

Abstract
Plexiform Neurofibromas (PN) are a common manifestation of the genetic disorder neurofibromatosis type 1 (NF1). These benign nerve sheath tumors often cause significant morbidity, with treatment options limited historically to surgery. There have been tremendous advances over the past two decades in our understanding of PN, and the recent regulatory approvals of the MEK inhibitor selumetinib are reshaping the landscape for PN management. At present, there is no agreed upon PN definition, diagnostic evaluation, surveillance strategy, or clear indications for when to initiate treatment and selection of treatment modality. In this review, we address these questions via consensus recommendations from a panel of multidisciplinary NF1 experts.
Keywords: MEK inhibitor, neurofibroma, neurofibromatosis 1, plexiform, review
Plexiform neurofibromas (PN) are histologically benign nerve sheath tumors that occur commonly in individuals with the tumor predisposition syndrome neurofibromatosis type 1 (NF1). They are a significant cause of morbidity and, until recently, no effective medical therapies were available. PN arise within nerves and consist of multiple cell types, including Schwann cells, fibroblasts, perineural cells, mast cells, and macrophages.1 While PN development requires biallelic loss of NF1 in Schwann cells,2,3 the NF1+/− microenvironment also contributes to PN tumorigenesis.1,4 The recent regulatory approvals (including United States, Europe, Brazil) of the MEK inhibitor (MEKi) selumetinib for children with NF1 and symptomatic, inoperable PN, as well as promising results from other clinical trials, have changed the clinical landscape with the potential to create a paradigm shift in the management of PNs. This manuscript represents consensus generated across the multidisciplinary authorship team reached after review and discussion of available peer-reviewed literature for the management of NF1-associated PN (see Supplementary Material).
PN Definitions: Clinical, Pathologic, and Imaging
There is no agreed upon PN definition; however, several PN classification systems have been proposed previously using histopathologic, clinical, and imaging findings (Table 1). In the past two decades, with the advent of clinical trials for PN, clinicians have taken a broad view on defining PN based on clinical and imaging findings. Here we propose a clinically relevant, MRI-based classification system of PN that incorporates aspects of these previous systems (Figure 1). We classify PN based on: 1) morphology or internal structure, 2) depth, and 3) relationship to adjacent tissues (Figure 2, Supplementary Figure S1). This classification system may not be fully applicable to some tumors, such as paraspinal PN (which can extend from the spinal nerve root causing diffuse nerve thickening) and discrete neurofibromas (Supplementary Figure S1).
Table 1.
Published Definitions of Plexiform Neurofibromas (PN) Based on Histopathologic, Clinical, and Imaging Characteristics
| Terminology | Description | Reference | |
|---|---|---|---|
| Histopathologic |
Neurofibroma: Benign Schwann cell neoplasm with thin, often wavy nuclei, wispy cell processes, and a myxoid to collagenous (“shredded carrots”) matrix. PN: Diffusely enlarging and replacing a nerve, often involving multiple nerve fascicles. |
18 | |
| Histopathologic |
Neurofibromas: Neurofibromas are characterized by cytologically bland spindle cells with thin, wavy nuclei representing the neoplastic Schwann cell, immersed in a variably loose myxoid stroma. Stromal collagen is characteristic, colorfully likened in classic pathology descriptions to shredded carrots. A variety of other cells are also identifiable in neurofibroma, including perineurial and perineurial-like cells, fibroblasts, and mast cells. PN: Plexiform neurofibroma is defined by its involvement of multiple nerve fascicles, each surrounded by perineurium. It most often involves a large nerve or plexus, imparting a bag-of-worms or ropy gross appearance. |
19 | |
| Clinical description | A proliferation of cells in the nerve sheath extending across the length of a nerve and involving multiple nerve fascicles The term …‘plexiform’ does not imply involvement of a nerve plexus…rather a network-like growth of neurofibroma involving multiple fascicles of a nerve, leading to a diffuse mass of thickened nerve. |
101 | |
| Imaging | Nodular | Confined to the nerve | 102 |
| Diffuse | Impinging on surrounding soft tissue | ||
| Imaging | Circumscribed | Locally circumscribed | 6 |
| Plexiform | Invasive or involved multiple nerves | ||
| Imaging | Superficial/deep | Above/below the muscle fascia | 103 |
| Fascicular-nodular | Collection of smaller components that were tubular or spherical or both | ||
| Diffuse | Lack of any definable geometry | ||
| Imaging | Fascicular | Tubular or rope-like configuration | 12 |
| Nodular | Small, round lesions of different diameter | ||
| Diffuse | No definable geometry | ||
| Superficial | Located cutaneously or subcutaneously respecting epifascial membrane and not penetrating into muscle with no clear demarcating borders | ||
| Displacing | Multinodular smoothly defined borders compressing adjacent structures, primarily along main nerves | ||
| Invasive | Conglomerating tumors which could not be divided from each other and which penetrate into muscle, fascia, joints, and surrounding tissue |
Fig. 1.

Proposed plexiform neurofibroma classification schema. For each tumor, determine classification in each category (A–C, D optional).
Fig. 2.

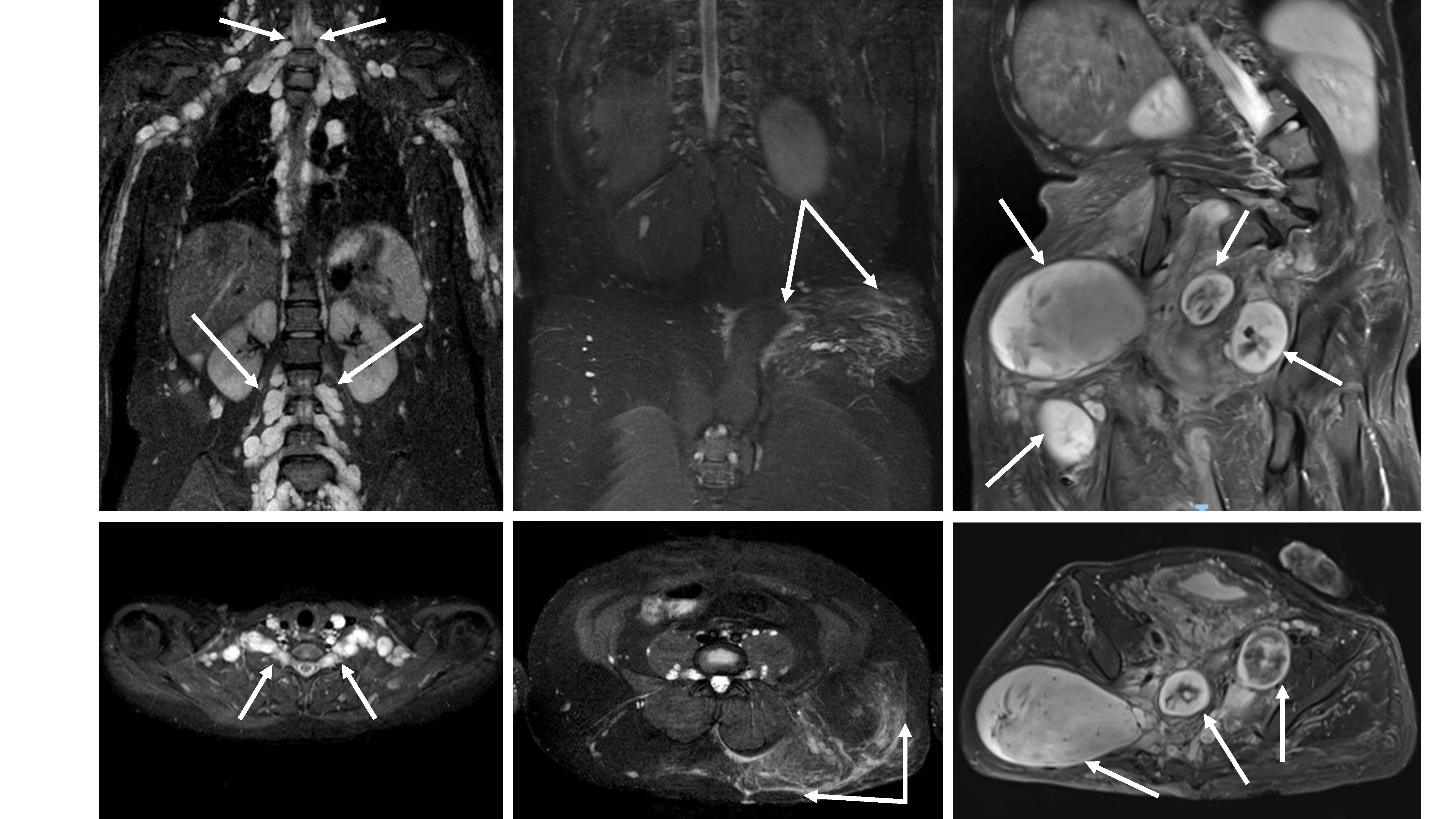
MRI examples of plexiform neurofibromas (PN) demonstrating tumor characteristics used in the proposed classification schema. All images are fat suppressed with STIR (short tau inversion recovery) technique. (A) The internal structure can be homogeneous without notable architectural elements (left panel, solid arrow), appear as conglomerate of small nodules (right panel, dotted arrows), or show a combination of both features (middle panel). (B) Any portion of peripheral nerves may be affected by PN. Proximal nerve segments give rise to deep internal PN (left panel, dotted arrows), superficial PN (right panel, solid arrows) are associated with terminal nerve branches, but many lesions have components of both (middle panel). (C) The interface between PN and surrounding tissues can range from interdigitating and intricately connected (left panel, solid arrows) to sharply defined and well separated (right panel, dotted arrows), with most lesions falling in between those two extremes (middle panel).
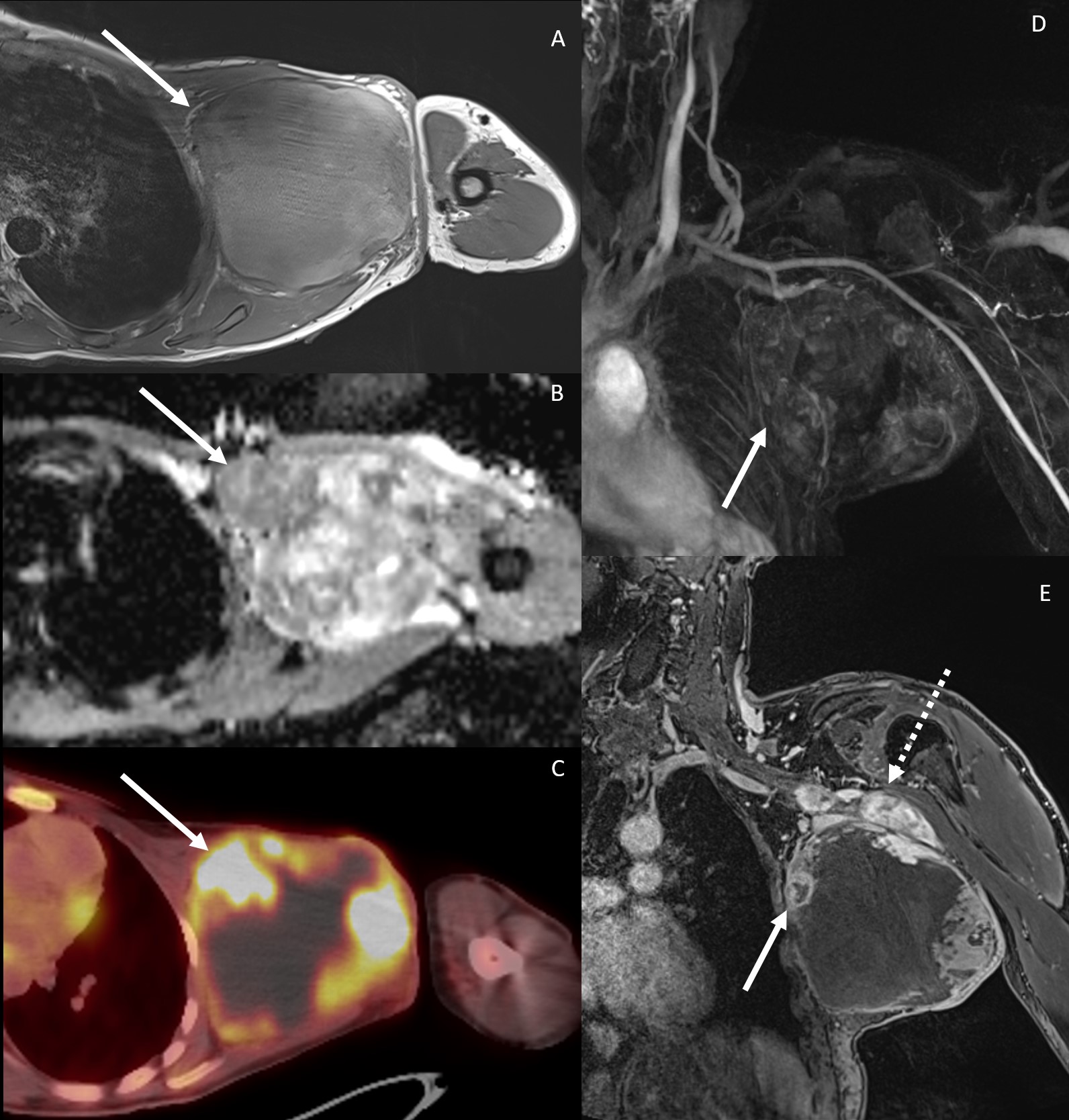
The term “distinct nodular lesion” (DNL) has been used in more recent literature to describe peripheral nerve sheath tumors with a characteristic MRI appearance (Figure 3). These lesions are well-demarcated, appear encapsulated, are ≥3 cm, lack the central target sign characteristic of classic PN, and can be present within or outside of a PN.
Fig. 3.

Coronal (top row) and axial (bottom row) STIR MRI examples of distinct nodular lesions (DNL) in patients with NF1. The left panels show a DNL (arrow) arising from the left sciatic nerve. In the middle panels, plexiform neurofibroma (PN) can be seen along the brachial plexus on both sides, with a prominent nodule present on the right (arrow). On the right, the DNL (arrow) stands out from the background of a large neck, shoulder, and chest PN.
Epidemiology, Clinical Presentation, Work-up and Screening, and Genotype–Phenotype Correlations
Epidemiology
Clinically-detectable PN are seen in approximately 30% of individuals with NF1.5 Case series utilizing whole-body MRI (WB-MRI) estimate the PN prevalence is 50–60% in individuals with NF1 (median PN number of 1.4–3).6–9 Many PN are diagnosed before 5 years of age suggesting that they may be congenital or develop early in childhood.10 PN can arise throughout the entire body, but are more common in the craniofacial area, neck, pelvis, and lower extremities.6,9,11–14
Clinical Presentation
Asymptomatic and symptomatic PN often coexist within the same individual. Tumor location, size, nerve involvement, and age may all impact symptoms and potential for complications (see “Morbidity” section). In children, >60% of symptomatic PNs are located in the head and neck; in contrast, PN in the thorax and abdomen usually remain asymptomatic.9,14 Among adults, PN involving the abdominopelvic region, brachial plexus, and lumbosacral plexus carry a high risk of morbidity.12,15 Dermatologic features can offer clues to PN presence, including thickening of the dermis or the presence of coarse hair or hyperpigmentation (Supplementary Figure S2). Rarely hemorrhage can occur in PN and can be life-threatening.16,17
PN may transform to atypical neurofibromas (AN)/atypical neurofibromatous neoplasm of uncertain biological potential (ANNUBP), characterized by at least two of the following features: cytological atypia, hypercellularity, loss of neurofibroma architecture, and an increased mitotic index.18,19 These tumors are potentially premalignant lesions with unique biology (frequently characterized by heterozygous or homozygous loss of CDKN2A/B)20 and growth characteristics.11,21 AN/ANNUBP may be asymptomatic, or present with pain and functional deficit. Individuals with AN/ANNUBP are at higher risk for the development of malignant peripheral nerve sheath tumors (MPNST), with a 33% incidence in one study versus the cumulative MPNST risk of 15.8% in the general NF1 population.18,21,22
Work-up and Screening
All individuals with NF1 should be assessed for the presence of PN by careful examination and monitored for PN growth. Standard evaluation includes history, physical, and neurological exam. Regional or WB-MRI is the imaging modality of choice for identification and characterization of PN. This is generally indicated in individuals with symptoms suggesting PN or with visible PN to assess size and impingement on critical structures. These findings inform appropriate management (observation and surveillance intervals, additional diagnostic evaluations, or possible treatment).23 Some practitioners advocate for baseline WB-MRI, particularly in late adolescence or early adulthood when transitioning from pediatric to adult care. This practice is based on reports that total body PN burden is correlated with lifetime risk of MPNST8,24 and data suggesting that if PN are not present by early adulthood they are unlikely to develop.25,26 In the future, screening of children with WB-MRI to detect asymptomatic PN may become warranted, if prospective studies identify factors predictive of progression and demonstrate an improved outcome with earlier identification. A biopsy to ascertain the histologic diagnosis of a PN is not needed unless concerning clinical or imaging findings suggest atypical behavior or malignant transformation.
Genotype–Phenotype Correlations
Attempts to correlate NF1 germline gene variants with specific clinical features have been largely unsuccessful. However, there are some clinically relevant genotype–phenotype correlations predictive of mild27–33 or more aggressive29,33,34 phenotypes (Supplementary Table S1)
Natural History
The growth of PN varies between tumors both within and across individuals; however, growth rates remain relatively constant within a PN for prolonged periods of time.11,25,35 Younger age correlates with more rapid tumor growth (Figure 4A).11,25,35,36 In children, PN growth rate exceeds increase in body weight over time, suggesting that PN growth is not related solely to normal growth during childhood.11,35,36 PN growth rate ≥20% per year by volume is unusual after adolescence,11 and preliminary data in adults (median age 42 years) suggest <5% of PN grow at that rate.26 Most PNs in older adolescents and adults grow slowly or not at all.11,25,26
Fig. 4.

Tumor growth rate plotted against patient’s age at initial MRI for plexiform neurofibromas (PN) (A) and distinct nodular lesions (DNL) (B). A moderate negative correlation was observed for PN, whereas only a weak association was noted for DNL. Adapted from Akshintala S, et al. Neuro Oncol. 2020.6Reprinted with permission.
Spontaneous PN shrinkage over time has been reported in some individuals,11,25,26 but mainly in adults. In one study, 59% of tumors in 26 adults demonstrated a spontaneous decrease in volume on MRI of ≥20% over nine years.26 By contrast, in a study of individuals ≤35 years old, a volume decrease of ≥ 10% was seen in only 8.8% of tumors with a median decrease of 3.6% per year.11 Importantly, spontaneous PN volume decreases ≥20% per year have not been reported.11,25,26
No significant differences in estimated PN growth rates have been noted in relation to tumor location, patient sex, race, concurrent pregnancy, or hormonal changes associated with puberty.11,37,38 In addition, there are no prospective data on the impact of hormone medications such as oral contraceptive medications on PN growth rate.
DNL show key differences in growth patterns compared to classic PN, suggesting biological differences. Some DNL have faster growth rates (≥20% per year may be seen even in adulthood) and develop at later ages compared to PN (Figure 4B).11 In addition, AN/ANNUBP frequently show a distinct nodular appearance on MRI.21 Additional studies are required to determine if DNL are an imaging correlate for AN/ANNUBP and if they warrant closer surveillance than PN. Growth of single nodular lesions within a PN or rapid tumor growth of ≥20% per year in patients ≥15 years of age should raise concern for tumor transformation to AN/ANNUBP or MPNST.11
Tumor Imaging and Measurement
Imaging plays a vital role in the management of people with PN (Supplementary Table S2) as a screening tool at baseline (see “Work-up and Screening” above), for surveillance (in individuals with known PN), to evaluate treatment response, and for preoperative assessment for surgical planning.
Conventional MRI sequences with short tau inversion recovery (STIR) and T2-weighted sequences with fat saturation visualize PN optimally, can be performed without the administration of intravenous contrast material, and are preferred over computed tomography (CT).39,40 Regional MRI and WB-MRI have different advantages. Regional MRI allows evaluating a specific PN in greater detail by optimizing the field of view (FOV) and is recommended for clinical trials. WB-MRI is advantageous in patients with multiple tumors or very large PN that cross traditional anatomic planes. Supplementary Table S2 summarizes MRI protocols and indications.
Imaging for Surveillance
For surveillance of known PN, regional MRI is utilized more often than WB-MRI.23 Currently, there is no data to support nor consensus on appropriate intervals for monitoring known PN, with intervals ranging from 3 to 24 months in a survey of 30 NF1 practitioners23; and clinical practices may vary beyond this. Factors to consider when selecting scanning intervals include: age of patient, tumor location, presence of PN-associated morbidity, imaging appearance, and whether growth of the PN is known from prior imaging. Imaging intervals may be extended for those with clinically or radiographically stable PN over time, or shortened if there is a change in imaging appearance or new symptoms.
For PN concerning for malignant transformation (Supplementary Figure S3 and Supplementary Table S2), metabolic imaging using18F-fluorodeoxyglucose (FDG) PET with CT (FDG-PET/CT) has high diagnostic accuracy for detection of MPNST arising in the background of PN (sensitivity: 89–100%; specificity: 72–94%) using semi-quantitative markers such as standard uptake values (SUV) and tumor to liver ratio.41 However, AN/ANNUBP frequently also have avid uptake of FDG on PET imaging.21 Further, there is a wide range of SUVmax that can be associated with MPNST or AN/ANNUBP and benign PN. Hence, awareness of the risk of a false positive result of FDG-PET in people with NF1 is important, and targeted biopsy is often needed to confirm the histology. Of note, differences in software and methodology may impact quantitative SUV analysis and limit generalization across institutions. There is emerging interest in WB FDG-PET/MRI for NF1 to reduce ionizing radiation from CT.42 More recently, diffusion weighted MR imaging and dynamic contrast enhanced sequences have been evaluated and shown to detect MPNST with high diagnostic accuracy (sensitivity: 92–100%; specificity: 94–98%).43,44 To date, these advanced MRI techniques have been piloted by only a few institutions. Prospective studies are underway to compare advanced MRI with FDG-PET for this purpose. Last, WB FDG-PET/CT, WB FDG-PET/MR, and diffusion weighted imaging can be used to target the site of biopsy for a suspicious PN component, and to detect local and/or distant metastases in patients with MPNST.45,46
Imaging for Treatment Response
For clinical care, regional or WB-MRI is performed prior to initiating a new therapy. For assessing treatment response, standard one-dimensional (1D) or 2D measurements may be sufficient, although given the complex shape and potential extensive size of PN, linear measurements may be difficult to reliably measure and track. In the setting of a clinical concern such as suspected treatment failure or treatment-related toxicity, 3D volumetric analysis may have value in the risk-benefit assessment of continuing treatment47; however, access to 3D imaging tools is currently limited.
For clinical trials, 3D volumetric tumor analysis is recommended, as it reproducibly detects minor size changes.47,48 Accurate longitudinal assessment of PN requires consistent image acquisition and 3D measurement tools (eg 3DQI-NF, MINT, BRAIN Lab, MEDex) (Figure 5). Imaging on clinical trials and when treating PN is performed at baseline and serially (typically every four months for the first year with increased intervals subsequently).47
Fig. 5.

Longitudinal assessment of plexiform neurofibroma (PN) size using volumetric MRI analysis. While the longest diameter of the left flank lesion remained almost the same over time, the bulk of the PN increased visibly between 4.9 years and 6.6 years of age, at which point the patient started selumetinib therapy resulting in PN shrinkage. Volumetric analysis is performed by identifying tumor contours as shown by the outlines in the bottom panels. This technique allows to detect small changes sensitively and reproducibly.
Preoperative Imaging Strategy
For PN undergoing resection, localized MRI with FOV encompassing the entire tumor is recommended. CT can assess osseous or pulmonary involvement and, in some cases, can be used to place a preoperative marker to localize a suspicious component that is surrounded by PN. Both CT and MR angiography can assess the intratumoral course of important blood vessels and the need for preoperative embolization.
Morbidity
Impact on Function and Appearance
Depending on their location, PN can cause a wide variety of symptoms including visual or hearing impairments, airway obstruction, speech and swallowing difficulties, motor dysfunction, bowel or bladder dysfunction, disfigurement, and other symptoms. PN-related morbidities are primarily caused by direct impact of the tumor on surrounding structures, and may be life-threatening when they compress vital organs. In one retrospective review of children with PN, the most common PN-related symptoms were pain and motor dysfunction.49 In another series, the most common symptoms that lead to PN-directed surgical interventions were neurologic deficits, disfigurement, orthopedic symptoms, and airway difficulties.13
Notably, children with NF1 and symptomatic PN have a higher mortality rate (3.2%) compared with children without PN or with asymptomatic PN (0.5%).13 Tumor size impacts the severity of PN-related symptoms, with symptomatic PN and those causing motor impairment tending to be larger.10,49 These functional impairments have a negative impact on overall quality of life (QOL) for individuals with NF1,50 emphasizing the need to monitor for improvement in these areas to detect the clinical impact of treatment.
There are few validated functional outcome measures for NF1-related complications; however, progress is being made. For evaluation of motor function, there is good inter- and intra-rater reliability for the Functional Reach, Timed Up and Go, and 10-Meter Walk tests in adults with NF1.51 In addition, the Response Evaluation in Neurofibromatosis and Schwannomatosis (REiNS) international collaboration has published evidence-based, consensus recommendations for assessment of various functional measures for PN-related morbidities, such as airway52 and vision.53 The phase 2 study of selumetinib for children with symptomatic, inoperable PN used prospective functional outcome measures and demonstrated objective improvement in strength, range of motion, and pulmonary function.54 These results were crucial to the recent regulatory approvals of selumetinib for PN, supporting the inclusion of functional evaluations in future clinical trials for PN.
Impact on Pain
Pain in individuals with NF1-PN can be episodic, chronic, or both; localized to the site of a PN or diffuse; and can range widely in functional impact.55 The prevalence of pain in individuals with NF1-PN is not well established. Based on natural history studies, only a minority of patients (11–30%) experience significant PN-related pain.10,36,56 The rate is higher (31–37%) for those enrolled in clinical trials.54,57 In contrast to PN, pain is often the predominant and/or presenting symptom in most individuals with AN and MPNST21,58,59; thus, progressive, severe pain in a PN should raise clinical suspicion for malignant transformation.59,60
The mechanisms by which PN cause pain are not well understood and may be neuropathic, visceral, bony, and inflammatory. Pain may be independent of direct tumor effect and may be potentiated by mechanisms leading to hyperalgesia.61 For example, dysfunction of RAS can result in alteration of pain pathways.62
Pharmacologic and nonpharmacologic interventions can reduce PN-related pain (Supplementary Table S3). Recent PN clinical trials reported a reduction of pain, as measured by pain intensity and pain interference, in individuals with tumor response.54,63,64 There are no NF1- or PN-specific trials using traditional pharmacological agents to treat neuropathic pain, such as GABAergic medications, antidepressants, opioid analgesics, anti-inflammatory drugs, and cannabinoids.
Acceptance and Commitment Therapy (ACT), a cognitive behavioral therapy (CBT) with effectiveness in individuals with chronic pain,65 significantly decreased pain interference in a recent randomized controlled trial of adolescents and adults with NF1 and PN-related pain.66 Traditional CBT and mindfulness-based therapies have potential for treating PN pain, but more rigorous research is needed in the NF1 population.67,68 Additional, potential nonpharmacological interventions for PN pain include heart rate variability biofeedback,69 acupuncture,70 and transcutaneous electrical nerve stimulation (TENS).71
Impact on Psychosocial Functioning and Quality of Life
Natural history studies indicate that 1/3 of children and adolescents with NF1-PN experience parent-reported social-emotional difficulties such as anxiety, depression, and social withdrawal, with greater disease severity associated with worse internalizing problems72 and worse QOL.73 PRO measures completed by pediatric54 and adult patients74 enrolling on PN clinical trials documented reduced QOL, with greater PN volume correlated with worse QOL in the adults. Patients of all ages share concerns regarding the impact of PN on social and emotional health, which manifests differently across age groups, reinforcing the importance of assessing psychosocial functioning across the lifespan.55,75 Medical treatments for PN have resulted in both parent- and child-reported improvements in emotional and/or social aspects of QOL.54,76 Additionally, e-health interventions that address coping with NF1 symptoms and stress resulted in improvements in QOL77 and may be relevant for individuals with PN.
Management
Indications for PN-directed Medical or Surgical Treatment
Careful selection of treatment versus observation for PN is important to maximize benefit and minimize risk. Several factors should be considered, including the age of the patient, and whether the PN is causing or at risk for causing morbidity, or demonstrates progressive growth. In most cases, the goal of treatment is improvement or prevention of PN-associated morbidity. The presence of morbidity, especially when refractory to symptomatic treatment, is of paramount importance. In addition, individuals with PN adjacent to or compressing structures such as the spinal cord or airway may not be symptomatic, but are at risk for future morbidity should the tumor grow.
Understanding if the PN is growing is helpful, as the larger the tumor gets, the more likely it may cause morbidity.49 However, PN growth must be assessed in the context of its growth rate and present/impending morbidity. For example, a PN with rapid growth (>20% increase in volume in the prior year) with impending morbidity is likely a candidate for intervention, while a tumor that is slowly increasing in size (eg 5% per year) with no actual or impending morbidity may warrant observation. Age of the patient is important, especially in cases where PN growth rate is unknown because prior imaging is unavailable. In such cases, age may influence decision-making, as young children are more likely to have growing PN.11,35 In sum, PN that are causing morbidity, or are growing and associated with impending morbidity, should be considered for treatment. In contrast, in most cases, stable tumors not causing morbidity should be observed, as they may never progress or cause symptoms.
If treatment is indicated, selection of surgical versus medical management should be evaluated with input from a multidisciplinary team including surgeons (general surgery, neurosurgery, orthopedic surgery, plastic surgery, etc.) and NF experts in medicine/pediatrics (oncology/neuro-oncology, neurology, etc.). In general, surgery is the optimal choice if the PN can be resected without significant morbidity. Unfortunately, as PN are invested with the nerve, this is challenging in most cases. Although there are no robust prospective studies on outcomes, the largest retrospective series suggest that complete tumor excision can be achieved in only 15% of cases,78 with PN re-growth occurring in 43% of those who underwent partial or subtotal resection,13,78 and permanent sequelae (mostly neurologic) in 5–18% of patients.13,78 Other considerations include whether resection can be achieved in a single versus multiple surgeries, what recovery may entail, and the urgency of need for tumor reduction. For example, at present, none of the medical therapies approved or tested shrink PN rapidly; thus, for a PN with spinal cord compression causing new and progressive neurological dysfunction, surgery (even subtotal resection) may be indicated to decompress the spinal cord and prevent permanent neurologic disability. When considering medical treatment (such as a MEKi or enrollment on a clinical trial), specific contraindications or preexisting conditions (eg certain cardiac and ophthalmologic conditions) or concerns about using a particular therapy (ie logistics, medication formulation, compliance, long-term safety, etc.) should be assessed.
Assessing for AN and/or MPNST
Prior to starting therapy for a PN, clinicians must be confident that there has not been malignant transformation of the PN. Change in tumor growth rate along with new onset or recent change of PN-related pain should prompt a careful consideration for possible malignancy prior to the initiation of therapy for PN, as the treatment of MPNST is substantively different. Work-up may include additional imaging (see “Imaging for Surveillance” above) and/or tumor biopsy. As many PN are heterogeneous, biopsy should target the area of most concern based on imaging (eg the region with the highest SUV or restricted diffusion); multiple core samples are encouraged in consultation with surgery as applicable. As AN often appear as DNL on MRI either within or separate from a typical PN,21 biopsy of rapidly growing DNL should be considered. In addition, patients receiving medical treatment for PN remain at risk for MPNST.
Treatment
Surgery
Decisions regarding indications for and scope of surgery need to be tailored to the tumor’s extent, location, growth rate, radiologic features, and within the context of the individual patient’s overall health. Indications for surgical intervention include actual or impending neurologic compromise or impingement on vital structures. Relative indications for surgical intervention may include pain, disfigurement, and aim to improve activities of daily living. In addition, as MPNST generally arise within preexisting PN and are not always easily distinguished from benign lesions, biopsy plays an important role in distinguishing these diagnoses. The risk of tumor re-growth following surgery is influenced by age and PN location. Retrospective studies13,78 suggest that tumor control is most difficult in young children and in those whose tumors involve the head, neck, and trunk.
PN in two specific locations, orbital–periorbital plexiform neurofibroma (OPPN) and paraspinal PN, deserve special attention. Although 10–22% of patients with OPPNs suffer vision loss from strabismic amblyopia, no studies exist to support early surgical treatment of strabismus or of the OPPN itself. Given the complex anatomy of OPPNs, which often extend into the orbital muscles, nasolacrimal duct, and the face, a consensus panel recommended nonoperative therapy in the absence of significant tumor growth.79 Debulking surgery can be considered for progressive tumors that might compromise critical structures or lead to functional decline or disfigurement.
Paraspinal PN with extension into the epidural space can lead to spinal cord compression and progressive radiculo-myelopathy. Two small series suggest that the intraspinal component of paraspinal PNs generally involves multiple levels and is amenable to surgical intervention with good functional outcomes and only a small likelihood of recurrence. In one study of 13 patients with cervical cord compression (11 with multi-level cord compression), weakness resolved in 45% of those who underwent subtotal resection of the intraspinal component of the PN, and 18% had no further progression of neurologic abnormalities.80 A separate study reported 10 patients with PN-related progressive myelopathy (8 with multi-level involvement) or cauda equina dysfunction; gross total resection of the intraspinal PN component was achieved for nine patients. Nine patients had complete recovery of neurological function and the other had significant improvement.81 Of note, PN that compress the spinal cord without associated symptoms or neurological findings do not necessarily require treatment.
For patients with PN many unanswered questions remain regarding the role of tissue sampling, prophylactic surgery (ie, to prevent malignant transformation), or more aggressive resections to address disfigurement (including face transplantation). For the recently described AN, marginal resection by an experienced surgeon should be considered if feasible without significant morbidity.82
Medical Treatment
The emerging understanding of PN pathogenesis, along with the development of preclinical models that mimic patients’ tumors in terms of histology and location, has contributed to new precision oncology approaches targeting the tumorigenic Schwann cell and/or the tumor microenvironment.83 Since the late 1990s, more than 20 clinical trials of targeted therapies for NF1-PN were launched (Table 2). Efficacy trials mostly used progression free survival (PFS) or partial response (PR) as primary objectives, adapting each study’s endpoint to the study population enrolled and goals of the study. Several advances have both improved and accelerated the development of clinical trials for PN. The establishment of 3D volumetric MRI analysis of PN provided a more sensitive and reproducible measurement of PN growth and response than 1D or 2D analysis, and has been incorporated into many efficacy trials using volume change as a primary endpoint.47 Recent trials include patient reported and functional outcome assessments as key secondary endpoints, as the importance of demonstrating clinical benefit is increasingly recognized. Finally, the REiNS international collaboration has helped define standard outcome measures for clinical trials. Additionally, in 2007, the NF Clinical Trials Consortium (NFCTC), sponsored by the Department of Defense NF Research Program, was formed with the goal of advancing clinical trial research for patients with NF. The NFCTC includes 25 clinical centers in the U.S. and Australia (https://www.uab.edu/nfconsortium) and has launched four PN studies (Table 2).
Table 2.
Clinical Trials for NF1 Plexiform Neurofibromas
| Phase 1 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Enrollment Dates | Agent (Route) | Mechanism of Action | Trial Design | Median Age (Range) (Years) | N | Median Baseline PN Volume (Range) (ml) | MTD | DLT | Response Data | Reference |
| 1997–1999 | Thalidomide (po) | Inhibit angiogenesis, anti-inflammatory (TFN-alpha) | 4 cohorts of 5 patients; dose-escalation | 17.5 (6–41)a | 20 | NA | 4 mg/kg/d (max 200 mg/d) continuous | None | 4 with minor response (2D) | 104 |
| 1998–2000 | Tipifarnib (po) |
Farnesyl transferase inhibitor | 3 + 3 | 6 (4–16) | 42 (17 w/NF1) | NA | 200 mg/m2/dose BID for 21 of 28 day cycle | Myelosuppression, rash, GI toxicity | NA (2D) |
105 |
| 2001–2006 | Peginterferon alpha-2b (sc) |
Inhibit proliferation and angiogenesis | 3 + 3 | 9.3 (1.9–34.7) | 30 | NA | 1 μg/kg/wk | Fatigue, behavior changes, neutropenia, myoclonus, ↑ AST/ALT | 1/17 pts (3D) | 106 |
| 2003 | Pirfenidone (po) |
Antibiotic- modulates cytokine action Anti fibrotic |
Pharmacokinetically-guided dose-escalation | 10.5 (3–19) | 16 | NA | 500 mg/ m2/dose TID continuous | Diarrhea, nausea and vomiting | No PR (3D) |
107 |
| 2008 | Photodynamic Therapy (iv, implantable light source) |
LS11 photosensitizer + light leads to vascular occlusion, thrombosis | 3 + 3 | NA | NA | NA | NA (study halted early due to equipment unavailability) | NA | NA | NCT 00716469 |
| 2008–2011 | Sorafenib (po) |
Inhibitor of CRAF, BRAF, RTK (VEGFR-2,3, PDGFR-β, c-kit, Flt3) | 3 + 3 | 8 (6–12) | 9 | 443 (5–10,162) | Unable to determine; intolerable | Tumor pain, rash, mood alteration | No PR (3D) |
108 |
| 2011–2014 | Selumetinib (po) |
MEK inhibitor | 3 + 3 | 10.9 (3–18.5) | 24 | 1205 (29–8,744) | 25 mg/ m2/dose BID continuous dosing | Elevated CPK, cellulitis, urticaria, decreased LVEF, mucositis, rash | PR 17/24 (3D) |
86 |
| 2015–2017 | Pexidartinib (PLX3397) (po) |
Microenvironment | Rolling six, 3 dose levels | 16 (4–21) | 16 (3 w/NF1) | NA | No MTD, highest dose level tolerated: 800 mg/m2/dose continuous dosing | None | No PR for NF1 patients | 109 |
| Phase 2 | ||||||||||
| Dates |
Agent
(Route) |
Target
(Tumor Cell, Microenvironment, Both) |
Trial Design | Median Age (Range) (Years) | N | Median Baseline PN Volume (Range) (ml) | Median TTP (mo)/N with PR (PN Volume ≥ 20%↓) | Max PN Volume Decrease | Activity (Yes/No) | Reference |
| 2001–2007 | Placebo | Placebo | Randomized, placebo controlled, double blinded, cross-over (TTP) | 8.5 (3–21.5) | 29 | 316 (39.6–4896) | TTP = 10.6 mo/0 PR | 7% | NA | 76 |
| Tipifarnib | Tumor | 31 | 572 (20.5–5573) | TTP = 19.2 mo/0 PR | 11% | No | ||||
| 2000–2004 | Pirfenidone- Adults (po) |
Microenvironment | Open label, single arm, | 30 (±13.5)a | 24 | NA | NA | >30% | 7/24 PR (≥15% PN decrease 3D) | 110 |
| 2004–2010 | Pirfenidone- Children and young adults (po) |
Microenvironment | Open label, single arm | 8.9 (3–18.8) | 36 | 349 (15–5629) | TTP = 13.2 mo/0 PR | 12% | No | 84 |
| 2006–2009 | Imatinib (po) |
Both | Open label, single arm | 13 (3–52) | 36 | NA | 6 PR | 38% | Yes | 111 |
| 2006–2014 | Peginterferon alpha-2b (sc) |
Both | Stratum 1: Asymptomatic PN | 12.4 (2.8–20.5) | 27 | 440 (9.4–6370) | 1 PR (not confirmed) |
NA | No | 85 |
| Stratum 2: Orbital PN, PN with pain, or PN with decrease in performance status | 9.4 (1.7–21.1) | 26 | Orbital PN 178 (34–283) Pain PN 503 |
|||||||
| (95–13,327) Performance status PN 615 (9.7–1,018) |
1 PR | NA | No | |||||||
| Stratum 3: Progressive PN | 7.1 (1.6–17.6) | 29 | 288 (14–3102) | TTP = 29.4 mo | NA | Yes | ||||
| 2008–2014 | Sirolimus (po) |
Tumor | Stratum 1: Progressive PN | 7.9 (3–45.4) | 49 | 186 (13–4808) | TTP = 15.4 mo 0 PR |
17% | Yes | 112 |
| Stratum 2: Nonprogressive PN | 16 (3–35) | 12 | 784 (23–2476) | 0 PR | 7.4% | No | 113 | |||
| 2011–2014 | Everolimus (RAD001) (po) |
Tumor | Open label, single arm | 31.6 (19.5–46.4)a | 23 | 54.5 (9–453.8) | 0 PR | NA | No | 114 |
| 2014–2017 | Mirdametinib (PD-0325901) (po) |
Tumor | Open label, single arm | 24 (16–39) | 19 | 797.8 (NA) | 8 PR | 28% | NA | 63 |
| Phase 2 | ||||||||||
| Dates |
Agent
(Route) |
Target
(Tumor Cell, Microenvironment, Both) |
Trial Design | Median Age (Range) (Years) | N | Median Baseline PN Volume (Range) (ml) | Median TTP (mo)/N with PR (PN Volume ≥ 20%↓) | Max PN Volume Decrease | Activity (Yes/No) | Reference |
| 2014-present | Cabozantinib (XL184) (po) |
Both | Stratum 1: Adults | 22 (16–34) | 23 | 557 (57–2954) | 8 PR (of 19 evaluable) | 38% | Yes | 64 |
| Stratum 2: Pediatric | NA | NA | NA | NA | NA | NA | NCT 02101736 | |||
| 2015–present | Selumetinib (po) |
Tumor | Stratum 1: PN morbidity | 10.2 (3.5–17.4) | 50 | 487 (5–3820) | 37 PR | 55.1% | Yes | 54 |
| Stratum 2: No PN morbidity | 12.3 (4.5–18.1) | 25 | 381(12–3159) | 18 PR | 46.3% | Yes |
115
NCT 01362803 |
|||
| 2015–present | Trametinib (po) |
Tumor | Phase 1/2 dose escalation, disease expansion | 5.5 (1–16) | 26 | NA | 12 PR | NA | Yes |
90
NCT 02124772 |
| 2016–present | Selumetinib (po) |
Tumor | Open label, single arm (adults) | 33 (18–60) | 23 | NA | 16 PR | 41% | Yes |
87
NCT 02407405 |
| 2017–present | Binimetinib (MEK162) (po) |
Tumor | Stratum 1: Adults | 23 (18–55) | 25 | 410 (7–3129) | 13 PR (of 20 evaluable) | 35.2% | Yes |
88,89
NCT 03231306 |
| Stratum 2: Pediatric | 12 (2–16) | 20 | 326 (8–6661) | 14 PR | 54% | Yes | ||||
| 2019–present | Mirdametinib (PD-0325901) (po) |
Tumor | Stratum 1: Adults | NA | NA | NA | NA | NA | NA | NCT 03962543 |
| Stratum 2: Pediatric | ||||||||||
Abbreviations: CPK, creatine phosphokinase; D, day; DLT, dose limiting toxicity; ED, erectile dysfunction; GI, gastrointestinal; iv, intravenous; LVEF, left ventricular ejection fraction; mo, months; MTD, maximum tolerated dose; N, number; NA, not available; PN, plexiform neurofibroma; po, oral; PR, partial response; sc, subcutaneous; TNF, tumor necrosis factor; TTP, time to progression; 2D, area; 3D, volumetric.
aMean age reported.
The majority of completed trials have not resulted in a clinically meaningful improvement in PFS or in PR (PN volume decrease ≥20%). Exceptions were a phase II trial of imatinib, which demonstrated PR in a subset of very small PN (<25 mL), and a phase 2 study of peginterferon-α-2b (INF- α), which demonstrated rare PR and substantially prolonged median PFS (29.4 months) compared to the placebo arm (10.6 months) of the previously conducted double-blind, randomized trial with tipifarnib.84,85
The recent success of MEKi, targeting a downstream effector of RAS, has changed the landscape for PN management. Phase 1/2 clinical trials of the MEKi selumetinib for children with inoperable symptomatic PN resulted in PR in 71% and 74% of patients, respectively.54,86 Importantly, patients also experienced less pain and improved function and quality of life while on treatment.54 Based on these findings, selumetinib became the first medical treatment approved by the FDA for the management of PN in children. Similar responses have been seen in an ongoing phase 2 trial of selumetinib for adults with symptomatic, inoperable PN (NCT02407405).87 Other MEKi’s show promise as well. Interim results from an ongoing phase 2 study of binimetinib for progressive or symptomatic PN (NCT03231306) reveal PR in 70% (14/20) of pediatric and 65% (13/20) of adult participants.88,89 Mirdametinib had a response rate of 42% (8/19) in adolescents (≥16 years of age) and adults with progressive or symptomatic PN,63 and a larger study (NCT03962543) in both children and adults is ongoing. Preliminary results of a phase 1/2a trial of trametinib in children with PN (NCT02124772) revealed PR’s in 46% (12/26)90; other studies using trametinib are in progress (NCT03363217, NCT03741101) (Table 2).
A recent phase 2 trial of cabozantinib, a small molecule tyrosine kinase inhibitor of c-Kit, VEGFR2, MET, RET, FLT3, and the TAM family receptors, for adolescents and adults with progressive or symptomatic, inoperable PN demonstrated a 42% (8/19) PR rate,64 and has completed enrollment of a stratum of children (3–15 years of age) (NCT02101736). Caution is advised in comparing response rates between studies, given the differences in age at enrollment, PN inclusion criteria, and study design.
A major challenge going forward will be the identification of individualized treatment schedules and/or therapeutic combinations that can provide the best outcomes for all patients who require treatment for PN. These new agents have known and potentially unknown toxicities. Practitioners considering prescribing should be familiar with the various adverse events, potential interventions for side effects, and need for close monitoring. Thus, these agents are best prescribed by providers who have an appropriate infrastructure to rapidly address issues that arise on treatment. Intermittent dosing of effective agents may be a means to reduce toxicity and improve tolerability in patients on long-term therapy.91,92 This is being evaluated in a clinical trial of selumetinib for patients with NF1-associated tumors (NCT03326388). At present, the ideal length of treatment, durability of response, and long-term adverse events of continuous targeted inhibition are unknown. In addition, some PN do not respond or maintain a durable response to monotherapy. Combination therapies may be needed to improve overall response rate, durability of response, and depth of response. Last, although most studies have focused on treatment of PN already associated with morbidity, preventative therapy (ie treating the PN before it causes morbidity) may be a way to improve overall outcomes for patients with PN. Studies are planned to evaluate if prophylactic MEKi treatment of asymptomatic PN in high-risk locations can prevent tumor progression and morbidity.
Radiation Therapy
Evidence for using radiation therapy (RT) for treatment of PN is limited to retrospective studies. Biologic rationale for RT use is extrapolated from treatment of similar benign tumors including schwannoma and meningioma. Series using stereotactic radiosurgery (SRS) for benign spinal tumors (meningioma, schwannoma, and neurofibroma) include mostly adults, only a few with NF1, and do not specify if patients with PN were included.93–95 Stereotactic approaches are highly conformal techniques used to deliver high doses of RT to focal, well-defined tumors. As PN often have indistinct borders, delineation of RT target volumes for highly conformal techniques can be difficult. The few case reports using RT specifically for PN include various techniques (both conventional fractionation and stereotactic approaches), have limited follow up, and are not sufficient to inform management recommendations.
Primary concerns with use of RT in NF1-PN are compounding of the baseline risk of RT-induced neoplasms in NF1, as well as the malignant degeneration of PN into MPNST.22,96–98 There is also theoretical concern that RT could exacerbate underlying vasculopathy in patients with NF1, who are high risk for vasculopathy. RT should be avoided for the treatment of PN, particularly in children given this known risk and unknown benefit. This recommendation does not supplant clinical judgement in individual circumstances if RT is considered for local control and symptom management when resection or systemic therapy are not options. Multidisciplinary input regarding all treatment options, including resection, systemic therapies, and clinical trials, and careful weighing of the risk-benefit ratio, should be considered prior to recommending the use of RT for PN.
Alternative Therapies
Little is known about the use of complementary and alternative medicine (CAM) in the prevention and treatment of PN. In a survey of 1489 individuals in the Children’s Tumor Foundation NF registry,99 approximately 25% of respondents with NF1 regularly take dietary supplements or nutraceuticals specifically to treat symptoms associated with their NF. PN were the second most common complication targeted by nutraceutical use. Vitamin D and Fish oil were the most commonly used nutraceuticals overall, and turmeric/curcumin and bee propolis were the most common supplements used to try to prevent or treat PN. Cannabis derivatives were the most commonly used nutraceutical to treat NF-related pain. Of the 64 separate nutraceuticals indicated in the survey results, there are few reports of preclinical or prospective clinical data to support their use. In one small series, 2 patients receiving a combination of the Mediterranean diet and Curcumin had improvement in cutaneous neurofibroma tumor burden and one patient had a reduction in PN size, although these results have not been validated in a larger population.100 Overall, although CAM and nutraceuticals are frequently used and discussed in the NF1 population, there is little data to support efficacy in treating NF1-PN at this time.
Conclusion
Many recent advances have been made in the management of NF1-PN. In addition to surveillance, symptomatic management, and surgery, effective targeted medical therapies such as MEKi have become available. The regulatory approvals of the MEKi selumetinib for children with symptomatic inoperable PN is an important advance. PN which are causing morbidity, or growing and at risk for impending morbidity, should be considered for treatment with the modality deemed most appropriate based on location, symptoms, and patient goals. Validated preclinical models and meaningful clinical trial designs and outcome measures are available to guide clinical development of novel therapies. The clinical implementation of therapies for NF1 requires careful consideration of multiple factors and should be done with the input of a multidisciplinary team experienced in NF1.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Contributor Information
Michael J Fisher, Division of Oncology, The Children’s Hospital of Philadelphia and the University of Pennsylvania Perelman School of Medicine, Philadelphia, Pennsylvania, USA.
Jaishri O Blakeley, Division of Neuro-Oncology, Department of Neurology, Johns Hopkins University School of Medicine, Baltimore, Maryland, USA.
Brian D Weiss, Division of Oncology, Cincinnati Children’s Hospital Medical Center, University of Cincinnati College of Medicine, Cincinnati, Ohio, USA.
Eva Dombi, Pediatric Oncology Branch, National Cancer Institute, Bethesda, Maryland, USA.
Shivani Ahlawat, Russell H. Morgan Department of Radiology & Radiological Science, Johns Hopkins University School of Medicine, Baltimore, Maryland, USA.
Srivandana Akshintala, Pediatric Oncology Branch, National Cancer Institute, Bethesda, Maryland, USA.
Allan J Belzberg, Department of Neurosurgery, Johns Hopkins University School of Medicine, Baltimore, Maryland, USA.
Miriam Bornhorst, Family Neurofibromatosis Institute, Center for Neuroscience and Behavioral Medicine,Children’s National Hospital, Washington, District of Columbia, USA.
Miriam A Bredella, Department of Radiology, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
Wenli Cai, Department of Radiology, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
Rosalie E Ferner, Neurofibromatosis Service, Department of Neurology, Guy’s Hospital, Guy’s & St. Thomas’ NHS Foundation Trust, London, UK.
Andrea M Gross, Pediatric Oncology Branch, National Cancer Institute, Bethesda, Maryland, USA.
Gordon J Harris, Department of Radiology, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
Robert Listernick, Department of Pediatrics, Ann & Robert H. Lurie Children’s Hospital of Chicago, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, USA.
Ina Ly, Stephen E. and Catherine Pappas Center for Neuro-Oncology, Massachusetts General Hospital, Boston, Massachusetts, USA.
Staci Martin, Pediatric Oncology Branch, National Cancer Institute, Bethesda, Maryland, USA.
Victor F Mautner, Department of Neurology, University Medical Center Hamburg-Eppendorf, Hamburg, Germany.
Johannes M Salamon, Department for Diagnostic and Interventional Radiology and Nuclear Medicine, University Medical Center Hamburg-Eppendorf, Hamburg, Germany.
Kilian E Salerno, Radiation Oncology Branch, National Cancer Institute, Bethesda, Maryland, USA.
Robert J Spinner, Department of Neurologic Surgery, Mayo Clinic, Rochester, Minnesota, USA.
Verena Staedtke, Division of Neuro-Oncology, Department of Neurology, Johns Hopkins University School of Medicine, Baltimore, Maryland, USA.
Nicole J Ullrich, Department of Neurology, Boston Children’s Hospital, Boston, Massachusetts, USA.
Meena Upadhyaya, Division of Cancer and Genetics, Cardiff University, Wales, UK.
Pamela L Wolters, Pediatric Oncology Branch, National Cancer Institute, Bethesda, Maryland, USA.
Kaleb Yohay, Grossman School of Medicine, Department of Neurology, New York, New York, USA.
Brigitte C Widemann, Pediatric Oncology Branch, National Cancer Institute, Bethesda, Maryland, USA.
Funding
None.
Conflict of interest statement. A.J.B.: paid consulting or advisory role for AstraZeneca; J.O.B.: unpaid consulting or advisory role for AstraZeneca and SpringWorks; B.D.W.: unpaid consulting or advisory role for Springworks; E.D.: unpaid consulting or advisory role for AstraZeneca and SpringWorks; M.B.: paid consulting or advisory role for AstraZeneca; W.C.: stockholder, IQ Medical Imaging LLC; R.E.F.: paid consulting or advisory role for AstraZeneca; M.J.F.: unpaid consulting or advisory role for AstraZeneca and SpringWorks, research support from AstraZeneca, Array BioPharma (subsidiary of Pfizer) and Exelixis; A.M.G.: unpaid consulting or advisory role for AstraZeneca and SpringWorks; G.J.H.: member, Novometrics LLC and IQ Medical Imaging LLC, Advisor, Fovia Inc; R.L.: paid consulting or advisory role for AstraZeneca; I.L.: unpaid consulting or advisory role for SpringWorks; V.F.M.: paid consulting or advisory role for AstraZeneca; N.J.U.: paid consulting or advisory role for AstraZeneca; B.C.W.: unpaid consulting or advisory role for AstraZeneca and SpringWorks; P.L.W.: unpaid consulting or advisory role for SpringWorks; K.Y.: paid consulting or advisory role for AstraZeneca. The other authors report no potential conflicts.
Authorship statement. Conception and design: M.J.F., B.C.W., J.O.B., B.D.W. Manuscript writing and final approval: All authors contributed and approved.
References
- 1. Staser K, Yang FC, Clapp DW. Pathogenesis of plexiform neurofibroma: tumor-stromal/hematopoietic interactions in tumor progression. Annu Rev Pathol. 2012; 7:469–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kluwe L, Friedrich RE, Mautner VF. Allelic loss of the NF1 gene in NF1-associated plexiform neurofibromas. Cancer Genet Cytogenet. 1999; 113(1):65–69. [DOI] [PubMed] [Google Scholar]
- 3. Serra E, Rosenbaum T, Winner U, et al. . Schwann cells harbor the somatic NF1 mutation in neurofibromas: evidence of two different Schwann cell subpopulations. Hum Mol Genet. 2000; 9(20):3055–3064. [DOI] [PubMed] [Google Scholar]
- 4. Yang FC, Ingram DA, Chen S, et al. . Nf1-dependent tumors require a microenvironment containing Nf1+/-- and c-kit-dependent bone marrow. Cell. 2008; 135(3):437–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Huson SM, Harper PS, Compston DA. Von Recklinghausen neurofibromatosis. A clinical and population study in south-east Wales. Brain. 1988; 111(Pt 6):1355–1381. [DOI] [PubMed] [Google Scholar]
- 6. Plotkin SR, Bredella MA, Cai W, et al. . Quantitative assessment of whole-body tumor burden in adult patients with neurofibromatosis. PLoS One. 2012; 7(4):e35711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jett K, Nguyen R, Arman D, et al. . Quantitative associations of scalp and body subcutaneous neurofibromas with internal plexiform tumors in neurofibromatosis 1. Am J Med Genet A. 2015; 167(7):1518–1524. [DOI] [PubMed] [Google Scholar]
- 8. Mautner VF, Asuagbor FA, Dombi E, et al. . Assessment of benign tumor burden by whole-body MRI in patients with neurofibromatosis 1. Neuro Oncol. 2008; 10(4):593–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nguyen R, Kluwe L, Fuensterer C, et al. . Plexiform neurofibromas in children with neurofibromatosis type 1: frequency and associated clinical deficits. J Pediatr. 2011; 159(4):652–5.e2. [DOI] [PubMed] [Google Scholar]
- 10. Waggoner DJ, Towbin J, Gottesman G, Gutmann DH. Clinic-based study of plexiform neurofibromas in neurofibromatosis 1. Am J Med Genet. 2000; 92(2):132–135. [PubMed] [Google Scholar]
- 11. Akshintala S, Baldwin A, Liewehr DJ, et al. . Longitudinal evaluation of peripheral nerve sheath tumors in neurofibromatosis type 1: growth analysis of plexiform neurofibromas and distinct nodular lesions. Neuro Oncol. 2020; 22(9):1368–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mautner VF, Hartmann M, Kluwe L, Friedrich RE, Funsterer C. MRI growth patterns of plexiform neurofibromas in patients with neurofibromatosis type 1. Neuroradiology. 2006; 48(3):160–165. [DOI] [PubMed] [Google Scholar]
- 13. Prada CE, Rangwala FA, Martin LJ, et al. . Pediatric plexiform neurofibromas: impact on morbidity and mortality in neurofibromatosis type 1. J Pediatr. 2012; 160(3):461–467. [DOI] [PubMed] [Google Scholar]
- 14. Avery RA, Dombi E, Hutcheson KA, et al. . Visual outcomes in children with neurofibromatosis type 1 and orbitotemporal plexiform neurofibromas. Am J Ophthalmol. 2013; 155(6):1089–1094.e1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zacharia TT, Jaramillo D, Poussaint TY, Korf B. MR imaging of abdominopelvic involvement in neurofibromatosis type 1: a review of 43 patients. Pediatr Radiol. 2005; 35(3):317–322. [DOI] [PubMed] [Google Scholar]
- 16. Azhar AF, Bittle JSH, Kwarcinski TJ, Hinshelwood JR. Embolization of a hemorrhaging abdominal plexiform neurofibroma. Proc (Bayl Univ Med Cent). 2020; 33(3):448–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Feng Y, Yang ZG, Chen T, Wang Q, Deng W. Giant plexiform neurofibroma with hemorrhage in cranio-maxillofacial region as depicted on CT and MRI. Eur J Med Res. 2010; 15(2):84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Miettinen MM, Antonescu CR, Fletcher CDM, et al. . Histopathologic evaluation of atypical neurofibromatous tumors and their transformation into malignant peripheral nerve sheath tumor in patients with neurofibromatosis 1-a consensus overview. Hum Pathol. 2017; 67:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rodriguez FJ, Reuss DE, Stemmer-Rachamimov AO. Neurofibroma. In: Cree IA, Lokuhetty D, Peferoen LAN, White VA, eds. WHO Classification of Tumours: Central Nervous System. 5th ed. Lyon: International Agency for Research on Cancer; 2021:265–268. [Google Scholar]
- 20. Pemov A, Hansen NF, Sindiri S, et al. . Low mutation burden and frequent loss of CDKN2A/B and SMARCA2, but not PRC2, define premalignant neurofibromatosis type 1-associated atypical neurofibromas. Neuro Oncol. 2019; 21(8):981–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Higham CS, Dombi E, Rogiers A, et al. . The characteristics of 76 atypical neurofibromas as precursors to neurofibromatosis 1 associated malignant peripheral nerve sheath tumors. Neuro Oncol. 2018; 20(6):818–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Uusitalo E, Rantanen M, Kallionpaa RA, et al. . Distinctive cancer associations in patients with neurofibromatosis type 1. J Clin Oncol. 2016; 34(17):1978–1986. [DOI] [PubMed] [Google Scholar]
- 23. Ahlawat S, Ly KI, Fayad LM, et al. . Imaging evaluation of plexiform neurofibromas in neurofibromatosis type 1: a survey-based assessment. Neurology. 2021; 97(7 Suppl 1):S111–S119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Evans DGR, Salvador H, Chang VY, et al. . Cancer and central nervous system tumor surveillance in pediatric neurofibromatosis 1. Clin Cancer Res. 2017; 23(12):e46–e53. [DOI] [PubMed] [Google Scholar]
- 25. Nguyen R, Dombi E, Widemann BC, et al. . Growth dynamics of plexiform neurofibromas: a retrospective cohort study of 201 patients with neurofibromatosis 1. Orphanet J Rare Dis. 2012; 7:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ly I, Thalheimer R, Cai W, Bredella M. Long-term follow-up of adult neurofibromatosis type 1 patients using whole-body MRI demonstrates dynamic changes in internal neurofibroma size. Eur J Neurol. 2020; 27(Suppl 1):97. [Google Scholar]
- 27. Pinna V, Lanari V, Daniele P, et al. . p.Arg1809Cys substitution in neurofibromin is associated with a distinctive NF1 phenotype without neurofibromas. Eur J Hum Genet. 2015; 23(8):1068–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rojnueangnit K, Xie J, Gomes A, et al. . High incidence of Noonan syndrome features including short stature and pulmonic stenosis in patients carrying NF1 missense mutations affecting p.Arg1809: genotype-phenotype correlation. Hum Mutat. 2015; 36(11):1052–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. van Minkelen R, van Bever Y, Kromosoeto JN, et al. . A clinical and genetic overview of 18 years neurofibromatosis type 1 molecular diagnostics in the Netherlands. Clin Genet. 2014; 85(4):318–327. [DOI] [PubMed] [Google Scholar]
- 30. Trevisson E, Morbidoni V, Forzan M, et al. . The Arg1038Gly missense variant in the NF1 gene causes a mild phenotype without neurofibromas. Mol Genet Genomic Med. 2019; 7(5):e616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Upadhyaya M, Huson SM, Davies M, et al. . An absence of cutaneous neurofibromas associated with a 3-bp inframe deletion in exon 17 of the NF1 gene (c.2970-2972 delAAT): evidence of a clinically significant NF1 genotype-phenotype correlation. Am J Hum Genet. 2007; 80(1):140–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Koczkowska M, Callens T, Gomes A, et al. . Expanding the clinical phenotype of individuals with a 3-bp in-frame deletion of the NF1 gene (c.2970_2972del): an update of genotype-phenotype correlation. Genet Med. 2019; 21(4):867–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Koczkowska M, Callens T, Chen Y, et al. . Clinical spectrum of individuals with pathogenic NF1 missense variants affecting p.Met1149, p.Arg1276, and p.Lys1423: genotype-phenotype study in neurofibromatosis type 1. Hum Mutat. 2020; 41(1):299–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Koczkowska M, Chen Y, Callens T, et al. . Genotype-phenotype correlation in NF1: evidence for a more severe phenotype associated with missense mutations affecting NF1 codons 844-848. Am J Hum Genet. 2018; 102(1):69–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dombi E, Solomon J, Gillespie AJ, et al. . NF1 plexiform neurofibroma growth rate by volumetric MRI: relationship to age and body weight. Neurology. 2007; 68(9):643–647. [DOI] [PubMed] [Google Scholar]
- 36. Tucker T, Friedman JM, Friedrich RE, et al. . Longitudinal study of neurofibromatosis 1 associated plexiform neurofibromas. J Med Genet. 2009; 46(2):81–85. [DOI] [PubMed] [Google Scholar]
- 37. Dagalakis U, Lodish M, Dombi E, et al. . Puberty and plexiform neurofibroma tumor growth in patients with neurofibromatosis type I. J Pediatr. 2014; 164(3):620–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Well L, Jaeger A, Kehrer-Sawatzki H, et al. . The effect of pregnancy on growth-dynamics of neurofibromas in Neurofibromatosis type 1. PLoS One. 2020; 15(4):e0232031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Filler AG, Maravilla KR, Tsuruda JS. MR neurography and muscle MR imaging for image diagnosis of disorders affecting the peripheral nerves and musculature. Neurol Clin. 2004; 22(3):643–682, vi-vii. [DOI] [PubMed] [Google Scholar]
- 40. Huang JH, Zhang J, Zager EL. Diagnosis and treatment options for nerve sheath tumors. Expert Rev Neurother. 2005; 5(4):515–523. [DOI] [PubMed] [Google Scholar]
- 41. Tovmassian D, Abdul Razak M, London K. The role of [(18)F]FDG-PET/CT in predicting malignant transformation of plexiform neurofibromas in neurofibromatosis-1. Int J Surg Oncol. 2016; 2016:6162182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Raad RA, Lala S, Allen JC, et al. . Comparison of hybrid 18F-fluorodeoxyglucose positron emission tomography/magnetic resonance imaging and positron emission tomography/computed tomography for evaluation of peripheral nerve sheath tumors in patients with neurofibromatosis type 1. World J Nucl Med. 2018; 17(4):241–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ahlawat S, Blakeley JO, Rodriguez FJ, Fayad LM. Imaging biomarkers for malignant peripheral nerve sheath tumors in neurofibromatosis type 1. Neurology. 2019; 93(11):e1076–e1084. [DOI] [PubMed] [Google Scholar]
- 44. Well L, Salamon J, Kaul MG, et al. . Differentiation of peripheral nerve sheath tumors in patients with neurofibromatosis type 1 using diffusion-weighted magnetic resonance imaging. Neuro Oncol. 2019; 21(4):508–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Brahmi M, Thiesse P, Ranchere D, et al. . Diagnostic accuracy of PET/CT-guided percutaneous biopsies for malignant peripheral nerve sheath tumors in neurofibromatosis type 1 patients. PLoS One. 2015; 10(10):e0138386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Macpherson RE, Pratap S, Tyrrell H, et al. . Retrospective audit of 957 consecutive (18)F-FDG PET-CT scans compared to CT and MRI in 493 patients with different histological subtypes of bone and soft tissue sarcoma. Clin Sarcoma Res. 2018; 8:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dombi E, Ardern-Holmes SL, Babovic-Vuksanovic D, et al. . Recommendations for imaging tumor response in neurofibromatosis clinical trials. Neurology. 2013; 81(21 Suppl 1):S33–S40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cai W, Steinberg SM, Bredella MA, et al. . Volumetric MRI analysis of plexiform neurofibromas in neurofibromatosis type 1: comparison of two methods. Acad Radiol. 2018; 25(2):144–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gross AM, Singh G, Akshintala S, et al. . Association of plexiform neurofibroma volume changes and development of clinical morbidities in neurofibromatosis 1. Neuro Oncol. 2018; 20(12):1643–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ferner RE, Thomas M, Mercer G, et al. . Evaluation of quality of life in adults with neurofibromatosis 1 (NF1) using the Impact of NF1 on Quality Of Life (INF1-QOL) questionnaire. Health Qual Life Outcomes. 2017; 15(1):34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mullin RL, Golding JF, Smith R, et al. . Reliability of functional outcome measures in adults with neurofibromatosis 1. SAGE Open Med. 2018; 6:2050312118786860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Plotkin SR, Davis SD, Robertson KA, et al. . Sleep and pulmonary outcomes for clinical trials of airway plexiform neurofibromas in NF1. Neurology. 2016; 87(7 Suppl 1):S13–S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fisher MJ, Avery RA, Allen JC, et al. . Functional outcome measures for NF1-associated optic pathway glioma clinical trials. Neurology. 2013; 81(21 Suppl 1):S15–S24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gross AM, Wolters PL, Dombi E, et al. . Selumetinib in children with inoperable plexiform neurofibromas. N Engl J Med. 2020; 382(15):1430–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jensen SE, Patel ZS, Listernick R, Charrow J, Lai JS. Lifespan development: symptoms experienced by individuals with neurofibromatosis type 1 associated plexiform neurofibromas from childhood into adulthood. J Clin Psychol Med Settings. 2019; 26(3):259–270. [DOI] [PubMed] [Google Scholar]
- 56. Creange A, Zeller J, Rostaing-Rigattieri S, et al. . Neurological complications of neurofibromatosis type 1 in adulthood. Brain. 1999; 122(Pt 3):473–481. [DOI] [PubMed] [Google Scholar]
- 57. Kim A, Gillespie A, Dombi E, et al. . Characteristics of children enrolled in treatment trials for NF1-related plexiform neurofibromas. Neurology. 2009; 73(16):1273–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Watson KL, Al Sannaa GA, Kivlin CM, et al. . Patterns of recurrence and survival in sporadic, neurofibromatosis Type 1-associated, and radiation-associated malignant peripheral nerve sheath tumors. J Neurosurg. 2017;126(1):319–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Valentin T, Le Cesne A, Ray-Coquard I, et al. . Management and prognosis of malignant peripheral nerve sheath tumors: the experience of the French Sarcoma Group (GSF-GETO). Eur J Cancer. 2016; 56:77–84. [DOI] [PubMed] [Google Scholar]
- 60. Stewart DR, Korf BR, Nathanson KL, Stevenson DA, Yohay K. Care of adults with neurofibromatosis type 1: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2018; 20(7):671–682. [DOI] [PubMed] [Google Scholar]
- 61. Moutal A, Cai S, Luo S, Voisin R, Khanna R. CRMP2 is necessary for Neurofibromatosis type 1 related pain. Channels (Austin). 2018; 12(1):47–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ji RR, Baba H, Brenner GJ, Woolf CJ. Nociceptive-specific activation of ERK in spinal neurons contributes to pain hypersensitivity. Nat Neurosci. 1999; 2(12):1114–1119. [DOI] [PubMed] [Google Scholar]
- 63. Weiss BD, Wolters PL, Plotkin SR, et al. . NF106: a neurofibromatosis clinical trials consortium phase II trial of the MEK inhibitor mirdametinib (PD-0325901) in adolescents and adults with NF1-related plexiform neurofibromas. J Clin Oncol. 2021; 39(7):797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Fisher MJ, Shih CS, Rhodes SD, et al. . Cabozantinib for neurofibromatosis type 1-related plexiform neurofibromas: a phase 2 trial. Nat Med. 2021; 27(1):165–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hughes LS, Clark J, Colclough JA, Dale E, McMillan D. Acceptance and commitment therapy (ACT) for chronic pain: a systematic review and meta-analyses. Clin J Pain. 2017; 33(6):552–568. [DOI] [PubMed] [Google Scholar]
- 66. Martin S, Allen T, Toledo-Tamula MA, et al. . Acceptance and commitment therapy for adolescents and adults with neurofibromatosis type 1, plexiform neurofibromas, and chronic pain: results of a randomized controlled trial. J Contextual Behav Sci. 2021; 22:93–101. [Google Scholar]
- 67. Ehde DM, Dillworth TM, Turner JA. Cognitive-behavioral therapy for individuals with chronic pain: efficacy, innovations, and directions for research. Am Psychol. 2014; 69(2):153–166. [DOI] [PubMed] [Google Scholar]
- 68. Hilton L, Hempel S, Ewing BA, et al. . Mindfulness meditation for chronic pain: systematic review and meta-analysis. Ann Behav Med. 2017; 51(2):199–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Palermo TM, Eccleston C, Lewandowski AS, de C Williams AC, Morley S. Randomized controlled trials of psychological therapies for management of chronic pain in children and adolescents: an updated meta-analytic review. Pain. 2010; 148(3):387–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Paley CA, Johnson MI. Acupuncture for the relief of chronic pain: a synthesis of systematic reviews. Medicina (Kaunas). 2019; 56(1):1–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gibson W, Wand BM, Meads C, Catley MJ, O’Connell NE. Transcutaneous electrical nerve stimulation (TENS) for chronic pain - an overview of Cochrane Reviews. Cochrane Database Syst Rev. 2019; 4(4):CD011890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Martin S, Wolters P, Baldwin A, et al. . Social-emotional functioning of children and adolescents with neurofibromatosis type 1 and plexiform neurofibromas: relationships with cognitive, disease, and environmental variables. J Pediatr Psychol. 2012; 37(7):713–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wolters PL, Burns KM, Martin S, et al. . Pain interference in youth with neurofibromatosis type 1 and plexiform neurofibromas and relation to disease severity, social-emotional functioning, and quality of life. Am J Med Genet A. 2015; 167A(9):2103–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wolters PL, Rosser T, Dombi E, Cutter G. Substantial Pain and reduced quality of life in adolescents and young adults with neurofibromatosis type 1 and plexiform neurofibromas enrolled in NF Consortium PN Clinical Trials. In: CTF Global Joint NF Conference, Paris, France; 2018. [Google Scholar]
- 75. Lai JS, Jensen SE, Patel ZS, Listernick R, Charrow J. Using a qualitative approach to conceptualize concerns of patients with neurofibromatosis type 1 associated plexiform neurofibromas (pNF) across the lifespan. Am J Med Genet A. 2017; 173(1):79–87. [DOI] [PubMed] [Google Scholar]
- 76. Widemann BC, Dombi E, Gillespie A, et al. . Phase 2 randomized, flexible crossover, double-blinded, placebo-controlled trial of the farnesyltransferase inhibitor tipifarnib in children and young adults with neurofibromatosis type 1 and progressive plexiform neurofibromas. Neuro Oncol. 2014; 16(5):707–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Vranceanu AM, Riklin E, Merker VL, et al. . Mind-body therapy via videoconferencing in patients with neurofibromatosis: an RCT. Neurology. 2016; 87(8):806–814. [DOI] [PubMed] [Google Scholar]
- 78. Needle MN, Cnaan A, Dattilo J, et al. . Prognostic signs in the surgical management of plexiform neurofibroma: the Children’s Hospital of Philadelphia experience, 1974-1994. J Pediatr. 1997; 131(5):678–682. [DOI] [PubMed] [Google Scholar]
- 79. Avery RA, Katowitz JA, Fisher MJ, et al. . Orbital/periorbital plexiform neurofibromas in children with neurofibromatosis type 1: multidisciplinary recommendations for care. Ophthalmology. 2017; 124(1):123–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Leonard JR, Ferner RE, Thomas N, Gutmann DH. Cervical cord compression from plexiform neurofibromas in neurofibromatosis 1. J Neurol Neurosurg Psychiatry. 2007; 78(12):1404–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Pollack IF, Colak A, Fitz C, Wiener E, Moreland M, Mulvihill JJ.. Surgical management of spinal cord compression from plexiform neurofibromas in patients with neurofibromatosis 1. Neurosurgery. 1998; 43(2):248–255; discussion 255-246. [DOI] [PubMed] [Google Scholar]
- 82. Nelson CN, Dombi E, Rosenblum JS, et al. . Safe marginal resection of atypical neurofibromas in neurofibromatosis type 1. J Neurosurg. 2019; 133(5):1516–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ratner N, Miller SJ. A RASopathy gene commonly mutated in cancer: the neurofibromatosis type 1 tumour suppressor. Nat Rev Cancer. 2015; 15(5):290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Widemann BC, Babovic-Vuksanovic D, Dombi E, et al. . Phase II trial of pirfenidone in children and young adults with neurofibromatosis type 1 and progressive plexiform neurofibromas. Pediatr Blood Cancer. 2014; 61(9):1598–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Jakacki RI, Dombi E, Steinberg SM, et al. . Phase II trial of pegylated interferon alfa-2b in young patients with neurofibromatosis type 1 and unresectable plexiform neurofibromas. Neuro Oncol. 2017; 19(2):289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Dombi E, Baldwin A, Marcus LJ, et al. . Activity of selumetinib in neurofibromatosis type 1-related plexiform neurofibromas. N Engl J Med. 2016; 375(26):2550–2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Coyne GHOS, Gross AM, Dombi E, et al. . Phase II trial of the MEK 1/2 inhibitor selumetinib (AZD6244, ARRY-142886 Hydrogen Sulfate) in adults with neurofibromatosis type 1 and inoperable plexiform neurofibromas. J Clin Oncol. 2020; 38(15_suppl):3612–3612. [Google Scholar]
- 88. Mueller S, Reddy A, Dombi E, et al. . MEK inhibitor Binimetinib shows clinical activity in children with neurofibromatosis type 1-associated plexiform neurofibromas: a report from PNOC and the NF Clinical Trials Consortium. In: International Symposium on Pediatric Neuro-Oncology, Karuizawa, Japan; 2020. [Google Scholar]
- 89. Reddy A, Fisher M, Dombi E, et al. . Binimetinib leads to radiographic response in adults with Neurofibromatosis Type 1 associated plexiform neurofibromas: a report from the NFCTC and PNOC. In: Paper presented at: Children’s Tumor Foundation Neurofibromatosis Virtual Conference; 2020. [Google Scholar]
- 90. McCowage GB, Mueller S, Pratilas CA, et al. . Trametinib in pediatric patients with neurofibromatosis type 1 (NF-1)–associated plexiform neurofibroma: a phase I/IIa study. J Clin Oncol. 2018; 36(15_suppl):10504–10504. [Google Scholar]
- 91. Reger de Moura C, Vercellino L, Jouenne F, et al. . Intermittent versus continuous dosing of MAPK inhibitors in the treatment of BRAF-mutated melanoma. Transl Oncol. 2020; 13(2):275–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Matter AV, Micaletto S, Urner-Bloch U, Dummer R, Goldinger SM. Long-term response to intermittent binimetinib in patients with NRAS-mutant melanoma. Oncologist. 2020; 25(11):e1593–e1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Chin AL, Fujimoto D, Kumar KA, et al. . Long-term update of stereotactic radiosurgery for benign spinal tumors. Neurosurgery. 2019; 85(5):708–716. [DOI] [PubMed] [Google Scholar]
- 94. Selch MT, Lin K, Agazaryan N, et al. . Initial clinical experience with image-guided linear accelerator-based spinal radiosurgery for treatment of benign nerve sheath tumors. Surg Neurol. 2009; 72(6):668–674; discussion 674-665. [DOI] [PubMed] [Google Scholar]
- 95. Gerszten PC, Burton SA, Ozhasoglu C, McCue KJ, Quinn AE. Radiosurgery for benign intradural spinal tumors. Neurosurgery. 2008; 62(4):887–95; discussion 895-886. [DOI] [PubMed] [Google Scholar]
- 96. Bhatia S, Chen Y, Wong FL, et al. . Subsequent neoplasms after a primary tumor in individuals with neurofibromatosis type 1. J Clin Oncol. 2019; 37(32):3050–3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Evans DG, Baser ME, McGaughran J, et al. . Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet. 2002; 39(5):311–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Sharif S, Ferner R, Birch JM, et al. . Second primary tumors in neurofibromatosis 1 patients treated for optic glioma: substantial risks after radiotherapy. J Clin Oncol. 2006; 24(16):2570–2575. [DOI] [PubMed] [Google Scholar]
- 99. Peires-Hughes M, Anstett K, Engelson C, Yohay K. The use of dietary supplements by persons with neurofibromatosis: a survey study. In: Children’s Tumor Foundation NF Conference, Washington, DC; 2017. [Google Scholar]
- 100. Esposito T, Schettino C, Polverino P, et al. . Synergistic interplay between curcumin and polyphenol-rich foods in the mediterranean diet: therapeutic prospects for neurofibromatosis 1 patients. Nutrients. 2017; 9(7):783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Korf BR. Plexiform neurofibromas. Am J Med Genet. 1999; 89(1):31–37. [DOI] [PubMed] [Google Scholar]
- 102. Ferner RE, Gutmann DH. Neurofibromatosis type 1 (NF1): diagnosis and management. Handb Clin Neurol. 2013; 115:939–955. [DOI] [PubMed] [Google Scholar]
- 103. Lim R, Jaramillo D, Poussaint TY, Chang Y, Korf B. Superficial neurofibroma: a lesion with unique MRI characteristics in patients with neurofibromatosis type 1. AJR Am J Roentgenol. 2005; 184(3):962–968. [DOI] [PubMed] [Google Scholar]
- 104. Gupta A, Cohen BH, Ruggieri P, Packer RJ, Phillips PC. Phase I study of thalidomide for the treatment of plexiform neurofibroma in neurofibromatosis 1. Neurology. 2003; 60(1):130–132. [DOI] [PubMed] [Google Scholar]
- 105. Widemann BC, Salzer WL, Arceci RJ, et al. . Phase I trial and pharmacokinetic study of the farnesyltransferase inhibitor tipifarnib in children with refractory solid tumors or neurofibromatosis type I and plexiform neurofibromas. J Clin Oncol. 2006; 24(3):507–516. [DOI] [PubMed] [Google Scholar]
- 106. Jakacki RI, Dombi E, Potter DM, et al. . Phase I trial of pegylated interferon-alpha-2b in young patients with plexiform neurofibromas. Neurology. 2011; 76(3):265–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Babovic-Vuksanovic D, Widemann BC, Dombi E, et al. . Phase I trial of pirfenidone in children with neurofibromatosis 1 and plexiform neurofibromas. Pediatr Neurol. 2007; 36(5):293–300. [DOI] [PubMed] [Google Scholar]
- 108. Kim A, Dombi E, Tepas K, et al. . Phase I trial and pharmacokinetic study of sorafenib in children with neurofibromatosis type I and plexiform neurofibromas. Pediatr Blood Cancer. 2013; 60(3):396–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Boal LH, Glod J, Spencer M, et al. . Pediatric PK/PD phase I trial of pexidartinib in relapsed and refractory leukemias and solid tumors including neurofibromatosis type I-related plexiform neurofibromas. Clin Cancer Res. 2020; 26(23):6112–6121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Babovic-Vuksanovic D, Ballman K, Michels V, et al. . Phase II trial of pirfenidone in adults with neurofibromatosis type 1. Neurology. 2006; 67(10):1860–1862. [DOI] [PubMed] [Google Scholar]
- 111. Robertson KA, Nalepa G, Yang FC, et al. . Imatinib mesylate for plexiform neurofibromas in patients with neurofibromatosis type 1: a phase 2 trial. Lancet Oncol. 2012; 13(12):1218–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Weiss B, Widemann BC, Wolters P, et al. . Sirolimus for progressive neurofibromatosis type 1-associated plexiform neurofibromas: a neurofibromatosis Clinical Trials Consortium phase II study. Neuro Oncol. 2015; 17(4):596–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Weiss B, Widemann BC, Wolters P, et al. . Sirolimus for non-progressive NF1-associated plexiform neurofibromas: an NF clinical trials consortium phase II study. Pediatr Blood Cancer. 2014; 61(6):982–986. [DOI] [PubMed] [Google Scholar]
- 114. Zehou O, Ferkal S, Brugieres P, et al. . Absence of efficacy of everolimus in neurofibromatosis 1-related plexiform neurofibromas: results from a phase 2a trial. J Invest Dermatol. 2019; 139(3):718–720. [DOI] [PubMed] [Google Scholar]
- 115. Glassberg B, Gross A, Dombi E, et al. . Selumetinib in children with clinically asymptomatic inoperable neurofibromatosis type 1 related plexiform neurofibromas. In: Paper presented at: Children’s Tumor Foundation Neurofibromatosis Virtual Conference; 2020. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.