

Abstract
Bacterial biofilms are associated with chronic infectious diseases and are highly resistant to conventional antibiotics. Antimicrobial bacteriocins are alternatives to conventional antibiotics and are characterized by unique cell‐killing mechanisms, including pore formation on cell membranes, nuclease activity, and cell wall synthesis inhibition. Here, we used cell‐free protein synthesis to rapidly evaluate the anti‐biofilm activities of colicins E1, E2, and E3. We found that E2 (with DNase activity) most effectively killed target biofilm cells (i.e., the K361 strain) while leaving nontargeted biofilms intact. We then engineered probiotic Escherichia coli microorganisms with genetic circuits to controllably synthesize and secrete colicin E2, which successfully inhibited biofilms and killed preformed indicator biofilms. Our findings suggest that colicins rapidly and selectively kill target biofilm cells in multispecies biofilms and demonstrate the potential of using microorganisms engineered to produce antimicrobial colicin proteins as live therapeutic strategies to treat biofilm‐associated infections.
Keywords: antimicrobial proteins, biofilms, cell‐free protein synthesis, colicins, synthetic genetic circuits
1. INTRODUCTION
Biofilms are microbial communities that form on biotic or abiotic surfaces and facilitate the development of antibiotic resistance. 1 Typically, conventional antibiotics disable bacterial growth by inhibiting replication, cell wall synthesis, or protein synthesis. Bacteria in biofilms are able to inhibit antibiotic activity and resist antibiotics more effectively than planktonic bacteria, 2 by producing extracellular polymeric substances, regulating biofilm‐specific gene expression, and inducing the formation of metabolically dormant persister cells. 3 Therefore, novel antibacterial strategies are required to combat biofilm‐associated infections.
Antimicrobial polypeptides known as bacteriocins are compelling alternatives to antibiotics because they are potent and effective against pathogenic bacteria, and they have low toxicity in the host. Bacteriocins may be produced in situ by probiotics and are easily bioengineered. 4 Colicins are bacteriocins produced by Escherichia coli that are characterized by various cell‐killing actions, including membrane pore formation and intracellular DNase and RNase activity. 5 Unlike conventional antibiotics that inhibit bacterial growth, colicins physically disrupt cellular components. We have demonstrated that colicins produced on a cell‐free protein synthesis (CFPS) platform rapidly kill bacteria in a cell growth‐independent manner. 6 We have also found that colicin production can be optimized to maximize activity. 7 Cells in biofilms grow slowly or become metabolically dormant, thus resulting in higher resistance to conventional antibiotic treatment. 2 However, colicins with growth‐independent cell‐killing activity may eradicate resilient biofilm cells more efficiently. Furthermore, because engineered colicins kill only nonhost E. coli, they may provide a method to specifically target and kill only one type of biofilm cell without affecting the other bacterial populations in multispecies biofilms. Thus, colicins have the potential to specifically kill undesired pathogenic biofilms while leaving nonpathogenic biofilms intact.
Advances in synthetic biology have enabled the development of living biotherapeutics, or cells that perform predefined functions in a controlled manner to treat disease. 8 Protein‐based drugs that are easily degraded by proteases in the host 9 can be incorporated into microorganisms, which then serve as factories that produce and secrete drugs at or near the sites of action. 10 Indeed, engineered bacterial therapeutics have already been developed to treat various diseases. For example, engineered Lactococcus lactis secreting human proinsulin and interleukin‐10 has been used to treat diabetes mellitus in mice 11 ; another L. lactis producing interleukin‐10 has been used to treat inflammatory bowel disease in a Phase I trial 12 ; Lactobacillus jensenii producing HIV‐1 entry inhibitor cyanovirin‐N has been used to treat HIV infection in macaques 13 ; and Salmonella typhimurium secreting flagellin B has been used to treat colon cancer in mice. 14 The engineered probiotic E. coli Nissle 1917 secreting pyocin S5 and anti‐biofilm enzyme DspB has been found to eliminate and prevent Pseudomonas aeruginosa gut infection in an animal model. 15 These findings demonstrate that live biotherapeutics based on engineered microorganisms are promising approaches to overcome the limitations of drug degradation. Live biotherapeutics facilitate drug delivery to the relevant sites of action and have the potential to specifically eradicate unwanted biofilm cells.
In this study, we hypothesized that colicin secretion from engineered microorganisms might overcome the mass‐transport limitations preventing colicins from reaching target biofilm cells, thus enhancing biofilm cell killing. We first used CFPS to rapidly characterize the effectiveness, kinetics, and specificity of colicins E1, E2, and E3. We found that colicin E2 provided the strongest and longest‐lasting cell killing and was able to specifically target a subset of bacteria within a multispecies consortium biofilm. On this basis, we developed engineered microorganisms with production and secretion of colicin E2 precisely controlled through genetic circuits, thus achieving inhibition of target biofilm growth and complete killing of preexisting biofilms. Our findings reveal new insights into antimicrobial‐producing microorganisms and provide exciting opportunities to develop live therapeutics to treat biofilm‐associated infections.
2. MATERIALS AND METHODS
2.1. Bacterial strains and plasmids
The bacterial strains and plasmids used in this study are listed in Table 1. The E. coli K361 strain 16 was used to determine the activity of colicins, the E. coli BL21 Star (DE3) strain was used for making crude extracts for CFPS, and E. coli Nissle 1917 ∆degP was used to produce engineered cells containing genetic circuits. Kanamycin (50 μg/ml), ampicillin (100 μg/ml), and streptomycin (100 μg/ml) were used to maintain plasmids as needed.
TABLE 1.
Strains and plasmids used in this study. AmpR, KanR, and StrR indicate ampicillin‐, kanamycin‐, and streptomycin‐resistant, respectively
| Strains and plasmids | Genotype/relevant characteristics | Source |
|---|---|---|
| Strains | ||
| E. coli K361 | W3110 strain, StrR | 16 |
| E. coli BL21Star (DE3) | F− ompT, hsdSB (rB −mB −), gal, dcm, rne131 (DE3) | Invitrogen |
| E. coli K964 | K91 containing pColE2 | 17 |
| E. coli TG1 | Strain containing colicin plasmid | 18 |
| E. coli DH5α | fhuA2, Δ(argF‐lacZ)U169, phoA, glnV44, Φ80 Δ(lacZ)M15, gyrA96, recA1, relA1, endA1, thi‐1, hsdR17 | NEB |
| E. coli Nissle 1917 ∆degP | Probiotic E. coli (EcN) lacking degP, KanR | 19 |
| Lactococcus lactis 11454 | Probiotic Lactococcus lactis subsp. Lactis ATCC strain (ATCC® 11454™) | ATCC |
| VSL#3 | Probiotic mixture | VSL3 Pharma |
| Plasmids | ||
| pKSJ331 | AmpR, ColE1 operon | 18 |
| pKSJ167 | AmpR, ColE3 operon | 20 |
| pJL1‐sfgfp | KmR, P T7 ::sfGFP, C‐terminal Strep‐tag, pY71‐sfGFP | 21 |
| pJL1‐E1 | KmR, P T7 ::colicin E1 and E1 immunity | This study |
| pJL1‐E2 | KmR, P T7 ::colicin E2 and E2 immunity | This study |
| pJL1‐E3 | KmR, P T7 ::colicin E3 and E3 immunity | This study |
| pBAD‐gfp | AmpR, P araBAD ::GFP | Addgene (#54764) |
| pBAD‐empty | AmpR | This study |
| pBAD‐E2 | AmpR, P araBAD ::colicin E2 and E2 immunity | This study |
| pBAD‐E2+lys | AmpR, P araBAD ::colicin E2 and E2 immunity, P tet ::E7 lysis, P proB ::tetR | This study |
2.2. Crude extract preparation
E. coli cell extracts were prepared from the BL21 Star (DE3) strain with a high‐throughput sonication method, as described previously. 6 Briefly, overnight cultures grown in Luria‐Bertani (LB) medium at 37°C 220 rpm were diluted 1000 times with 1 L of 2× YTPG medium (16 g/L tryptone, 10 g/L yeast extract, 5 g/L NaCl, 7 g/L K2HPO4, 3 g/L KH2PO4, and 18 g/L glucose; adjusted to pH 7.2 with KOH) and incubated at 37°C and 220 rpm until an optical density at 600 nm (OD600 nm) of 0.5 was reached. T7 RNA polymerase was then induced by the addition of 1 mM of isopropyl β‐D‐1‐thiogalactopyranoside during incubation. Cell cultures were harvested after an OD600 nm of 3.0 was reached. Cell pellets were suspended in 1 ml of S30 buffer per gram of cells and lysed on ice with a Q125 sonicator (Qsonica). Cell debris and insoluble components were removed by two rounds of centrifugation for 10 min at 14,000 × g at 4°C, and supernatants were filtered through a 0.2‐μm sterile syringe filter (Corning). The crude extracts were stored at −80°C until use.
2.3. CFPS reaction
CFPS reactions were used to produce colicins as described previously. 6 Briefly, 15 μl of CFPS reactions in 1.5‐ml microcentrifuge tubes were prepared by mixing the following components: 130 mM potassium glutamate; 10 mM ammonium glutamate; 12 mM magnesium glutamate; 33 mM phosphoenolpyruvate; 1.2 mM ATP; 0.85 mM each of GTP, UTP, and CTP; 34.0 μg/ml folinic acid; 200 ng DNA template; 2 mM each of 20 standard amino acids; 0.33 mM nicotinamide adenine dinucleotide; 0.27 mM coenzyme‐A; 1.5 mM spermidine; 1 mM putrescine; 4 mM sodium oxalate; and 4 μl of crude extract. The samples were incubated for 20 h at 30°C.
2.4. Autoradiogram
Radioactive 14C‐Leu was used in the CFPS reaction at a final concentration of 10 μM. Total and soluble fractions of each reaction were heat‐denatured and reduced by dithiothreitol and electrophoresed on 4%–12% NuPAGE sodium dodecyl sulfate‐polyacrylamide gels in MOPS buffer (Invitrogen). The gels were stained with InstantBlue Coomassie stain (Expedeon) for 1 h, destained overnight in dH2O, soaked in Gel Drying Solution (Bio‐Rad) for 30 min, fixed with cellophane films, dried overnight in a GelAir Dryer (Bio‐Rad), and exposed for 72 h on Storage Phosphor Screens (GE Healthcare Biosciences). The autoradiograms were scanned with a Typhoon FLA7000 Imager (GE Healthcare Biosciences). To determine the proportion of colicins to immunity proteins, we followed Davarinejad's protocol in the ImageJ software. 22 Briefly, the band intensity determined by ImageJ was used to calculate the mass fraction. The molar fraction was calculated as the mass fraction and molar mass. The average molecular weight and average number of leucines were calculated based on the molar fraction of each protein (Table S1). These average values were used separately to quantify colicins and immunity proteins in each radioactive sample measured with 14C‐leucine radioactive scintillation counting (Table 2).
TABLE 2.
Total and soluble colicin concentration. Total and soluble yields of cell‐free synthesized colicins, quantified with radioactive 14C‐Leu scintillation counting
| Colicin | Total protein (nM) | Soluble protein (nM) |
|---|---|---|
| E1 | 1550 ± 30 | 1420 ± 70 |
| E2 | 4200 ± 70 | 3900 ± 300 |
| E3 | 1090 ± 90 | 810 ± 70 |
2.5. Radioactive 14C‐Leu assays
Total and soluble protein yields were measured on the basis of radioactive 14C‐Leu incorporation. 23 Briefly, triplicate CFPS reactions were supplemented with 10 μM 14C‐leucine (PerkinElmer) and incubated at 30°C for 20 h. Soluble and insoluble proteins were separated by centrifugation at 12,000 × g for 15 min at 4°C. Proteins were precipitated and washed three times with 5% trichloroacetic acid, then washed in 100% ethanol. The radioactivity of the proteins was counted with a Microbeta2 liquid scintillation counter (PerkinElmer). To quantify the total and soluble protein concentrations in a sample, we used the average molecular weights and the average number of leucines calculated from the autoradiogram gels according to a previously reported method. 23 After calculating the total and soluble mass concentration of proteins, we calculated the concentrations of colicins separated from immunity proteins by multiplying the mass fraction of colicins (Table S1) by the mass concentrations of proteins and then converting to molar concentrations (Table 2).
2.6. Biofilm cell killing assays with cell‐free synthesized colicins
Biofilm formation assays were conducted according to a previously described method 19 with modifications. Briefly, overnight cultures were adjusted to an OD600 nm of 0.01 (approximately 5 × 106 CFU/ml) with LB medium, and then 1.5 ml of adjusted culture was added to polypropylene culture tubes (Falcon PN352006, Corning). For dual‐ and multispecies biofilm formation, equal numbers of cells (approximately 5 × 106 CFU/ml) from two or more bacterial strains grown overnight were inoculated into 1.5 ml of LB medium in polypropylene culture tubes (Falcon PN352006, Corning). After incubation at 37°C for 24 h without shaking, a cell‐free product (no plasmid control or colicins) was added to the culture tubes and incubated at 37°C for various time intervals. Then, the planktonic cultures were discarded and the culture tubes containing biofilm were rinsed three times with 3 ml of 0.85% NaCl solution. Biofilm cells were carefully collected with cotton swabs 24 and resuspended in 3 ml of 0.85% NaCl solution by vortexing for 30 s. Biofilm cells were serially diluted (101–106 dilution), and 10 μl was applied on LB agar plates and incubated at 37°C.
2.7. Plasmid construction
All primers used for cloning are listed in Table S2. Plasmids containing genes encoding colicins E1, E2, or E3 with corresponding immunity protein genes were constructed with pJL1‐plasmid as the backbone. E1 and its immunity genes were amplified from pKSJ331 plasmids with E1‐F. E1imm‐R primers, E2, and its immunity genes were amplified from the K964 strain with E2‐F and E2imm‐R primers. E3 and its immunity genes were amplified from the pKSJ167 plasmid with E3‐F and E3imm‐R primers. The amplified fragments were inserted into a pJL1‐backbone via the NdeI and SalI restriction sites. The resulting plasmids were pJL1‐E1, pJL1‐E2, and pJL1‐E3. The plasmid pBAD‐E2 (circuit A in Figure 3A) contains col and imm under the control of the L‐arabinose inducible araBAD promoter. To construct this plasmid (circuit A), we amplified col and imm from pJL1‐E2 with NdeI‐F and HindIII‐R primers. The PCR fragment was cloned into the pBAD vector via the NdeI and HindIII restriction sites by replacing gfp in the pBAD‐gfp plasmid. The plasmid pBAD‐E2+lys (circuit B in Figure 3A) contains col and imm under the control of an L‐arabinose‐inducible araBAD promoter, tetR under the control of a constitutive proB promoter, and lys under the control of an anhydrotetracycline‐inducible tet promoter. This plasmid was constructed with a gBlock DNA fragment (Table S2) containing tetR and lys with corresponding RBS and promoters, synthesized by Integrated DNA Technology, and amplified with lys‐F and tetR‐R primers. The PCR fragment was cloned into pBAD‐E2 via the SalI and HindIII restriction sites after imm. The plasmid pBAD‐empty was constructed by removal of the gfp sequence from the plasmid pBAD‐gfp via inverse PCR with pBAD‐empty‐F and pBAD‐empty‐R primers. Both ends of the PCR fragment were digested with the HindIII restriction enzyme and ligated to form pBAD‐empty.
FIGURE 3.
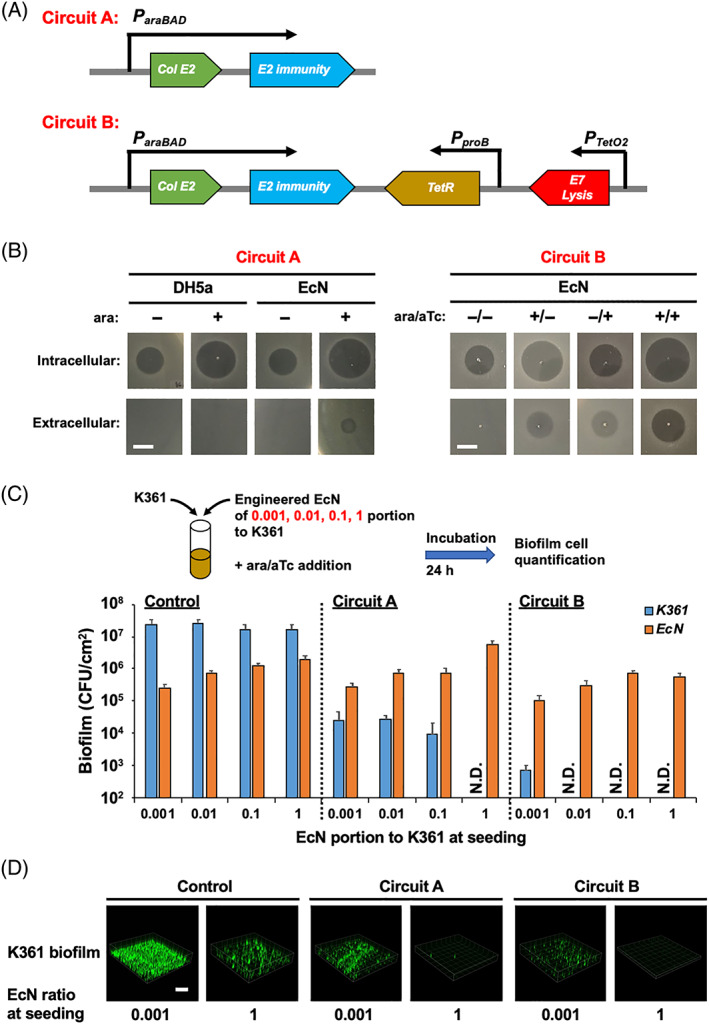
Design and validation of colicin‐producing genetic circuits. (A) Genetic circuit for colicin E2 and its immunity gene expression controlled by L‐arabinose (circuit A). Improved genetic circuit with the addition of E7 lysis gene expression induced by anhydrotetracycline; tetR gene was added to circuit A (or circuit B). (B) For circuit A, colicin E2 was produced with 0.1% L‐arabinose (ara) induction and secretion. For circuit B, colicin E2 was produced with 0.1% ara, and lysis protein was produced with 500 nM of anhydrotetracycline (aTc). K361 inhibition zones were formed at 37°C for 24 h with cell‐free supernatants containing colicin E2, with sonication (intracellular) and without sonication (extracellular). The scale bar indicates 0.5 cm. (C) Dual‐species biofilm formation of engineered EcN with circuit A (pBAD‐E2), circuit B (pBAD‐E2+lys), or control (pBAD‐gfp) with K361 indicator cells in various proportions (0.001, 0.01, 0.1, and 1 ratios of EcN to the K361 population) at seeding. Biofilms were formed at 37°C for 8 h and incubated for an additional 16 h with 0.1% ara or 500 nM of aTc. Viable biofilm cells are quantified in colony‐forming units per area (CFU/cm2). Error bars indicate the standard deviation from two independent cultures with three replicates. (D) K361 biofilm images under the same conditions as in (C). The scale bar indicates 200 μm
2.8. Arabinose optimization
Overnight cultures of EcN/pBAD‐gfp and DH5α/pBAD‐gfp were reinoculated in LB medium and grown to early exponential phase (OD600 nm ~ 0.3). Various L‐arabinose concentrations (0.02%, 0.1%, or 0.5%) were added for 4 h at 37°C and 220 rpm to induce green fluorescent protein (GFP) fluorescence. GFP production was quantified by fluorescence (in arbitrary units: a.u.) measured with a Synergy HTX plate reader with excitation at 485 nm and emission at 528 nm, and a cutoff at 510 nm, in 96‐well half‐area black plates (Costar 3694; Corning Incorporated).
2.9. Cell growth measurement
Overnight cultures of EcN/pBAD‐E2+lys were adjusted to OD600 nm 0.3 with fresh LB medium supplemented with chloramphenicol, and then 200 μl of cell suspension was added to 96‐well plates. After the addition of various concentrations of anhydrotetracycline (4, 20, 100, or 500 nM), cell growth was measured at OD600 nm every 20 min at 37°C on a Synergy HTX plate reader (Biotek) in fast shaking mode. Each data point was obtained from three replicate wells with two independent cultures.
2.10. Inhibition zone assays
Inhibition zones formed by colicin activity were measured with a previously reported method with modifications. 25 Briefly, 5 × 106 CFU/ml of K361 cells in 10 ml of precooled (44°C) 1% LB agar solution was poured into 100‐mm diameter Petri dishes. After the plates fully solidified, cell‐free produced colicins or cell‐free supernatants containing colicins (5 μl each) were deposited on the plate and incubated at 37°C for 24 h.
2.11. Biofilm cell killing assays with colicin‐producing EcN
For biofilm formation with genetic circuits, various concentrations of EcN (5 × 103, 5 × 104, 5 × 105, and 5 × 106 CFU/ml) were mixed with K361 (5 × 106 CFU/ml) (i.e., 0.001, 0.01, 0.1, and 1 ratios of engineered EcN to K361 cells) into 1.5 ml of LB medium supplemented with antibiotics in polypropylene culture tubes (Falcon PN352006, Corning). After incubation at 37°C for 8 h without shaking, 0.1% L‐arabinose or 500 nM of anhydrotetracycline was added to the culture to induce colicin production, and the cultures were incubated at 37°C for an additional 16 h. The planktonic cultures were then discarded, and the culture tubes containing biofilms were rinsed three times with 3 ml of 0.85% NaCl solution. Biofilm cells were carefully collected with a cotton swab 24 and resuspended in 3 ml of 0.85% NaCl solution via vortexing for 30 s. Cells were diluted serially (101–106 dilution), and 10 μl was applied on LB agar plates with different antibiotic markers, then incubated at 37°C.
2.12. Preformed biofilm cell killing assays with colicin‐producing EcN
Preformed K361 biofilms were killed through three different approaches. First, overnight cultures of K361/pBAD‐gfp were adjusted to an OD600 nm of 0.01 (approximately 5 × 106 CFU/ml) with LB medium supplemented with chloramphenicol, and then 1.5 ml of adjusted culture was added to polypropylene culture tubes (Falcon PN352006, Corning) and incubated at 37°C for 24 h without shaking. The planktonic cultures were then discarded, and the culture tubes containing biofilms were rinsed three times with 3 ml of 0.85% NaCl solution. Then 5 × 107 CFU/ml of exponential phase EcN/pBAD‐E2+lys (circuit B) or EcN/pBAD‐gfp (control) was added into preformed K361 biofilm culture tubes with 0.1% L‐arabinose and 500 nM anhydrotetracycline to induce colicin production and cell lysis, respectively. The cells were incubated for 1, 4, 8, or 24 h. K361 biofilms were formed after incubation for 24, 48, or 72 h. Exponential phase EcN/pBAD‐E2+lys (circuit B) or EcN/pBAD‐gfp (control) (5 × 107 CFU/ml) was added into preformed K361 biofilm culture tubes with 0.1% L‐arabinose and 500 nM anhydrotetracycline, and incubated for 4 h. Finally, engineered EcN with circuit B (5 × 105, 5 × 106, or 5 × 107 CFU/ml) was added to the 24 h‐preformed K361 biofilms and incubated for 2 h with or without 0.1% L‐arabinose to induce colicin production and to allow engineered EcN to attach to the surface. The cultures were then incubated an additional 2 h with or without 500 nM of anhydrotetracycline to induce colicin secretion via cell lysis. Additionally, planktonic cells after detachment for 4 h were quantified to determine the number of viable cells.
2.13. Confocal microscopy analysis of biofilm inhibition and detachment
To observe biofilm inhibition, we cultured K361/pBAD‐gfp and EcN engineered cells with pBAD‐empty (control), pBAD‐E2 (circuit A), and pBAD‐E2‐lys (circuit B) in LB medium with 0.1% ampicillin to retain the pBAD‐based plasmids. Overnight K361/pBAD‐gfp cultures were diluted to an OD at 600 nm of 0.01 and mixed with the EcN engineered cells in different ratios (0.001 and 1) in LB medium. Subsequently, 200 μl of the mixture was transferred to each well of a 96 well plate (Costar 3370, Corning) and incubated for 8 h at 37°C. Then, 0.1% L‐arabinose and 500 nM of anhydrotetracycline were added, and the cells were incubated for an additional 16 h. To observe biofilm detachment, we diluted overnight K361 cultures to an OD at 600 nm of 0.01 with LB medium, transferred them to a 96‐well plate, and incubated them for 24 h at 37°C. After removal of planktonic cells, EcN engineered cells in LB medium with 0.1% L‐arabinose and 500 nM of anhydrotetracycline were introduced to the K361 biofilms in the 96‐well plate, and then incubated an additional 4 h at 37°C. Planktonic cultures were removed by careful rinsing of the biofilms three times with 0.85% NaCl solution. GFP images were collected with a Nikon A1 confocal microscope (Nikon, Japan), and 3D biofilm images were constructed in NIS‐Element AR software (Nikon, Japan). Six different biofilm locations were randomly examined, and one representative image per each condition was chosen.
3. RESULTS AND DISCUSSION
3.1. Cell‐free produced colicin E1, E2, and E3 kill biofilm cells
Our previous study has shown that cell‐free‐produced colicin E1 and E2 effectively kill metabolically dormant persister cells. 6 Because persister cells are enriched in biofilms 26 and colicins kill persister cells, 6 we reasoned that colicins with growth‐independent killing activity might be used to eradicate biofilm cells. We first produced colicins E1, E2, and E3 through CFPS with E. coli BL21 Star (DE3) extract. Colicins and immunity proteins were produced together to maintain activity during the production stage, because we have found that E3 immunity is required to maintain colicin E3 activity. 7 Colicins were successfully produced, on the basis of the observation of their expected protein sizes (E1 at 57.2 kDa, E2 at 61.6 kDa, and E3 58.0 at kDa) on 14C‐Leu autoradiogram gels (Figure S1). The concentrations of soluble E1, E2, and E3 in completed CFPS reactions were determined by radioactive 14C‐Leu scintillation counting to be 1420, 3900, and 810 nM (Table 2), respectively.
Next, we exposed biofilms formed by a colicin‐sensitive E. coli K361 strain to cell‐free synthesized colicins. Specifically, a K361 biofilm was formed at 37°C for 24 h and was then exposed to 3.5 nM of E1, E2, and E3 colicins for 8 h (Figure 1). We monitored biofilm cell survival after colicin treatment at the 5 min, 1, 4, and 8 h time points. In a control experiment, we observed that biofilm levels in the absence of colicin treatment remained stable over time (approximately 5 × 107 CFU/cm2). Colicins E1, E2, and E3 killed biofilm cells, thus resulting in a 3‐, 2‐, and 1‐log reduction, respectively, at 5 min. The biofilms treated with E1 and E3 colicins maintained approximately similar levels of surviving cells between the 5 min and >8‐h time points. However, colicin E2 continued to kill the biofilm cells until 4 h, eventually resulting in a nearly 5‐log reduction in surviving cells (Figure 1). Immediate biofilm cell‐killing was expected, on the basis of our previously reported observation of an approximately 6‐log reduction of planktonic cells after just 3 min of colicin treatment. 6 These findings suggested that rapid and substantial biofilm cell killing was achieved by colicin treatment, especially with colicin E2. These results are quite promising, given the robust nature of the biofilm matrix.
FIGURE 1.
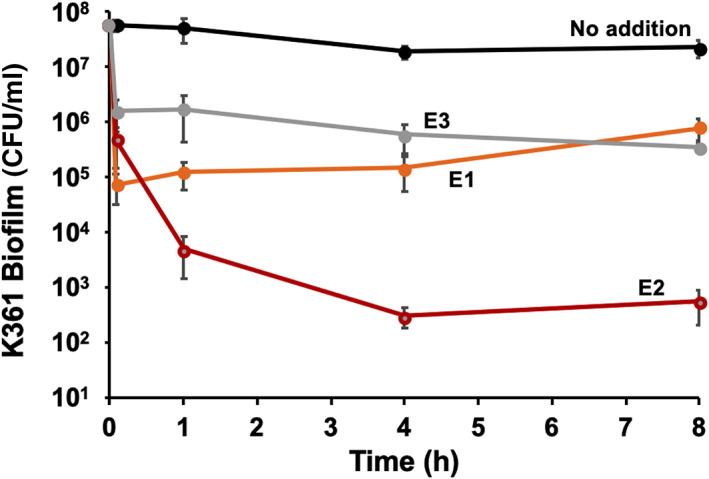
Biofilm cell killing with cell‐free produced colicins. Time‐course of killing of Escherichia coli K361 biofilm cells with the addition of cell‐free produced colicins E1, E2, and E3. Overnight K361 cultures were adjusted to an OD600 nm of 0.01 (approximately 5 × 106 CFU/ml) in LB medium, incubated at 37°C for 24 h to form biofilms, and exposed to each cell‐free produced colicin (3.5 nM) for 5 min, 1 h, 4 h, and 8 h at 37°C. Viable biofilm cells are quantified in colony‐forming units per area (CFU/cm2). Error bars indicate the standard deviation from two independent cultures with three replicates
3.2. Colicins kill target E. coli biofilm cells without affecting other bacterial populations in multispecies biofilms
Whereas most biofilm studies have focused on single‐species biofilms, various microorganisms exist as multispecies consortia in nature. 27 Some microorganisms are beneficial or commensal to the human host, whereas others are deleterious and cause diseases. Colicins recognize specific receptors on target cells during killing 5 and thus have the potential to be used as target‐specific antimicrobial agents. Hence, we investigated dual‐species biofilm formation with cell‐free produced colicins to measure target‐specific cell‐killing by colicins in the presence of other bacterial species in biofilms.
E. coli K361 and the probiotic strain L. lactis ATCC 11454 were cocultured to form biofilms in LB medium at 37°C for 24 h. Then, cell‐free produced colicins E1, E2, and E3 at a final concentration of 3.5 nM were added into the culture tubes, where biofilms were formed and incubated for an additional 1 h. The biofilm levels of L. lactis were not affected by maintaining approximately 5 × 105 CFU/cm2 cell survival, regardless of colicin treatment (Figure 2A). However, all colicins effectively killed E. coli K361 biofilm cells, with a 5‐log reduction in the presence of E2 colicin compared with the untreated control (Figure 2A). Next, we extended our investigation by examining multispecies biofilms including more bacterial species. We cocultured E. coli K361 with a commercially available probiotic mixture (VSL#3) containing eight probiotic species to form biofilms in LB medium at 37°C for 24 h. We then incubated the cells for an additional 1 h with cell‐free colicin. The level of probiotic mixture biofilms was not affected by colicin treatment. However, the K361 biofilm cells were killed (Figure 2B). These results suggested that colicins selectively kill target cells in multispecies biofilms without affecting other bacterial populations.
FIGURE 2.
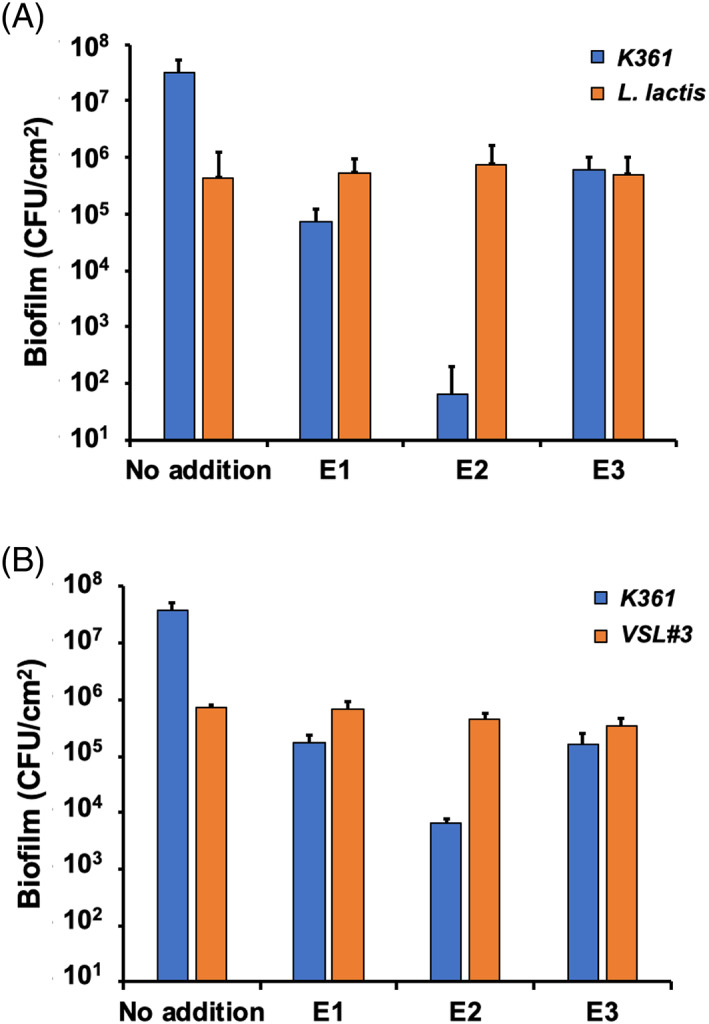
Dual‐ and multispecies biofilms of Escherichia coli K361 and probiotics with cell‐free produced colicins. (A) Lactococcus lactis (5 × 106 CFU/ml) and (B) probiotic mixture VSL#3 (5 × 106 CFU/ml) were seeded together with the K361 strain (5 × 106 CFU/ml) to form dual‐species biofilms in polypropylene culture tubes in LB medium at 37°C for 24 h. The biofilms were treated with cell‐free produced colicins (3.5 nM) for 1 h. Viable biofilm cells are quantified in colony‐forming units per area (CFU/cm2). Error bars indicate the standard deviation from two independent cultures with three replicates
3.3. Engineered E. coli with colicin‐producing capability kills target biofilm cells during dual‐species biofilm formation
Whereas colicins kill persisters 6 and biofilm cells (Figure 1A) in a rapid and target‐specific manner, external colicin delivery to biofilms might still limit the ability to kill biofilm cells. We hypothesized that using microorganisms with colicin‐producing capability would address such challenges because these engineered cells can integrate into multispecies biofilms and produce colicins within the biofilm to effectively kill target species. To this end, we constructed a colicin‐producing genetic circuit for the controlled production of E2 colicin after induction by L‐arabinose (circuit A, Figure 3A). The immunity protein was required to protect the host strain while producing the colicin. We then integrated circuit A into a laboratory strain (E. coli DH5α) and a probiotic strain (EcN) that has been used to treat inflammatory bowel diseases, 28 cancer detection, 29 and gut mucosal healing. 30 We determined that 0.1% L‐arabinose was optimal for inducing protein production with a GFP reporter in both strains (Figure S2A). On the basis of the inhibition zones formed by cell lysates, we determined that active E2 colicin production increased in both DH5α and EcN cells after induction (intracellular, Figure 3B). However, studies with intact cells showed that although colicin E2 remained in the DH5α host, some of the produced colicin was released from the EcN host strain, thus forming a small inhibition zone (extracellular, Figure 3B). The release of colicin from EcN without cell lysis implied that EcN can secrete proteins outside cells. This hypothesis is additionally supported by our previous finding that EcN can secrete the DegP protein, thereby inhibiting pathogenic E. coli biofilm formation. 19 Therefore, we chose EcN as a host for colicin production and secretion.
We performed a dual‐species biofilm experiment with engineered EcN producing colicin E2 (EcN/circuit A) and a K361 indicator strain with different ratios of initial seeding populations: 0.001, 0.01, 0.1, and 1 ratios of engineered EcN to K361 (Figure 3C). The K361 populations in biofilms containing EcN cells engineered to produce colicin E2 were more than 1000‐fold lower than those in the control biofilms with EcN cells that did not produce E2. Increasing the initial concentration of EcN/circuit A cells further decreased the K361 biofilm levels, so that no viable K361 cells were detected in biofilms when equal numbers of EcN/circuit A and K361 cells were introduced (Figure 3C). These results suggest that the generation of active colicins from engineered microorganisms in biofilms may completely kill target biofilm cells, which is difficult to achieve using the external colicin addition approach.
However, the killing of K361 biofilm cells was not efficient when the EcN/circuit A population was low (Figure 3C). This finding might be explained by the limited protein secretion capability of EcN without controlled lysis (Figure 3B). To achieve better control of colicin secretion, we added cell lysis machinery to EcN by integrating the E7 lysis gene 31 into the colicin‐producing genetic circuit (circuit B, Figure 3A). The new circuit was expected to lyse the host cells after anhydrotetracycline induction, thus enhancing colicin secretion. We determined that 500 nM of anhydrotetracycline was required to lyse the host cells in a growth experiment with various concentrations of anhydrotetracycline (Figure S2B). The expression of the lysis gene increased colicin secretion, and the enhanced secretion of colicins from engineered EcN with circuit B was confirmed by the appearance of larger inhibition zones with the extracellular supernatants obtained from the EcN/circuit B culture with the addition of L‐arabinose and anhydrotetracycline (Figure 3B). The controlled lysis and colicin production in EcN/circuit B cells resulted in a dramatic inhibition of K361 in biofilms containing both cell types (Figure 3C). With initial seeding at a 0.001 ratio of EcN/circuit B cells to K361 cells, the K361 levels were five orders of magnitude lower than the no‐colicin EcN control. When EcN/circuit B cells were seeded in ratios of 0.01, 0.1, or 1 with respect to the initial K361 population, we did not observe any viable K361 biofilm cells. The inhibition of K361 biofilms by the engineered EcN cells was confirmed by confocal microscopy (Figure 3D). These findings suggest that controlled release of colicin outside cells is essential to inhibit target biofilm formation.
Together, these genetic circuit results suggest that the production of colicin for killing target cells, immunity protein for protecting the host, and lysis protein for colicin secretion can be synchronized to optimize the activity of engineered microorganisms to yield maximum target biofilm cell killing.
3.4. Engineered EcN secreting colicin E2 kills preformed biofilms
Although killing biofilm cells during formation is helpful for prevention, the eradication of preformed biofilms is important from a therapeutic standpoint. To investigate whether engineered EcN producing colicin E2 might kill preformed biofilm cells, we applied EcN/circuit B cells to preformed K361 biofilms. When we exposed the K361 biofilms to EcN/circuit B for 1 h, K361 biofilm cells decreased by only 1‐log, as compared with the control that did not produce colicin (Figure 4A). This result suggests that more time is needed to induce colicin production and secretion to enhance biofilm cell‐killing. When we increased the treatment time to 4, 8, or 24 h, we observed complete killing of K361 preformed biofilm cells (Figure 4A), thus indicating that existing biofilm cells can be deactivated by the addition and induction of engineered microorganisms that can also form biofilms and secrete antimicrobial proteins. Next, we investigated whether engineered EcN might kill aged K361 biofilms. We formed K361 biofilms for 24, 48, or 72 h and exposed them to engineered EcN/circuit B for an additional 4 h to induce the release of colicin E2 (Figure 4B). Regardless of biofilm age, all K361 biofilm cells were killed by the colicin‐producing engineered EcN. We also observed that the preformed biofilm cells were decreased after introduction of the EcN/circuit B cells that produced and secreted colicin E2 (Figure 4C), thus indicating that the cell killing by colicin E2 induces detachment of the target biofilm cells. Biofilms contain elevated levels of persister cells that are not susceptible to conventional antibiotics. Our results in Figure 4B corroborated those of our previous report where colicins killed target cells and nongrowing persister cells in a growth‐independent manner. 6
FIGURE 4.
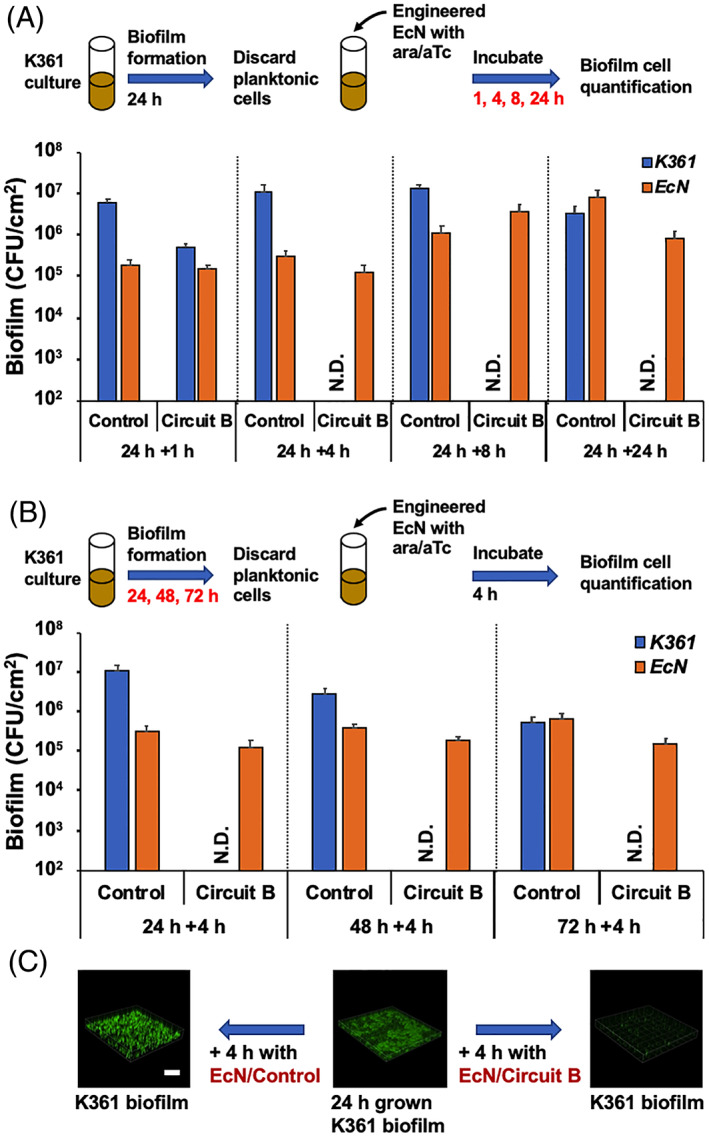
Preformed biofilm cell killing by engineered EcN. (A) K361 biofilms were formed in polypropylene culture tubes in LB medium supplemented with chloramphenicol at 37°C for 24 h. Engineered EcN containing circuit B (5 × 107 CFU/ml) harvested in exponential phase was added to the K361 biofilms with 0.1% L‐arabinose (ara) and 500 nM anhydrotetracycline (aTc) for 1, 4, 8, or 24 h. EcN containing pBAD‐gfp was used as a control. (B) K361 biofilms were formed in polypropylene culture tubes in LB medium supplemented with chloramphenicol at 37°C for 24, 48, or 72 h. Engineered EcN containing circuit B (5 × 107 CFU/ml) harvested in exponential phase was added into the K361 biofilms with 0.1% ara and 500 nM aTc for 4 h. Viable biofilm cells are quantified in colony‐forming units per area (CFU/cm2). Error bars indicate the standard deviation from two independent cultures with three replicates. (C) K361 biofilm images after introduction of the engineered EcN for 4 h. The scale bar indicates 200 μm
Finally, we investigated whether the destruction of preformed K361 biofilms might be precisely controlled by separate induction of the production and release of colicin E2 with various seeding populations of EcN/circuit B (Figure 5). Without the induction of colicin production and secretion, K361 biofilm cells were scarcely killed in all three EcN/circuit B seeding populations. Colicin production through the addition of L‐arabinose increased K361 biofilm cell killing by four orders of magnitude with a low seeding population (5 × 105 CFU/ml) of engineered EcN, thus resulting in complete K361 biofilm cell killing by increasing the engineered EcN seeding populations. With both production and lysis‐assisted release of colicin E2 (by the addition of L‐arabinose and anhydrotetracycline), no viable K361 biofilm cells were detected with all three conditions of EcN seeding populations (Figure 5). In addition, the detached K361 cells from the biofilms were completely killed by the induction of colicin E2 production and secretion from the EcN/circuit B cells (Figure S3). These results suggest that precise control of engineered EcN is possible by optimizing the production and secretion of antimicrobial colicin in the host.
FIGURE 5.
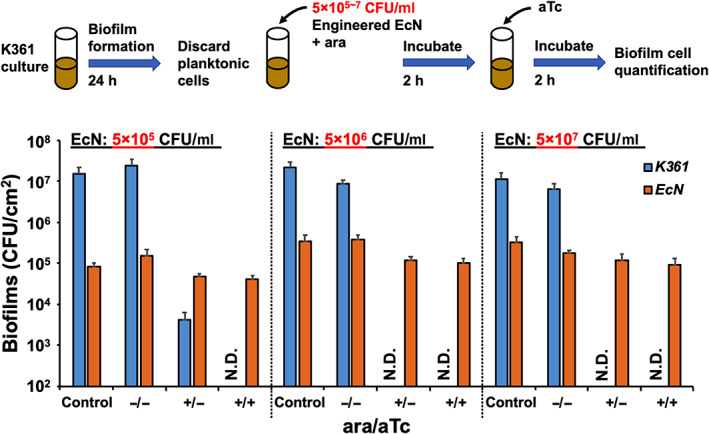
Preformed biofilm cell killing by various concentrations of engineered EcN. K361 biofilms were formed in polypropylene culture tubes in LB medium supplemented with antibiotics at 37°C for 24 h. Various concentrations of engineered EcN with circuit B (5 × 105, 5 × 106, and 5 × 107 CFU/ml) were added into the K361 biofilms and incubated with or without 0.1% L‐arabinose (ara) to induce colicin production at 37°C for 2 h. The cultures were then incubated an additional 2 h with or without 500 nM of anhydrotetracycline (aTc) to induce colicin secretion. Viable biofilm cells are quantified in colony‐forming units per area (CFU/cm2). Error bars indicate the standard deviation from two independent cultures with three replicates
Eradicating bacterial biofilms is challenging because biofilm cells often resist conventional antibiotics by becoming dormant or blocking the mass transfer of externally introduced compounds into the biofilm. In the present study, we used CFPS to demonstrate that colicins can be used as antibiofilm agents to rapidly kill cells in biofilms (Figure 1) containing metabolically dormant persister cells. 3 We also found that colicins selectively killed target biofilm cells without affecting other biofilm populations (Figure 2). Utilizing the target‐specificity of antimicrobial proteins as a novel therapeutic agent is promising because it allows commensal or beneficial microbial communities to remain intact while deleterious populations are selectively eradicated.
To kill target cells, colicins must recognize them by binding and parasitizing a small group of outer membrane proteins (e.g., BtuB, Cir, FepA, and FhuA) that are normally used as nutrient transporters. 32 E colicins, including E2, use the vitamin B12 transporter BtuB as their high‐affinity primary receptor, are translocated from the outer membrane to the periplasm and inner membrane via TolA, B, Q, R translocation machinery, and subsequently kill the cells, depending on the activity of the cytotoxicity domain. For example, E2 kills cells through DNase activity in the cytoplasm. 33 E. coli strains lacking BtuB are killed much less efficiently by E colicins, 32 thus indicating that receptor binding of colicins is critical to killing target cells.
For a model system, we used the K361 strain to test target‐specific killing by colicins. 6 This approach can be expanded to other types of bacteriocins to eradicate pathogens. For example, pyocins and aureocins can be used to rapidly and selectively kill nonhost P. aeruginosa 34 and Staphylococcus aureus 35 in a similar manner to colicins, as illustrated in the present study. Protein engineering to combine domains of colicins with other bacteriocins may allow the engineered colicins to kill pathogens 36 other than E. coli. Thus, our results demonstrate the promise of using colicins as next‐generation agents to inhibit pathogenic biofilms. After we identified E2 colicin as a promising therapeutic candidate in cell‐free conditions, we showed that the controlled synthesis and the secretion of colicin E2 from engineered EcN completely inhibited target biofilm formation and killed cells in preformed biofilms (Figures 3, 4, 5). Because engineered cells form biofilms together with target biofilm cells, colicin production by engineered cells can bypass the mass‐transfer requirement for antimicrobial compounds to enter biofilms. However, delivering inducer molecules to produce colicins in engineered biofilm cells might not be practically viable. To address this challenge, the engineered cells could be developed to recognize biofilm‐specific signals such as quorum sensing (QS) molecules. For example, P. aeruginosa cells produce and secrete autoinducers (e.g., acylhomoserine lactone) as QS signals during biofilm formation. QS‐based genetic switches have been widely used in various studies 37 , 38 , 39 for applications including the control of consortial biofilm 40 and the prevention of P. aeruginosa biofilms in the gut. 15 If the engineered cells sense the autoinducers and trigger the production of P. aeruginosa‐specific toxins such as pyocin, the engineered cells can kill P. aeruginosa biofilm cells without a need for external inducers.
Although EcN has some protein secretion capability, 19 we successfully developed a method to enhance the release of colicins from this host strain by adding the E7 lysis gene to our genetic circuits (Figure 3). Using this method, we were able to control colicin production and secretion to achieve complete disruption of preexisting biofilms (Figures 4 and 5). Producing and secreting therapeutic compounds from the engineered host have an additional benefit in that therapeutic molecules can be generated continually or inducibly while cells are growing, thus potentially eliminating the requirement for repetitive dosing of antimicrobials to treat biofilms associated with diseases. Beyond biofilm killing and therapeutic applications, our target‐specific approach to rapid cell killing could be used to help solve the challenging problem of controlling populations of mixed species in biomanufacturing by using microbial consortia. 41 , 42 Genetic circuits might potentially be used to control colicin synthesis and secretion, similarly to the circuits developed here to control subpopulations within synthetic bacterial consortia or to trigger a change in consortium makeup by removing certain cells from the population at desired times, thus improving the maximal titer of the end‐product and the metabolic stability.
This study demonstrates that colicins may serve as a promising antimicrobial approach to eradicate target biofilms rapidly and selectively in multispecies biofilms. We also found that engineering microorganisms as antimicrobial protein producers may be a promising live therapeutic candidate to treat biofilm‐associated infections. In the future, we expect that increasing the utilization and complexity of synthetic genetic circuits will enable the development of beneficial microorganisms with diverse and novel functions for various biofilm‐control applications.
AUTHOR CONTRIBUTION
Xing Jin: Conceptualization (equal); data curation (lead); formal analysis (lead); investigation (lead); methodology (equal); visualization (lead); writing–original draft (lead), writing–review & editing (equal). Sungjun An: Data curation (supporting); investigation (equal); software (lead); validation (supporting); visualization (equal); writing–review & editing (supporting). Weston Kightlinger: Investigation (supporting); writing–review & editing (supporting). Jiacheng Zhou: Resources (supporting); validation (supporting). Seok Hoon Hong: Conceptualization (lead); data curation (lead); formal analysis (equal); funding acquisition (lead); investigation (equal); methodology (lead); project administration (lead); resources (equal); software (equal); supervision (lead); validation (equal); visualization (equal); writing–original draft (lead); writing–review & editing (lead).
Supporting information
Appendix S1: Supporting Information
Jin X, An S, Kightlinger W, Zhou J, Hong SH. Engineering Escherichia coli to produce and secrete colicins for rapid and selective biofilm cell killing. AIChE J. 2021;67(12):e17466. doi: 10.1002/aic.17466
Funding information National Institute of Allergy and Infectious Diseases, Grant/Award Number: R15AI130988
[The copyright line updated on 24 Nov 2021, after online publication.]
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Roberts AEL, Kragh KN, Bjarnsholt T, Diggle SP. The limitations of in vitro experimentation in understanding biofilms and chronic infection. J Mol Biol. 2015;427(23):3646‐3661. 10.1016/j.jmb.2015.09.002 [DOI] [PubMed] [Google Scholar]
- 2. Ciofu O, Rojo‐Molinero E, Macià MD, Oliver A. Antibiotic treatment of biofilm infections. Apmis. 2017;125(4):304‐319. 10.1111/apm.12673 [DOI] [PubMed] [Google Scholar]
- 3. Olsen I. Biofilm‐specific antibiotic tolerance and resistance. Eur J Clin Microbiol Infect Dis. 2015;34(5):877‐886. 10.1007/s10096-015-2323-z [DOI] [PubMed] [Google Scholar]
- 4. Cotter PD, Ross RP, Hill C. Bacteriocins – a viable alternative to antibiotics? Nat Rev Microbiol. 2013;11(2):95‐105. 10.1038/nrmicro2937 [DOI] [PubMed] [Google Scholar]
- 5. Cascales E, Buchanan SK, Duché D, et al. Colicin biology. Microbiol Mol Biol Rev. 2007;71(1):158‐229. 10.1128/MMBR.00036-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jin X, Kightlinger W, Kwon Y‐C, Hong SH. Rapid production and characterization of antimicrobial colicins using Escherichia coli‐based cell‐free protein synthesis. Synth Biol. 2018;3(1):ysy004. 10.1093/synbio/ysy004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jin X, Kightlinger W, Hong SH. Optimizing cell‐free protein synthesis for increased yield and activity of colicins. Methods Protoc. 2019;2(2):28. 10.3390/mps2020028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ausländer S, Ausländer D, Fussenegger M. Synthetic biology—the synthesis of biology. Angew Chemie Int Ed. 2017;56(23):6396‐6419. 10.1002/anie.201609229 [DOI] [PubMed] [Google Scholar]
- 9. McGregor DP. Discovering and improving novel peptide therapeutics. Curr Opin Pharmacol. 2008;8(5):616‐619. 10.1016/j.coph.2008.06.002 [DOI] [PubMed] [Google Scholar]
- 10. Riglar DT, Silver PA. Engineering bacteria for diagnostic and therapeutic applications. Nat Rev Microbiol. 2018;16(4):214‐225. 10.1038/nrmicro.2017.172 [DOI] [PubMed] [Google Scholar]
- 11. Takiishi T, Cook DP, Korf H, et al. Reversal of diabetes in NOD mice by clinical‐grade proinsulin and IL‐10‐secreting Lactococcus lactis in combination with low‐dose anti‐CD3 depends on the induction of Foxp3‐positive T cells. Diabetes. 2017;66(2):448‐459. 10.2337/db15-1625 [DOI] [PubMed] [Google Scholar]
- 12. Braat H, Rottiers P, Hommes DW, et al. A phase I trial with transgenic bacteria expressing interleukin‐10 in Crohn's disease. Clin Gastroenterol Hepatol. 2006;4(6):754‐759. 10.1016/j.cgh.2006.03.028 [DOI] [PubMed] [Google Scholar]
- 13. Lagenaur LA, Sanders‐Beer BE, Brichacek B, et al. Prevention of vaginal SHIV transmission in macaques by a live recombinant Lactobacillus . Mucosal Immunol. 2011;4(6):648‐657. 10.1038/mi.2011.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zheng JH, Nguyen VH, Jiang SN, et al. Two‐step enhanced cancer immunotherapy with engineered Salmonella typhimurium secreting heterologous flagellin. Sci Transl Med. 2017;9(376):eaak9537. 10.1126/scitranslmed.aak9537 [DOI] [PubMed] [Google Scholar]
- 15. Hwang IY, Koh E, Wong A, et al. Engineered probiotic Escherichia coli can eliminate and prevent Pseudomonas aeruginosa gut infection in animal models. Nat Commun. 2017;8:15028. 10.1038/ncomms15028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jakes KS. Translocation trumps receptor binding in colicin entry into Escherichia coli . Biochem Soc Trans. 2012;40(6):1443‐1448. 10.1042/BST20120207 [DOI] [PubMed] [Google Scholar]
- 17. Levengood‐Freyermuth SK, Click EM, Webster RE. Role of the carboxyl‐terminal domain of TolA in protein import and integrity of the outer membrane. J Bacteriol. 1993;175(1):222‐228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jakes KS. The colicin E1 TolC box: identification of a domain required for colicin E1 cytotoxicity and TolC binding. J Bacteriol. 2017;199(1):e00412‐16. 10.1128/JB.00412-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fang K, Jin X, Hong SH. Probiotic Escherichia coli inhibits biofilm formation of pathogenic E. coli via extracellular activity of DegP. Sci Rep. 2018;8(1):4939. 10.1038/s41598-018-23180-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Soelaiman S, Jakes K, Wu N, Li C, Shoham M. Crystal structure of colicin E3: implications for cell entry and ribosome inactivation. Mol Cell. 2001;8(5):1053‐1062. 10.1016/S1097-2765(01)00396-3 [DOI] [PubMed] [Google Scholar]
- 21. Hong SH, Ntai I, Haimovich AD, Kelleher NL, Isaacs FJ, Jewett MC. Cell‐free protein synthesis from a release factor 1 deficient Escherichia coli activates efficient and multiple site‐specific non‐standard amino acid incorporation. ACS Synth Biol. 2014;3(6):398‐409. 10.1021/sb400140t [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. de Chávez‐López M, Zúñiga‐García V, Castro‐Magdonel BE, et al. Eag1 gene and protein expression in human retinoblastoma tumors and its regulation by pRb in HeLa cells. Genes (Basel). 2020;11(2):119. 10.3390/genes11020119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Swartz JR, Jewett MC, Woodrow K. Cell‐free protein synthesis with prokaryotic combined transcription‐translation. Methods Mol Biol. 2004;267:169‐182. 10.1385/1-59259-774-2:169 [DOI] [PubMed] [Google Scholar]
- 24. Shao X, Fang K, Medina D, Wan J, Lee JL, Hong SH. The probiotic, Leuconostoc mesenteroides, inhibits Listeria monocytogenes biofilm formation. J Food Saf. 2019;40:e12750. 10.1111/jfs.12750 [DOI] [Google Scholar]
- 25. Schulz S, Stephan A, Hahn S, et al. Broad and efficient control of major foodborne pathogenic strains of Escherichia coli by mixtures of plant‐produced colicins. Proc Natl Acad Sci USA. 2015;112(40):E5454‐E5460. 10.1073/pnas.1513311112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hall CW, Mah T‐F. Molecular mechanisms of biofilm‐based antibiotic resistance and tolerance in pathogenic bacteria. FEMS Microbiol Rev. 2017;010:276‐301. 10.1093/femsre/fux010 [DOI] [PubMed] [Google Scholar]
- 27. Tan CH, Lee KWK, Burmølle M, Kjelleberg S, Rice SA. All together now: experimental multispecies biofilm model systems. Environ Microbiol. 2017;19(1):42‐53. 10.1111/1462-2920.13594 [DOI] [PubMed] [Google Scholar]
- 28. Scaldaferri F, Gerardi V, Mangiola F, et al. Role and mechanisms of action of Escherichia coli nissle 1917 in the maintenance of remission in ulcerative colitis patients: an update. World J Gastroenterol. 2016;22(24):5505‐5511. 10.3748/wjg.v22.i24.5505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Danino T, Prindle A, Kwong GA, et al. Programmable probiotics for detection of cancer in urine. Sci Transl Med. 2015;7(289):289ra84. 10.1126/scitranslmed.aaa3519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Praveschotinunt P, Duraj‐Thatte AM, Gelfat I, Bahl F, Chou DB, Joshi NS. Engineered E. coli Nissle 1917 for the delivery of matrix‐tethered therapeutic domains to the gut. Nat Commun. 2019;10(1):1‐14. 10.1038/s41467-019-13336-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Saeidi N, Wong CK, Lo T‐M, et al. Engineering microbes to sense and eradicate Pseudomonas aeruginosa, a human pathogen. Mol Syst Biol. 2011;7:521. 10.1038/msb.2011.55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jakes KS, Cramer WA. Border crossings: colicins and transporters. Annu Rev Genet. 2012;46(1):209‐231. 10.1146/annurev-genet-110711-155427 [DOI] [PubMed] [Google Scholar]
- 33. Sharma O, Yamashita E, Zhalnina MV, et al. Structure of the complex of the colicin E2 R‐domain and its BtuB receptor. The outer membrane colicin translocon. J Biol Chem. 2007;282(32):23163‐23170. 10.1074/jbc.M703004200 [DOI] [PubMed] [Google Scholar]
- 34. Behrens HM, Six A, Walker D, Kleanthous C. The therapeutic potential of bacteriocins as protein antibiotics. Emerg Top Life Sci. 2017;1(1):65‐74. 10.1042/ETLS20160016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. dos Nascimento JS, Coelho MLV, Ceotto H, et al. Genes involved in immunity to and secretion of aureocin A53, an atypical class II bacteriocin produced by Staphylococcus aureus A53. J Bacteriol. 2012;194(4):875‐883. 10.1128/JB.06203-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kageyama M, Kobayashi M, Sano Y, Masaki H. Construction and characterization of pyocin‐colicin chimeric proteins. J Bacteriol. 1996;178(1):103‐110. 10.1128/jb.178.1.103-110.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hooshangi S, Bentley WE. From unicellular properties to multicellular behavior: bacteria quorum sensing circuitry and applications. Curr Opin Biotechnol. 2008;19(6):550‐555. 10.1016/j.copbio.2008.10.007 [DOI] [PubMed] [Google Scholar]
- 38. Choudhary S, Schmidt‐Dannert C. Applications of quorum sensing in biotechnology. Appl Microbiol Biotechnol. 2010;86(5):1267‐1279. 10.1007/s00253-010-2521-7 [DOI] [PubMed] [Google Scholar]
- 39. Fang K, Park OJ, Hong SH. Controlling biofilms using synthetic biology approaches. Biotechnol Adv. 2020;40:107518. 10.1016/j.biotechadv.2020.107518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hong SH, Hegde M, Kim J, Wang X, Jayaraman A, Wood TK. Synthetic quorum‐sensing circuit to control consortial biofilm formation and dispersal in a microfluidic device. Nat Commun. 2012;3:613. http://www.nature.com/ncomms/journal/v3/n1/suppinfo/ncomms1616_S1.html [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. McCarty NS, Ledesma‐Amaro R. Synthetic biology tools to engineer microbial communities for biotechnology. Trends Biotechnol. 2018;37(2):181‐197. 10.1016/j.tibtech.2018.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Han Y, Zhang F. Control strategies to manage trade‐offs during microbial production. Curr Opin Biotechnol. 2020;66:158‐164. 10.1016/j.copbio.2020.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.