Insets show colocalization of MAP1LC3B with RECA-1+, ACTA2+ and PDGFRB+ cells. Scale bar: 50 μm. (E) Immunoblotting shows the expression of MAP1LC3B-II, BECN1 and SQSTM1 in vessels of the ipsilateral thalamus from the sham-operated and MCAO groups. (F-H) Quantitative analysis of MAP1LC3B-II, BECN1 and SQSTM1 levels relative to GAPDH. n = 4, data are expressed as median ± interquartile range. *P < 0.05, compared with the sham-operated group.
To evaluate autophagic flux, BECN1 and SQSTM1 (sequestosome 1) were detected in isolated thalamic vessels as indicators of autophagic induction and autolysosome degradation [22,23]. The results showed that both levels of BECN1 and SQSTM1 were significantly increased in vessels of the ipsilateral thalamus from MCAO-treated animals than those in the sham-operated thalamus (both P < 0.05, (Figure 1E, G, H)). This suggests that excessive autophagic activation might result from elevated autophagic induction and reduced autophagosome clearance in the thalamic vessels after cerebral infarction.
Spatial expression profiles of RTN4 and S1PR2 in the ipsilateral thalamus after cortical infarction
Seven days after MCAO, RTN4 was observed to be predominantly expressed in MAP2 (microtubule-associated protein 2)-labeled neurons and CLDN11/OSP (claudin 11)-positive oligodendrocytes in the ipsilateral thalamus. In contrast, we did not observe colocalization with GFAP (glial fibrillary acid protein)-positive astrocytes, AIF1/Iba1 (allograft inflammatory factor 1)-positive microglia or RECA-1 labeled endothelial cells. Spatially, RTN4-labeled neurons and oligodendrocytes were distributed adjacent to blood vessels, as identified by RECA-1 staining (Figure 2A). Cellular colocalization was further verified by confocal images (Figure S1E and F). Immunoblotting results showed a markedly elevated expression of RTN4 in the vessels within the ipsilateral thalamus after MCAO when compared to the sham-operated animals (P < 0.05, (Figure 2B-C)).
Figure 2.
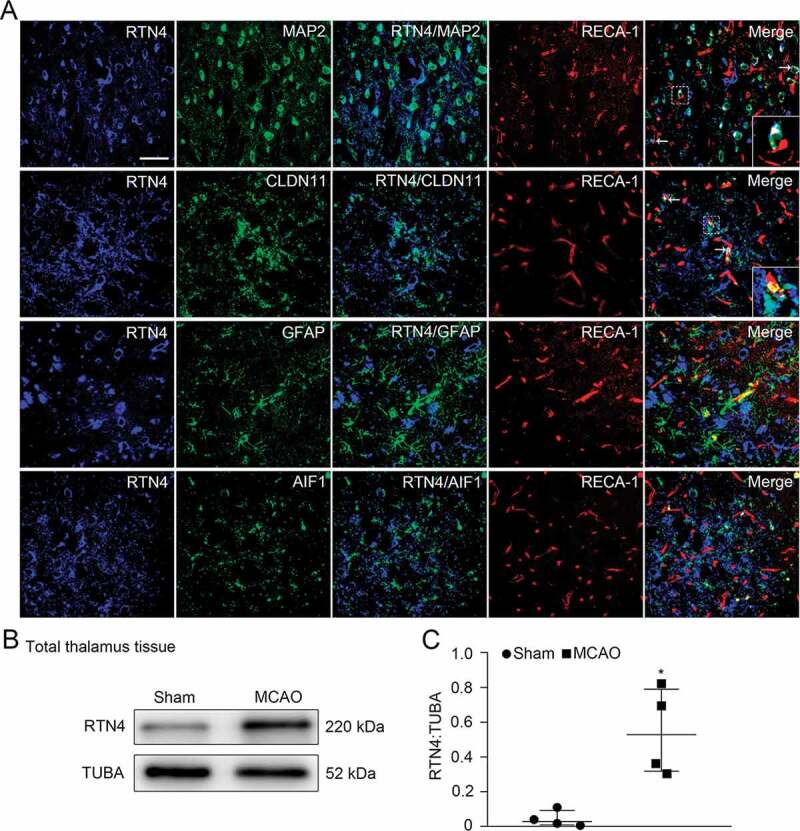
Colocalization of RTN4 on different cell types in the ipsilateral thalamus after cortical infarction. (A) Triple-staining of RTN4 (blue) with MAP2+ neurons, CLDN11+ oligodendrocytes, GFAP+ astrocytes and AIF1+ microglia (green) and their spatial distribution along RECA-1+ vessels (red) within the ipsilateral thalamus at 7 days after MCAO (arrows). Insets show RTN4-expressing neurons and oligodendrocytes adjacent to RECA-1-labeled vessels. Scale bar: 50 μm. (B) Immunoblotting shows RTN4 expression in the ipsilateral thalamus of the sham- and MCAO-operated animals. (C) Quantitative analysis of RTN4 level relative to TUBA. n = 4, data are expressed as median ± interquartile range. *P < 0.05, compared with the sham-operated controls.
We further checked the expression profiles of RTN4 receptors on the thalamic vessels and found that S1PR2 was colocalized with RECA-1, ACTA2 or PDGFRB ((Figure 3A); Figure S1G-I). Notably, S1PR2 staining appeared not to occupy entire RECA-1+ cells. The levels of S1PR2, together with its downstream molecules, including RHOA (ras homolog family member A) and ROCK1 (Rho-associated coiled-coil containing protein kinase 1), were significantly increased in vessels of the ipsilateral thalamus after MCAO when compared to the sham-operated group, respectively (all P < 0.05, (Figure 3B-E)). Altogether, these results suggest that neuronal or oligodendroglial RTN4 might exert angiogenic actions through its S1PR2 receptor in vessels of the ipsilateral thalamus after cerebral infarction.
Figure 3.
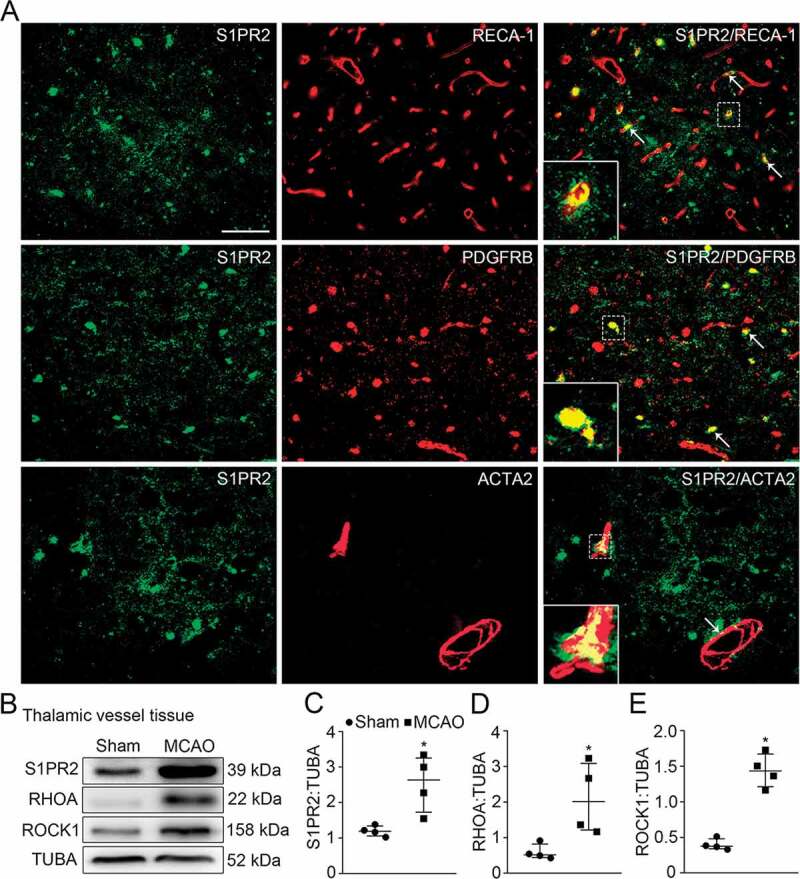
Expression profiles of S1PR2 and its downstream molecules in vessels of the ipsilateral thalamus after cortical infarction. (A) Co-staining of S1PR2 (green) with RECA-1+ endothelial cells, PDGFRB+ pericytes, or ACTA2+ smooth muscle cells (red) within the ipsilateral thalamus in the MCAO group (arrows). Scale bar: 50 μm. (B) Immunoblotting shows the expression of S1PR2, RHOA and ROCK1 in vessels of the ipsilateral thalamus from the sham- and MCAO-operated animals. (C-E) Quantitative analysis of S1PR2, RHOA and ROCK1 levels relative to TUBA. n = 4, data are expressed as median ± interquartile range. *P < 0.05, compared with the sham-operated controls.
Rtn4 knockdown inhibited vascular autophagy and enhanced angiogenesis in the ipsilateral thalamus after cortical infarction
Next, we determined the role of RTN4-S1PR2 signaling in autophagic flux in vessels and angiogenesis in the ipsilateral thalamus using siRNA-mediated downregulation of Rtn4. To ensure the efficacy of virus transfection, the Rtn4-siRNA or Scramble-siRNA lentivirus was stereotactically delivered to the unilateral thalamus of hypertensive animals at 2 weeks before MCAO surgery (Figure 4A). Seven days after MCAO, GFP-fluorescence tagged in the siRNA lentivirus was detected to be predominantly distributed within the ipsilateral thalamus (Figure 4B). GFP-tagged Rtn4-siRNA was predominantly found to colocalize with MAP2+ neurons (~51.4%) and CLDN11+ oligodendrocytes (~39.6%) in the ipsilateral thalamus (Figure 4C). Immunoblotting results showed that RTN4 expression was considerably reduced in the ipsilateral thalamus treated with Rtn4-siRNA relative to the Scramble-siRNA-treated thalamus (P < 0.05, (Figure 4D-E)). The levels of S1PR2, RHOA and ROCK1 were significantly decreased in the ipsilateral thalamic vessels of the Rtn4-siRNA group when compared to the Scramble-siRNA group (all P < 0.05, (Figure 5A-D)). These results suggest that Rtn4 knockdown considerably disrupted its interactions with S1PR2 in the vessels of the ipsilateral thalamus after cerebral infarction.
Figure 4.
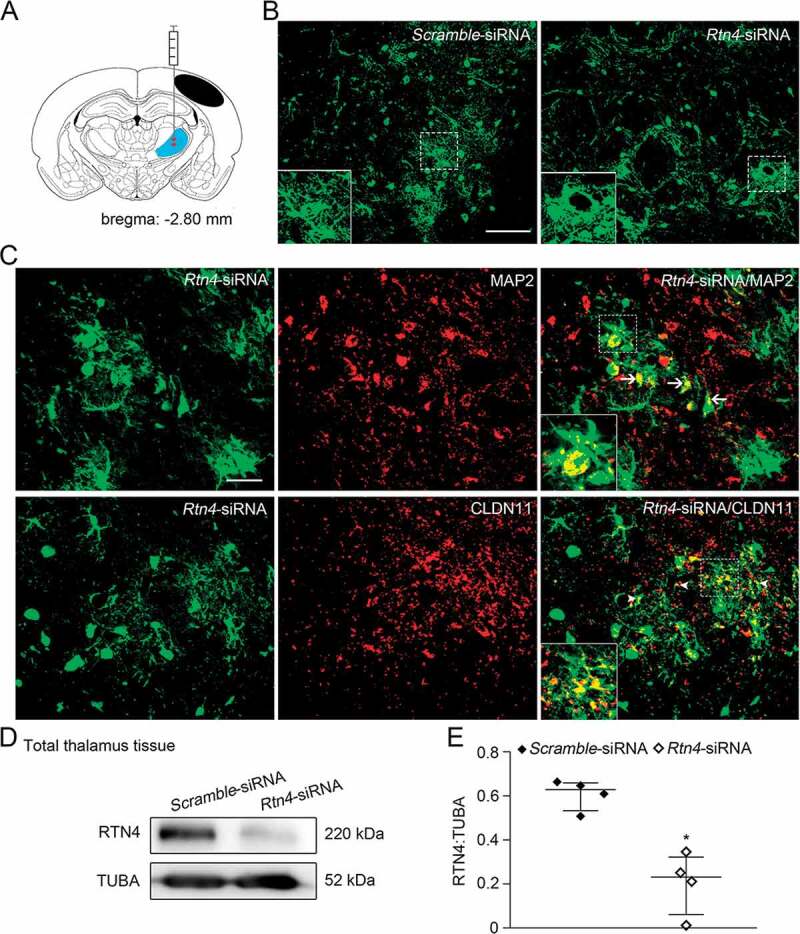
Knockdown of Rtn4 mediated by siRNA in the ipsilateral thalamus after cortical infarction. (A) Schematic diagram of brain section (−2.80 mm from bregma) shows the location of cortical infarction (black area) and the ipsilateral thalamus (blue area) where siRNA-lentivirus is delivered to four sites (red). (B) GFP-tagged Scramble- and Rtn4-siRNA were detected in the ipsilateral thalamus at 7 days after MCAO. Scale bar: 100 μm. (C) Immunostaining shows GFP-tagged Rtn4-siRNA to be expressed predominantly on MAP2+ neurons (arrows) and CLDN11+ oligodendrocytes (arrowheads). Scale bar: 50 μm. (D) Immunoblotting shows RTN4 expression in the ipsilateral thalamus from the Scramble- and Rtn4-siRNA groups at 7 days after MCAO. (E) Quantitative analysis of RTN4 level relative to TUBA. n = 4, data are expressed as median ± interquartile range. *P < 0.05, compared with the Scramble-siRNA group.
Figure 5.
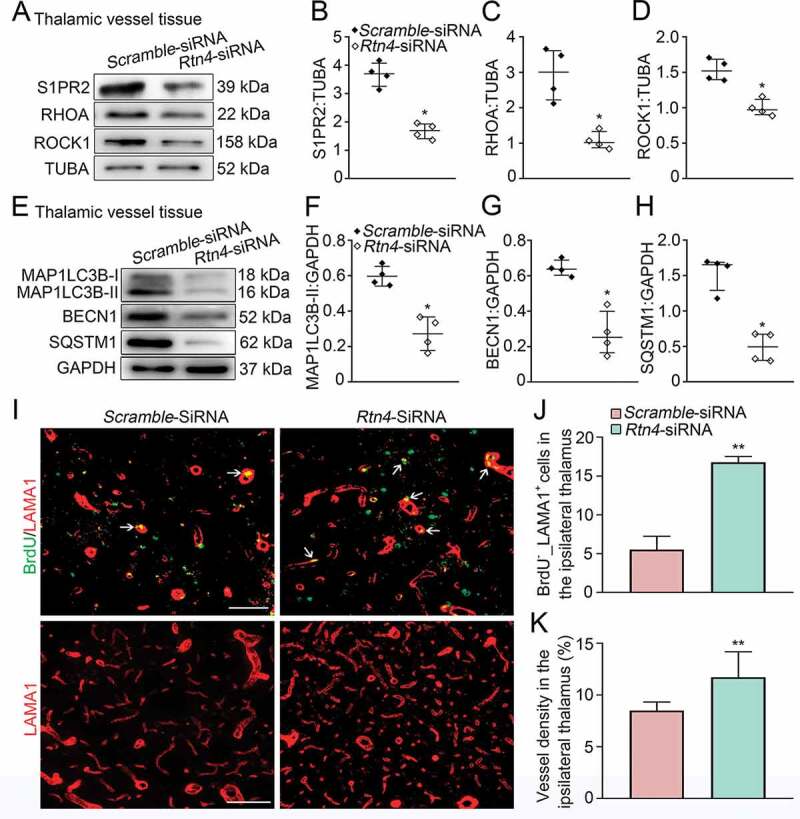
Rtn4 knockdown and vascular autophagic activation, as well as angiogenesis the ipsilateral thalamus after cortical infarction. (A) Immunoblotting shows the expression of S1PR2, RHOA and ROCK1 in vessels of the ipsilateral thalamus in the Scramble- and Rtn4-siRNA groups at 7 days after MCAO. (B-D) Quantitative analysis of S1PR2, RHOA and ROCK1 levels relative to TUBA. n = 4, data are expressed as median ± interquartile range. *P < 0.05, compared with the Scramble-siRNA group. (E) Immunoblotting shows the expression of MAP1LC3B-II, BECN1 and SQSTM1 in vessels of the ipsilateral thalamus in the Scramble- and Rtn4-siRNA groups. (F-H) Quantitative analysis of MAP1LC3B-II, BECN1 and SQSTM1 levels relative to GAPDH. n = 4, data are expressed as median ± interquartile range. *P < 0.05, compared with the Scramble-siRNA group. (I) Co-staining of BrdU (green) with LAMA1 (red) in the ipsilateral thalamus of the Scramble- and Rtn4-siRNA groups at 7 days after MCAO (arrows). Scale bar: 50 μm, 100 μm. (J and K) Quantitative analysis of BrdU+_LAMA1+ cells and vessel density. n = 6, data are expressed as median ± interquartile range. **P < 0.01, compared with the Scramble-siRNA group.
Immunoblotting analysis showed that there were significant reductions in MAP1LC3B-II conversion as well as expression of BECN1 and SQSTM1 in the ipsilateral thalamic vessels in the Rtn4-siRNA group when compared to the Scramble-siRNA group (all P < 0.05, (Figure 5E-H)), suggesting that blockade of RTN4-S1PR2 interaction restores autophagosome clearance and suppresses autophagic induction in the thalamic vessels. In parallel, the number of BrdU+_LAMA1+ cells in the ipsilateral thalamus of the Rtn4-siRNA-treated group were markedly increased compared with those in the Scramble-siRNA group (P < 0.01, (Figure 5I-J)). Additionally, the density of LAMA1-labeled vessels in the ipsilateral thalamus of the Rtn4-siRNA-treated animals was significantly higher than that in the Scramble-siRNA group (P < 0.01, (Figure 5I-K)). Taken together, these results indicate that Rtn4 knockdown suppresses excessive autophagic activation in vessels and enhances angiogenesis in the ipsilateral thalamus after cerebral infarction.
Activating RTN4-S1PR2 accelerated vascular autophagy and suppressed angiogenesis in the ipsilateral thalamus after cortical infarction
To further confirm the action of RTN4-Δ20-S1PR2 on autophagic activation in vessels and angiogenesis in the ipsilateral thalamus, recombinant RTN4-Δ20-Fc chimera was administered to activate S1PR2 signaling with the specific amino acids 544–725 domain of RTN4-Δ20 as previously reported [24]. The results showed that RTN4-Δ20-Fc treatment markedly increased the expression of S1PR2, RHOA and ROCK1 in the vessels of the ipsilateral thalamus at 7 days post-MCAO (Figure 6A). The relative levels of S1PR2, RHOA and ROCK1 in isolated vessels from the ipsilateral thalamus of the RTN4-Δ20-Fc group were up to 1.5 ~ 2 fold of the vehicle group respectively (all P < 0.05, (Figure 6B-D)). Moreover, RTN4-Δ20-Fc treatment significantly increased the conversion of MAP1LC3B-II, and levels of BECN1 and SQSTM1 in vessels of the ipsilateral thalamus compared to the vehicle group (all P < 0.05, (Figure 6E-H)), suggesting that RTN4-S1PR2 signaling can induce autophagic induction but also reduce the degradation of autophagosomes in the thalamic vessels after cerebral infarction.
Figure 6.
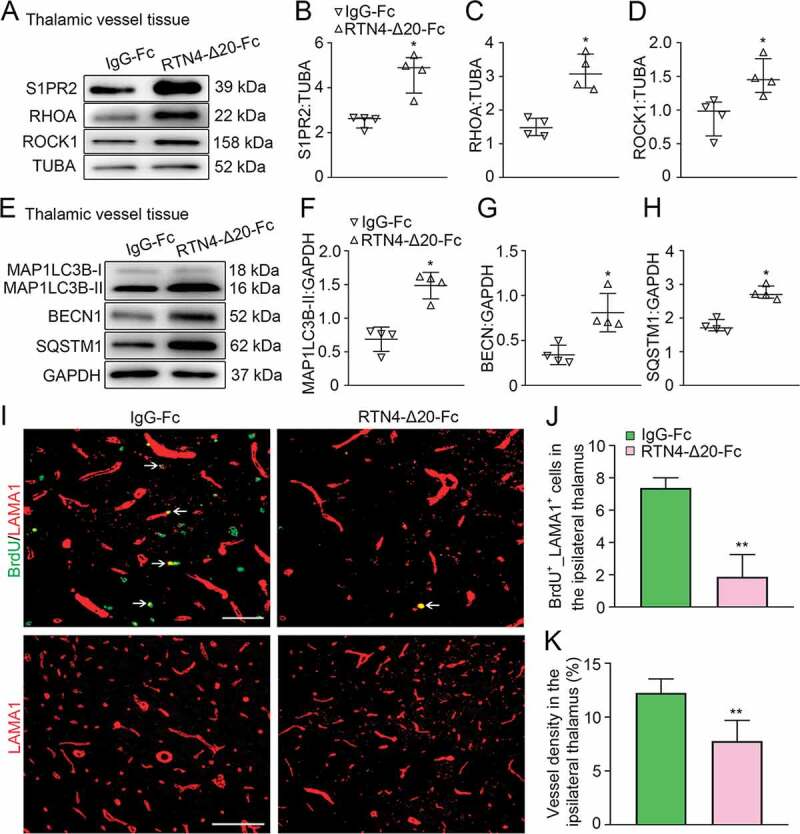
Activation of RTN4-S1PR2 and vascular autophagic activation, as well as angiogenesis in the ipsilateral thalamus after cortical infarction. (A) Immunoblotting shows the expression of S1PR2, RHOA, and ROCK1 in vessels of the ipsilateral thalamus in the IgG-Fc and RTN4-Δ20-Fc groups at 7 days after MCAO. (B-D) Quantitative analysis of S1PR2, RHOA and ROCK1 levels relative to TUBA. n = 4, data are expressed as median ± interquartile range. *P < 0.05, compared with the IgG-Fc group. (E) Immunoblotting shows the expression of MAP1LC3B-II, BECN1 and SQSTM1 in vessels of the ipsilateral thalamus in the IgG-Fc and RTN4-Δ20-Fc groups at 7 days after MCAO. (F-H) Quantitative analysis of MAP1LC3B-II, BECN1 and SQSTM1 levles relative to GAPDH. n = 4, data are expressed as median ± interquartile range. *P < 0.05, compared with the IgG-Fc group. (I) Co-staining for BrdU (green) and LAMA1 (red) within the ipsilateral thalamus in the IgG-Fc and RTN4-Δ20-Fc group at 7 days after MCAO (arrows). Scale bar: 50 μm, 100 μm. (J and K) Quantitative analysis of BrdU+_LAMA1+ cells and vessel density. n = 6, data are expressed as median ± interquartile range. **P < 0.01, compared with the IgG-Fc group.
In concert with the marked autophagic activation, RTN4-Δ20-Fc treatment resulted in considerable decreases in BrdU+_LAMA1+ cells in the ipsilateral thalamus when compared to the vehicle group at 7 days after MCAO (P < 0.01, (Figure 6I-J)). Additionally, the vessel density of the ipsilateral thalamus was significantly decreased in response to RTN4-Δ20-Fc treatment (P < 0.01, (Figure 6I-K)). These results indicate a negative relationship between RTN4-S1PR2-induced vascular autophagic activation and angiogenesis in the ipsilateral thalamus after cerebral infarction.
Vascular autophagy and angiogenesis in the ipsilateral thalamus after cortical infarction
To confirm that the induction of autophagy affects angiogenesis in the ipsilateral thalamus, two autophagic inhibitors, 3-MA and spautin-1, were employed. The results showed that 3-MA treatment at 24 h after MCAO markedly reduced MAP1LC3B-II conversions in vessel tissues of the ipsilateral thalamus at 7 days after MCAO compared to the corresponding vehicle group (P < 0.05, (Figure 7A-B)). This was accompanied by marked increases in the numbers of BrdU+_LAMA1+ cells and the density of LAMA1-labeled vessels in the ipsilateral thalamus (both P < 0.05, (Figure 7C-E)). Consistently, spautin-1 treatment significantly decreased the level of MAP1LC3B-II in the ipsilateral thalamic vessels as compared with that in the vehicle group at 7 days after MCAO (P < 0.05, (Figure 7F-G)). In parallel, the number of BrdU+_LAMA1+ cells and vessel density were markedly increased in the ipsilateral thalamus in the spautin-1 group when compared to the vehicle group (both P < 0.05, (Figure 7H-J)).
Figure 7.
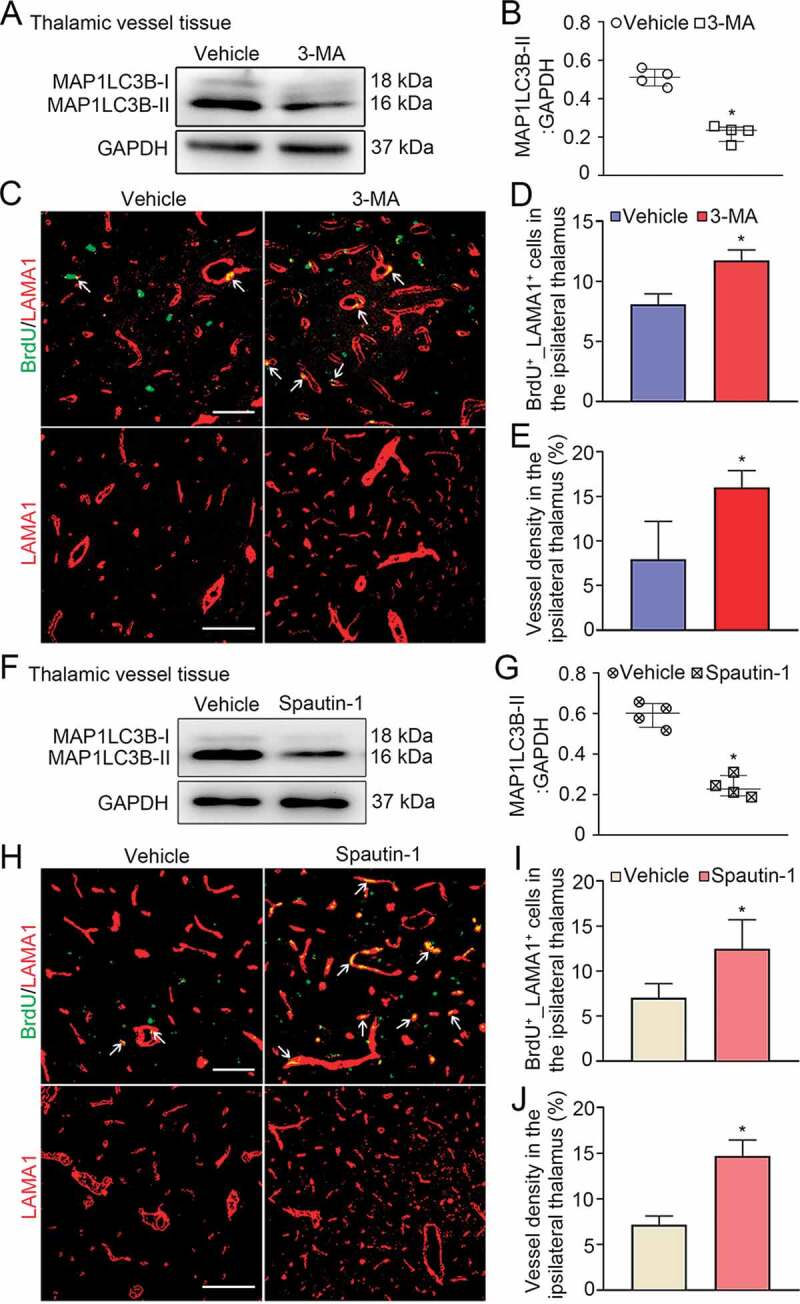
Treatment with 3-MA or spautin-1 enhanced angiogenesis in the ipsilateral thalamus after cortical infarction. (A) Immunoblotting shows the expression of MAP1LC3B-II in vessels of the ipsilateral thalamus in the vehicle and 3-MA groups at 7 days after MCAO. (B) Quantitative analysis of MAP1LC3B-II expression relative to GAPDH. n = 4, data are expressed as median ± interquartile range. *P < 0.05, compared with the vehicle group. (C) Co-staining for BrdU (green) and LAMA1 (red) within the ipsilateral thalamus of the vehicle and 3-MA groups at 7 days after MCAO (arrows). Scale bar: 50 μm, 100 μm. (D and E) Quantitative analysis of BrdU+_LAMA1+ cells and vessel density. n = 6, data are expressed as median ± interquartile range. *P < 0.05, compared with the vehicle group. (F) Immunoblotting shows MAP1LC3B-II expression in vessels of the ipsilateral thalamus in the vehicle and spautin-1 groups at 7 days after MCAO. (G) Quantitative analysis of MAP1LC3B-II expression relative to GAPDH. n = 4, data are expressed as median ± interquartile range. *P < 0.05, compared with the vehicle group. (H) Co-staining of BrdU (green) with LAMA1 (red) in the ipsilateral thalamus of the vehicle and spautin-1 groups at 7 days after MCAO (arrows). Scale bar: 50 μm, 100 μm. (I and J) Quantitative analysis of BrdU+_LAMA1+ cells and vessel density. n = 6, data are expressed as median ± interquartile range. *P < 0.05, compared with the vehicle group.
To further examine the possible impact of lysosomal degradation function on angiogenesis, bafilomycin A1 (BafA1), a drug that suppresses the fusion of autophagosomes with lysosomes and thus prevent autophagic degradation [25], was used. The results showed that BafA1 treatment significantly enhanced the extent of MAP1LC3B-II conversion and SQSTM1 accumulation compared to the vehicle group at 7 days after MCAO (both P < 0.05, (Figure 8A-C)). This effect was accompanied by marked decreases in BrdU+_LAMA1+ cells and LAMA1-labeled vessel density in the ipsilateral thalamus in the BafA1 group as compared with the vehicle group (both P < 0.01, (Figure 8D-F)), suggesting that inhibition of lysosomal function reduces angiogenesis. Collectively, our data suggest that either enhanced autophagic initiation or reduced lysosomal degradation influences angiogenesis in a negative manner.
Figure 8.
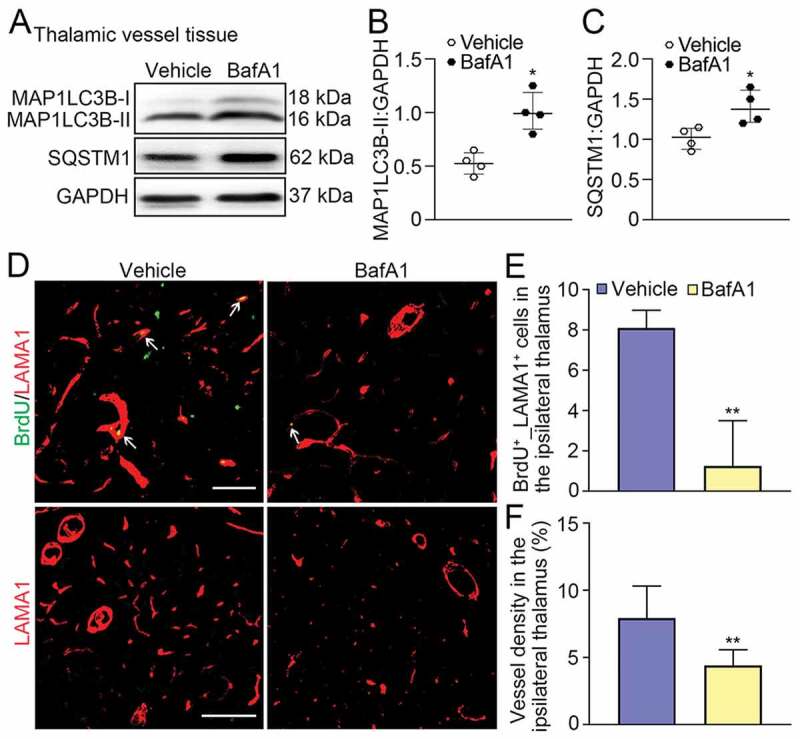
Treatment with BafA1 reduced angiogenesis in the ipsilateral thalamus after cortical infarction. (A) Immunoblotting reveals MAP1LC3B-II conversion and SQSTM1 accumulation in vessels of the ipsilateral thalamus in the vehicle and BafA1 groups at 7 days after MCAO. (B and C) Quantitative analysis of MAP1LC3B-II or SQSTM1 expression relative to GAPDH. n = 4, data are expressed as median ± interquartile range. *P < 0.05, compared with the vehicle group. (D) Co-staining of BrdU (green) with LAMA1 (red) in the ipsilateral thalamus in the vehicle and BafA1 groups at 7 days after MCAO (arrows). Scale bar: 50 μm, 100 μm. (E and F) Quantitative analysis of BrdU+_LAMA1+ cells and vessel density. n = 6, data are expressed as median ± interquartile range. **P < 0.01, compared with the vehicle group.
Interaction of ROCK1 with BECN1 in vessels of the ipsilateral thalamus after cortical infarction
We further explored the potential mechanisms underlying RTN4-S1PR2 induced autophagic activation in the thalamic vessels after cerebral infarction. A possible interaction of the S1PR2 downstream effector ROCK1 with the autophagic initiation regulator BECN1, was determined in the thalamic vessels. The immunofluorescent analysis detected ROCK1 to be colocalized with BECN1 in isolated vessels of the ipsilateral thalamus after MCAO (Figure 9A). Co-immunoprecipitation analysis showed that both ROCK1 and BECN1 were detected in the pulldown protein fraction of vessels from the ipsilateral thalamus in both sham- and MCAO-operated animals (Figure 9B), suggesting an interaction of ROCK1 with BECN1 in vessels of the thalamus after cerebral infarction. To investigate the functional significance of ROCK1-BECN1 interaction, we further examined the phosphorylation of BECN1 at Thr119, a crucial phosphorylation site dependent on ROCK1 for metabolic stress-induced autophagy [26]. The results showed that the level of p-BECN1 (Thr119) was markedly elevated in vessels of the ipsilateral thalamus at 7 days after MCAO, as compared with that in the sham-operated controls (P < 0.05, (Figure 9C-F)). Knockdown of Rtn4 markedly suppressed the phosphorylation of BECN1 in the thalamic vessels when compared to the Scramble-siRNA group (P < 0.05, (Figure 9D-G)). In contrast, RTN4-Δ20-Fc treatment significantly increased the level of phosphorylated BECN1 in the thalamic vessels compared with that in the IgG-Fc group at 7 days after MCAO (Figure 9E-H). Altogether, these findings suggest that ROCK1 phosphorylation of BECN1 at Thr119 might play an import role in RTN4-S1PR2-induced vascular autophagy and angiogenesis in the thalamus after cerebral infarction.
Figure 9.
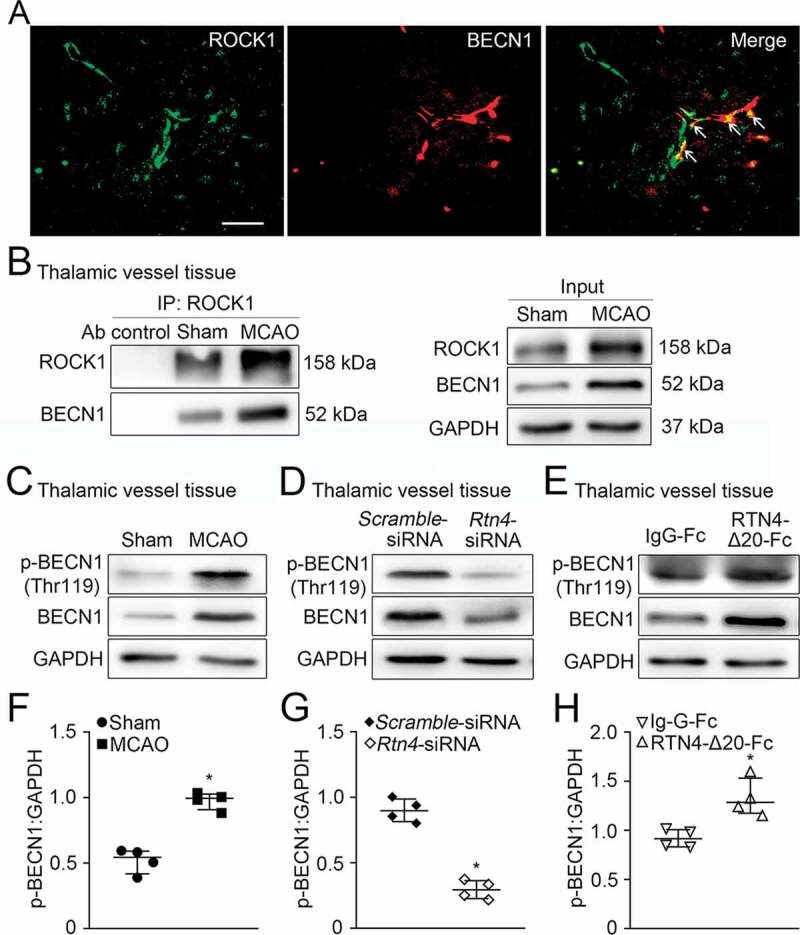
Interaction of ROCK1 with BECN1 in vessels of the ipsilateral thalamus after MCAO. (A) Double-staining shows ROCK1 (green) to be colocalized with BECN1+ cells (red) in vessels of the ipsilateral thalamus (arrows). Scale bar: 50 μm. (B) Co-immunoprecipitation was performed with vessels extracts of the ipsilateral thalamus in the sham- and MCAO-operated animals by using ROCK1-crosslinked agarose, and the resulting immunoprecipitation was blotted with the indicated antibodies. The relative levels of ROCK1 and BECN1 of input were detected by immunoblotting. n = 3. (C-E) Immunoblotting shows the levels of p-BECN1 (Thr119) and total BECN1 in the ipsilateral thalamic vessels of the sham-operated, MCAO, Scramble-siRNA, Rtn4-siRNA, IgG-Fc, and RTN4-Δ20-Fc groups at 7 days after MCAO, respectively. (F-H) Quantitative analysis of p-BECN1 expression relative to GAPDH in the ipsilateral thalamic vessels of different groups. n = 4, data are expressed as median ± interquartile range. *P < 0.05, compared with the respective control group.
Neurological function and secondary thalamic damage after cortical infarction
The Morris maze results showed that animals in the Rtn4-siRNA group had shorter escape latencies to seek hidden platforms compared to the Scramble-siRNA group on day 6 after MCAO (P < 0.01, (Figure 10A and B)). In contrast, animals treated with RTN4-Δ20-Fc had longer escape latencies relative to the IgG-Fc control group from days 4–6 after MCAO (P < 0.01, (Figure 10A and B)). As for the probe trial on day 7, animals treated with Rtn4-siRNA had longer stays in the target quadrant (P < 0.05, (Figure 10A and C)), while those treated with RTN4-Δ20-Fc had a shorter stay time when compared to their respective controls (P < 0.01, (Figure 10A and C)).
Figure 10.
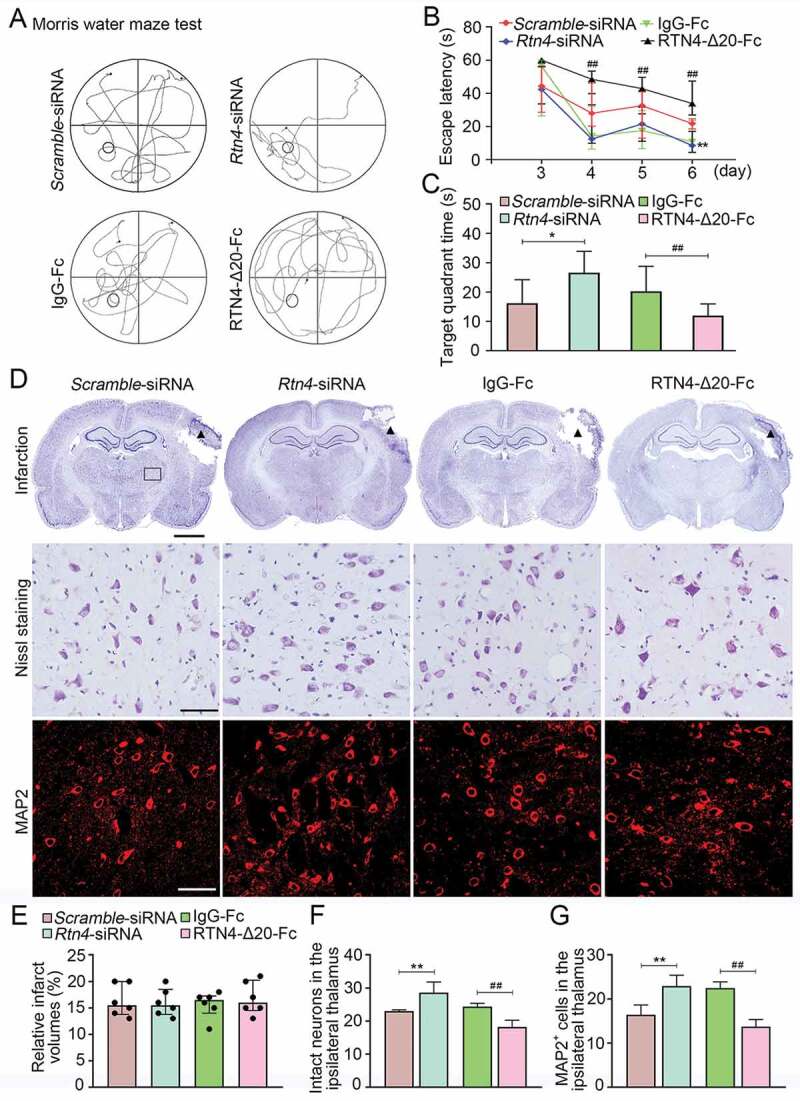
Cognitive function and secondary neuronal damage of the ipsilateral thalamus after cortical infarction. (A) The representative swimming path of the animals in the Scramble-siRNA, Rtn4-siRNA, IgG-Fc, and RTN4-Δ20-Fc groups at 7 days after MCAO. (B) Animals in the Rtn4-siRNA group showed a shorter escape latency to reach the platform on day 6 after MCAO than that in the Scramble-siRNA group. In contrast, animals treated with RTN4-Δ20-Fc had longer escape latency from day 4–6 after MCAO than those in the IgG-Fc group. **P < 0.01, compared with the Scramble-siRNA group, ##P < 0.01, compared with the IgG-Fc group. (C) On day 7 after MCAO, animals of the Rtn4-siRNA group spent longer target quadrant time than that in the Scramble-siRNA group while animals treated with RTN4-Δ20-Fc showed less time in the target quadrant compared to the IgG-Fc group. n = 8, data are expressed as median ± interquartile range. *P < 0.05, compared with the Scramble-siRNA group, ##P < 0.01, compared with the IgG-Fc group. (D) Representative Nissl staining indicates the cortical infarction (black triangle) and neuronal damage within the ipsilateral thalamus (black rectangle) of the Scramble-siRNA, Rtn4-siRNA, IgG-Fc, and RTN4-Δ20-Fc groups. Immunostaining analysis of MAP2+ neurons within the ipsilateral thalamus across the different groups. Scale bar: 2 mm, 50 μm. (E-G) Quantitative analysis of the relative infarct volumes, and the number of intact neurons and MAP2+ cells. n = 6, data are expressed as median ± interquartile range. **P < 0.01, compared with the Scramble-siRNA group, ##P < 0.01, compared with the IgG-Fc group.
No significant differences in infarct volumes were found across the Scramble-siRNA, Rtn4-siRNA, IgG-Fc and RTN4-Δ20-Fc groups at 7 days after MCAO (all P > 0.05, (Figure 10D and E)). In the ipsilateral thalamus, there was an approximately 43% reduction of neuron loss in the Rtn4-siRNA group in relative to the Scramble-siRNA group, as evidenced by Nissl staining and MAP2 immunofluorescence (all P < 0.01, (Figure 10D, F, G)). Conversely, RTN4-Δ20-Fc treatment resulted in a marked increase in neuron loss when compared with that in the IgG-Fc control group (all P < 0.01, (Figure 10D, F, G)). Additionally, there was a significant positive relationship between the number of BrdU+_LAMA1+ cells and those of intact neurons (r = 0.82, P < 0.001) or MAP2+ cells (r = 0.79, P < 0.001) in the ipsilateral thalamus across the four groups by controlling for the respective infarct volumes. A significant relationship was also detected between the number of BrdU+_LAMA1+ cells in the ipsilateral thalamus and shorter escape latencies when controlling for the infarct volumes in the above groups (r = 0.60, P = 0.006). These results suggest that blocking RTN4-S1PR2 interaction enhances angiogenesis and rescues secondary neuronal damage of the thalamus; thereby, improving cognitive function after cerebral infarction.
RTN4-S1PR2 regulated autophagic flux on endothelial cells
To confirm our in vivo findings, we further investigated autophagic flux in primary rat brain microvascular endothelial cells (BMVECs) and human umbilical vein endothelial cells (HUVECs). As showed in (Figure 11A-C), immunoblotting results showed that RTN4-Δ20 markedly increased MAP1LC3B-II conversion and accumulation of the lysosomal degradation marker SQSTM1 in BMVECs. 3-MA, an inhibitor of autophagic initiation or BafA1, a lysosome inhibitor, was used to distinguish whether the increase of MAP1LC3B-II level is due to elevated autophagy initiation or declined autophagy degradation. BafA1 treatment further augmented the extent of MAP1LC3B-II and SQSTM1 accumulation induced by RTN4-Δ20 alone in BMVECs, indicating that RTN4-Δ20 partially blocks lysosomal degradation of autophagosomes. In contrast, addition of 3-MA significantly abrogated an increase in MAP1LC3B-II and SQSTM1 accumulation by RTN4-Δ20, indicating a possible involvement of RTN4-Δ20 in autophagic initiation. Additionally, inhibition of S1PR2 signaling by JTE-013 markedly ablated the accumulation of MAP1LC3B-II and SQSTM1 in RTN4-Δ20 co-cultured BMVECs. These results were further confirmed in HUVECs (Figure S2A-C).
Figure 11.
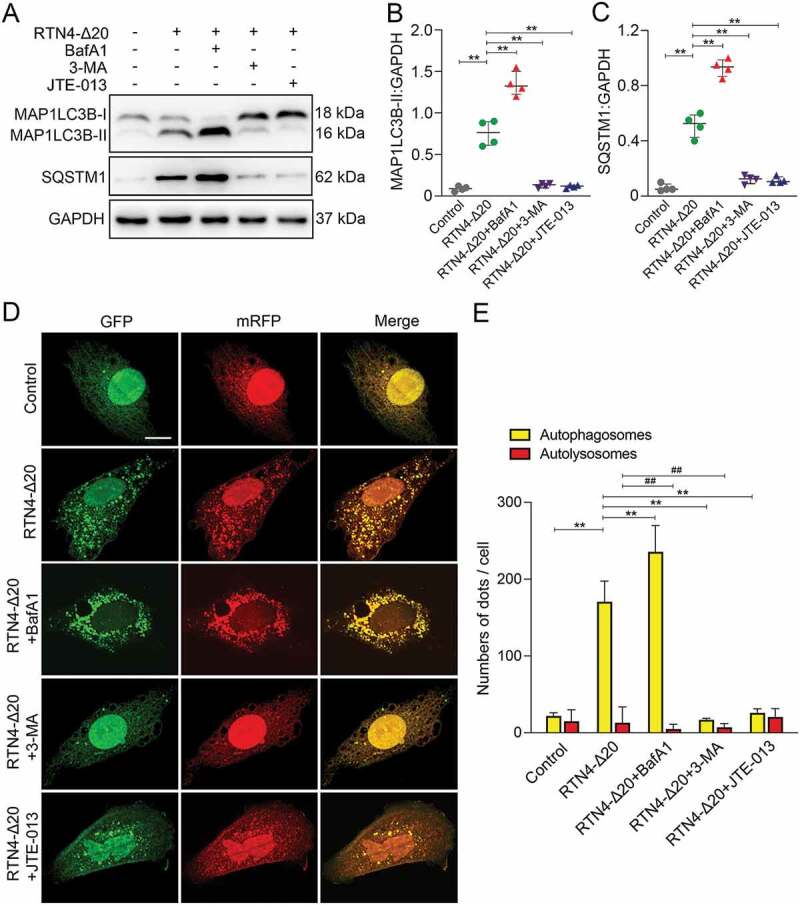
RTN4-S1PR2 induced autophagy in BMVECs. (A) Immunoblotting analysis shows the expression of MAP1LC3B and SQSTM1 in BMVECs in the absence or presence of RTN4-Δ20 individual, or in combination with BafA1, 3-MA or JTE-013. (B and C) Quantitative analysis of MAP1LC3B-II and SQSTM1 levels relative to GAPDH. n = 4 from four independent experiments, data are expressed as median ± interquartile range. **P < 0.01, one-way ANOVA. (D) Representative confocal microscopic images of LC3B puncta in BMVECs transfected with mRFP-GFP-LC3 in the absence or presence of RTN4-Δ20 alone or in combination. Autophagosomes are indicated by yellow dots with both GFP and RFP, and autolysosomes by free red dots with only RFP in merged images. Scale bar: 20 μm. (E) Quantitative analysis of autophagosomes and autolysosomes per cell. n = 20–30 cells of four independent experiments, data are expressed as median ± interquartile range. **P < 0.01 (autophagosomes), ##P < 0.01 (autolysosomes), one-way ANOVA.
The observation obtained by immunoblot was validated in cells transfected with an exogenous mRFP-GFP-LC3 construct. In this assay, yellow puncta with both GFP and RFP correspond to autophagosomes whereas free red puncta with only RFP are indicative of autolysosomes [27]. As showed in (Figure 11D and E), RTN4-Δ20 treatment markedly increased the number of yellow puncta but not free red puncta in BMVECs. This effect was further augmented in the presence of BafA1 while markedly ablated by addition of 3-MA. In addition, co-incubation of cells with RTN4-Δ20 and the S1PR2 antagonist JTE-013 markedly reduced the number of yellow puncta in BMVECs. Similar results were obtained in HUVECs (Figure S2D and E). Taken together, these results suggest that RTN4-S1PR2 signaling induces autophagosome formation and partially suppresses lysosomal degradation, thus influencing autophagic flux.
Discussion
The present study showed the specificity of RTN4 up-regulation in neurons or oligodendrocytes and S1PR2 in vessels of the ipsilateral thalamus after cerebral infarction. Knockdown of Rtn4 mediated by siRNA markedly downregulated MAP1LC3B-II conversion and the expression of BECN1 and SQSTM1, coinciding with increased angiogenesis, reduced neuronal loss of the ipsilateral thalamus and improved cognitive function on water maze test following cerebral infarction. In contrast, the activation of S1PR2 signaling with RTN4-Δ20 led to a converse effect. The causal relationship between vascular autophagy and angiogenesis in the thalamus was verified by inhibiting autophagic initiation or blocking autophagic degradation. Furtherly, the control of autophagic flux by RTN4-Δ20-S1PR2 signaling was confirmed by our in vitro experiments. Mechanistically, we found an interaction of S1PR2 downstream effector ROCK1 with the autophagy-related protein BECN1, along with increased phosphorylation of BECN1 at Thr119 in vessels of the thalamus after cerebral infarction. Knockdown of Rtn4 reduced BECN1 phosphorylation while activating S1PR2 increased its phosphorylation in vessels. Taken together, our data suggest that RTN4-S1PR2 signaling negatively regulates angiogenesis through enhancing autophagic initiation and partially blocking lysosomal degradation of autophagosomes in the thalamic vessels after stroke. Moreover, ROCK1 phosphorylation of BECN1 might play a role in the regulation of vascular autophagy in the thalamus.
Ischemic stroke results in secondary neuronal damage of the ipsilateral thalamus, free from primary insults and delayed cognitive recovery [4,28]. In the present study, focal cortical infarction within the MCA territory was induced based in hypertensive animals, which mimics human stroke etiology and restricts infarcts in cortical regions with the ipsilateral thalamus intact [29]. Consistent with previous studies [19–21], we found that BrdU_LAMA1 double-positive cells and vessel density increased in the ipsilateral thalamus at 7 days after distal cerebral infarction, suggesting compensatory endogenous vascular repair in addition to progressive secondary neurodegeneration in the thalamus following cerebral infarction. However, the control of neovascular remodeling in this regard remains unclear.
The neurite growth inhibitor RTN4 has been shown to inhibit vascularization in the developing central nervous system and postnatal brain [19,20]. Recently, it has been demonstrated that ablation of Rtn4 promotes vascular remodeling in the peri-infarct zone in cerebral ischemic mice [21]. In the present study, RTN4 was found to be up-regulated specifically in neurons and oligodendrocytes adjacent to blood vessels in the ipsilateral thalamus, remote from the ischemic regions. Knockdown of Rtn4 mediated by siRNA led to an increased number of BrdU+_LAMA1+ cells and density of blood vessels in the ipsilateral thalamus, suggesting an inhibitory effect of RTN4 expressed on neurons or oligodendrocytes on angiogenesis in the ipsilateral thalamus at the early stage of ischemic stroke. In agreement with previous studies [21,30,31], we found that RTN4 exerted antiangiogenic function via its corresponding Δ20 receptor – S1PR2 expressed in blood vessels in the ipsilateral thalamus, as evidenced by marked decreases in angiogenic parameters with recombinant RTN4-Δ20-Fc infusion. Activating the S1PR2 signaling is suggested to suppress sprouting and migration of vascular endothelial cells in vitro [19]. In contrast, we found that S1PR2 was also expressed in vascular smooth muscle cells and pericytes, in addition to endothelial cells in the ipsilateral thalamus after cerebral infarction. Given the essential involvement of vascular smooth muscle cells or pericytes in vascular remodeling or maturation [32,33], we speculate a similar inhibitory action of neuronal or glial RTN4 on vascularization by activating the S1PR2 receptor expressed on these supportive mural cells in an intercellular communication manner.
We showed the activation of autophagy in vessels of the ipsilateral thalamus after cerebral infarction as evidenced by increased MAP1LC3B-II, which is known to correlate with autophagosome formation [34]. The role of autophagy in the regulation of angiogenesis remains unclear under this condition. Previous studies have revealed that inhibition of autophagy reduces angiogenesis in endothelial cells [35,36], while enhanced endothelial autophagy promotes angiogenic sprouting to stabilize the cardiac microvasculature [37]. In contrast to these findings, the present results showed that Rtn4 knockdown inhibited autophagic activation in vessels and promoted angiogenesis in the ipsilateral thalamus, whereas activating S1PR2 signaling led to the reverse effects. This suggests a negative relationship between RTN4-S1PR2 induced autophagy and angiogenesis in this regard. Our findings are supported by other studies showing that Becn1-deficiency largely promotes hypoxia-induced tumor angiogenesis [14], while enhanced autophagic activation inhibits angiogenesis by deleting the gastrin-releasing peptide receptor in a vascular neuroblastoma cell model [15]. BECN1 forms phosphatidylinositol 3-kinase complexes together with the core lipid kinase PIK3C3/VPS34 and the regulatory component PIK3R4/VPS15 to induce autophagosome formation and maturation [22,38]. In contrast, SQSTM1 acts as a receptor protein that links MAP1LC3B-II with ubiquitin moieties on misfolded proteins during autophagy, and thus its accumulation reflects deficits in autophagosomes clearance [23,39]. Using these markers, we found that Rtn4 knockdown reduced the levels of BECN1 and SQSTM1 in vessels of the ipsilateral thalamus whereas activation of S1PR2 led to a converse effect, suggesting the possible actions of RTN4-S1PR2 signaling on the regulation of both autophagic initiation and lysosomal degradation. This effect is further confirmed by our in vitro observations showing that RTN4-Δ20 induced autophagosome formation but also partially blocked the lysosomal degradation of autophagosomes in a S1PR2–dependent manner. Additionally, the detrimental effect of vascular autophagy was evident in the fact that inhibition of autophagic formation by 3-MA or spautin-1 enhanced angiogenesis in the thalamus whereas blocking autophagy degeneration by BafA1 resulted in a converse effect. Taken together, these results imply that blockade of RTN4-S1PR2 interaction would have the potential benefit of reducing autophagic initiation and alleviating dysfunction of autophagic degradation in angiogenesis of the thalamus after stroke.
The action of S1PR2 signaling requires intracellular activation of the RHOA-ROCK pathway [20]. ROCK1 activation can lead to autophagic activation through its interaction with and phosphorylation of BECN1 in response to metabolic stress [26]. Similarly, another study has shown that activated ROCK1 facilitates intracellular autophagosome formation via its interaction with BECN1 during Alzheimer disease progression [40]. Consistently, we identified a direct interaction between ROCK1 and BECN1 along with increased phosphorylation of BECN1 at Thr119 in vessels of the ipsilateral thalamus after cerebral infarction. Further neutralization of Rtn4 by genetic knockdown markedly reduced the levels of phosphorylated BECN1, while activation of S1PR2 signaling led to a reverse effect, suggesting RTN4-S1PR2 facilitates ROCK1 phosphorylation of BECN1 at Thr119 in the thalamic vessels after cerebral infarction. ROCK1 phosphorylation of BECN1 at Thr119 allows for dissociation of BECN1-BCL2 complex, thus leading to the formation of autophagosomes [26,41]. Together with these previous findings, our data suggest that RTN4 might trigger autophagic activation in the thalamic vessels by activating the S1PR2-ROCK1 pathway and subsequently phosphorylating BECN1. Nevertheless, this observation needs to be interpreted with caution. BECN1 may coordinate autophagosome maturation with fusion to the lysosomes and endocytic trafficking by targeting UVRAG complex [22,42]. It is still unclear whether RTN4-S1PR2 signaling affects autophagy, angiogenesis and secondary nerve damage through the ROCK1-BECN1 pathway. Several potential mechanisms have been proposed to explain the regulation of ROCK1 in autophagy. For example, ROCK1 has been shown to activate the MAPK/JNK (mitogen-activated protein kinase) signaling pathway to induce the activation of autophagy by phosphorylating BCL2, which allows for the release of BECN1 from their complex [43]. Further studies are needed to elucidate the additional mechanisms by which S1PR2 regulates vascular autophagy. It should be noticed that there is a lack of using knockout mice for Rtn4, S1pr2 or Becn1 to support the conclusions in the present study. Although we have identified casual correlates of vascular autophagy with angiogenesis by siRNA and pharmacological approaches, the findings need to be further confirmed in knockout mice for Rtn4, S1pr2 or Becn1.
Strategies to promote angiogenesis have been attempted to improve functional improvement. In the present study, we showed that Rtn4 knockdown promoted angiogenesis, coinciding with decreased neuronal loss and improved spatial learning function. These neural and behavioral benefits likely relate to either a direct effect on neurons and oligodendrocytes or vessel-mediated indirect effect by Rtn4 knockdown. It has been showed that Rtn4 loss-of-function by knockout promotes spatial learning by enhancing neuronal plasticity in the mouse hippocampus [44]. Similarly, blockade or ablation of neuronal Rtn4 or its receptor Rtn4R enhances activity-dependent structural and functional synaptic plasticity in the hippocampus [45,46]. Further, our previous studies have demonstrated the detrimental effects of RTN4 on the neuronal regeneration in the thalamus and function outcomes following stroke [6,18]. Together with these findings, the strong correlation between neuronal damage and angiogenic effects in the present study might indicate a critical role of vascular repair in Rtn4 knockdown-mediated nerve cell rescue as well as functional improvement. Additionally, we demonstrated a strong positive relationship between angiogenic and functional measures, possibly implicating its dependence on vascular repair by knockdown of Rtn4. Supporting this, a recent study has showed that enhanced motor improvements by anti-RTN4 treatment are abrogated when angiogenesis is inhibited in the peri-infarct areas [21]. Similarly, the behavioral improvements induced by dilvery of angiogenic biomaterials are abolished by blocking revascularization in the infarct regions following stroke [47]. Taken these findings together, we speculate that the mutual effects of angiogenesis and neuronal repair coordinately contribute to post-stroke cognitive function improvement by Rtn4 knockdown.
In summary, the present study revealed vascular autophagy as a novel mechanism by which RTN4 regulates angiogenesis in the ipsilateral thalamus after cerebral infarction. This also suggests that autophagy may represent a new target for RTN4 intervention in conjunction with vascular repair and cognitive function secondary to ischemic stroke.
Materials and methods
Animal models and experimental design
The stroke-prone renovascular hypertensive rats (RHRSP) were established from male Sprague-Dawley (SD) rats weighting 90–110 g using a two-kidney two-clip method, as described in our previous studies [29]. After 12 weeks, RHRSP with systolic blood pressure > 180 mmHg and without any stoke symptoms were selected according to the following design. The experimental protocols were approved by the local ethics committee for animal research, and all procedures involving animals were conducted according to the institutional guidelines.
In the first part, a group of RHRSPs were randomly assigned to receive the MCAO or sham operation (17 animals per group). MCAO was introduced as previously described, with minor modifications [48]. Briefly, under anesthesia with 10% chloral hydrate (3 mg/kg, i.p.), the right middle cerebral artery was exposed and then occluded distal to the origin of the striatal branches with a bipolar electrocoagulation device, which resulted in a permanent cortical infarction in the right hemisphere. Animals for the sham operation were treated similarly, except for occlusion of the right MCA. In the second part, RHRSPs were randomly selected to receive stereotactically guided intrathalamic injections of Rtn4-siRNA or Scramble-siRNA lentivirus at 10 weeks after renovascular hypertension surgery and then underwent MCAO at 2 weeks after the injection operation (14 animals per group). In the third part, animals with successful MCAO were randomly selected for intraventricular delivery of recombinant RTN4-Δ20-Fc or IgG-Fc (10 animals per group) 24 h after MCAO. Additionally, animals underwent MCAO were randomly chosen to receive 3-MA, spautin-1, BafA1 or the respective vehicle at 24 h after MCAO (10 animals per group). A total of 18 animals were excluded from the study due to failure to establish MCAO (eight animals), intracranial hemorrhage during operations (six animals), and death due to anesthesia during intracranial injection (four animals).
SiRNA preparation and intrathalamic delivery
Four siRNA sequences targeting rat Rtn4 (Genbank, NM_031831) and a negative control sequence were constructed by Genechem (Shanghai, China). The separate sequences were synthesized and inserted into GV248 lentiviral vector containing a CMV-driven EGFP receptor gene and an Rnu6 promoter upstream of the restriction sites (AgeI and EcoRI). All constructs were confirmed by sequence analysis. Recombinant lentivirus was produced by transfecting 293 T cells with Lipofectamine 2000 (Invitrogen, 11,668–500) according to the standard protocol. Virus titers, expressed as transducing units (TU) per milliliter, were determined by measuring GFP expression in 293 T cells transduced with different dilutions of lentivirus. The best performed Rtn4-siRNA sequence was determined as GTGATCCAGGCTATCCAGAAA, and the negative control Scramble-siRNA sequence was TTCTCCGAACGTGTCACGT. The final titer was 1.5 × 109 TU/ml. Lentivirus preparations were injected stereotactically into the right thalamus, as we previously described [7,9]. RHRSPs were placed on a stereotactic apparatus under anesthetized as described above. Lentivirus preparations were delivered into four separate sites within the right thalamus using a 15-μl syringe at a dose of 3 μl for each injection with an infusion rate of 0.5 μl/min, following the coordinates: −2.6 mm anteroposterior, 2.6 mm rightwards, −6.4 or −6.6 mm dorsoventral; −3.0 mm anteroposterior, 2.8 mm rightwards, −6.2 or −6.4 mm dorsoventral relative to the bregma. Fourteen days after injection, animals were subjected to MCAO surgery to enable sufficient efficiency of gene knockdown.
Intraventricular delivery of agencies
Recombinant rat RTN4 Fc chimera corresponding to the specific amino acids 544–725 domain of RTN4 (R&D Systems, 2445-NG) was administered stereotactically into the right lateral ventricle to activate RTN4-Δ20-S1PR2 signaling [24]. Recombinant human IgG Fc (R&D Systems, 110-HG) was used as a control. In brief, under anesthetized, RTN4-Δ20-Fc or IgG-Fc was dissolved in 0.01 M phosphate-buffered saline (PBS; Gibco, C10010500BT; pH 7.4) and delivered to the right lateral ventricle at a dose of 100 μl (40 μg/kg) using osmotic minipumps (Alza Scientific Products, Alzet 1003D) at 24 h after MCAO to avoid potential effects on the primary infarction. The minipump-connected microtubule was inserted into the right lateral ventricle under a stereotaxic apparatus (−1.0 mm anteroposterior, 1.4 mm mediolateral, −4.0 mm dorsoventral relative to the bregma). The osmotic minipump was designed to administer RTN4-Δ20-Fc or IgG-Fc for 3 consecutive days.
To determine the correlation between vascular autophagy and angiogenesis, 3-MA and spautin-1 were employed to inhibit the early-stage autophagy by suppressing autophagy initiation. Spautin-1 suppresses autophagosome formation by specifically inhibiting USP10 and USP13 that target the BECN1 subunit of PIK3C3/VPS34 complexes [49]. In addition, BafA1 was used to suppress the lysosomal degradation function by blocking the fusion of autophagosomes with lysosomes [25]. 3-MA (Sigma-Aldrich, M9281) or BafA1 (Selleck, S1413) was dissolved in 2% DMSO (Sigma-Aldrich, D2650), and spautin-1 (Selleck, S7888) was dissolved in 40% DMSO plus 40% PEG300 (MedChemExpress, 85,948). The drug doses of administration were selected based on published studies [7,50,51]. At 24 h after MCAO, a volume of 10 μl 3-MA (60 mM), spautin-1 (44 mM), BafA1 (10 nM) or the respective vehicle was delivered stereotactically to the right lateral ventricle using a 10-μl syringe with an infusion rate of 0.5 μl/min following the coordinates as described above. After infusion, the syringe was left in place for 5 min to allow for drug diffusion.
In vivo BrdU labeling
To label the newly generated endothelial cells, BrdU (Sigma-Aldrich, B5002) was dissolved in 0.9% sodium chloride and intraperitoneally injected (50 mg/kg) twice daily at a 12-h interval for 6 consecutive days from 24 h after MCAO. This allows to identify the cumulative proliferated cells in the thalamus [9].
Cognitive function assessment
Eight rats were randomly selected from the Scramble-siRNA, Rtn4-siRNA, IgG-Fc and RTN4-Δ20-Fc groups from day 2 to 7 after MCAO for the cognitive assessment using the Morris water maze test as previously described [52]. The test consists of adaptive training, spatial acquisition training and probe trials. On day 2 after MCAO, the animals were allowed to freely swim in the pool for 3 min for adaptive excises. From day 3–6, spatial acquisition trials were performed with a 10-cm diameter platform under the water in the southwest quadrant. Each animal was placed in the water facing the pool wall and allowed to start from a position to climb onto the platform within 60s. This session was repeated four times each day. The time spent seeking the platform (referred as to escape latency) and swimming distance were recorded to measure spatial learning ability. On day 7 post-MCAO, the probe trial was performed with the platform removed. The time spent in the target quadrant and the frequency of crossing the original location of the platform within 60s were respectively recorded to assess the degree of spatial memory.
Tissue preparation
Seven days after MCAO, six rats from each group were randomly selected for euthanasia. Under deep anesthesia with 10% chloral hydrate (5 mg/kg, i.p.), transcardiac perfusion was performed using 0.9% sodium chloride at 4°C followed by 4% paraformaldehyde in 0.1 M PBS (pH 7.4). Afterward, the brains were separately removed, kept in the same fixative, and then immersed sequentially in 20% and 30% sucrose solutions in 0.1 M PBS overnight at 4°C. Coronal sections of 10-μm thickness were cut on a cryostat (Leica, CM1900) between +4.7 to −5.2 mm relative to bregma for histological analysis as follows.
Nissl staining
A series of coronal sections were chosen from each group for Nissl staining to detect primary infarct volume and neuronal damage in the ipsilateral thalamus as previous described [53]. Standard Nissl staining was conducted with 0.3% cresyl violet (Sigma-Aldrich, C5042) and images were captured with a computer connected microscope (Olympus, BX51). The primary infarct volume was determined by calculating the percentage of infarct volume relative to the respective contralateral hemisphere volume.
Immunofluorescent staining for in vivo experiments
Another series of coronal sections between −2.8 and −4.4 mm relative to bregma were selected from each group to perform immunofluorescence analysis as follows. Sections were rinsed in 0.01 M PBS (pH 7.4) and blocked with 5% normal donkey or goat-derived serum (Solarbio, SL050, SL038) at 24–26°C for 60 min. Thereafter, the sections were incubated at 4°C overnight with the respective primary antibodies: mouse anti-BrdU (1:250; Sigma-Aldrich, B2531), rabbit anti-LAMA1 (1:500; Sigma-Aldrich, L9393), mouse anti-RECA-1 (1:400; Abcam, ab9774), mouse anti-ACTA2 (1:200; Abcam, ab7817), rabbit anti-PDGFRB (1:100; Abcam, ab32570), mouse anti-MAP1LC3B (1:200; Cell Signaling Technology, 83506S), rabbit anti-MAP1LC3B (1:200; Abgent, AP1802a), mouse anti- BECN1 (1:200; Santa Cruz Biotechnology, sc-48,341), goat anti-RTN4 (1:200; R&D Systems, AF3098-SP), rabbit anti-RTN4 (1:400; Abcam, ab62024), chicken anti-MAP2 (1:1000; Abcam, ab5392), rabbit anti-CLDN11 (1:200; Abcam, ab53041), chicken anti-GFAP (1:1500; Abcam, ab4674), rabbit anti-AIF1 (1:400; Wako, 019–19,741), rabbit anti-S1PR2 (1:200; Novus, NBP2-26,691), mouse anti-S1PR2 (1:200; Santa Cruz Biotechnology, sc-365,589), rabbit anti-ROCK1 (1:100; Abcam, ab134181). Negative control sections were incubated with 0.01 M PBS instead of the primary antibody. The sections were then rinsed in 0.01 M PBS and then incubated with the respective following secondary antibodies at 24–26°C for 60 min: Alexa Fluor 555-conjugated goat anti-mouse IgG (1:500; Cell Signaling Technology, 4409s), Alexa Fluor 488-conjugated goat anti-mouse IgG (1:500; Cell Signaling Technology, 4408s), Alexa Fluor 555-conjugated goat anti-rabbit IgG (1:500; Cell Signaling Technology, 4412s), Alexa Fluor 488-conjugated goat anti-rabbit IgG (1:500; Cell Signaling Technology, 4413s), Alexa Fluor 488-conjugated goat anti-chicken IgY (1:1000; Abcam, ab150173), Alexa Fluor 405-conjugated goat anti-rabbit IgG (1:500; Abcam, ab175652) and Alexa Fluor 405-conjugated donkey anti-goat IgG (1:500; Abcam, ab175665). Fluorescent signals were detected using a fluorescence microscope (Olympus, BX51). To verify colocalization of those double-stained cells in the ipsilateral thalamus after MCAO, high-magnification images were acquired using a confocal microscopy (Nikon Eclipse Ni-E) under a Plan-Apochromat 63 × 1.4 Oil λS objective lens with a z-step of 0.5 μm.
Isolation of thalamic vessels
To investigate the vascular specificity of autophagy, blood vessels were isolated from the thalamus as previously described [54] with minor modifications. Briefly, seven animals from the sham-operated or MCAO groups and four from the other groups were randomly selected for sacrificed at 7 days after MCAO under deep anesthesia as described above. The animals then underwent transcardially perfusion with 0.9% sodium chloride at 4°C. The ipsilateral thalamus was rapidly dissected and homogenized in 2 ml of cold MCDB131 medium (Thermo Fisher Scientific, 10,372,019). The homogenate was centrifuged at 2,000 × g for 5 min at 4°C. The supernatant was discarded and the pellet was resuspended in 1 ml 15% (wt:vol) cold dextran-Dulbecco’s phosphate-buffered saline (DPBS) (dextran: ~70,000 Da, Sigma-Aldrich, 31,390; DPBS: Thermo Fisher Scientific, 14,190,250). The pellet was gently sluiced and centrifuged at 10,000 × g for 15 min at 4°C. After carefully removing the supernatant, the pellet was resuspended in dextran-DPBS. These steps were repeated twice to obtain the final vessel-containing pellet for further analysis.
To confirm the purity of vessel extracts, immunofluorescence analysis was conducted to detect cell components of vessels using the following markers: rabbit anti-PECAM1/CD31 (1:100; Abcam, ab281583), rabbit anti-PDGFRB, mouse anti-ACTA2, chicken anti-GFAP and mouse anti-AQP4 (1:400; Abcam, ab9512). Additionally, immunoblotting analysis were performed on the vessel preparations and whole-thalamus tissues with specific markers for vessels (rabbit anti-PECAM1, 1:1000), neurons (chicken anti-MAP2, 1:10,000), astrocytes (chicken anti-GFAP, 1:2000) or myelin (mouse anti-MBP2, 1:1000; Abcam, ab11159). Consistent with the previous study [54], the vessel fragments contained endothelial cells, pericytes, vascular smooth muscle cells, and astrocytes end-feet components but not neurons or myelin (Figure S3), suggesting a high degree of purity in isolated vessels with the current protocols.
Immunoblotting analysis for in vivo experiments
To investigate the expression of RTN4 in the ipsilateral thalamus, four animals from the sham-operated, MCAO, Scramble-siRNA or Rtn4-siRNA group were randomly selected for sacrificed at 7 days after MCAO. The respective ipsilateral thalamus was separated as above and then homogenized in RIPA lysis buffer (Cell Signaling Technology, 9806) with cocktail protease inhibitor (Millipore, 539,132–1SETCN) on ice. After sonication on ice, the homogenate was centrifuged at 12,000 × g for 30 min at 4°C and the supernatant was collected for immunoblotting analysis. Additionally, the isolated vessel-containing pellets from four animals across groups were treated following the above procedures. Protein concentration was determined using BCA protein Assay Kit (Thermo Scientific, 23,250).
Eighty micrograms of protein from the total thalamus or ten micrograms of protein from the isolated vessels in the respective groups were separated using 6–12% gradient Tris-glycine sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred onto immublio-P membranes (Millipore, ISEQ00010). The membranes were blocked with 5% nonfat milk (Bio-Rad Laboratories, 1,610,175) for 60 min at 24–26°C and then incubated with the following primary antibodies at 4°C overnight: mouse anti-MAP1LC3B (1:1000), rabbit anti-BECN1 (1:1000; Abcam, ab62557), rabbit anti-SQSTM1 (1:1000; Cell Signaling Technology, 5114S), mouse anti-RTN4 (1:1000; BD, 612,238), mouse anti-S1PR2 (1:1000), rabbit anti-RHOA (1:1000; Abcam, ab86297), rabbit anti-ROCK1 (1:1000), rabbit anti-p-BECN1 Thr119 (1:300; Abcepta, AP3765a), rabbit anti-GAPDH (1:1000; Cell Signaling Technology, 2118S) or rabbit anti-TUBA/ɑ-tubulin (1:1000; Cell Signaling Technology, 2125S). Afterward, the membranes were washed with Tris-buffered saline (Solarbio, T1080) plus Tween-20 (0.1%; Beyotime, ST825; TBST, pH 7.4) and incubated with the respective secondary antibodies: horse anti-mouse IgG (1:2000; Cell Signaling Technology, 7076) or goat anti-rabbit IgG (1:2000; Cell Signaling Technology, 7074) for 60 min at 24–26°C. Membranes were then washed with TBST and visualized using an enhanced-chemiluminscence system (Millipore, WBKLS0100).
Immunoprecipitation analysis for in vivo experiments
Three isolated vessel-containing pellets of the sham-operated and MCAO groups were lysed in cell lysis buffer (Beyotime, P0013) with a cocktail protease inhibitor (Millipore, 539,132–1SETCN). The lysates were centrifuged at 12,000 × g for 30 min at 4°C, and the supernatant were collected as thalamic vessel lysates. A volume of 50 μl protein G magnetic beads (MagnaBindTM; Invitrogen, 10007D) were coated with 5 μg of ROCK1 antibody or normal rabbit IgG (Cell Signaling Technology, 2729) at 4°C for 2 h. Next, 100 μl of isolated lysates were added to antibody-coated beads for immunoprecipitation overnight at 4°C. Afterward, the beads were washed with washing buffer for three times with the assistance of a magnetic rack. Finally, the washed beads were resuspended in an elution buffer to separate the proteins, and the supernatant was collected with the assistant of the magnetic rack. The supernatant was denaturized with 5 × sodium dodecyl sulfate-containing loading buffer for 5 min at 100°C, and then immunoblotted for ROCK1 and BECN1 as described above.
BMVECs isolation and culture
Primary BMVECs were isolated from ten brain tissues of P6 laboratory SD rats according to previous publication [55]. Briefly, the rat cortical tissues were dissected, removed the meninges and large vessels, and BMVECs were isolated as described. BMVECs were cultured in the endothelial cell medium (ECM; ScienCell, 1001) containing a supplemental mix of 10% fetal bovine serum (ScienCell, 0025), 1% endothelial cell growth supplement (ScienCell, 1052) and 1% penicillin-streptomycin solution (ScienCell, 0503) at 37°C in a humidified 5% CO2 atmosphere. The purity of BMVECs was identified to be ≥ 90% by immunofluorescent analysis of PECAM1. BMVECs were used for the present experiments at passage 1–3.
HUVECs culture
HUVECs were purchased from ScienCell (8000) and cultured in the endothelial-cell growth medium as described above. HUVECs were used before passage 8 in the present study.
Confocal microscope assay for in vitro experiment
MRFP-GFP-LC3 adenoviral vectors were purchased from HanBio Technology (AP21071905) and employed to detect autophagic flux as previously described [27,56]. BMVECs and HUVECs were plated in 24-well plates and allowed to reach 50% – 70% confluence. Cells were co-cultured with the adenoviruses containing mRFP-GFP-LC3 construct in ECM for 8 h at 37°C according to the manufacturer’s protocol. To explore the impacts of RTN4-S1PR2 signaling on autophagic flux, cells were then treated with RTN4-Δ20 (0.8 μg/ml) alone or in combination with the autophagy inhibitors BafA1 (100 nM), 3-MA (10 mM), or the S1PR2 antagonist JTE-013 (1 μM; Selleck, S7182) for 24 h. The indicated final concentrations were chosen according to published literatures [21,30,57]. LC3B-puncta were detected with FV3000 confocal microscope (Olympus) fitted with a Plan-Apochromat 60 × 1.42 Oil λS objective lens with a z-step of 0.5 μm.
Immunoblotting analysis for in vitro experiments
After treatment, cells were lysed in RIPA lysis buffer containing a protease inhibitor cocktail and the protein concentration was measured as described in tissue preparation in vivo. Samples containing 20 μg protein extracts were electrophoresed through SDS-PAGE, and the densities of immunoblot for autophagic markers MAP1LC3B, SQSTM1 or GAPDH were analyzed. The experiments were repeated for four times.
Statistical analysis
To determine the volume of the primary cortical infarction, every 12th Nissl-stained section from bregma +4.7 mm to −5.2 mm was analyzed as described previously [18]. For quantitative analysis of Nissl staining or immunostaining in the thalamus, sections between −2.8 and −4.4 mm relative to bregma were randomly selected. The immunoreactive cells were counted within three non-overlapping fields (400 × magnification) in the thalamus of each section. For quantitative analysis of autophagosomes and autolysosomes in vitro experiments, numbers of yellow and red dots per cell were separately calculated under 600 × magnification. For immunoblotting analysis, the ratios of band densities for RTN4, S1PR2, RHOA, ROCK1, BECN1, p-BECN1, MAP1LC3B-II and SQSTM1 relative to TUBA or GAPDH were calculated. All quantitative analyses were implemented using ImageJ software (Bethesda). Data are presented as median ± interquartile ranges. The difference between two groups were analyzed by Mann-Whitney U test and multiple comparisons were analyzed by one-way variance (ANOVA) test using GraphPad Prism (version 8.0). In a post-hoc analysis, partial correlations were analyzed using SPSS (version 25.0), to determine the associations between angiogenesis and neuronal loss in the ipsilateral thalamus or cognitive sores across the Rtn4-siRNA, Scramble-siRNA, RTN4-Δ20-Fc and IgG-Fc groups by controlling for infarct volumes. Statistical significance was set at P < 0.05.
Supplementary Material
Acknowledgments
The study was supported by the National Key R&D Program of China (2017YFC1307500), the National Nature Science Foundation of China (81971118, 82171281), the Natural Science Foundation of Guangdong Province, China (2021A1515010094), the Science and Technology Planning Project of Guangzhou, China (201904010313), the Southern China International Cooperation Base for Early Intervention and Functional Rehabilitation of Neurological Diseases, China (2015B050501003), Guangdong Provincial Engineering Center for Major Neurological Disease Treatment, Guangdong Provincial Translational Medicine Innovation Platform for Diagnosis and Treatment of Major Neurological Disease, and Guangdong Provincial Clinical Research Center for Neurological Diseases, China.
Funding Statement
This work was supported by the National Key R&D Program of China [2017YFC1307500]; the National Natural Science Foundation of China [81971118, 82171281]; the Natural Science Foundation of Guangdong Province, China [2021A1515010094]; the Science and Technology Planning Project of Guangzhou, China [201904010313];; the Southern China International Cooperation Base for Early Intervention and Functional Rehabilitation of Neurological Diseases [2015B050501003]; Guangdong Provincial Engineering Center for Major Neurological Disease Treatment; Guangdong Provincial Translational Medicine Innovation Platform for Diagnosis and Treatment of Major Neurological Disease; Guangdong Provincial Clinical Research Center for Neurological Diseases, China.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Tamura A, Tahira Y, Nagashima H, et al. Thalamic atrophy following cerebral infarction in the territory of the middle cerebral artery. Stroke. 1991;22(5):615–618. [DOI] [PubMed] [Google Scholar]
- [2].Freret T, Chazalviel L, Roussel S, et al. Long-term functional outcome following transient middle cerebral artery occlusion in the rat: correlation between brain damage and behavioral impairment. Behav Neurosci. 2006;120(6):1285–1298. [DOI] [PubMed] [Google Scholar]
- [3].Iizuka H, Sakatani K, Young W.. Neural damage in the rat thalamus after cortical infarcts. Stroke. 1990;21(5):790–794. [DOI] [PubMed] [Google Scholar]
- [4].Zhang J, Zhang Y, Xing S, et al. Secondary neurodegeneration in remote regions after focal cerebral infarction: a new target for stroke management? Stroke. 2012;43(6):1700–1705. [DOI] [PubMed] [Google Scholar]
- [5].Block F, Dihné M, Loos M. Inflammation in areas of remote changes following focal brain lesion. Prog Neurobiol. 2005;75(5):342–365. [DOI] [PubMed] [Google Scholar]
- [6].Wang F, Liang Z, Hou Q, et al. Nogo-A is involved in secondary axonal degeneration of thalamus in hypertensive rats with focal cortical infarction. Neurosci Lett. 2007;417(3):255–260. [DOI] [PubMed] [Google Scholar]
- [7].Xing S, Zhang Y, Li J, et al. Beclin 1 knockdown inhibits autophagic activation and prevents the secondary neurodegenerative damage in the ipsilateral thalamus following focal cerebral infarction. Autophagy. 2012;8(1):63–76. [DOI] [PubMed] [Google Scholar]
- [8].Ling L, Zeng J, Pei Z, et al. Neurogenesis and angiogenesis within the ipsilateral thalamus with secondary damage after focal cortical infarction in hypertensive rats. J Cereb Blood Flow Metab. 2009;29(9):1538–1546. [DOI] [PubMed] [Google Scholar]
- [9].Xing S, Pan N, Xu W, et al. EphrinB2 activation enhances angiogenesis, reduces amyloid-β deposits and secondary damage in thalamus at the early stage after cortical infarction in hypertensive rats. J Cereb Blood Flow Metab. 2019;39(9):1776–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Galluzzi L, Baehrecke EH, Ballabio A, et al. Molecular definitions of autophagy and related processes. EMBO J. 2017;36(13):1811–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Nussenzweig SC, Verma S, Finkel T. The role of autophagy in vascular biology. Circ Res. 2015;116(3):480–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Jiang F. Autophagy in vascular endothelial cells. Clin Exp Pharmacol Physio. 2016;43(11):1021–1028 [DOI] [PubMed] [Google Scholar]
- [13].Tasaki T, Kim ST, Zakrzewska A, et al. UBR box N-recognin-4 (UBR4), an N-recognin of the N-end rule pathway, and its role in yolk sac vascular development and autophagy. Proc Natl Acad Sci U S A. 2013;110(10):3800–3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lee SJ, Kim HP, Jin Y, et al. Beclin 1 deficiency is associated with increased hypoxia-induced angiogenesis. Autophagy. 2011;7(8):829–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kim KW, Paul P, Qiao J, et al. Enhanced autophagy blocks angiogenesis via degradation of gastrin-releasing peptide in neuroblastoma cells. Autophagy. 2013;9(10):1579–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Chen MS, Huber AB, van der Haar ME, et al. Nogo-A is a myelin-associated neurite outgrowth inhibitor and an antigen for monoclonal antibody IN-1. Nature. 2000;403(6768):434–439. [DOI] [PubMed] [Google Scholar]
- [17].Schwab ME. Functions of Nogo proteins and their receptors in the nervous system. Nat Rev Neurosci. 2010;11(12):799–811. [DOI] [PubMed] [Google Scholar]
- [18].Xu W, Xiao P, Fan S, et al. Blockade of Nogo-A/Nogo-66 receptor 1 (NgR1) inhibits autophagic activation and prevents secondary neuronal damage in the thalamus after focal cerebral infarction in hypertensive rats. Neuroscience. 2020;431:103–114. [DOI] [PubMed] [Google Scholar]
- [19].Wälchli T, Pernet V, Weinmann O, et al. Nogo-A is a negative regulator of CNS angiogenesis. Proc Natl Acad Sci U S A. 2013;110(21):E1943–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wälchli T, Ulmann-Schuler A, Hintermüller C, et al. Nogo-A regulates vascular network architecture in the postnatal brain. J Cereb Blood Flow Metab. 2017;37(2):614–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Rust R, Grönnert L, Gantner C, et al. Nogo-A targeted therapy promotes vascular repair and functional recovery following stroke. Proc Natl Acad Sci U S A. 2019;116(28):14270–14279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Funderburk SF, Wang QJ, Yue Z. The Beclin 1-VPS34 complex–at the crossroads of autophagy and beyond. Trends Cell Biol. 2010;20(6):355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ichimura Y, Kominami E, Tanaka K, et al. Selective turnover of p62/A170/SQSTM1 by autophagy. Autophagy. 2008;4(8):1063–1066. [DOI] [PubMed] [Google Scholar]
- [24].Huo Y, Yin XL, Ji SX, et al. Amino-Nogo inhibits optic nerve regeneration and functional recovery via the integrin αv signaling pathway in rats. Cell Physiol Biochem. 2015;35(2):616–626. [DOI] [PubMed] [Google Scholar]
- [25].Klionsky DJ, Elazar Z, Seglen PO, et al. Does bafilomycin A1 block the fusion of autophagosomes with lysosomes? Autophagy. 2008;4(7):849–850. [DOI] [PubMed] [Google Scholar]
- [26].Gurkar AU, Chu K, Raj L, et al. Identification of ROCK1 kinase as a critical regulator of Beclin1-mediated autophagy during metabolic stress. Nat Commun. 2013;4:2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yu T, Guo F, Yu Y, et al. Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell. 2017;170(3):548–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ong LK, Zhao Z, Kluge M, et al. Chronic stress exposure following photothrombotic stroke is associated with increased levels of Amyloid beta accumulation and altered oligomerisation at sites of thalamic secondary neurodegeneration in mice. J Cereb Blood Flow Metab. 2016;37(4):1338–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zeng J, Zhang Y, Mo J, et al. Two-kidney, two clip renovascular hypertensive rats can be used as stroke-prone rats. Stroke. 1998;29(8):1708–1713. [DOI] [PubMed] [Google Scholar]
- [30].Kim GS, Yang L, Zhang G, et al. Critical role of sphingosine-1-phosphate receptor-2 in the disruption of cerebrovascular integrity in experimental stroke. Nat Commun. 2015;6:7893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kempf A, Tews B, Arzt ME, et al. The sphingolipid receptor S1PR2 is a receptor for Nogo-a repressing synaptic plasticity. PLoS Biol. 2014;12(1):e1001763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kang TY, Bocci F, Jolly MK, et al. Pericytes enable effective angiogenesis in the presence of proinflammatory signals. Proc Natl Acad Sci U S A. 2019;116(47):23551–23561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Buschmann I, Schaper W. Arteriogenesis versus angiogenesis: two mechanisms of vessel growth. News Physiol Sci. 1999;14:121–125. [DOI] [PubMed] [Google Scholar]
- [34].Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140(3):313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Du J, Teng RJ, Guan T, et al. Role of autophagy in angiogenesis in aortic endothelial cells. Am J Physiol. Cell Physiol. 2012;302(2):C383–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Sachdev U, Cui X, Hong G, et al. High mobility group box 1 promotes endothelial cell angiogenic behavior in vitro and improves muscle perfusion in vivo in response to ischemic injury. J Vasc Surg. 2012;55(1):180–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gogiraju R, Hubert A, Fahrer J, et al. Endothelial leptin receptor deletion promotes cardiac autophagy and angiogenesis following pressure overload by suppressing Akt/mtor signaling. Circ Heart Fail. 2019;12(1):e005622. [DOI] [PubMed] [Google Scholar]
- [38].Matsunaga K, Saitoh T, Tabata K, et al. Two beclin 1-binding proteins, Atg14l and rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol. 2009;11(4):385–396. [DOI] [PubMed] [Google Scholar]
- [39].Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282(33):24131–24145. [DOI] [PubMed] [Google Scholar]
- [40].Hu YB, Zou Y, Huang Y, et al. ROCK1 is associated with Alzheimer’s disease-specific plaques, as well as enhances autophagosome formation but not autophagic Abeta clearance. Front Cell Neurosci. 2016;10:253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Pattingre S, Tassa A, Qu X, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122(6):927–939. [DOI] [PubMed] [Google Scholar]
- [42].Liang C, Lee JS, Inn KS, et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat Cell Biol. 2008;10(7):776–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Wei Y, Pattingre S, Sinha S, et al. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30(6):678–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Zagrebelsky M, Lonnemann N, Fricke S, et al. Nogo-A regulates spatial learning as well as memory formation and modulates structural plasticity in the adult mouse hippocampus. Neurobiol Learn Mem. 2017;138:154–163. [DOI] [PubMed] [Google Scholar]
- [45].Lee H, Raiker SJ, Venkatesh K, et al. Synaptic function for the Nogo-66 receptor NgR1: regulation of dendritic spine morphology and activity-dependent synaptic strength. J Neurosci. 2008;28(11):2753–2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Raiker SJ, Lee H, Baldwin KT, et al. Oligodendrocyte-myelin glycoprotein and Nogo negatively regulate activity-dependent synaptic plasticity. J Neurosci. 2010;30(37):12432–12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Nih LR, Gojgini S, Carmichael ST, et al. Dual-function injectable angiogenic biomaterial for the repair of brain tissue following stroke. Nat Mater. 2018;17:642–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Brint S, Jacewicz M, Kiessling M, et al. Focal brain ischemia in the rat: methods for reproducible neocortical infarction using tandem occlusion of the distal middle cerebral and ipsilateral common carotid arteries. J Cereb Blood Flow Metab. 1988;8(4):474–485. [DOI] [PubMed] [Google Scholar]
- [49].Liu J, Xia H, Kim M, et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell. 2011;147(1):223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Shehata M, Abdou K, Choko K, et al. Autophagy enhances memory erasure through synaptic destabilization. J Neurosci. 2018;38(15):3809–3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ryan F, Khodagholi F, Dargahi L, et al. Temporal pattern and crosstalk of necroptosis markers with autophagy and apoptosis associated proteins in ischemic hippocampus. Neurotox Res. 2018;34(1):79–92. [DOI] [PubMed] [Google Scholar]
- [52].Vorhees CV, Williams MT. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc. 2006;1(2):848–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].He M, Xing S, Yang B, et al. Ebselen attenuates oxidative DNA damage and enhances its repair activity in the thalamus after focal cortical infarction in hypertensive rats. Brain Res. 2007;1181:83–92. [DOI] [PubMed] [Google Scholar]
- [54].Lee YK, Uchida H, Smith H, et al. The isolation and molecular characterization of cerebral microvessels. Nat Protoc. 2019;14(11):3059–3081. [DOI] [PubMed] [Google Scholar]
- [55].Wachtel M, Bolliger M, Ishihara H, et al. Down-regulation of occludin expression in astrocytes by tumour necrosis factor (TNF) is mediated via TNF type-1 receptor and nuclear factor kappaB activation. J Neurochem. 2001;78(1):155–162. [DOI] [PubMed] [Google Scholar]
- [56].Zhou C, Zhong W, Zhou J, et al. Monitoring autophagic flux by an improved tandem fluorescent-tagged LC3 (mTagRFP-mWasabi-LC3) reveals that high-dose rapamycin impairs autophagic flux in cancer cells. Autophagy. 2012;8(8):1215–1226. [DOI] [PubMed] [Google Scholar]
- [57].Zhang L, Qiang P, Yu J, et al. Identification of compound CA-5f as a novel late-stage autophagy inhibitor with potent anti-tumor effect against non-small cell lung cancer. Autophagy. 2019;15(3):391–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.