

Abstract
The heterogeneity of bacterial growth and replicative rates within a population was proposed a century ago notably to explain the presence of bacterial persisters. The term “growth rate” at the single-cell level corresponds to the increase in size or mass of an individual bacterium while the “replicative rate” refers to its division capacity within a defined temporality. After a decades long hiatus, recent technical innovative approaches allow population growth and replicative rates heterogeneity monitoring at the single-cell level resuming in earnest. Among these techniques, the oldest and widely used is time-lapse microscopy, most recently combined with microfluidics. We also discuss recent fluorescence dilution methods informing only on replicative rates and best suited. Some new elegant single cell methods so far only sporadically used such as buoyant mass measurement and stable isotope probing have emerged. Overall, such tools are widely used to investigate and compare the growth and replicative rates of bacteria displaying drug-persistent behaviors to that of bacteria growing in specific ecological niches or collected from patients. In this review, we describe the current methods available, discussing both the type of queries these have been used to answer and the specific strengths and limitations of each method.
Review of the current available methods to monitor at the single-cell level the bacterial growth and replicative rates, discussing both the type of queries these have been used to answer and the specific strengths and limitations of each method.
Introduction
To an historical perspective, at the beginning of the 20th century, scientists had already emphasized the importance of measuring the bacterial replicative rate at the single cell level (Barber 1908, Reichenbach 1911, Wilson 1922, Kelly and Rahn 1932). It correlated with the concomitantly wide development of techniques to isolate single microorganisms as reviewed by Hildebrand in 1938 (Hildebrand 1938). Marshall Barber developed an elegant mechanical method to isolate single cell within microdrops involving capillary micropipettes that he combined with manual time-lapse microscopy (Barber 1904). Helped by this method Barber investigated the replicative rates at the single-cell level of Bacillus coli at different temperatures and further demonstrated that homologous immune antipneumococcic serum had no effect on the Pneumococcus replicative rate of single isolated bacterial pairs (Barber 1908, 1919). Meanwhile, bacteriologists questioned the accuracy of population measurements, especially when specific subsets were found unable to grow on agar plates (Reichenbach 1911, Wilson 1922). In 1922, Wilson highlighted the frequent discrepancy of colony forming unit measurements and direct microscopic counts (Wilson 1922). This was assigned to a subset of bacteria within an actively growing population that would not grow on agar plates but could still be seen looking down a microscope. He proposed that this subpopulation corresponded to dead bacteria (Wilson 1922). Before that, Reichenbach had proposed that a certain proportion of the actively growing bacteria became dormant at each new generation and that, therefore, this population increased with age explaining the viable but non-culturable bacterial subset observed (Reichenbach 1911). Later, Kelly and Rahn tested these hypotheses by studying the replicative rate of single isolated bacteria via manual time-lapse microscopy (Kelly and Rahn 1932). Interestingly, they showed that all cells continue to multiply once they have started to do so and that no progenies cease growing, become dormant or die. Results must be counterbalanced by the temporal limitation: tracking was not possible beyond the fourth generation. Moreover, few cases were recorded where a cell did not divide in the new medium as previously shown by Barber (Barber 1908, Kelly and Rahn 1932). A decade later, Hobby and Bigger both separately working on the mechanism of action of penicillin toward streptococci and staphylococci showed that it killed the actively growing bacteria (Hobby et al. 1942, Bigger 1944). Bigger discovered and employed for the first time the term persisters to name a small subset of antibiotic-insensitive, drug-persistent bacteria within a population of replicating cells (Bigger 1944). Persisters were supposed to persist penicillin pressure owing to their arrest in a temporally dormant, non-dividing phase, corroborating Reichenbach's assumption. After a decades long hiatus, the study of growth and replicative bacterial rates at the single-cell level resumed in earnest only in the last decades (Balaban et al. 2004, Godin et al. 2010, Helaine et al. 2010, Wang et al. 2010; Wakamoto et al. 2013, Claudi et al. 2014, Ursell et al. 2014, Flannagan and Heinrichs 2018, Table 1).
Table 1:
Summary of the different methods available to monitor at the single-cell level the growth and replicative rates.
| Methods | Schemes | Purposes | Number of generations tracking | Strengths | Limitations | ||
|---|---|---|---|---|---|---|---|
| Mains methods | Monitoring of colony appearance and size |
|
The first replicative step and the bacterial lag time. Colony growth rates. Viable but non-cultivable bacteria ratio. | 1 | Rapid and inexpensive user-friendly method. | Indirect method. Requires a known number of bacteria spread. | |
| Time-lapse microscopy and microfluidics |
|
Growth and replicative rates. Inter and intra lineages comparisons. Mother cell and/or progenies tracking | From 4 to hundreds | Long generations tracking. Combined measurement of growth and replicative rates. Works with concomitant genes fluorescent reporter. | Constant microscopic tracking requirement. End-point monitoring failed. | ||
| Dyes dilution |
|
Replicative rates | Up to 8 | Tremendous events analyze at a time by FACS. Fit with in cellulo and in situ analysis. | Mismatch lineages and growth rates monitoring. Limited generations tracking. Previous genetic engineering or dyes labeling required. | ||
| Fluorescent proteins dilution |
|
Replicative rates | Up to 10 | Tremendous events analyze at a time by FACS. Report on metabolic behavior following replicative monitoring. Fit with in cellulo and in vivo analysis. | Mismatch lineages and growth rates monitoring. Limited generations tracking. Previous genetic engineering or dyes labeling required. | ||
| Fluorescent proteins dilution and maturation kinetic |
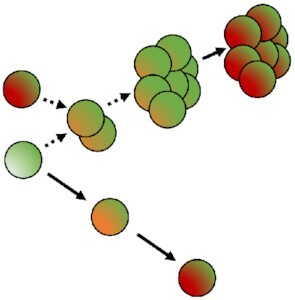
|
Replicative rates | Not tested, theoretically infinite | Tremendous events analyze at a time by FACS. Overcome generations tracking limitation. Fit with in cellulo and in vivo analysis. Does not require inducer. | Mismatch lineages and growth rates monitoring. Long time required to detect replicative rates switching. DsRed works best with aerobic condition. Poor discrimination of the extremes replicative rates. Previous genetic engineering required. | ||
| Structural markers | Septum and regrowth delay-body |
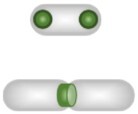
|
Regrowth delay-body (RDB): distinguishes non-growing subset; monitoring the bacterial lag time. Septum: replicative rates. | None | RDB: end-point analysis allowing discrimination based on the growth behavior. Septum: combined with time-lapse microscopy allows hundreds of generations tracking. | RDB: mismatch lineages. Manichean discrimination depending on the growth behavior. Septum: required time-lapse microscopy. | |
| Peptidoglycan and mycomembrane synthesis enlightenment |
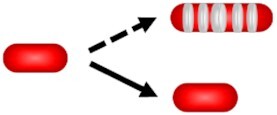
|
Bacterial and subcellular growth rates. Distinguishes dividing subset | None | Report on subcellular growth patterns. Fit with intracellular analysis. | Mismatch lineages. To our knowledge, not employed to discriminate bacterial subset depending on growth and replicative behavior despite its theoretical workability. | ||
| Emerging and/or sporadically used methods | Suspended microchannel resonator |
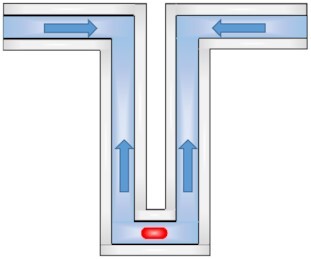
|
Growth rates | Few minutes | Precise growth rate measurement relying on buoyant mass. | Weak events at a time. Short tracking. System not broadly used. | |
| Chromosomal replication monitoring |
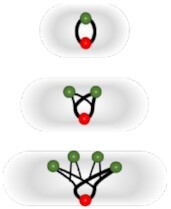
|
Distinguish respectively growing or replicating versus non-growing or non-replicating subsets | None | Fit with in vivo assays. Based for growth monitoring on replication initiation at conserved bacterial sized per chromosome. | Previous genetic engineering required. Manichean growth analysis depending on integer ratio. Manichean replicative monitoring based on time-lapse microscopy control. Work with mass doubling times < 60 minutes. Mismatch lineages. | ||
| Stable isotope probing |
|
Bacterial and subcellular growth rates | None | Report on the isotope incorporation notably via anabolic activity. Allowed monitoring of clinical isolates without prior treatment. Short incubation time ex vivo. Inter and intra-bacterial isotope incorporation monitoring. | Based on (population) empirical relationship. Required maintenance turnover measurement. Mismatch lineages and dividing assessment. | ||
| rRNA content |
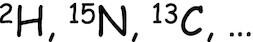
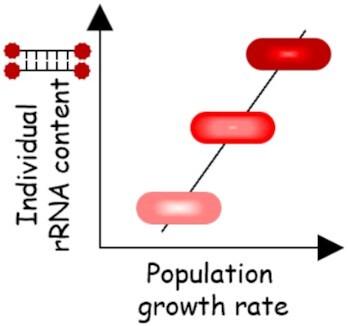
|
Linking single-cell rRNA level with population growth rates | None | Allowed monitoring of clinical isolates without prior treatment. Short incubation time ex vivo. | Based on in vitro standard curve correlating single-cell rRNA content with population growth rate. Mismatch lineages and dividing assessment. | ||
The bacterial growth rate at a population level commonly refers to the increase of bacterial number in the batch. At the single-cell level, the growth rate corresponds to the increase in size or mass of an individual bacterium. Thus, the growth rate at the single-cell level had to be distinguished from the replicative rate referring to the division capacity of a single bacterium into two daughter cells within a defined temporality. Actually, a single bacterium can growth without dividing, thus forming a filament while it cannot divide without having previously grown.
Time-lapse microscopy is at our knowledge the oldest method to monitor the bacterial replicative rate at the single-cell level (Barber 1908, Kelly and Rahn 1932). Conceptually it is based on microscopic tracking over-time of bacteria and their progenies division time and size. Thus, the created lineages of microcolonies informed on the replicative and growth rates at the single-cell level. Combination with (i) microfluidics allow almost instantaneous medium variation while keeping tracking and (ii) micropatterning allows trapping of bacteria improving the temporal resolution of the method especially by removal of the daughter cells (Balaban et al. 2004, Wakamoto et al. 2005, Wang et al. 2010). Owing to the improvement of microfluidics and single-cell level microscopy, Balaban et al. (2004) were able to demonstrate for the first time the hypotheses of Bigger 60 years later. Rare Escherichia coli survivors that resumed growth after drug pressure removal were indeed initially non-growing or slow growing. The best-suited approaches currently available to monitor the replicative rate are methods involving the tracking of fluorescence dilution by flow cytometry and microscopy (Roostalu et al. 2008, Helaine et al. 2010, Flannagan and Heinrichs 2018). These emerged from advances in fluorescent dyes and materials, preceded by the discovery of fluorescent proteins. In concept, it is based on the assumption that the total bacterial content is halved at each replicative step. Labeling of at least a part of this bacterial content or the use of fluorescent proteins permits monitoring of bacterial replicative rates and generation rank at the single cell level in vitro and in vivo. Combination with slow sequentially maturing fluorescent proteins whose maturation time are longer than the generation time overcome the fluorescence dilution sensitivity threshold (Claudi et al. 2014, Schulte et al. 2021). De facto, other methods are employed to determine the growth and replicative rates at the single cell level such as structural markers monitoring (Table 1, Kuru et al. 2012, Santi et al. 2013, Ursell et al. 2014, Yu et al. 2019). Emerging and/or as of yet sporadically used but elegant methods allow monitoring of growth rates at the single-cell level. Amongst others, the suspended microchannel resonator (SMR) enables growth rates measurement via buoyant mass monitoring (Godin et al. 2010, Cermak et al. 2016). Moreover, chromosomal replication marker monitoring and stable isotope probing give insight into single-cell growth rate (Kopf et al. 2016, Haugan et al. 2018). Individual rRNA content monitoring via FISH is linked to population growth rates via a standard curve (Poulsen et al. 1993, Kragh et al. 2014). Finally, population level monitoring such as bacterial colony appearance informs indirectly on the ability of a single bacterium to perform at least one replicative step as well as its extrapolated lag time (Levin-Reisman et al. 2010, Bär et al. 2020).
Several other methods, such as plasmid or fluorescent particle dilution, rely on single-cell level analysis to inform on the population growth rate (Gill et al. 2009, Adams et al. 2011, Myhrvold et al. 2015). Plasmid and particle dilution methods are based on the fact that a single copy of a plasmid or a particle will be dispatched within only one of the two resulting daughter cells. Thus, diluting by half the bacterial population carrying the plasmid or particle at each replicative step. Monitoring at the single-cell level the remaining proportion of the population bearing the plasmid, or the particle allows measurement of the population growth rate and is, therefore, not discussed here.
In this review, we discuss the different available methods to monitor bacterial growth and replicative rates at the single-cell level in vitro and in vivo (Table 1). Furthermore, we review the findings obtained using concomitant techniques such as antibiotics susceptibility and intracellular bacterial localization, to assess their appropriateness and likely increasing importance in the future.
Monitoring of colony appearance and size: not true single cell methods, even indirectly
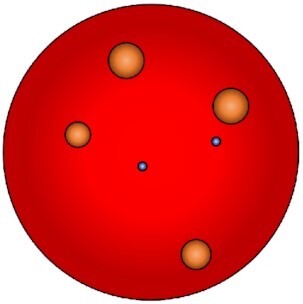
End-point monitoring of bacterial colonies on agar plate inform on the ability of a single bacterial ancestor to perform at least one division (Fig. 1; Table S1, Supporting Information). Then, the replicative information at the single cell level is lost. Immediately after the first replication step, two hypothesis are in conflict: (i) both daughter cell could continue to divide or (ii) one can stop while the other continues to replicate. Thus, to indirectly measure the percentage of bacteria having the capability to perform at least one replication cycle versus the percentage of non-growing also called viable but non-cultivable (VBNC) bacteria, a known number of bacteria must be spread on agar plate. Then, colonies are recordable when they reach a sufficient area or volume named the detection threshold, which is a major limitation of this indirect method. It depends on the image resolution and quality, depending amongst other elements on the lighting system. Indeed, if the progeny of a replicative bacterium enters an arrested growth phase before that the detection threshold of the method is reached, it cannot be positively counted and becomes a false negative as well as colony having a delayed appearance.
Figure 1.

Principle of the colonies monitoring methods to record indirectly bacterial first replicative step at the single cell level. A known number of bacteria are spread onto agar plates (left panel). Then acquisition device, either a scanner or a digital camera, record plates at intervals during the experiment to perform real-time monitoring or at the end of the experiments to perform end-point monitoring. Using both methods, colonies are detected when it reaches the detection threshold of the systems (middle panel). End-point monitoring allows determining at a specific time point the ratio of bacteria having done at least one replicative step versus VBNC (right panel). Real-time monitoring allows determining colony lag time and/or subsequent growth rates of colonies. Using live microscopic control, the colonies calculated appearance delays could be extrapolated as bacterial lag time.
Monitoring of colony appearance and size using real-time systems to capture and analyze images over time brings new parameters such as the lag time of the colonies appearance. It is defined at the single cell level as the time needed by each bacterium to perform its first replicative step. However, this indirect population level method measures the lag time as the time required by the resulting colonies to be detected by the system giving only an estimation of the accurate single cell lag time. Quantitative microscopic single cell tracking must be done to discriminate between both hypotheses (i) a lag time before the first replication or (ii) a slow growth of the colony. Besides, the growth rates, sizes, shapes, and colors of the colonies could be monitored allowing among other segregation depending on the surface and, thus highlighting of small colony variants. Another limitation of this method is that clumped bacteria or inhomogeneous spreading methods could lead to single colonies actually deriving not from a bacterium but from an uninterpretable cluster of them. Further, conventional methods for spreading on plates using glass balls or a rake could conduct to uneven spreading leading to different neighbor effects affecting the growth rate by nutrient competition and colonies crosstalk using quorum sensing. Finally, several other issues inherent to the method could lead to artefacts such as imperfection in the plate, low image resolution, light reflectance distortions and temperature variation. Of note, some software were developed to analyze other types of colonies such as eukaryotic (Shah et al. 2007, Bewes et al. 2008, Cai et al. 2011). However, although the switch from yeast to bacterial colonies seems to be reliable, others eukaryotic colonies can present some difficulties. Thus, due to the wide range of software and systems available we cannot suggest a specific one to the user. Their choice must rely on the purpose and the combination of agar and colony colors, image resolution, and so on.
End-point colony monitoring
The user could do end-point colony monitoring manually. However, it is time consuming and suffers from variability between one person to another. Thus, image analysis devices and software were developed allowing high-throughput assessment. Automatic colony counters are commercially available such as Scan from Interscience, Easycount from Biomérieux as well as Premium 90 HR from VWR.
A less expensive solution consists of plates pictures using a digital camera or scanner and analysis using end-user software homemade. Despite some variation, image analysis algorithms either on MATLAB, ImageJ, or CellProfiler follow the same pipeline. It consists of images preprocessing to remove noise and improve images, thresholding and optional watershed or edge segmentation algorithms leading to isolated colonies count (Bewes et al. 2008, Clarke et al. 2010, Cai et al. 2011, Geissmann 2013, Choudhry 2016, Khan et al. 2018). Furthermore, in the last decade few applications were developed to acquire and process images using smartphone helped by the improvement of their camera resolution (Wong et al. 2016, Austerjost et al. 2017). This paves the ways of scientific apps development, which could displace well-established laboratory devices.
Monitoring of colony appearance and size
As for end-point monitoring the user can do monitoring of colony appearance and size by repeating tedious end-point monitoring. However, end-point monitoring devices are generally outside temperature-controlled chambers and repeated temperature variation could impact the results. Thus, real-time colony counting incubator was developed. It is commercially available at Interscience and named ScanStation allowing to monitor up to 300 petri dishes at 1-hour intervals automatically.
Several high-throughput homemade solutions were designed combining an automated acquisition device and image analysis software, rather similar to monitor colony parameters such as count, appearance, size, or volume and physical characteristic (Michel et al. 2008, Levin-Reisman et al. 2010, Takeuchi et al. 2014). These colony growth dynamics parameters could inform on the lag of colony appearance, the growth rates of the colony, and the maximal size reached. The system is predominantly composed of an array of scanners placed in a temperature-controlled room. It was employed to monitor resistant bacteria arising across a 2D matrix of drug concentration (Michel et al. 2008), to show that starvation resulted in more bacteria remaining dormant for longer periods (Levin-Reisman et al. 2010) and to demonstrate growth defects of single-gene protein synthesis knockouts E. coli mutants (Takeuchi et al. 2014). Besides, to confirm that the ScanLag method monitors the lag time of single cells before the first replicative step and not subsequent events, data were compared with single cell microscopy and support that the delay of colonies monitoring is due to bacterial lag time and not to slow growth of the colony (Levin-Reisman et al. 2010). Of note, this control should be repeated for each bacterial species and strains studied. Further, the main limitation of the Colony-live system is due to the spreading technique; grown colonies were spotted into agar plate using short pins (Takeuchi et al. 2014). A colony evidently arise from more than one bacterium and imped with indirect single cell first replication monitoring. A method to bypass this issue could be to spot single bacteria using fluorescence-activated cell sorting (FACS). Moreover, it was showed that measuring the colony mass of the center of the colony minimized the neighbor effect (Takeuchi et al. 2014). A limitation of this method is that periodic scanning of the plates induces strong light exposure and temperature gradients, which could be sensed by the bacteria and might impact the growth. An optional module which turns on the electronics components that heat up only when an acquisition is done could be implemented (Levin-Reisman et al. 2010).
Bär et al. (2020) created a software called ColTapp supporting both standard endpoint analysis and time-lapse images analysis, thus filling a gap within previous existing image analysis tools. This application allows extraction of parameters such as colony lag time, growth rate, size, and further morphology descriptors as well as spatial metrics. Moreover, the software includes an end-point framework allowing estimation of colony lag time.
Time-lapse microscopy and microfluidics-based methods
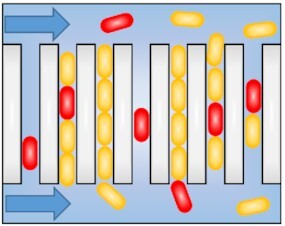
Single cell tracking overtime using time-lapse microscopy is a powerful tool to monitor the bacterial growth and replicative rates highlighting non-replicative bacteria for experiments with long durations (Fig. 2). Conceptually it relies on basic photonic, later fluorescent, time-lapse imaging of fields and manually tracking over-time of the non-replicating or replicating bacteria and their progenies within microcolonies, thus creating lineages. To our knowledge it is the first reported method employed to monitor the bacterial replicative rates at the single cell level (Barber 1908). Later, automated microscopes and design of homemade macros working within analysis images software improve this tedious task by implementing automated analysis. Next, microfluidics allows applying almost instantaneous medium variation without loss of the bacteria tracked. Moreover, microfluidics by providing fresh medium renewal and waste removal allows for the first time long-term monitoring of single-cell dynamics. The bias to the time-lapse microscopy method of 2D microcolonies is that analysis of a large number of single cell events at a time is technically challenging. Furthermore, after a defined number of replication events, the mother cell can no longer be linked to its progenies. Actually, the microcolonies can exceed the dimensional limits of the field and the bacteria tend to create stack impeding analysis. For this reason, it is a prerequisite of the method that the system holds the bacteria in a single focal plane. Later, micropatterning connectable with microfluidics allows trapping of the bacteria and increase the time-span resolution of the method, notably by sequentially removing the daughter cells via shear force. Moreover, due to the intrinsic properties of the method, historical growth and replicative rates from intracellular or in vivo recovered bacteria cannot been evaluated in contrast with the fluorescence dilution method. The growth rates are obtained by fitting to an exponential curve the size of an individual bacterium over-time. Subsequent potential division between two daughter cells can be highlighted by rapid drop and scission of the curve or by manual tracking.
Figure 2.

Patterns of the time-lapse microscopy method to monitor growth and replicative rates. Schemes representing methods for time-lapse microscopy 2D microcolonies (A) and (B), (top left), linear microcolonies (C)–(F), (top right and bottom left), and individual bacterium monitoring (G), (bottom right). (A) 2D monolayer microcolonies method; bacteria are spread on a pad and allowed to grow and divide in two dimensions. (A)–(G) Optionally, medium is flowed in via microfluidics to feed the microcolonies. (B) Turbidostat method; bacteria are caught on a trap formed by two large neighboring cavities (above and below). Laminar fluidic shear force remove and flow away the bacterial ancestor and its progenies having fallen into the opposite openings via growth and dividing spatial force. (C) Narrow grooves method, bacteria are caught on linear traps and allowed to grow and divide, thus forming linear microcolonies. (D) Successive interconnected chambers method; bacteria are seeded inside the first chamber of the array. Ancestors enter within narrow grooves, growth and dividing spatial force push half of the linear microcolonies inside the next chamber. The successive chambers have thinner grooves than the first it is connect to. (E) Mother machine method; individual bacteria are seeded at the dead end of narrow grooves. Growth and dividing spatial force pushes progenies toward the opened end. Bacteria are then flowed away by laminar fluidic shear force. Mother bacteria with the oldest pole remain at the bottom of the channels. (F) Chemostat method; bacteria are seeded inside narrow grooves opened at both sides. Growth and dividing spatial force pushes progenies and ancestor outside of the channels. Shear force flows away bacteria having fallen. (G) Individual bacteria monitoring method; adherent bacteria are spread on a pad and allowed to grow and divide. Laminar fluidic shear force removes the non-adhering progenies while the ancestors remain anchored. Furthermore, ancestor and progenies can be isolated or trashed via optical tweezers able to penetrate inside micropattern (not represented).
Time-lapse microscopy of microcolonies
Since the first part of the 20th century, time-lapse microscopy method to monitor the bacterial replicative rates at the single cell level was reported (Barber 1908, Kelly and Rahn 1932). Helped by sequential isolation of progeny every few generations within microdrops, Barber elucidate the replicative rates at the single cell level of B. coli over different temperatures by manual time-lapse microscopy (Barber 1908). Later, Kelly and Rahn isolated single cell of Bacterium aerogenes, Bacillus cereus, and Saccharomyces ellipsoideus using a method derived from the one developed by Ørskov in 1922 using spreading of bacteria onto agar (Ørskov 1922). The authors demonstrated that if any bacterium divided once, all its progenies maintained replication over four generations corresponding to the limit of the method since after that the progenies could not been linked to their mother cells. Of note, bacterial lag time before the first replicative step was in some cases reported.
In the last decades, improvement of the microscopic device and analysis software allowed automated acquisition of time-lapse single cell tracking and data analysis. Liquid culture spotted on a coverslip could be covered with a semipermeable membrane and a micropatterned polydimethylsiloxane (PDMS) chip allowing microfluidic medium switches or a polyacrylamide gel leading to sparsely platted bacteria (Wakamoto et al. 2013, Manina et al. 2015). Then phase and fluorescence image were recorded at intervals. Analysis of the images series records bacterial planar area over time as well as their fluorescence intensity and specifies their position in a lineage tree (Wakamoto et al. 2013, Manina et al. 2015). This method allowed monitoring nine generations of lineages representing about 500 cell cycles (Kiviet et al. 2014). Growth rates were obtained by sequentially fitting cell length or area with exponential curves and divisions were highlighted by shift in the curve. The division rates were calculated from the number of division events normalized by the total cell number at each time point divided by the time-lapse interval. Wakamoto et al. (2013) employed the time-lapse microscopy method to analyze the growth and replicative rates of Mycobacterium smegmatis framing and during isoniazid drug challenge. Helped by the time-lapse method, it was demonstrated that M. smegmatis persistence against isoniazid is not correlated with single-cell growth rates but linked with the isoniazid-activating enzyme catalase-peroxidase KatG dynamics. Moreover, the authors showed that isoniazid inhibits growth faster than division. Later, Kiviet et al. (2014) employed time-lapse microscopy method to investigate the growth rates of E. coli at the single cell level and quantify time-resolved cross-correlations with expression of lac genes. Cells were first manually selected and then automatic followed by microscopy. It was shown that the growth rates varied in time depending on the medium employed in the gel, as an example, the more the IPTG concentration increased the more the growth rate increased. Moreover, lac genes expression was monitored by GFP fusion into the lac operon allowing quantifying the lac production rate and concentration at the single cell level. This allowed highlighting that lac expression fluctuations positively correlated later growth fluctuations and that it then propagates back to disturb expression. Manina et al. (2015) combined quantitative time-lapse microscopy and fluorescent reporters to assess both bacterial growth rates and ribosomal RNA expression concomitantly. Single-cell dynamics of ribosomal RNA of Mycobacterium tuberculosis was monitored using a genetically encoded fluorescent reporter consisting of a destabilized green fluorescent protein (GFP) inserted at the rrn locus. The half-life of the radiolabeled GFP destabilized was several hours depending on the growth phase of the bacteria. Single-cell tracking by time-lapse microscopy of M. tuberculosis was done framing and during isoniazid intoxication which was done by intermittent pulses using the microfluidic device to mimic the pharmacokinetic profile in patients. Moreover, bacteria were stained with Sytox blue, a reporter of membrane damaging. This combined method highlighted three undamaged membrane bacterial subsets after the isoniazid removal (i) cells that resumed growth and recovered fluorescence (ii) non-growing but metabolically active cells that contain bursts of fluorescence and (iii) non-growing cells with equal fluorescence. Interestingly, single-cell time-lapse analysis showed no exclusive correlations between growth rates prior to drug intoxication and the bacterial fates after drug removal. Furthermore, bacteria explanted from the lungs of chronically infected immune-competent mice were inoculated into the permissive condition of the microfluidic device. The majority of the recovered bacteria did not resume growth despite the fact that a substantial fraction was physically intact and metabolically active as demonstrated by rRNA locus de novo expression monitored by fluorescence intensity. Next, Goormaghtigh and Van Melderen (2019) used single-cell time-lapse method coupled with SOS induction and DNA content reporters to characterize exponential phase E. coli persisters to ofloxacin . The bacteria were design to express a psulA::gfp reporter monitoring the SOS response induction and an HU-GFP reporter to record nucleoids. Using this system, they showed that prior to ofloxacin treatment, persistent bacteria are not necessarily non or slow growing. Actually, the authors observed growing and dividing bacteria that only ceased replicating during drug intoxication while resuming growth upon drug removal, subsequently forming long polynucleoïde bacterial filaments prior to eventual division and nucleoid segregation. Moreover, persisters and sensitive cells showed similar level of SOS induction during ofloxacin intoxication. Suggesting that the growth rate and the SOS response cannot be employed as a marker of persisters. Of note, they defined persisters by the ability to regrow after drug exposure, which put aside possible long term non regrowth persisters cells.
Thus, improvement of the systems required for the time-lapse method simplified monitoring of bacterial growth rate and replication at the single cell level. Moreover, microfluidic devices permit growth rates monitoring under rapidly various successive medium conditions without loss of the bacteria of interest. Time-lapse microscopy method was parallelized with genes expression monitoring via fluorescent reporter allowing to establish such a cross-correlation in vitro and using in vivo explanted bacteria at the single-cell level. Moreover, due to the lack of bacterial growth history, end-point analysis failed, and prior in vivo growth and replicative rates cannot be recorded or extrapolated. Finally, bacterial growth rates and division at the single-cell level was monitored before, during and after antibiotics treatment helped by microfluidics.
The microfluidics had improved the temporal resolution of the time-lapse microscopy method via constant medium input, as previously discussed, but also via bacterial progenies removal. Actually, Ullman et al. (2013) designed a microfluidic turbidostat for single-cell growth monitoring and high-throughput gene expression analysis. The micropattern consists of a trap limited by two walls and two openings. Bacteria are caught in the traps and allowed to grow until covering the entire surface as 2D microcolony. Then the acquisition is started, the daughter cells reaching the openings are removed into the surrounding cavity and carried away via medium flow. Each experiment allows monitoring of around 3000 complete replication steps. The authors demonstrated that length of bacteria in rich media vary more than generation time at birth while the reverse was observed in poor media. Further, the dynamics of synthesis and localization of the lactose repressor were investigated using a LacI-Venus fluorescent construction expressed from native promoter in E. coli. Single fluorescent molecules detection using short excitation light pulses showed an increase in the expression at the birth. Later, Wallden et al. (2016) employed the microfluidic turbidostat to decipher the chromosome replication cycle to the replication rate of E. coli via labeled DNA replication components. The authors labeled the epsilon subunit of the DNA polymerase III named DnaQ allowing monitoring of single replisomes via single-molecule fluorescence imaging. Using this single-cell method they demonstrated that the initiation of the chromosome replication is launched at a fixed volume per chromosome independently of growth rates and bacterial volume at birth. Furthermore, it was showed that the generation time and division size was dependent to the growth rate.
Thus, removal of the bacteria from the 2D microcolonies increase the temporal and spatial resolution of the time-lapse microscopy method. Coupling it with replisomes detection and localization allow both chromosome and cell cycle correlation. However, large individual bacterial shifting within the 2D microcolonies can induces loss of cells and lineages end.
Overall, the oldest method to monitor at the single-cell level the bacterial growth and replicative rates was improved by the breakthrough of automation and software analysis (Fig. 2). Time-lapse method allows monitoring the bacterial growth and replicative rates at the single-cell level by analyzing the individual's cellular size and their drop within 2D microcolonies. A strength of the method is the cross-correlation with fluorescent reporter and medium switches with microfluidics. The main limitation relies on the bacterial tracking to create among other the lineages. Actually, the temporal limit of the methods to measure replicative rates within a 2D microcolonies depend on the robustness of the analysis to link daughters cells to their mothers, which mainly depends on the size of the microcolonies and individual bacterial shifting. Microfluidic turbidostat development improves the temporal resolution of the method by sequential removal of the bacteria from the trap via neighboring cavities, thus improving the spatial resolution. Furthermore, due to the intervals tracking principle of the method, end-point analysis is not possible. Indeed, bacterial growth history before starting acquisition cannot be recordable nor extrapolated.
Time-lapse microscopy of linear microcolonies
Channel-design allows long-term single-cell tracking of a unique lineage from a single ancestor tacking over the spatial limitation of the 2D microcolonies method (Fig. 2). It relies on linear narrow grooves mainly made in PDMS, which trap bacteria allowing only 1D motility. The grooves are slightly taller than the cell to avoid cell stranding and allow optimal and equal nutrition all along the channel. Linear colonies simplified tracking and lineage construction compared to 2D microcolonies. Of note, this method is, however, only suitable for rod-shaped bacteria.
Balaban et al. (2004) employed the channel-design combined to time-lapse microscopy to investigate the single-cell growth rate of E. coli during antibiotic challenge. The micropattern device used consisted of a stack of layers made by linear grooves in PDMS, a cellulose membrane and a flow channel in PDMS. The bacteria were seeded into the PDMS channels and allowed several cell divisions before exposure to ampicillin treatment using microfluidics. The growth rates of the daughter cells were derived from the length of the newly formed linear microcolonies. The authors showed that rare non-growing or slow growing bacteria could survive antibiotic treatment and then regrow and divide after drug removal. Thus, a channel-type design micropattern combined with microfluidics allow for the easiest microcolony tracking, by limiting spatial diffusion of the bacteria during longer periods of time, as well as antibiotic growth rates susceptibility monitoring.
Männik et al. (2009) engineered a submicron channel containing microfluidic device to monitor the bacterial growth and replicative rates as well as motility at the single-cell level. The micropattern consisted of successive chambers interconnected by progressively narrower channels ranging in width from 5 to 0.3 µm while the bacterial diameter used ranged from 0.7 to 1.1 µm. The swimming bacterial ancestor enters the channel by itself or pressed by the other bacteria. Helped by this time-lapse microscopy method, the authors showed that E. coli and Bacillus subtilis escaped channels exceeding only marginally their diameters by swimming. In smaller grooves, although the bacterial motility capacity was lost, the bacterial escaping ability was maintained. Actually, within narrower channels the bacterial dispersal is driven by the growth and replication pushing themselves. Half of the population was pushed forward into the next chamber while the other half was pushed backward toward the originating chamber. Escherichia coli was still able to pass through channels smaller than its diameter by a factor two resulting in a variety of anomalous cell shapes. Later, this model was employed to demonstrate that despite irregular morphologies E. coli maintains its ability to divide into two equally sized daughter cells (Mannik et al. 2012). Moreover, inhibition of the Min system and of nucleoid occlusion are largely dispensable regarding the accuracy of central divisions in these bacteria with anomalous shapes. Of note, minC deletion impacts the divisome placement within normal rod-shaped bacteria. Thus, an interconnected successively narrower channels design allows monitoring of growth and replicative rates depending on constrictions forces at the single-cell level via time-lapse microscopy.
Wang et al. (2010) improved the temporal resolution range of the narrow grooves method by removing the daughter cells at one side of the channels via constant flow allowing monitoring of the mother cell growth and replicative rates for hundreds of generations. It allows monitoring of a large number of bacteria at a defined reproductive age simultaneously. A series of single bacterially seeded narrow grooves are oriented at right angles toward a mainstream channel carrying the growth medium at a constant flow, which results to both bacterial nutrition and removal of progenies emerging from the growth grooves. The bacterium, i.e. confined at the end of the groove is the mother cell, which donates one of its poles to the growing bacterial lineage and which never leaves channel. In contrast, after the channel fills up, subsequent growth pushes the earliest progenies out and they are removed by the flowing medium. The authors called the method “mother machine” since it allows monitoring of cells inheriting the same pole over many generations. Using this method, they showed that the growth rates of the mother cell and its immediate progenies did not change over time. The cell length of 105 individual cells was fitted to a time curve, and then interval between birth and division was fitted to exponential function giving the growth rate at each replicative age. Thus, the mother machine improves the temporal resolution range of the narrow grooves method to monitor growth rates of the mother cell for hundreds of generations as it is gradually getting old, and of its direct progenies.
The mother machine method was broadly used to monitor bacterial growth rates and divisions at the single-cell level. Transient oscillations in E. coli initial cell size, which could extend over several generations was highlighted (Tanouchi et al. 2015). The authors demonstrated negative feedback on the cell size control, i.e that bacteria with an initially higher cell size tend to divide earlier and inversely. Using the mother machine, it was shown depending on studies that a progeny taller than its sister tends to grow slower or that the growth rate is independent of initial cell size (Tanouchi et al. 2015, Kohram et al. 2021). Fluorescent reporters were broadly combined with the phenotypically analysis of the mother machine. Virulence genes fluorescent reporter in Salmonella typhimurium was correlated with drug intoxication showing that virulence expression promotes survival by reducing growth (Arnoldini et al. 2014). Fluorescent reporter and the mother machine were further associated with end-point live/dead assay showing that both VBNC and persisters cells, here defined as having the ability to growth after drug removal, shared similar cell length before drug intoxication as well as similar tnaC level (Bamford et al. 2017). Of note, tnaC is part of the tnaCAB operon responsible for tryptophan metabolism and was half of those reported in susceptible cells prior to drug intoxication and can, therefore, be considered as new biomarkers. Later, fluorescent reporter of the main multidrug efflux pump AcrAB-TolC in E. coli showed that it was unevenly allocated between daughters cells since it accumulates at the old pole (Bergmiller et al. 2017). Furthermore, a growth difference was highlighted between successive generations under subinhibitory tetracycline challenge. The mother machine was further combined with a library of CRISPR interference knockdowns to highlight genes involved in the regulation of chromosome replication initiation (Camsund et al. 2020). The initiation of replication was simultaneously monitored via a seqA–yfp fusion allowing tracking of the chromosomal replication machinery. After phenotypic monitoring, bacteria were fixed, and genotype monitoring was done by sequential FISH to a barcode. The authors thus identified genes as required in the regulation of chromosome replication initiation at a defined volume per chromosome. Recently, Manuse et al. (2021) employed an ATP concentration reporter to characterize the persisters physiology. The reporter consists of a genetically modified ATP synthase binding subunit which absorbs at two different wavelengths between both free and ATP linked states and emits at one. The ratio between fluorescent signals from the two excitations wavelengths reports on the ATP concentration. Interestingly, the authors showed that even prior to antibiotic treatment persisters did not grow or grew very slowly at low ATP levels and resumed growth heterogeneously after drug removal. Thus, time-lapse microscopy methods employing a mother machine-type micropattern were combined with fluorescent reporters informing on genes expressions and efflux pump localization. Further combined with drug intoxication via microfluidics and ATP concentration reporter.
Norman et al. (2013) improved the mother machine method to decipher the B. subtilis switch between motile and sessile connected chains during the exponential phase of growth. The length of the narrow grooves must accommodate two parameters, cell feeding and cell retention. Longer grooves impeded uniform feeding but better retained the progenies. The authors overcame this limitation by adding shallow side grooves surrounding the bacteria allowing optimal growth medium diffusion all along the channels. They created 75 µm long channels working with the sessile chains’ lifestyle of the bacteria. Motile cells were recorded via the fluorescent reporter of the flagellin gene while the chains were highlighted by fluorescent reporter encoding copy of the matrix tapA gene. Using these combined methods, they demonstrated that the motile lifestyle is memoryless while the time spent as sessile bacteria is tightly controlled. Later, Baltekin et al. (2017) engineered the mother machine by adding an opening at the end of each narrow groove. Its size prevents bacterial exit but allows medium flow through the channels. The authors employed it to assess rapid antibiotic susceptibility of clinical samples. Thus, improvement of the mother machine design allows monitoring of long cell filaments and rapid detection of antibiotic susceptibility.
The narrow grooves can also have both of their ends opened toward two parallel mainstream channels (Moffitt et al. 2012, Long et al. 2013). It allows feeding and escaping of the bacterial mothers and their progenies at both sides, thus avoiding aging altogether. Actually, due to the dual opening of the grooves, bacteria including the mother cells are constantly removed, thus creating a linear microcolony of bacteria of similar age. Because the bacteria are maintained in a chemostatic environment Moffitt et al. (2012) termed their device the single cell chemostat. The chemostat was made of agarose, a porous material allowing cross microcolony communication. Using this device, the authors showed that the division time, the growth rate, and the length at division of E. coli remained constant throughout the lineage. The bacteria leave the grooves at a rate that increased proportionally to the distance from the center and is constant in time. Thus, the temporal limit of tracking depends amongst other parameters on the length of the grooves. They investigated the growth rates of a synthetic microbial community including two E. coli auxotrophs that shared amino acids in co-culture. Time-lapse microscopy analysis showed that each strain of the mixed community grew while a single community did not. Moreover, increasing the flow rate decreased the growth rate of the microcolonies. At the single-cell level all the bacteria of the mixed community grew with widely different rates. Subsequently, Long et al. (2013) developed a PDMS-based chemostat to monitor the bacterial replicative rates and validated it for track fluorescent reporters such as chromosomal loci over-time. Thus, the chemostat design improves the temporal resolution range of the narrow grooves method by allowing tracking of successive lineages over 30–40 generations and avoiding bacterial aging. However, the porous agarose-based pads complicate medium changes inside the channels due to agarose saturation and are, therefore, best reserved for experiments that do not require changes in medium and microcolonies isolation.
Overall, linear 1D-restricted micropattern devices improved temporally the basic time-lapse microscopy method using 2D microcolonies monitoring (Fig. 2). The channel design allows long-term monitoring of a unique lineage from a single trapped ancestor beyond the temporal resolution range of the 2D microcolonies, which rely on the spatial limit. Besides, the mother machines allow monitoring for hundreds of generations the aging of mother cells trapped at the dead-end, as well as of their direct progenies sequentially remove from the channel. Then, chemostat design created linear colony composed of bacteria sharing similar age via constant bacterial released at both side of the channel including the mother cells. Such as the basic previously described time-lapse method, the channel design techniques impeded with end-point analysis. Moreover, the mechanical restricted linear growth allows only mother to daughter cells contact thus avoiding population direct and diverse talk using as example quorum sensing. Furthermore, it was showed that the channel spatial characteristic such as width and length interfere with the mother cell growth and replication by mechanical forces (Yang et al. 2018).
Time-lapse microscopy of individual bacteria
Elfwing et al. (2004) developed a flow chamber allowing monitoring of growth and division events of the mother cells attached to a transparent solid surface via removal of any daughter cells by fluidic shear force which fed bacteria. Later, Iyer-Biswas et al. (2014) diverted the physiological properties of Caulobacter crescentus, which divides into two phenotypically distinct daughter cells, one is motile, and the other is adherent. They design a strain bearing the holdfast synthesis A (hfsA) gene controlling the surface adhesion under an inducible promoter. Thereby, before starting monitoring, the gene was expressed in order to adhere bacteria at the glass surface. Then, the inducer is removed, and subsequent daughter cells are unable to adhere and are removed via the medium flow. This method allows monitoring of more than one hundred generation from an individual mother cell. Helped by this system, it was showed that the individual lag time, which is referred as the time required by E. coli and Listeria innocua to double its size increased respectively with the salt concentrations and sublethal heat shock (Elfwing et al. 2004). Further, it was demonstrated that the bacteria divide upon reaching a critical multiple of their initial sizes and that the mean division time decreases as the temperature increases (Iyer-Biswas et al. 2014). Thus, bacterial attachment prior to shear force on solid surface allows monitoring of individuals mother cells growth and division rates.
Wakamoto et al. (2005) developed a microchamber cultivation array under fluidics flow to evaluate the length and replication time of individuals E. coli over generations. The micropattern is composed of four chambers connected to two ending areas allowing nutrition and discarding of the progenies via medium flow. In concept, the method which consist of repeated isolation steps is similar to that developed by Barber (1908). From a unique ancestor the four vacant microchambers were filled by progenies via optical tweezers. Then, at each replicative step, four randomly selected progenies were picked up and discarded. This method allows monitoring of four single bacteria from a unique ancestor for more than 10 generations. The authors showed that the initial and final length were correlated with those of proximal generations. In contrast, the division time had no correlation with that of the consecutive generations. Thus, microchamber pattern combines with time-lapse microscopy and manual progenies isolation allows monitoring of length and division of single successive isolated bacteria.
Overall, sequential removal of daughter cells using optical tweezers or shear force allow monitoring of growth and replication of individual isolated bacterium over respectively 10 to more than 100 generation by time-lapse microscopy (Fig. 2). It allows monitoring of bacterial parameters under various conditions such as salt concentrations and temperatures ranges. The main limitation of this method is that the tracked bacteria remain alone preventing investigation of colonies behaviors. Furthermore, isolation of single bacteria at each replicative step is tedious. On the other hand, the removal of progenies allows tracking a few individual bacteria from the same lineage or mother cells over generations under a single lifestyle.
Dyes dilution-based methods
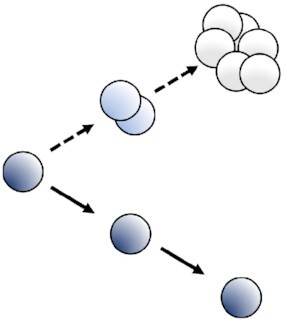
In concept, the dyes dilution-based methods rely on the total initial bacterial content dilution by a factor of two at each replicative step between the two resulting daughter cells (Fig. 3). The different commercially available dyes separate into two mains classes depending on the location where they are retained by the bacteria: (1) reactive compounds forming covalent bonds with free amines; (2) lipophilic compounds intercalating with lipids of the plasma membrane. Another class corresponding to (3) labeled artificial surface receptors involving among other fluorescent oligodeoxynucleotide (ODN) small molecule conjugates was recently tested to fit with the dyes dilution-based model. It should be noted that we present here only a selection of dyes dilutions assays, those that we felt were most suited to our comparative analysis of this type of methodology. The subset listed in Table S2 (Supporting Information) is, therefore, not representative of the diversity of dyes available. A prerequisite to the workability of the dye staining is that the labeling employed must be neither metabolized nor otherwise removed from the bacteria during the course of the study (Flannagan and Heinrichs 2018). Indeed, only then is it safe to assume, that the dyes are diluted proportionally by a factor two at the outset of each bacterial division, relying either on total protein or cell wall synthesis and then dilution. Bacterial subsets undergoing replication should dilute the fluorescent dye equally between daughter cells at each replication step while non-dividing bacteria should retain all of their initial fluorescent labeling. Thus, the dye dilution-based methods shed light on bacterial replicative rates at the single-cell level over time. Nevertheless, this fluorescence method cannot be used to track bacterial replication over a long period of time since the signal will decrease to near background, reaching the sensitivity threshold, after only a few bacterial divisions, commonly about 4–8. A further weakness of the method is that dividing or non-dividing states that happen after a first replicative period over the limit of detection of the model cannot be recorded (Fig. 4). Furthermore, in contrast with time-lapse microscopy methods, the dyes dilution-based method did not report on bacterial growth rates at the single-cell level. The covalent binding of dyes to proteins; as well as the expression of fluorescent proteins presented later; may affect the physiological behaviors of the bacteria and lead to misinterpretation of results. Thus, as far as possible controls must be done using others approaches to corroborate the correctness of the replicative rates measured with fluorescence dilution methods.
Figure 3.

Principles of the different fluorescence dilutions methods to study bacterial replicative rates at the single cell level. Dyes(bottom and bottom right) either label-free amines or intercalate in the lipid membrane(s) of bacteria. At each replicative step the initially labeled content of the bacteria will be diluted by a factor two resulting in halving of the fluorescence intensity recorded. When pre-expressed, single inducible fluorescent proteins(top left) will be similarly diluted during bacterial proliferation. Meanwhile bacteria that do not undergo any replication will remain as labeled initially. Dual reporter methods combine a single inducible fluorescent protein with either a constitutive fluorescent protein (top) or another inducible fluorescent protein (top right). Dual-reporters constructed on the basis of two independently inducible fluorescent proteins improve the detection range of the method by successive removal of each inducer. The TIMER method employing mainly DsRed protein(bottom left) relies on both the global initial bacterial content reduction at each replicative step and the sequential maturing fluorescent proteins. One fluorescent protein matures faster than the other does and can, thus accumulate within the replicating bacterial population whereas, in contrast, the slower maturing cannot. Moreover, within proliferation arrested bacteria both differentially maturing proteins can accumulate. In contrast, when replicating arrested bacteria enter a replicative phase the slowly maturing protein will be diluted by a factor two at each replicative step.
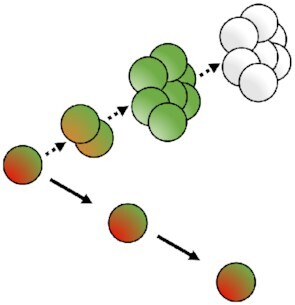
Figure 4.

Limits of the fluorescence dilution methods. In total, four classes of limits are here represented: (1) a proliferation arrested phase occurring before the detection limit of the method is reached after a first period of replication cannot be detected without bacterial tracking, (2) any dividing, or (3) dividing-arrested phases outside of the dilution detection limit will not be recordable. (1), (2), and (3) represent limits of dyes, single inducible fluorescent proteins,anddual-reporters tools(top, left, and bottom). Finally; and specific to the TIMER method (right), (3) rapid dividing state changes will not be detectable due to the slow maturing protein. Amine labeling dye combined with constitutively expressed fluorescent protein as well as dual-reporter constructions carrying both a constitutive and an inducible fluorescence reporter allow bacterial tracking outside of the dilution limit via microscopy.
Reactive compounds forming covalent bonds with free amines
Carboxifluorescein succinimidyl ester
A fluorescent dye dilution-based method was developed with the carboxifluorescein succinimidyl ester (CFSE) probe to monitor the various replicative rates of a liquid culture or intracellular bacteria at the single cell level (Ueckert et al. 1997, Atwal et al. 2016, Wong et al. 2019). The non-fluorescent compound 5- (and 6-) carboxyfluorescein diacetate succinimidyl ester (CFDA-SE) can freely diffuse throughout the cell membrane and reach the cytoplasm (Bronner-Fraser 1985). Once intracellular, both acetate group of the CFDA-SE molecules are cleaved by esterases resulting in the highly photostable and membrane impermeable CFSE fluorescent compound. Of note, the permeability of Gram-negative bacteria was increased with EDTA or Triton X-100 to improve the staining (Diaper and Edwards 1994, Hoefel et al. 2003, Wong et al. 2019). The succinimidyl group of the CFSE compound then reacts covalently with primary amines. Successive rounds of centrifugation and resuspension with medium containing amines quenches any unreacted fluorophore. Using this approach, the decreases in CFSE dilution tend to correlate with the known division time of Acinetobacter baumannii and Orientia tsutsugamushi supporting the strength of the method to follow bacterial division at the single-cell level (Moffatt et al. 2010, Antunes et al. 2011, Giengkam et al. 2015). Interestingly, Atwal et al. (2016) showed that the CFSE and its variant CellTrace FarRed could not be properly retained by O. tsutsugamushi after liquid culture fixation with paraformaldehyde or acetone. However, it appeared that neither dye had any impact on the phenotypic state of the bacteria or on the infection cycle. Further, cultures of non-dividing O. tsutsugamushi, kept their fluorescence intensity demonstrating that as expected the dilution of fluorescence was due to the bacterial replication state rather than caused by compromised fluorescence probes.
Since 1997, Ueckert et al. (1997) pointed out the vast interest of replicative rate measurement at the single-cell level compared to the conventional average population data. Lactobacillus plantarum was colabeled with CFSE and propidium iodide, a non-permeant compound that enters only inside bacteria harboring damaged membranes. A total of three replicative bacterial subsets were detected corresponding to: a non-growing state and two groups of slow-growing bacteria, having each undergone 1 or 2 cell division, respectively. After nisin treatment, an antimicrobial peptide which forms pores within the membrane, the surviving bacteria, i.e. those that were propidium iodide negative, were able to divide without a lag phase and the replicative steps were tracked for up to eight generations by flow cytometry. CFSE-labeled bacteria could be further monitored in vitro during the early events of cells invasion by live confocal microscopy (Atwal et al. 2016). Quantification of bacterial CFSE fluorescence intensity recorded was done by measuring the highest pixel within single bacteria. A fast decrease of the fluorescence intensity average was recorded, highlighting a population bacterial replication tendency. Of note, a small subset of bacteria was still highly fluorescent at the end of the monitoring indicating that they were non-dividing or dead. Indeed, dead bacteria are labeled if the death happens after CFDA-SE maturation into CFSE and that CFSE is not eliminated. The CFSE dye dilution-based method was also employed to track both the bacterial propagation and the replicative rates in situ of Comamonas sp and Acidovorax sp in aquifers and aquifer sediments (Mailloux and Fuller 2003). Microcosm experiments revealed that the method gave similar division rates as bacterial counting and that these rates were independent of cell concentration. Furthermore, data from a field-scale assay of Comamonas sp using CFSE labeling showed that a slight and consistent decrease of the recorded fluorescence intensity happened over time and distance traveled giving a doubling time of 15 days (Fuller et al. 2000, Mailloux and Fuller 2003). Recently, flow cytometry analysis of CFSE-labeled A. baumannii cultures allowed monitoring replications for up to 2 hours representing approximately four generations (Wong et al. 2019). Concomitantly, live and dead bacteria was assessed by propidium iodide incorporation. Rifampin alone and in combination with polymyxin-B led to a sustained non-replicating state despite some slight resumption of replication. Minimal bacterial killing was recorded suggesting that antibiotics pressure leads to drug-persistent non-replicating A. baumannii, which regrow upon antibiotics removal determined by CFSE dilution as the parental strain. Of note, CFSE monitoring and CFU counting were similar without antibiotics pressure but appeared largely different under antibiotic treatment, consistent with the previous finding showing that only a part of the drug-persistent bacteria are able to resume proliferation (Helaine et al. 2014, Wong et al. 2019). This points to a serious limitation of the broadly used CFU counting method, which does not consider the VBNC bacteria.
Overall, the widespread CFSE fluorescent dilution-based method allows monitoring at the single cell level of the replicative rate of both Gram-negative and Gram-positive bacteria in liquid culture or during cell infection by live confocal microscopy and flow cytometry (Fig. 3). It is compatible with in situ study to relate bacterial lifestyle such as progeny travel and concomitant bacterial labeling in liquid culture, notably therefore informing on the survival upon bacterial injury (Table S2, Supporting Information). Moreover, single-cell CFSE fluorescence intensity monitoring by flow cytometry allows the distinguishing of four to eight cell divisions depending on the bacterial species until that fluorescence reaches the background level (Fig. 4). Due to the dilution of the dye out of the detection limit it cannot highlight dividing or non-dividing bacteria after a first period of proliferation. A limitation to the microscopy study is that the bacteria not colabeled with a constitutively expressed fluorescent protein, which underwent replication became undetectable over the experimental time course.
eFluor-670 cell proliferation dye
Flannagan and colleagues developed a fluorescent dilution approach based on the cell proliferation fluorophore eFluor-670 to distinguish replicating and non-dividing intracellular bacteria at the subcellular level in vitro (Flannagan et al. 2016, Flannagan and Heinrichs 2018). This non-specific fluorescent marker is an amine reactive compound that will, thus label any bacterial proteins or structures containing primary amines. As for CFSE, successive rounds of centrifugation and resuspension with medium containing amines quenched any unreacted fluorophore. Bacteria constitutively expressing a fluorescent reporter protein allow tracking the replicative subset beyond the dilution threshold limit. As control, labeled bacteria were rendered unable to grow by killing with paraformaldehyde fixation. Once fixed, bacteria maintained their initial staining in liquid culture and inside macrophages demonstrating that eFluor-670 dye dilution occurs only during bacterial replication.
Using this approach, Staphylococcus aureus was shown to begin intracellular replication within membranes vacuoles of macrophages at around 12 hours post-infection (Flannagan et al. 2016). Confocal immunofluorescence microscopy demonstrated that non-growing as well as replicating bacteria, i.e. those lacking eFluor labeling while expressing GFP, were contained either within or outside phagosomal LAMP1-positives membranes. Later, the intracellular replicative fates of Staphylococcus lugdunensis was investigated using the eFluor-670 fluorescent dye dilution method corroborating its effectiveness within others species (Flannagan et al. 2018). This method works with both Gram-positive and Gram-negative bacteria during cell infection as demonstrated by staining protocol adaptation toward Yersinia pseudotuberculosis, Citrobacter rodentium, and E. coli despite induced species variations (Flannagan and Heinrichs 2018).
Overall, eFluor-670 fluorescent dye dilution-based method allows distinction between bacterial replicative states during eukaryotic cell infection using fluorescent microscopy from fixed or live cells. Moreover, due to its compatibility with others cellular fluorescent probes it enables characterization of the bacterial subcellular localization accurately at the single cell level (Table S2, Supporting Information). However, any attempt to sort the infected eukaryotic cells depending on the intracellular replicative rate of the bacteria by flow cytometry have at this time failed due to the various dividing rates observed within any given macrophage in such populations. Yet, analysis of the terminal stage of infected cells, which often contain only replicating or non-replicating bacteria was achieved (Saliba et al. 2017). As previously, it cannot highlight dividing states after a first period of replication although constitutively expressed fluorescent protein allows tracking bacteria beyond the dilution limit.
Lipophilic compounds intercalating with lipids of the plasma membrane
The stable lipophilic PKH fluorescent dyes, which will incorporate their aliphatic part within the exposed membrane lipid bilayer were used to monitor the bacterial replicative rates at the single-cell level (Raybourne and Bunning 1994, Sturm et al. 2011). It should efficiently work solely for Gram-negative bacteria having at their surface a lipid membrane fully accessible to the dye. Of note, Sturm et al. (2011) used the bacterial strain wbaP- lacking the LPS-O side chain to permit PKH efficient labeling via reducing steric hindrance. The labeling was done at the onset of the assay and the excess dye was removed with serial washing steps. A linear and inverse correlation was done between the fluorescence intensity recorded by flow cytometry, of PKH-2-labeled S. typhimurium and Listeria monocytogenes, and the CFU counting over-time, confirming the soundness of the method (Raybourne and Bunning 1994). Furthermore, viability of labeled and unlabeled bacteria was assessed demonstrating identical results. Of note, bacterial PKH dyes fixation and live microscopy compatibility were investigated during Borrelia burgdorferi interactions with different tick cell lines (Teixeira et al. 2016). Further, replicative penalty of Salmonella caused by ttss-1 virulence factor expression was highlighted by combining gene fluorescent reporter with PKH26 dye dilution method at the single-cell level (Sturm et al. 2011).
Overall, the PKH dyes fluorescence dilution-based method permits monitoring of bacterial dividing rates at the single level by flow cytometry (Fig. 3). It could be combined with another discriminant fluorescent bioreporter, itself genomically expressed (Table S2, Supporting Information). The main limitation of the method is the requirement for a fully accessible bacterial lipid bilayer.
Labeled artificial surface receptors
Lahav-Mankovski et al. (2020) developed a fluorescent dynamic artificial receptor system affecting the bacterial properties such as surface adhesion and cells interaction. To this end, a hexa-histidine tag was fused to an outer membrane protein C of E. coli under inducible promoter. Then conjugate of ODN and trinitrilotriacetic acid-nickel (NTA) complexes incubated with the bacteria bind to the hexa-histidine tag at the surface in presence of Ni2+ ions via the NTA part. Of note, suitable metal chelators such as EDTA reverse the binding by chelating Ni2+ ions and remove the artificial receptors from the surface on demand. ODN can also be decorated with the following dyes Cy5, TAMRA, and FAM emitting respectively in far red, yellow, and green. Moreover, unlabeled attached ODN can be hybridize with a labeled complementary strand named ODN-2. The dual ODN method enable rapid strand displacement by adding a complementary strand named ODN-3 since ODN-2 bear a short overhang site. It allows, as example, the newly free unlabeled ODN to bind another ODN-2 carrying a different dye. Moreover, this method thwarts the synthetic difficulty of a single strand bearing both dyes and NTA. Using this synthetic fluorescent receptor method, the authors demonstrated by time-lapse microscopy that the individual fluorescence of each bacteria decrease overtime while the number of labeled bacteria increases.
Overall, the labeled artificial bacterial surface receptors involving ODN-dyes conjugates allows monitoring of the bacterial dye dilution overtime at the single cell level and should, therefore, highlight the replicative rate. It could be conjugated with biomimetic synthetic receptors carrying distinct motif such as a thiol group, thus modulating bacterial adhesives properties. The initial fluorescence signal relies on the initial quantity of outer membrane protein C that needs to be tightly calibrated to avoid bacterial disturbance due to the artificial receptor's complexes.
Fluorescent proteins dilution and maturation kinetic-based methods
Fluorescent protein dilution methods are all conceptually based on the same principle as dye dilution methods (Fig. 3). The fluorescence signal intensity recorded, provides information on the historical replicative state of individual bacteria. Of note, the bacterial division time must be verified to be smaller than the half-life of the fluorescent protein. This is to ensure that the decrease of fluorescence is overwhelmingly due to the dilution generated by replication events and not to reporter protein degradation. Further, the dilution-based approach may be extended into the maturation kinetic method when the sequential maturation rate of two fluorescent proteins, one maturing faster than the other does, are analyzed in parallel (Fig. 3). The replicating bacterial subset accumulates greater level of the rapidly maturing protein whereas the other, slow maturing one, cannot sufficiently build up before the dilution induced by bacterial division. Meanwhile, the non-replicating subpopulation accumulates firstly the rapid maturing proteins before containing a mixed of both mature fluorescent proteins, i.e. the rapid and the slow maturing. Based on the same concept, a pool of preformed and constitutively expressed fluorescent proteins will be diluted at each replicative step and reaccumulates once replicating phase stops. Obviously, the fluorescence intensity must be recordable all along the experiment despite potential huge drops. For the system to work, the maturation kinetics of the slower maturing fluorescent protein must also be slower than the bacterial replicative rate. Thus, fluorescent protein dilution and differential maturation kinetics will shed light on the bacterial dividing rate at single cell level. Furthermore, in contrast with the time-lapse microscopy methods and consistent with the dyes dilutions-based method, the fluorescent proteins dilution and maturation kinetic-based methods did not report on bacterial growth rates at the single-cell level.
Dilution of a single fluorescent inducible protein
Bacterial cell division leading to dilution of a single protein was numerically formulated as part of a theoretical model of GFP accumulation within single bacterial cells (Leveau and Lindow 2001). It predicts that the initial GFP content without de novo synthesis will dilute from dividing cells at a rate equal to growth rate. This was supported by experimental observation in which the fluorescent protein constitutively expressed content of Pseudoalteromonas cells was reduced when grown in rich medium compared to minimal medium (Stretton et al. 1998).
The single-cell inducible fluorescent protein dilution method to detect and sort bacterial subset according to dividing rates in liquid cultures was first reported by Roostalu et al. (2008). Escherichia coli was engineered to express the highly stable isoform, GFPmut2, under the control of homoserine lactone (HSL; Roostalu et al. 2008). After removal of the inducer, the fluorescent signal intensity was sequentially monitored using flow cytometry. This experimental approach demonstrated that an exponential phase culture of E. coli contained homogeneously dividing bacteria, while a stationary phase culture was made of homogeneously non-dividing bacteria. Furthermore, stationary phase cultures diluted in fresh medium resulted in two subsets of live cells, one replicating as observed by chromophore fluorescence intensity decreasing and one non-replicating recognized by stable chromophore fluorescence intensity. This resulting culture was then treated with ampicillin which did not affect the non-dividing subset but largely killed the dividing population. The replicative resumption kinetics from a stationary phase culture was further investigated using an E. coli strain expressing GFPmut2 under the control of an isopropyl-b-D-thiogalactopyranoside (IPTG) inducible promoter (Jõers et al. 2010). Briefly, bacteria grown in presence of the inducer until stationary phase were diluted into either fresh rich or poor medium. As expected, the kinetics of bacterial awakening were dependent on the medium used since replicating bacteria having reduced GFPmut2 content were firstly recorded by flow cytometry within rich medium. Then, E. coli was engineered to carry a chromosomally inserted T5p-mCherry cassette under the control of IPTG, thereby eliminating plasmid copy number variation (Orman and Brynildsen 2013). The authors focused on the replicative state of drug persistent bacteria formed prior to antibiotic exposition. Interestingly, approximatively 1% of sorted non-growing bacteria via FACS persisted antibiotic pressure since regrowing on LB agar plate, while approximatively 0.01% of sorted dividing bacteria could do so, even when those sorted were rapidly dividing. This could be due to a non-recordable replicative arrested phase occurring after a first period of proliferation (Fig. 4). Further, it was described that sorted bacterial subset with high redox activity had more non-growing bacteria in newly fresh media and that respiration inhibition impaired formation of persisters from stationary phase (Orman and Brynildsen 2015). In the meantime, stationary phase of E. coli having a pool of pre-induced fluorescent GFPmut2 proteins were de novo cultured without inducer and challenged with serum (Putrinš et al. 2015). Interestingly, the complement system eradicated a large proportion of dividing cells without affecting the small non-dividing and rapidly replicating bacterial subsets. Of note, only the non-dividing subset survived during antibiotic challenge in combination with serum treatment. Recently, E. coli persisters resuscitation was determine to occur within 1 hour upon transfer to fresh media regardless of antibiotic treatment times (Mohiuddin et al. 2020). Thus, the single inducible fluorescent protein dilution method allows monitoring of bacterial replicative states using flow cytometry from different growth phases. Moreover, it permits the linking of drug persistence to bacterial replication behavior or rather lack, thereof as well as the resuscitation patterns. Further, the redox activity of the different sorted replicative subsets could be concomitantly investigated using flow cytometry (Table S2, Supporting Information).
Single fluorescent protein dilution method was next extended to monitor intracellular bacterial replicative fates (Khandekar et al. 2018, Peyrusson et al. 2020). To this end, human monocytes were infected with previously opsonized Pseudomonas aeruginosa expressing the GFP under the control of arabinose and phagocytosis allowed for 2 hours in presence of the inducer (Khandekar et al. 2018). Infected monocytes were then incubated with antibiotics for 24 hours after arabinose removal, intracellular bacteria then gathered were analyzed by flow cytometry. For both wild-type and DnpA, part of the LPS biosynthesis cluster, mutant strains no or only a slight decrease of fluorescence intensity was noticed using meropenem or amikacin indicating that most surviving bacteria recorded were not dividing. On the other hand, using fluoroquinolones a part of the dividing population survived and was higher with the mutant. Further, the method was extend to intracellular S. aureus transformed with a plasmid encoding the gfp gene reporter under the tetracycline-inducible promoter (Peyrusson et al. 2020). To confirm the single fluorescent protein dilution-based method, a parallel with the optical density at 600 nm of the culture was done. Identical growth curve and doubling times were recorded for five generations for both techniques. Previously opsonized S. aureus was internalized within eukaryotic cells for 30 minutes, the inducer was present from the overnight liquid bacterial culture until the post-phagocytosis washing step. The infected cells were incubated with antibiotics and either imaged using confocal microscopy or the intracellular bacterial content was collected and analyzed by flow cytometry. This demonstrated that intracellular S. aureus challenged with low antibiotic pressure gave rise to an equilibrium between killing and replication plus a subpopulation that rapidly entered a non-growing state. In contrast, high antibiotic pressure conducted to the killing of dividing S. aureus, leading to a homogeneous population of non-dividing, antibiotic-persistent bacteria at the end of the assay. Furthermore, the non-replicating, antibiotic-persistent bacteria could revert to a normal phenotype in terms of proliferation and antibiotics susceptibility. Thus, the single inducible fluorescent protein dilution method enables to monitor intracellular bacterial replication under antibiotics pressure using microscopy and flow cytometry (Table S2, Supporting Information).
Remus-Emsermann and Leveau then engineer a fluorescent bioreporter for individual-based microbial ecology, which they named CUSPER (reverse of REPSUC, standing for REProductive SUCcess; Remus-Emsermann and Leveau 2010). They engineered two Erwinia herbicola—also called Pantoea agglomerans—to carry a chromosomal mini-Tn5-Km insertion coding for either GFPmut3 or DsRed under promoter controlled by IPTG (Remus-Emsermann and Leveau 2010, Remus-Emsermann et al. 2013). Reproductive success, i.e. bacterial replication, was monitored in a heterogeneous environment represented by the leaf surface of bean plants (Remus-Emsermann and Leveau 2010). Clear different subsets regarding replicative rates were recorded by microscopy when the bacteria were released onto the phyllosphere, supporting the notion that local conditions determine the abundance and replication dynamics of bacteria on leaf surfaces (Remus-Emsermann and Leveau 2010). Further, it was showed that pre-colonization of the leaf had an impact on the proliferation rate of a secondary bacterial immigrant using both previously described E. herbicola (Remus-Emsermann et al. 2013). Measurements of fluorescence intensity demonstrated that the average of the reproductive success of the second colonizer was reduced when the population level of the first colonizer increased. Thus, the use of single fluorescent proteins having different spectrum across mixed bacterial populations allows concomitant replicative rate assessment on heterogeneous substrates via dilution-based method.
Overall, the dilution of a single pre-expressed fluorescent protein allows monitoring of the bacterial replicative state at single-cell level in different niches such as the surface plant leaves or inside eukaryotic cells, by flow cytometry and microscopy, and further combined with concomitant assays (Fig. 3; Table S2, Supporting Information). However, as for the dyes dilution-based methods, it cannot be employed to track replicating bacteria over long periods of time and non-replicative state happening beyond the dilution limit (Fig. 4). Moreover, bacteria outside the resolutive time frame of the method cannot be detected by microscopy without tracking, highlighting the needs of a second fluorescent protein constitutively expressed.
The dual-reporter fluorescent proteins dilution-based method
The protein fluorescence dilution method to detect and sort subsets of bacteria with different replicative rates was considerably enhanced by Helaine et al. (2010) with the addition of another chromophore, either expressed constitutively or induced (Fig. 3). They developed for Salmonella dual fluorescence reporter plasmids expressing (i) the DsRed protein under arabinose promoter and (ii) the EGFP protein either constitutively or upon induction with IPTG. Based on the dilution of the pre-induced DsRed fluorescent protein these constructs allow monitoring of bacterial replication for up to six generations. Meanwhile, the constitutively expressed EGFP fluorescence remains stable and permits bacterial tracking outside of this detectable dilution range. On the other hand, the system with both inducible fluorescent proteins improves the temporal range of bacterial replication detection up to 10 generations by sequential induction of the two promoters. However, bacterial tracking by microscopy using this last dual-reporter version is evidently lost after ten generations (Fig. 4). Results obtained with this dilution method paralleled those with CFU and allowed estimation of the number of cell replication based on the ratio of the geometric mean fluorescence intensity recorded by flow cytometry (Helaine et al. 2010). This enhanced method corroborates the previous finding of homogeneous replication in liquid culture by flow cytometry using S. typhimurium (Roostalu et al. 2008, Helaine et al. 2010, Peyrusson et al. 2020).
The dual-reporter method was employed to shed light on both the intracellular and in vivo bacterial replicative rates (Helaine et al. 2010, Figueira et al. 2013, Mouton et al. 2016). In contrast with reported studies using the single fluorescent protein dilution-based method, the inducer present in the liquid bacterial culture was not maintained during the early step of the in cellulo infection allowing internalization process to occur. Gathered S. typhimurium from infected macrophages showed heterogeneity of bacterial replication among the population by flow cytometry and confirmed by microscopy highlighting different bacterial dividing rates even in the same eukaryotic cell (Helaine et al. 2010). Of note, a large proportion of non-replicating S. typhimurium bacteria were monitored both in vitro and in vivo inside recovered splenic macrophages from infected mice. To gain insight into their subcellular localization, immunolabeling revealed that dividing and replicative-arrested bacteria were contained within vacuoles decorated by LAMP1. Figueira and colleagues modified the fluorescent dual-reporter construction to express mCherry, which matures faster than DsRed, under a constitutive promoter, and GFP expression was put under an arabinose inducible promoter (Shaner et al. 2004, Figueira et al. 2013). Using mutant strains carrying this dual-reporter plasmid they determined that approximatively 30% of individual type III secretion effectors of the Salmonella pathogenicity island-2 system were implicated to intramacrophage replication of S. typhimurium (Figueira et al. 2013). Further, the turboFP635, a far-red protein also named Katushka, was expressed under theophylline-inducible promoter (Shcherbo et al. 2007, Mouton et al. 2016). It was assessed for its high stability even in acidic conditions, confirming its suitability to report on replicative rate inside the vacuolar environment (Mouton et al. 2016). Combined with GFP expression under a constitutive promoter, this fluorescence dilution technique allowed monitoring of mycobacterial replication for up to five generations corroborated by CFU counting. Analysis of far-red fluorescence intensity by flow cytometry of M. smegmatis and M. tuberculosis gathered from infected macrophages, highlighted a heterogeneous mycobacterial replicative state owing to wide distribution of the far-red fluorescence intensity (Mouton et al. 2016). Thus, in contrast with dyes labeling methods, the dual-reporters method was also employed to study the bacterial replicative fates in vivo (Table S2, Supporting Information). Combined with mutant strains it allows determining indispensable proteins in the bacterial replicative process.
The dual-reporter method was next combined with antibiotic pressure to monitor drug-persistent behavior of bacteria (Helaine et al. 2014, Mouton et al. 2016). Mice were infected per os by S. typhimurium carrying the modified dual-reporter plasmid developed by Figueira and colleagues and treated with enrofloxacin in drinking water (Figueira et al. 2013, Helaine et al. 2014). All the gathered bacteria were non-dividing, as showed by lack of fluorescence dilution recorded by flow cytometry. The culture of this sorted population into Luria–Bertani medium indicated resumption of proliferation through reduction of green fluorescence intensity. Further, antibiotic challenged M. tuberculosis-infected macrophages demonstrated an enrichment of high-intensity inducible fluorescent bacteria compared to untreated condition (Mouton et al. 2016). Thus, during antibiotic challenges, dual-reporters method allows characterization of the bacterial replicative rates of survivors, in vitro and in vivo.
Saliba et al. (2017) combined the dual-reporter method with single-cell RNA-sequencing analysis. Macrophages infected by Salmonella were isolated by FACS depending on the intracellular bacterial replicative states, focusing on macrophages containing exclusively non-dividing or dividing bacteria (Fig. 6). As bacteria proliferate, the constitutive signal intensity of single macrophage increases while macrophage containing exclusively non-dividing bacteria display constant both inducible and constitutive intensities over time. The sorted cells were then subjected to RNA-seq analysis. These combined methods demonstrated that macrophages carrying non-dividing, bacterial bystander cells adopted a proinflammatory M1 polarization state while macrophages carrying replicating bacteria harbored anti-inflammatory, M2-like state. Later, Stapels et al. (2018) reanalyzed the single cell data showing bimodality of M1 and M2 genes expression in macrophages having non-replicating bacteria. Further, dual-RNAseq showed that Salmonella persisters, non-dividing but metabolically active, used Salmonella pathogenicity island 2 type 3 secretion system to reprogram macrophages. Thus, dual-reporters-based method allows sorting of infected cells depending on the intracellular bacterial replicative status by FACS and subsequent single RNAseq or dual-RNAseq performing. This provided highly novel information on the phenotypic characteristics of drug persistent bacteria and the RNA status of host cells. Single bacterium RNAseq depending on the replicative rate could be interesting to deeply understand the persisters characteristic and point out drugs target (Imdahl and Saliba 2020).
Figure 6.

Dual-reporters plasmids concomitant assessment. (A) Metabolic assessment (top) and (B) host cell sorting depending on bacterial intracellular replicative content (bottom).
Further, the dual-reporter plasmid thus engineered expressing both fluorescent proteins under inducible promoters allows investigation of metabolic and viability states concomitantly to replicative rates (Helaine et al. 2010, 2014; Fig. 6). Expression of the first inducible fluorescent protein at the onset of the infection informed on the replicative state while addition to the culture medium of the second inducer revealed the responsiveness of the bacteria via the second fluorescent protein expression. These concomitant analyses performed by time-lapse microscopy revealed heterogeneity of metabolic activity and viability of the non-dividing bacterial subset at the single cell level (Helaine et al. 2010). Later, this experiment showed that 40% of drug-persistent non-dividing bacteria remaining were metabolically active. Interestingly, only 5% of them resumed intracellular proliferation when incubated with naïve macrophages, as evidenced by dilution of both fluorescent signals (Helaine et al. 2014). Thus, the dual-inducible fluorescent proteins reporter permits to monitor concomitantly the bacterial dividing rates with the bacterial viability and the metabolic activity.
Overall, the previous single fluorescent protein dilution-based method was enhanced by the expression of another fluorescent protein either under constitutive or inducible promoter (Fig. 3). Beyond the increase in monitoring range from five to six generations up to 10 brought by the dual inducible fluorescent proteins, the strongest advantage of this method is that it allows tracking of bacteria beyond dilution limit of the method via the constitutive fluorescence using real-time microscopy. Further, Gram-positive and Gram-negative bacteria carrying dual-reporter plasmid were employed successfully for both in vitro and in vivo infection with concomitant assays (Table S2, Supporting Information). Finally, dual-reporter plasmids with two inducible proteins inform concomitantly to replicative rates, on the viability and metabolic status of the bacteria. However; as for dyes and single fluorescent proteins dilution method; tracking replicative bacteria over long periods of time as well as replicative-arrested phase occurring after a first period of proliferation beyond the dilution limit cannot be recorded, highlighting the needs of the combination with maturation kinetics method (Fig. 4).
Combination of fluorescent proteins dilution and maturation kinetics: TIMER-based method
Another fluorescence approach based on both the dilution and the sequential maturation kinetics of fluorescent proteins, called TIMER, was developed to monitor the replicative rate of individual bacteria (Fig. 3). It relies on the different maturation kinetics of two fluorophores, e.g. a GFP, which matures and emits fluorescence signal faster than a red fluorescent protein that takes more time to mature and produce fluorescence. Theoretically, the dividing bacterial subset contains mostly GFPs than the red chromophore. The latter matures more slowly and cannot sufficiently accumulates before the dilution of intra-bacterial content at each replicative step. The non-replicating bacteria accumulates firstly the green chromophore and then the slowly maturing red fluorescent protein. This means that the dividing bacteria show a green fluorescence while the non-dividing present first green and then red fluorescence. Furthermore, so long as the maturation kinetics of the proteins are slower than the replicative rates, if the slower maturing fluorescent protein is presynthesized and constitutively expressed, upcoming bacterial proliferation will dilute this fluorescent protein at each replicative step. Next, if the growing bacteria enter a non-replicative state, the proteins will reaccumulate. This could overcome the main limitation of dyes, single fluorescent proteins and dual-reporters dilution methods which cannot monitor a dividing or a non-dividing state occurring outside of the detection area (Fig. 4).
TIMER-based method was employed to study the replicative rates of S. typhimurium at the single-cell level during macrophages and mice infections (Claudi et al. 2014). The efficiency of the method; relying on DsRed variant called TIMERbac (Fig. 7); was assessed by comparison with both the plasmid dilution and the dual-reporter method (Helaine et al. 2010, Claudi et al. 2014). The thermosensitive, single copy plasmid pVE6007 cannot replicates at 37°C and it is, thus transmitted to only one daughter bacterium at each replicative step, meaning that in effectively replicating bacterial population the plasmid dilution is higher compared to slow or non-growing bacterial subsets (Fig. 5). A close correlation was observed between the calculated bacterial replicative rates obtained from both the plasmid dilution method and the TIMER-based approach. These confirm the appropriateness of the technique to monitor and sort single bacteria depending on their specific replicative rate from a heterogeneous population. Various intracellular Salmonella replicative subsets with mainly low green/orange ratio representing slow and non-growing bacteria was highlighted using this method. Interestingly, a large non-dividing subset was obtained using inoculae from stationary phase culture in contrast to more homogeneous replicative rates with exponential phase culture. Besides, in vivo analysis demonstrated that fast dividing Salmonella subsets dominated during the course of the disease progression. Further, when the TIMERbac method was correlated with fluoroquinolones treatment, the fluorescent proteins were retained for at least 1 hour and the intensity remained stable, thus informing on the pre-treatment replicative rate. It is due to both the slow maturation kinetics of the protein and to the mechanism of action of the antibiotic breaking double stranded DNA ultimately causing cell lysis (Hooper 2001). Within infected mice, enrofloxacin daily treatment led to a low survival of fast-growing subset while slow and non-growing better survived but were poorly represented. The large moderate growing subsets i.e., with one to four divisions per day, dominated throughout therapy. Of note, in vitro proteome analysis was accomplished indicating distinct proteomes for the different bacterial replicative rates subsets. Recently, Luk et al. (2021) designed a plasmid that they called Salmonella Intracellular Analyzer (SINA). It combines the TIMERbac fluorescent reporter with promoters reporting on the subcellular localization of the bacteria, either vacuolar or cytosolic. Using this method, they highlighted a dormant persisters subset within a vesicular compartment distinct from the conventional Salmonella-containing vacuoles in epithelial cells. The TIMERbac-based method was also adapted for Legionella pneumophila allowing replicative rates assessment of both intracellular and sessile lifestyle (Personnic et al. 2019, 2021). As previously, the TIMERbac fluorescence intensity ratios were mathematically correlated to the bacterial dividing rates. Then, the bacterial replicative rates were highlighted within amoebae, biofilms and microcolonies showing the coexistence of various dividing rates inside eukaryotic cells and biofilms (Personnic et al. 2019). Further investigation of intracellular non-replicating Legionella employed fluorescent fusion protein, propidium iodide incorporation, fluorescence recovery after photobleaching (FRAP) method and MitoTracker labeling, which is a staining revealing the presence of bacterial membrane potential. Besides demonstrating that the nondividers could reside within vacuole, were metabolically active and viable it accommodates the compatibility of others staining and FRAP approach with the TIMERbac-based method. Further, the Legionella increased drug efflux or decreased drug uptake might explain the higher drug-persistence observed within the non-growing subset. To test these hypothesis, an ethidium bromide accumulation technique was performed showing that the non-growing subset accumulated lower level of compound than other subpopulation (Personnic et al. 2019, 2021). Then, using strains carrying mutants on the quorum sensing Lqs system and LvbR, a transcription factor, the authors showed that the heterogeneity of the Legionella replicative rates subsets was controlled by both (Personnic et al. 2019, 2021). Thus, the TIMER-based method reports on bacterial replicative rates both in vitro and in vivo. It could be combined with several concomitant assays such as proteomes analysis of sorted bacteria, allowing in-depth characterization of the different dividing bacterial rates subpopulations (Table S2, Supporting Information).
Figure 7.

Models for the DsRed maturation pathways. Mutational engineering shed light on the E5 DsRed mutant regarding its fluorescence that change over time (Terskikh 2000). During the maturation kinetics, a green fluorescence appears early and declines as red fluorescence appears as shown in the Conventional model (Terskikh 2000; top). However, even after a prolonged period of maturation, the chromophore sample was composed of equally distributed green and red fluorescence (Garcia-Parajo et al. 2001, Gross et al. 2000), suggesting a coexistence of both chromophore, which makes null and void the Conventional model. The Irreversible deprotonation model inserts an intermediate blue species within the maturation path (Verkhusha et al. 2004; middle). It gives by competition either the green chromophore by deprotonation of the phenolic group or the red chromophore by oxidation, explaining the final coexistence of both. Of note, within the irreversible deprotonation model, intratetramer fluorescence resonance energy transfer (FRET) occurred from green to red fluorescent proteins, decreasing green fluorescence while increasing red fluorescence (Strack et al. 2010). Strack et al. (2010) proposed a novel branched pathway model (bottom). In this model, a branch point intermediate is the place of two competing reactions. Dehydration opens the way to the green branch, while final oxidation creates a blue species, which itself may give rise to a red and non-absorbing species.
Figure 5.
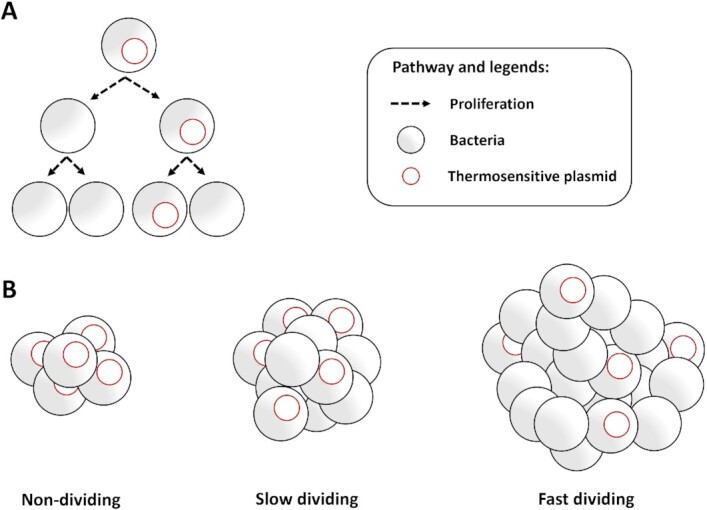
Thermosensitive plasmid method to analyze population-dividing rates from single cell data level. (A) Plasmid dilution principle (top) (B) Schematic representation of plasmid dilution ratio within Non-dividing, Slow dividing and Fast dividing bacterial population (bottom).
Recently, the replicative rates of intracellular S. typhimurium and in vivo Y. pseudotuberculosis at the single cell level were studied using a simplified TIMER-based method (Patel et al. 2021, Schulte et al. 2021). Firstly, correlation of the dividing rates and the fluorescence dilution of a set of fluorescent proteins; displaying various maturation times; comprising DsRed, tagRFP-T, mCherry, mCherry2, or sfGFP was done using flow cytometry (Schulte et al. 2021). DsRed demonstrated the strongest growth phase-dependent reduction of x-median relative fluorescence intensity, which remained sufficiently high to monitor the bacteria and discriminate them from debris in crude-infected host cells lysates. Of note, none of the tested constitutively expressed fluorescent reporters affected proliferation, and all were sufficiently well-tolerated at the expression level used. As expected, drastic drop of the pre-expressed DsRed fluorescence intensity was noticed during the exponential phase while bacteria entering the stationary phase showed continuously increasing fluorescence intensity. Further confirming effectiveness of the method, the DsRed fluorescence intensity profile varied depending on culture conditions and mutant strains known to yield various replicative rates. This simplified TIMER-based method records only the red fluorescence of the DsRed protein putting aside the GFP fluorescence intensity to record another fluorescent reporter. To this end, plasmids were engineered to express DsRed under constitutive promoter and sfGFP under promoters of genes involved in periplasmic or cytosolic stress response. When replicating, the preformed pool of DsRed is diluted while if a stress response is engaged the sfGFP proteins will be expressed. Murine macrophages were infected by stationary phase culture of S. typhimurium highlighting distinct intracellular subsets showing various levels of stress response and proliferation. Over the time course of the infection, non-replicating bacteria were detected, while a replicating and a faster-replicating subsets appeared after different bacterial lag times. Interestingly, the slow-dividing and proliferation arrested bacteria had reduced responses to intracellular stress compared to the moderately fast-replicating subset. In the meantime, Patel et al. (2021) engineered a DsRed42 derivative accumulating only red but not green fluorescence in stationary phase of Y. pseudotuberculosis and E. coli. It is best suited for quickly replicating bacterial species since it maturates faster than the original TIMER-based method allowing investigation of slow replicating Y. pseudotuberculosis subset into deep tissue mouse infection model with concomitant green fluorescent stress reporter assessment. Thus, TIMER-based methods employing DsRed proteins was simplified by focusing on the red fluorescence intensity variation only. Subsequently, the green channel could be dedicated to the concomitant study of bacterial fluorescent reporter such as stress response.
Besides, the TIMER-based method was engineered with two chromosomally inserted fluorescent proteins having different maturation rates and applied to P. aeruginosa (Xia et al. 2018). This constitutively expressed tandem fluorescent protein is composed of sfGFP and Tdimer2, which matures more slowly. The strength of the method was assessed via modulation of the Fe3+ concentration; which impacts the population growth rate; and correlation with the ratio of single-cell fluorescence intensities recorded. The reported ratio was, thus linearly related with the replicative rates. This TIMER-based approach demonstrated that the subsets composed of non-dividing or slow-dividing bacteria increased during tobramycin treatment while the fast-dividing bacterial subset concomitantly diminished. Thus, TIMER-based methods were developed using tandem fluorescent proteins to monitor the bacterial replicative rates at the single-cell level. Careful to the maturing time and spectrum of the selected proteins must be done.
Overall, the combination of the dilution-based method with the sequential and slower maturation rate of fluorescent proteins allows monitoring of bacterial replicative status at the single cell level (Fig. 3). The main strength of this method resides in its long time-span resolution allowing sequential identification of a non-dividing bacterial subset happening after a first period of proliferation without inducer addition requirement. Moreover, TIMER-based method distinguishes the different phenotypic replicative rates via microscopy and flow cytometry from in vitro and in vivo infection with concomitant assays (Table S2, Supporting Information). The recent improvement of the technique permits via focusing on the red fluorescence to release the green channel for concomitant assessment of bacterial green fluorescent reporters. The time required to detect the color change between switches from one division rate to another taking several hours; mainly due to the slow maturation rate combined with bacterial division dilution of the fluorescent proteins; evidently impede the monitoring of short-term fluctuations (Fig. 4). Moreover, the TIMER-based method does not distinguish slow replicating bacteria from non-dividing subset, and it is not best suited for very fast replicating bacteria due to its sensitivity threshold and slow maturation time. Thus, this method is useful for a limited range of replicative rates, since it has a poor resolution of the extremes. Furthermore, in vitro, the O2 concentration impacts the maturation kinetics of DsRed and GFP fluorescent proteins ratio and could skew the replicative rate assessments (Strack et al. 2010). This is a major limitation of the method using DsRed as bioreporter for in vitro and in vivo study due to subcellular compartment and organ diversity regarding oxygenation levels (Liu et al. 2011).
Structural markers-based methods
The structural markers-based methods rely on the microscopic observation of fluorescently labeled bacterial structures informing on either growth or replication status at the single-cell level. Bacterial growth implies peptidoglycan synthesis; formed by glycan strands cross-linked via peptide chains; to extend the cell wall allowing elongation prior to division. Thus, illumination of nascent peptidoglycans synthesis via fluorescent derivatives of antibiotics, lectins, and amino acids or via a pulse-chase assay inform on the growth rate and actively dividing status of the bacteria (Fig. 8). Short pulse labeling using specifics dyes highlights the nascent peptidoglycan regions and long pulse labeling uniformly labels growing bacterial subset with active peptidoglycan synthesis. Besides, unspecific peptidoglycan dyes label the entire peptidoglycan region of growing and non-growing bacteria. The pulse-chase method; working with uniformly labeled bacteria; identify via fluorescence intensity dilution due to new incorporation of unlabeled peptidoglycan moieties the growth rates of entire single cells and even subcellular regions. Moreover, a drop in fluorescence intensity at the septum of dividing bacteria was reported when they were labeled using fluorescent tripeptide entering the recycling pathway. Although the peptidoglycan synthesis enlightenment methods report on growth rates and division status, to our knowledge, these methods have not yet been used to identify bacterial subsets depending on growth and replicative behavior despite this being theoretically possible. Furthermore, these methods so far were mainly employed to decipher the peptidoglycan synthesis patterns rather than to study growth and replicative rates. Moreover, based on the same concept, mycomembrane synthesis enlightenment reports on the growth pattern of mycobacteria.
Figure 8.

Principles of the peptidoglycan biosynthesis highlighting methods to monitor growth rate and dividing behavior. The nascent and total bacterial peptidoglycan regions can be labeled using antibiotics, lectins, and amino acids derivatives. (A) Peptidoglycan synthesis dyes during short pulse labeled the nascent peptidoglycan sites while long pulse uniformly labeled growing bacteria. Of note, non-growing bacterial subset remains unlabeled. (B) Bacteria uniformly labeled with either nascent peptidoglycan dyes during long pulse or total peptidoglycan dyes will incorporated unlabeled new materials during the chase period. Time-lapse microscopy monitoring allows measuring the bacterial and the subcellular growth rate based on unlabeled region extension. A non-growing bacterial subset should remain uniformly labeled at the chase time. (C) Short sequential pulse-chase using different fluorophores label the nascent peptidoglycan sites sequentially. End-point analysis creates a growing map over-time. (D) Continuous pulse enable real-time measurement of the nascent peptidoglycan sites kinetics of growing bacteria using time-lapse microscopy. Growing labeled bacteria via the recycling pathway highlight dividing step due to fluorescence intensity drop at the septum. (C) and (D) Short sequential pulse-chase and continuous pulse should, therefore, discriminate the non-growing unlabeled subset and highlighted the growth rate.
The bacterial replication event is characterized by the creation of a ring septum composed of several known proteins at the division site. Basically, the septum allows the division of the mother cell into two daughter cells by membrane invagination. Monitoring of septum proteins fluorescently tagged over-time allows measurement of replicative rates via successive formation and dissolution of the ring structure. Besides, the regrowth-delay body is a structure forming two granular poles within bacteria in growth-arrested phase. Monitoring of the fluorescently tagged regrowth-delay body allows identification of the non-growing bacterial subset. Nevertheless, while the first method required time-lapse monitoring to measure replicative rates, the second failed to highlight growth and replicative rates, discriminating only the non-growing subset.
Septum and regrowth-delay body
Time-lapse microscopy method and fluorescent septation marker was combined to identify M. smegmatis division events (Santi et al. 2013). The essential late-division Wag31 protein localizes at the septum and remains associated with both newly created daughter poles. Fusion of Wag31 with GFP or mCherry allowed monitoring of the protein density over the time course of the experiment at single-cell level. The beginning of the septation was monitored via invagination of the bacterial membrane labeled with FM4-64 dye preceding Wag31-GFP structural apparition. The authors defined and monitored the interdivision time as the time ranging from the apparition of the septal Wag31-GFP structure at birth to the next apparition at the subsequent replication event. As previously described, the growth rate was measured via monitoring of the cell length over-time. Using this method, they showed that although mother cells performed asymmetric division, the unequally sized progenies grown at similar rates and tolerate antibiotics equally well. Later, time-lapse phase contrast images and fluorescent segmentation marker were employed to study the growth and replicative rates of E. coli within the turbidostat device (Wallden et al. 2016). Time-course of fluorescence intensity highlighted accumulation of the cell division protein FtsQ tagged with GFP at the septum before its disassembly and vanishing at the replicative step. The division time obtained with both the phase contrast division classifier based on bacterial contours and the fluorescent segmentation marker methods was correlated with a difference of 2 minutes, thus corroborating each other. Thus, via time-lapse microscopy method, the structural segmentation markers, either Wag31 or FtsQ, allowed monitoring of the replicative rates at the single-cell level.
Further, Yu et al. (2019) fortuitously discovered a reversible subcellular structure composed of two pole-granules that they named the regrowth-delay body highlighting the non-growing bacterial subset at the single-cell level. The FtsZ cell division protein, fused with the mNeonGreen, was localized at the Z-ring structure during exponential growth phase while, interestingly, it was observed via microscopy as two bacterial pole-granules during late non-growing stationary phase of E. coli cultures (Ma et al. 1996, Yu et al. 2019). Moreover, a relocation of the fused protein from the regrowth-delay body of late stationary phase to the Z-ring within bacteria resuming growth in fresh medium was observed after a lag phase (Yu et al. 2019). Furthermore, older stationary phase cultures with aged regrowth-delay bodies better tolerate the selective pressure imposed by ofloxacin or ampicillin addition. Of note, during regrowth culture assay all the bacteria keeping the regrowth-delay body structures, the non-growing bacterial subset, survived the ampicillin treatment while the bacteria with dissolved pole-granules, the growing subset, died. Thus, the regrowth-delay body is a structural marker that allows monitoring of non-growing bacteria at the single-cell level as well as the bacterial lag time.
Overall, structural markers consisting of the septum and regrowth-delay body enable motoring of the non-growing subset and the replicative rate at the single-cell level. The septum kinetics required time-lapse microscopy monitoring while the regrowth-delay body can be analyzed using end-point microscopy. Antibiotic pressure combined with septum and regrowth-delay body monitoring allowed discrimination of the persistent bacterial subset based on their growth behavior. The main limitation of septum vanishing tracking is inherent to the time-lapse method, which impedes end-point measurements. The progressive formation over the cell division cycle of the regrowth-delay body impedes early-stage monitoring. Moreover, although this method allowed monitoring of the bacterial regrowth lag time it impedes growth and replicative rates measurements.
Highlighting peptidoglycan synthesis
Antibiotics-based probes
The peptidoglycan synthesis pattern of Gram-positive bacteria were probed and investigated using vancomycin and ramoplanin antibiotics fluorescent derivatives (Daniel and Errington 2003, Tiyanont et al. 2006, Kang et al. 2008, Aldridge et al. 2012). Vancomycin linked with the terminal D-alanine-D-alanine dipeptide and ramoplanin linked with the reducing end of the glycan chain at the initiation site of lipid II incorporation during peptidoglycan synthesis (Hammes and Neuhaus 1974, Tiyanont et al. 2006). Daniel and colleagues suggested the rapid protection of this terminus from vancomycin binding by processing in mature peptidoglycan (Höltje 1998, Daniel and Errington 2003). As a result, fluorescent derivatives of vancomycin should bind to nascent peptidoglycan precursors not yet processed and, thus highlighting the pattern of newly peptidoglycan insertion. To confirm this hypothesis, bacteria were engineered with the murE gene encoding UDP-N-acetylmuramyl tripeptide synthetase involves in earliest step of synthesis under IPTG promoters (Daniel and Errington 2003). As a result, lack of the inducer leads in reduction in overall staining. Of note, vancomycin fluorescent molecules also bind in a smaller proportion the mature peptidoglycan due to peptide motifs that have not been cross-linked. Further, both antibiotic fluorescent derivatives showed dose-dependent labeling and ramoplanin worked at a lower concentration than vancomycin (Tiyanont et al. 2006). Using this labeling method, the pattern of B. subtilis peptidoglycan insertion in the cylindrical portion of the bacteria was showed to be helical (Daniel and Errington 2003, Tiyanont et al. 2006). Besides, Streptococcus pneumoniae showed a middle insertion pattern while Streptomyces coelicolor showed nascent peptidoglycan precursors at poles (Daniel and Errington 2003). Further, vancomycin fluorescent derivate demonstrated a correlation of the nascent peptidoglycan sites with Wag31 fluorescent intensity at the poles of M. smegmatis (Kang et al. 2008). Later, it illustrated that the nascent peptidoglycan regions were preferentially located at the old pole over the new pole of M. smegmatis highlighting unipolar fashion elongating pattern (Aldridge et al. 2012).
Overall, vancomycin and ramoplanin fluorescent derivatives targeting the nascent peptidoglycan precursor combined with fluorescence microscopy allowed monitoring of the peptidoglycan synthesis pattern at the single-cell level (Fig. 8). Since nascent peptidoglycan synthesis occurs only within the growing bacterial subset, the antibiotics-based probes method should, therefore, allow distinguishing between growing labeled and non-growing unlabeled single-cell. Furthermore, the antibiotic concentration and time of treatment require precise adjustment to balance both suitable staining and avoid cell damage. Main limitation of this method is the requirement for fully accessible bacterial wall such as within Gram-positive bacteria or Gram-negative mutants with a compromised outer membrane.
Lectin wheat germ agglutinin pulse-chase-based probe
Ursell et al. (2014) investigated the subcellular growth rates, geometry and cytoskeletal organization of E. coli using time-lapse microscopy and pulse-chase labeling of the cell wall. The authors reasoned that if bacterial growth was heterogeneous along the surface, thus a uniform peripheral labeling would be spread apart by the addition of new unlabeled material. To this end, E. coli were unexpectedly labeled with a fluorescent analog of the lectin wheat germ agglutinin (flWGA) followed by excess dye removal before growth monitoring. Wheat germ agglutinin was known to label the N-acetylglucosamine motif of the peptidoglycan suggesting that flWGA diffuse through or binds to a specific component of the outer membrane. Time-lapse imaging after excess WGA removal revealed the transition from a uniform peripheral labeling to a punctate staining. Moreover, surface fractional elongation rates were calculated relying on the difference in spatial and temporal position between two adjacent flWGA fluorescence intensity peaks. Growth map computing the rate of spreading between adjacent peaks highlighted heterogeneity. Some subcellular regions had higher rates than the calculated bacterial growth rate. Concomitantly, the MreB actin-like protein, essential to maintain the bacterial rod-like shape, preferentially localized and targeted bacterial elongation; showed by negative correlation with flWGA staining; at subcellular negative cell wall curvature regions leading to cell straightening.
Overall, the bacterial wall flWGA pulse-chase labeling combined with time-lapse microscopy allows monitoring of both subcellular and total bacterial growth rates at the single-cell level (Fig. 8). The main limitation of the pulse-chase method relies on the detection threshold of unlabeled growing regions. Actually, these regions must have been spaced far enough from the two adjacent fluorescent puncta to overcome the optical limitation of the microscope. Moreover, wheat germ agglutinin preferentially labels Gram-positive bacteria, despite some exception as presented here. This method should therefore discriminate at single-cell level between growing subset heterogeneously labeled and non-growing uniformly labeled subset.
Amino acids-based probes
The L-alanyl-γ-D-glutamyl-L-lysine tripeptide was labeled with N-7-nitro-2,1,3-benzoxadiazol-4-yl and supplemented in the bacterial medium to investigate the peptidoglycan synthesis in E. coli (Olrichs et al. 2011). The authors showed that the labeled peptide reached the cytoplasm and was incorporated within the peptidoglycan via the bacterial-wall-recycling pathway. This method demonstrated that elongating bacteria were uniformly labeled while dividing bacteria had a significant decrease in fluorescence at the septum linked with the FtsZ presence and AmiC amidase elevated activity. Thus, this fluorescent tripeptide method sheds light on the peptidoglycan-recycling pathway and should, therefore, discriminate between dividing and non-dividing bacteria. However, the bacterial growth was inhibited after four generations in the presence of the labeled tripeptide, thus limiting the usefulness of the method.
Further, a series of fluorescently labeled D-amino acids responsible for the cross-link of glycan strands and thus incorporating into nascent peptidoglycan sites were engineered (Kuru et al. 2012, 2015). The fluorophores 7-hydroxycoumarin-3-carboxylic acid, 4-chloro-7-nitrobenzofurazan, 5 (and 6-)-carboxytetramethylrhodamine succinimidyl ester, and FITC were linked to the 3-amino-d-alanine backbone resulting in various fluorescent D-amino acids HADA, NADA, TDL, and FDL probes, respectively. The authors validated the strengthening of the probes to incorporate and label nascent peptidoglycan sites via mass spectrometry analysis demonstrating the incorporation of the dye within peptidoglycan peptide stems. Of note, probes based on the enantiomer L-amino acids failed to label growing bacteria. As expected, long-term staining labeled uniformly the bacterial wall while short-term staining probed uniquely the peptidoglycan synthesis regions of the growing bacterial subset. Pulse-chase and time-lapse microscopy experiments combined with long-term staining highlighted the labeled growing subset while the non-growing unlabeled subset should, therefore, be distinguished. Subsequent culture without probes highlighted the growing labeled subset loosing progressively, as their growth progressed, the labeling intensity via incorporation of unlabeled new materials leading to fluorescence dilution. The potentially non-growing subset after a first period of growth during the labeling pulse should retain fluorescence intensity at similar level and, therefore, be distinguished. Using this long-term method, the authors demonstrated that growing E. coli and B. subtilis ΔdacA retained signal at the mother poles and lost it at the lateral walls. The short-term method highlighted diverse patterns of peptidoglycan synthesis regions within broad species. Furthermore, sequential pulse-chase assay using different spectral D-amino acid probes allows peptidoglycan nascent sites formation tracking via end-point microscopy informing on the chronological incorporation of the probes during their respective labeling periods. However, it required long washing steps between pulses creating a delay impeding with this highly dynamic process. Of note, the developed probes were used to label a broad array of Gram-positive and Gram-negative bacteria appearing to potentially be universal. Thus, labeled D-amino acids allowed monitoring of nascent peptidoglycan regions using short-term labeling. Pulse-chase experiments highlighted growing subsets by fluorescence dilution via news unlabeled materials incorporation and should, therefore, distinguish the non-growing uniformly labeled subset.
Click chemistry were concomitantly developed using D-amino acids probes allowing to monitor the peptidoglycan nascent region of intracellular bacteria (Kuru et al. 2012, 2015, Siegrist et al. 2013, Liechti et al. 2014). D-amino acids analogue; termed either ethynyl-D-alanine and azido-D-alanine or R-propargylglycine and R-2-amino-3-azidopropanoic acid; can be conjugated to any molecules carrying the complementary functional group as azides and alkynes fluorophores (Kuru et al. 2012, 2015, Siegrist et al. 2013). The two latter were putted in contact with growing planktonic and intracellular bacteria before reaction with azides and alkynes fluorophores, subsequent fluorescent intensities were recorded by microscopy (Siegrist et al. 2013). These methods labeled the nascent peptidoglycan regions of growing intracellular L. monocytogenes at septa after a short pulse. Later, Liechti et al. (2014) employed the D-alanine-D-alanine dipeptide click chemistry probes to label the peptidoglycan of intracellular Chlamydia trachomatis. Again, the labeling localized to the septal part of dividing subset. Thus, since the eukaryotic cells did not generally produce D-amino acids, the clickable fluorescent D-amino acids-based probes allow monitoring of nascent peptidoglycan regions of single bacteria and should, therefore, distinguish the unlabeled non-growing intracellular bacteria.
Hsu and colleagues developed a rotor-fluorogenic D-amino acids to study peptidoglycan synthesis in real-time with higher temporal resolution than the previously described sequential pulse-chase method (Kuru et al. 2012, Hsu et al. 2019). Fluorescent molecular rotors exist in two stages depending on the spatial hindrance. In low spatial hindrance, the excited molecule had a twisted intramolecular charge transfer state resulting in nonradiative relaxation. In high spatial hindrance, the twisted intramolecular charge transfer state is inhibited resulting in fluorescence emission of illuminated fluorescent molecular rotors. It was showed that the peptidoglycan environment induced sufficient spatial hindrance to activate fluorescent molecular rotors coupled with D-amino acids, thus reporting on peptidoglycan synthesis in real-time. Of note, due to the intrinsic properties of the fluorescent molecular rotors, this enables end-point analysis without washing steps. Short-pulses highlighted nascent peptidoglycan at the poles of Streptomyces venezuelae and long-pulses involving B. subtilis resulted in homogeneous labeling as previously described. Moreover, real-time imaging of S. venezuelae showed fluorescent signal appearing first at poles and then at the division septum. Although the labeling fit well with Gram-positive bacteria, outer membrane permeabilization of Gram-negative bacteria is mandatory. Thus, the rotor-fluorogenic D-amino acids allowed monitoring of peptidoglycan synthesis in real-time at the single level and should, therefore, distinguish non-growing bacteria.
Overall, amino acids labeling informs on the peptidoglycan lifecycle of the growing and dividing bacteria possibly intracellularly and in real-time and should, therefore, discriminate non-growing subset (Fig. 8). Of note, the D-amino acids probes developed by Kuru et al. (2012) are more broadly used due to their compatibility with Gram-positive and Gram-negative bacteria as compared to others. To our knowledge amino acids labeling has not been employed to monitor any of the following: bacterial growth, replicative rate, or discrimination between both growing and non-growing bacteria despite its theoretical workability.
Mycomembrane synthesis enlightenment
Hodges et al. (2018) designed a quencher–trehalose–fluorophore (QTF) probe to report on the mycolyltransferases activity of mycobacteria in real-time. The antigen 85 complex (Ag85), which is a mycolyltransferase of M. tuberculosis, elaborates the mycolic acid membrane. Trehalose monomycolate is processed to trehalose dimycolate or mycolyl-arabinogalactan and inserted into the bilayer-like membrane. The developed QTF probe is an analog of the trehalose monomycolate and bears the fluorescent dye BODIPY-FL appended to the fluorescence quencher DABCYL. When QTF is processed by nucleophilic attack of Ag85, the DABCYL part is removed, thus separating the FRET pair and enabling fluorescence emission. Combination of QTF probe with time-lapse microscopy method allows monitoring of dividing M. smegmatis over four generations. The authors demonstrated that the resulting QTF fluorescence was localized to the septa and both poles, with higher intensity at the older poles. Of note, co-incubation with the HADA probe showed similar localization as QTF. Moreover, the three mycolyltransferases of M. smegmatis were fused to mCherry and colocalized with the QTF signal.
Overall, QTF probe highlights the mycomembrane synthesis by reporting the mycolyltransferase activity at the single-cell level in real-time. The QTF fluorescence intensity colocalizes with nascent peptidoglycan regions and should, therefore, discriminate between growing and non-growing subset. The main limitation of this method is that the QTF probe worked only with mycolyltransferase-producing bacteria. Actually, fluorescence was recorded at the surface of M. smegmatis and C. glutamicum in contrast to E. coli and B. subtilis.
Emerging and sporadically used methods
Some emerging and/or sporadically used methods were employed to monitor the growth and replicative rates at the single-cell level. Highly precise detection of the individual buoyant mass of bacteria over-time using a SMR combining cantilever and microfluidics informs on the bacterial growth rates (Godin et al. 2010, Cermak et al. 2016). Later, based on the bidirectional chromosomal replication pattern and initiation at a conserved bacterial sized per chromosome, oriC:terC ratio allows discrimination of subsets depending on growth behaviors (Haugan et al. 2018). Further, stable isotope probing report among other parameters on the anabolic activity of bacteria detected via secondary ion mass spectrometry (SIMS) or Raman spectrometry (Kopf et al. 2016, Taylor et al. 2017). The increase in mass or shift in spectra detected were translated into growth rate via empirical equations. Finally, rRNA content calculated via FISH of individual bacteria was linked to population growth rate helped by a standard curve, which must be previously carried out (Poulsen et al. 1993). Strengths and strong limitations of these methods were summarized in Table 1.
SMR measuring bacterial buoyant mass
Time-lapse microscopy-based methods previously described rely mainly on the measured cell length or area as a reporter of the cell growth. However, continuous accumulation of mass and volume measurement of a single bacteria report on the individual creation of new biomass, also termed growth rate. Techniques involving resonating microelectromechanical system allowing measurement of bacterial buoyant mass with femtogram precision over-time were developed (Godin et al. 2010, Cermak et al. 2016). The buoyant mass of the bacteria corresponds to their own mass minus the mass of the fluid it displaces. Bacteria transit into a SMR consisting of a vacuum packed hollow cantilever beam containing an embedded fluidic microchannel. Upon bacterial detection via the SMR, fluidic flow changes its direction, thus reintroducing the bacteria within the cantilever and allowing repeated measures over-time of the same single-cell (Godin et al. 2010). A shift in the resonance frequency of the system was measured at each passage and converted into buoyant mass. Growth rate was obtained from linear fits to the buoyant mass representing increasing amplitude of the peaks. A rapid drop by a factor two of the magnitude of the frequency shifts suggest that a replication step had occurred and a progeny escaped the trap. Escherichia coli and B. subtilis could be trapped on average for 300–500 seconds, although few cells were trapped over the entire cell-cycle. Later, the method was engineered by serially connecting cantilevers, thus improving the throughput of the technique from one single bacterium at a time to 150 bacteria per hour (Cermak et al. 2016). An array of SMRs is fluidly connected in series and spaced by serpentine delay channels allowing the growth of the bacteria between each cantilever during the crossing. The design permits monitoring of several individuals’ bacteria at a time during 4 minutes via a fluidic bacterial chain crossing 10–12 resonant mass sensors distributed along the length of the microfluidic. The authors demonstrated by plotting the growth rates against the initial buoyant mass that heavier bacteria grow faster than lighter ones (Godin et al. 2010). The long duration trap of B. subtilis covering a full cell cycle confirm the data obtained with shorter measurement via an exponential fit of the growth rates. Cermak et al. (2016) demonstrated that after kanamycin addition to an E. coli culture, the growth rates dropped rapidly reaching near 0 in 30 minutes showed by almost null mass accumulation. However, there is variation in the timescale of switch to growth arrest phase at the single cell level, non-growing and normally growing bacteria coexist at 20 minutes post-intoxication.
Overall, the SMR method allows monitoring of the bacterial growth rates at the single-cell level relying on high-precision buoyant mass measures over-time. This technique enables precise monitoring of growth rates notably under drugs challenges. The main limitation of this method is that in contrast to the time-lapse microscopy method the duration of measurement lasts less than the bacterial cycle covering only a short snapshot of the single-cell growth. Moreover, the bacterial replication cannot be directly recorded. Furthermore, the SMR array is currently not suitable for measuring growth of adhering bacteria. Time-lapse microscopy methods are more broadly employed than the SMR system due to its wider availability and easier operation.
Chromosomal replication monitoring via foci detection
Haugan et al. (2018) monitored, via fluorescence microscopy, chromosomal replication markers reporting on the bacterial growth status at the single-cell level. The DNA replication of the single circular chromosome of E. coli starts from a single origin of replication (oriC) and spreads bidirectionally to reach the opposite replication terminus region named terC, allowing the progenies to bear a unique copy of the chromosome. Under optimal conditions, few initiations of replication can occur concomitantly per division cycle resulting in progenies with overlapping oriC copies at birth. Moreover, the chromosomal replication starts at a fixed cell mass per origin meaning that the cell mass increases proportionally to the oriC copies number. Based on this principle, the copy number ratio of oriC to terC reports on the growth behavior. A ratio of 1 reports on non-replicating bacteria with a unique oriC while a ratio superior to 1 reports on growing bacteria bearing at least 2 oriC with ongoing chromosomal replication. To this purpose, P1-ParB and pMT-ParB proteins fluorescently labeled report respectively to origins and termini via foci detection by microscopy. A clear correlation between cell size and growth behaviors, derived from the chromosomal replication marker monitoring method, support its effectiveness. Moreover, during the exponential phase of growth, a mass doubling time was deduced from the oriC:terC ratio and was similar to the one deduced from the population optical density measurements. Stationary phase culture diluted in fresh media presented first homogenous non-growing bacteria; with oriC:terC around 1; and then heterogenous growing bacteria each with staggered ongoing chromosomal replications. Later, a tendency toward growth homogeneity was detected with ratio egal to 4 until reach once again a homogenous population of non-growing bacteria. In contrast, complete cessation of chromosome replication was not observed at any point during mice infection. Later, the chromosomal replication monitoring method was employed to assess the antibiotics efficacy as a function of growth rate in vitro and in vivo (Haugan et al. 2019). Sukumar et al. (2014), based on the chromosomal replication concept monitored the replicative behavior of in vivo M. smegmatis via monitoring of the single stranded binding protein (SSB) component of the replisome fused to GFP. The authors showed via time-lapse microscopy that the dynamics of SSB-GFP foci defined cell cycle timing and discriminated replicating from non-replicating subsets.
Overall, the chromosomal replication monitoring method discriminates bacterial subset depending either on their growth or replicative behaviors at the single-cell level. An oriC:terC ratio of 1 correspond to non-growing bacteria and the higher the number of copies, the higher the size and mass. The main limitation of the method is that it requires a previous proteins labeling to highlight oriC and terC chromosomal foci. Moreover, overlapping fluorescent foci impede accurate monitoring of oriC:terC ratio. Furthermore, physiological dysregulation between replication and division coupling will impede interpretation. Importantly, an oriC:terC ratio of 1 and a lack of SSB foci may correspond either respectively to a non-growing or non-replicating bacterium, or to a growing or replicating bacterium between active stage of chromosomal replication.
Stable isotope tracer incorporation monitoring
The stable heavy water 2H2O isotope was employed to measure the anabolic activity of S. aureus from cystic fibrosis patient and to calculate the related growth rate and mass doubling times at the single-cell level (Kopf et al. 2016). The 2H incorporation and removal from bacterial population are governed by the population growth rate, the turnover rate and the death/degradation rate. It was linked to the growth rate taking into account the limitation of non-instantaneous water exchange, the hydrogen metabolism of S. aureus and the maintenance turnover. The latter was approximately calculated from S. aureus chemostat culture at near null replicative rate. Thus, the authors calculated and estimated the single-cell growth rate based on a population level equation, which does not take into account known heterogeneity of growth within a population (Balaban et al. 2004, Gangwe Nana et al. 2018, Haugan et al. 2018). To this end, expectorations of sputum were recovered and allowed to grow in presence of 2H2O before samples fixation. During the labeling period, only active cells incorporated the isotope tracers in contrast to dormant or dead bacteria. The isotopic 2H bulk membrane mass enrichment was measured at the single-cell level via secondary ion mass spectrometry (NanoSIMS). Coupled fluorescence in situ hybridization (FISH) method allowed specific highlighting of S. aureus or P. aeruginosa. Using this method, large growth rates heterogeneity was demonstrated. The calculated mass doubling times ranged from 256 days to 6 hours and should explain antibiotic failure. Later, 13C isotope tracer probing method was developed in individual photoautotrophic bacteria to calculate the growth rate via quantitative Raman spectrometric measurement (Taylor et al. 2017). Assimilation of 13C-enriched dissolved inorganic carbon was quantified at the single-cell level from red shifted wavenumber in resonance Raman spectral peaks emanating from carotenoid pigments. The detected isotopic assimilation was then converted into single-cell level growth rate via an empirical relationship. Calculated Synechococcus sp. growth rate showed heterogeneity at the single-cell level and was similar to the instantaneous population growth rate independently measured. Further, Synechococcus sp. and Thalassiosira pseudonana growing at different rates were mixed and single-cell Raman spectrometry, combined with FISH phylogenetic identification, could distinguish among them. Recently, the method was extended to individual heterotrophic E. coli growing in mixed microbial assemblages on complex 13C carbon sources (Weber et al. 2021). Further, 15N isotope incorporation monitoring by NanoSIMS into steady state liquid culture of E. coli and B. subtilis was employed to calculate the growth rate at both population and single-cell levels (Gangwe Nana et al. 2018). The authors estimated the mass doubling times of single cell assuming conservation of nitrogen, individual exponential growth as well as no significant changes in the media throughout the assay. The population growth rate calculated based on 15N isotope incorporation was similar to the one calculated with the optical density supporting the rightness of the method. Moreover, control for the basal 15N isotope incorporation by a non-growing culture is missing. Otherwise, results were much less erratic, and hence appeared more trustworthy with labeling period above 32 minutes giving a mass doubling time of 65 minutes. Inter-bacterial 15N isotope incorporation reflecting heterogeneity of the mass doubling times of individual bacteria was demonstrated using this method. Interestingly, asymmetric incorporation within bacteria was shown indicating an intra-bacterial growth rate heterogeneity, which was more pronounced after a labeling period covering two mass doubling times. It could be explained by the combination of the semi-conservative nature of DNA replication and the strand-specific phenotype model explaining metabolic activities segregation to one DNA strand into one resulting daughter cell (Meselson and Stahl 1958, Rocha et al. 2003).
Overall, isotope tracer incorporation reporting on the anabolic activity informed on the approximated growth rate of bacteria at the single-cell level. Strength of this method is that it does not require any prior treatment such as genetic modification, and thus report on the quasi-instantaneous growth rates of clinically isolated pathogens after a short incubation time ex vivo, which should not significantly disrupt the in vivo physiology. Combination with FISH phylogenetic identification allows highlighting of bacteria of interest within a community. Nevertheless, it is based on population empirical relationship, which does not take into account heterogeneity at the single-cell level. Further, basal incorporation of the isotope must be characterized from in vitro viable non-replicating culture.
Individual rRNA content estimating the culture-like growth rate
The correlation between the S. typhimurium rRNA content and the population growth rate has long been recognized (Schaechter et al. 1958). DeLong et al. (1989) developed a method based on rRNA single-cell quantification to approximate the culture derived growth rate of E. coli. rRNA was quantified using FISH with a universal oligonucleotide probe in media supporting different growth rates. The resulting fluorescent intensity informing of the rRNA content varies linearly with the population growth rate. Later, specific 16S rRNA fluorescent probe linked to the population growth rate showed that the average doubling time was longer in established biofilm than in young biofilm of E. coli (Poulsen et al. 1993). Moller et al. (1995) confirmed the workability of the method by correlating the rRNA content of individual Pseudomonas putida with specific culture growth rate. Further, the metachromatic dye acridine orange labeling single- and double-stranded nucleic acid in red and green fluorescence respectively showed that total DNA and RNA measure at single-cell level via flow cytometry were related with the culture growth rate. The whole-cell hybridization method were then employed to monitor the rRNA content of E. coli and S. typhimurium gathered from large intestine of streptomycin-treated mice (Poulsen et al. 1995, Licht et al. 1996). It was converted to specific culture growth rates helped by an in vitro standard curve showing overall high intestinal growth rate, which appeared incompatible with the size of the population and the fecal excretion colony forming unit rate. Precursor-16S rRNA probe highlighted two population coexisting with different apparent growth rates (Licht et al. 1999). Later, the FISH rRNA monitoring to decipher the correlated culture growth rate of in vivo individual P. aeruginosa was employed (Yang et al. 2008, Kragh et al. 2014). The individual rRNA bacterial level from cystic fibrosis patient mostly correlated with heterogeneous in vitro culture growth while few bacteria correspond to a non-growing stationary phase subset (Yang et al. 2008). Further, a negative correlation was highlighted between the neighboring polymorphonuclear leukocytes concentration and the culture-like growth rate of individual bacteria within biofilm aggregates from lungs of patients and mice (Kragh et al. 2014). It is due to induced O2 limitation and alleviated by denitrification. Of note, at low growth rate, individual rRNA content did not linearly correlate with the optical density of the culture. Furthermore, since a correlation can return negative growth rates the authors considered that in these cases growth rate was null. Later, a weak growth of Stenotrophomonas maltophilia in sputum; calculated from FISH estimated ribosomal content; correlated with the inability of S. maltophilia isolates to perform denitrification (Hansen et al. 2014, Kolpen et al. 2015).
Overall, FISH method allowed monitoring of the bacterial rRNA content immediately after patient gathering without pre-treatment requirement at the single-cell level, which correlated linearly to the global population culture growth rate. Although the method is based on single cell rRNA measurements, it correlates with population growth rate measurement methods, which does not consider growth heterogeneity within a culture. Furthermore, a weakness of the method is that it required an in vitro standard curve; linking rRNA content of individual bacteria with the culture growth rate; evidently obtained from distinct condition than in vivo samples. Moreover, considering the global rRNA content without processing step distinction induces errors. Of note, rRNA content could be decoupled from the growth rate in some species.
Concluding remarks
Over a century ago, heterogeneity of growth and replicative rates within a bacterial population was first hypothesized and then studied thanks to technological innovations. Among the diversity of the tools available to determine the bacterial replicative rate at the single-cell level, fluorescence dilution methods are becoming increasingly used because they almost exclusively overcome limitations, such as mandatory tracking, by giving insight into replicative history. It allows very rapid sequential analyses of single cells using flow cytometry from in vitro, in situ, and in vivo samples although requiring pre-treatment such as dyes labeling. A less costly method is based on colony monitoring on agar plates, but it reports only on the ability of an individual bacteria to perform at least one replicative step with its associated bacterial lag time. The bacterial growth rate at the single-cell level was broadly analyzed using time-lapse microscopy methods notably due to the lack of alternative user-friendly methods. It allows concomitantly monitoring of the replicative rate and required constant tracking making it impractical for historical or instantaneous evidence, especially from in vivo samples. Furthermore, in vitro structural markers-based methods such as peptidoglycan synthesis and regrowth-delay bodies enable mainly end-point monitoring of inter- and intra-bacterial growth or replicative behavior, but the corresponding rates rely on time-lapse microscopy methods. Strength of this method as well as stable isotope probing technique is that it reports on intracellular growth pattern. Moreover, the sporadically used buoyant mass monitoring via SMR calculates precisely the growth rate for a few minutes, and hence mismatch with replicative rate monitoring. Furthermore, chromosomal replication marker-based methods enable only to distinguish single-cell bacteria depending on their growing behavior. Some others emerging and/or sporadically used approaches such as stable isotope probing and rRNA content monitoring allow calculation of the instantaneous growth rate of individual bacteria gathered directly from patient. However, the apparent growth rate was either empirically based on a population equation with short incubation time ex vivo or based on standard curve linking single-cell measurement to population growth rate. Furthermore, these two latest methods cannot be used to monitor concomitantly the replicative rate. Thus, there is an urge need for a precise end-point method combining both growth and replicative single-cell historical rates monitoring from in vitro, in situ and especially in vivo samples which could allow better understanding on the underlying mechanism of both rates in vivo potentially implicated to therapeutic treatment failures.
The methods presented in this review are in constant evolution with fluorescent proteins and dyes advances as well as monitoring device resolution increase. Therefore, these methods should continue to increase our knowledge and understanding on single bacteria growth and replicative rates within broad conditions from a heterogeneous and/or drugs challenged in vitro population to clinical isolates for years to come.
Authors' contributions
F.M. discussed and planned the manuscript with F.L., J.J., and A.B.; F.M. did all the bibliography and wrote, then revised the manuscript; J.J. and A.B. each reviewed and edited the manuscript.
Contributor Information
Florian C Marro, Evotec ID Lyon, In Vitro Biology, Infectious Diseases and Antibacterials Unit, Gerland, 69007 Lyon, France; CIRI – Centre International de Recherche en Infectiologie, Inserm, U1111, Université Claude Bernard Lyon 1, CNRS, UMR5308, Ecole Normale Supérieure de Lyon, Univ Lyon, F-69007 Lyon, France.
Frédéric Laurent, CIRI – Centre International de Recherche en Infectiologie, Inserm, U1111, Université Claude Bernard Lyon 1, CNRS, UMR5308, Ecole Normale Supérieure de Lyon, Univ Lyon, F-69007 Lyon, France; Institut des Sciences Pharmaceutiques et Biologiques (ISPB), Université Claude Bernard Lyon 1, Lyon, France; Centre de Référence pour la prise en charge des Infections ostéo-articulaires complexes (CRIOAc Lyon; www.crioac-lyon.fr), Hospices Civils de Lyon, Lyon, France; Laboratoire de bactériologie, Institut des Agents Infectieux, French National Reference Center for Staphylococci, Hospices Civils de Lyon, Lyon, France.
Jérôme Josse, CIRI – Centre International de Recherche en Infectiologie, Inserm, U1111, Université Claude Bernard Lyon 1, CNRS, UMR5308, Ecole Normale Supérieure de Lyon, Univ Lyon, F-69007 Lyon, France; Institut des Sciences Pharmaceutiques et Biologiques (ISPB), Université Claude Bernard Lyon 1, Lyon, France; Centre de Référence pour la prise en charge des Infections ostéo-articulaires complexes (CRIOAc Lyon; www.crioac-lyon.fr), Hospices Civils de Lyon, Lyon, France.
Ariel J Blocker, Evotec ID Lyon, In Vitro Biology, Infectious Diseases and Antibacterials Unit, Gerland, 69007 Lyon, France.
Funding
F.M. holds a CIFRE PhD fellowship cofunded by the French ANRT and EVOTEC ID Lyon (convention no. 2019/0813); F.L. is a full-time employee of UCBL1 and HCL; J.J. is a full-time employee of UCBL1; and A.B. is a part-time employee of EVOTEC ID Lyon.
References
- Adams KN, Takaki K, Connolly LE. et al. Drug tolerance in replicating mycobacteria mediated by a macrophage-induced efflux mechanism. Cell. 2011;145:39–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldridge BB, Fernandez-Suarez M, Heller D.et al. Asymmetry and aging of mycobacterial cells lead to variable growth and antibiotic susceptibility. Science. 2012;335:100–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antunes LCS, Imperi F, Carattoli A. et al. Deciphering the multifactorial nature of Acinetobacter baumannii pathogenicity. PLoS ONE. 2011;6:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnoldini M, Vizcarra IA, Peña-Miller R. et al. Bistable expression of virulence genes in Salmonella leads to the formation of an antibiotic-tolerant subpopulation. Balaban N (ed.). PLoS Biol. 2014;12:e1001928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwal S, Giengkam S, VanNieuwenhze M. et al. Live imaging of the genetically intractable obligate intracellular bacteria Orientia tsutsugamushi using a panel of fluorescent dyes. J Microbiol Methods. 2016;130:169–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austerjost J, Marquard D, Raddatz L.et al. A smart device application for the automated determination of E. coli colonies on agar plates. Eng Life Sci. 2017;17:959–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban NQ, Merin J, Chait R. et al. Bacterial persistence as a phenotypic switch. Science. 2004;305:1622–5. [DOI] [PubMed] [Google Scholar]
- Baltekin Ö, Boucharin A, Tano E. et al. Antibiotic susceptibility testing in less than 30 min using direct single-cell imaging. Proc Natl Acad Sci. 2017;114:9170–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamford RA, Smith A, Metz J.et al. Investigating the physiology of viable but non-culturable bacteria by microfluidics and time-lapse microscopy. BMC Biol. 2017;15:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bär J, Boumasmoud M, Kouyos RD.et al. Efficient microbial colony growth dynamics quantification with ColTapp, an automated image analysis application. Sci Rep. 2020;10:16084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber MA. A new method of isolating micro-organisms. J Kans Med Soc. 1904;4:487. [Google Scholar]
- Barber MA. A study by the single cell method of the influence of homologous antipneumococcic serum on the growth rate of pneumococcus. J Exp Med. 1919;30:569–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber MA. The rate of multiplication of Bacillus coli at different temperatures. J Infect Dis. 1908;5:379–400. [Google Scholar]
- Bergmiller T, Andersson AMC, Tomasek K. et al. Biased partitioning of the multidrug efflux pump AcrAB-TolC underlies long-lived phenotypic heterogeneity. Science. 2017;356:311–5. [DOI] [PubMed] [Google Scholar]
- Bewes JM, Suchowerska N, McKenzie DR.. Automated cell colony counting and analysis using the circular Hough image transform algorithm (CHiTA). Phys Med Biol. 2008;53:5991–6008. [DOI] [PubMed] [Google Scholar]
- Bigger JW. Treatment of staphylococcal infections with penicillin by intermittent sterilisation. Lancet North Am Ed. 1944;244:497–500. [Google Scholar]
- Bronner-Fraser M. Alterations in neural crest migration by a monoclonal antibody that affects cell adhesion. J Cell Biol. 1985;101:610–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z, Chattopadhyay N, Liu WJ.et al. Optimized digital counting colonies of clonogenic assays using ImageJ software and customized macros: comparison with manual counting. Int J Radiat Biol. 2011;87:1135–46. [DOI] [PubMed] [Google Scholar]
- Camsund D, Lawson MJ, Larsson J. et al. Time-resolved imaging-based CRISPRi screening. Nat Methods. 2020;17:86–92. [DOI] [PubMed] [Google Scholar]
- Cermak N, Olcum S, Delgado FF. et al. High-throughput measurement of single-cell growth rates using serial microfluidic mass sensor arrays. Nat Biotechnol. 2016;34:1052–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhry P. High-throughput method for automated colony and cell counting by digital image analysis based on edge detection. Simpson MJ (ed.). PLoS ONE. 2016;11:e0148469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke ML, Burton RL, Hill AN.et al. Low-cost, high-throughput, automated counting of bacterial colonies. Cytometry Part A. 2010;77A:790–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claudi B, Spröte P, Chirkova A.et al. Phenotypic variation of Salmonella in host tissues delays eradication by antimicrobial chemotherapy. Cell. 2014;158:722–33. [DOI] [PubMed] [Google Scholar]
- Daniel RA, Errington J.. Control of cell morphogenesis in bacteria: two distinct ways to make a rod-shaped cell. Cell. 2003;113:10. [DOI] [PubMed] [Google Scholar]
- Delong EF, Wickham GS, Pace NR.. Phylogenetc stains: ribosomal RNA-based probes for the identification of single cells. Science. 1989;243:1360–3. [DOI] [PubMed] [Google Scholar]
- Diaper JP, Edwards C.. The use of fluorogenic esters to detect viable bacteria by flow cytometry. J Appl Bacteriol. 1994;77:221–8. [Google Scholar]
- Elfwing A, LeMarc Y, Baranyi J.et al. Observing growth and division of large numbers of individual bacteria by image analysis. Appl Environ Microbiol. 2004;70:675–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueira R, Watson KG, Holden DW. et al. Identification of Salmonella pathogenicity island-2 Type III secretion system effectors involved in intramacrophage replication of S. enterica Serovar Typhimurium: implications for rational vaccine design. Sansonetti PJ (ed.). mBio. 2013;4:e00065–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannagan RS, Heinrichs DE.. A fluorescence based-proliferation assay for the identification of replicating bacteria within host cells. Front Microbiol. 2018;9:3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannagan RS, Heit B, Heinrichs DE.. Intracellular replication of Staphylococcus aureus in mature phagolysosomes in macrophages precedes host cell death, and bacterial escape and dissemination: S. aureus replicates in mature phagolysosomes in macrophages. Cell Microbiol. 2016;18:514–35. [DOI] [PubMed] [Google Scholar]
- Flannagan RS, Watson DW, Surewaard BGJ.et al. The surreptitious survival of the emerging pathogen Staphylococcus lugdunensis within macrophages as an immune evasion strategy. Cell Microbiol. 2018;20:e12869. [DOI] [PubMed] [Google Scholar]
- Fuller ME, Streger SH, Rothmel RK. et al. Development of a vital fluorescent staining method for monitoring bacterial transport in subsurface environments. Appl Environ Microbiol. 2000;66:4486–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangwe Nana GY, Ripoll C, Cabin-Flaman Aet al. Division-based, growth rate diversity in bacteria. Front Microbiol. 2018;9:849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Parajo MF, Koopman M, van Dijk EMHP. et al. The nature of fluorescence emission in the red fluorescent protein DsRed, revealed by single-molecule detection. Proc Natl Acad Sci. 2001;98:14392–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissmann Q. OpenCFU, a new free and open-source software to count cell colonies and other circular objects. Merks RMH (ed.). PLoS ONE. 2013;8:e54072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giengkam S, Blakes A, Utsahajit P. et al. Improved quantification, propagation, purification and storage of the obligate intracellular human pathogen Orientia tsutsugamushi. Walker DH (ed.). PLoS Negl Trop Dis. 2015;9:e0004009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill WP, Harik NS, Whiddon MR. et al. A replication clock for Mycobacterium tuberculosis. Nat Med. 2009;15:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godin M, Delgado FF, Son S.et al. Using buoyant mass to measure the growth of single cells. Nat Methods. 2010;7:387–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goormaghtigh F, Van Melderen L.. Single-cell imaging and characterization of Escherichia coli persister cells to ofloxacin in exponential cultures. Sci Adv. 2019;5:eaav9462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross LA, Baird GS, Hoffman RC. et al. The structure of the chromophore within DsRed, a red fluorescent protein from coral. Proc Natl Acad Sci. 2000;97:11990–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammes WP, Neuhaus FC.. On the mechanism of action of vancomycin: inhibition of peptidoglycan synthesis in Gaffkya homari. Antimicrob Agents Chemother. 1974;6:722–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen N, Rasmussen AKI, Fiandaca MJ.et al. Rapid identification of Stenotrophomonas maltophilia by peptide nucleic acid fluorescence in situ hybridization. New Microbes New Infect. 2014;2:79–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haugan MS, Charbon G, Frimodt-Møller N.et al. Chromosome replication as a measure of bacterial growth rate during Escherichia coli infection in the mouse peritonitis model. Sci Rep. 2018;8:14961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haugan MS, Løbner-Olesen A, Frimodt-Møller N.. Comparative activity of ceftriaxone, ciprofloxacin, and gentamicin as a function of bacterial growth rate probed by Escherichia coli chromosome replication in the mouse peritonitis model. Antimicrob Agents Chemother. 2019;63:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helaine S, Cheverton AM, Watson KG. et al. Internalization of Salmonella by macrophages induces formation of non-replicating persisters. Science. 2014;343:204–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helaine S, Thompson JA, Watson KG.et al. Dynamics of intracellular bacterial replication at the single cell level. Proc Natl Acad Sci. 2010;107:3746–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand EM. Techniques for the isolation of single microorganisms. Botan Rev. 1938;4:627–64. [Google Scholar]
- Hobby GL, Meyer K, Chaffee E.. Observations on the mechanism of action of penicillin. Exp Biol Med. 1942;50:281–5. [Google Scholar]
- Hodges HL, Brown RA, Crooks JA. et al. Imaging mycobacterial growth and division with a fluorogenic probe. Proc Natl Acad Sci. 2018;115:5271–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoefel D, Grooby WL, Monis PT.et al. A comparative study of carboxyfluorescein diacetate and carboxyfluorescein diacetate succinimidyl ester as indicators of bacterial activity. J Microbiol Methods. 2003;52:10. [DOI] [PubMed] [Google Scholar]
- Höltje JV. Growth of the stress-bearing and shape-maintaining murein sacculus of Escherichia coli. Microbiol Mol Biol Rev MMBR. 1998;62:181–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper DC. Mechanisms of action of antimicrobials: focus on fluoroquinolones. Clin Infect Dis. 2001;32:S9–15. [DOI] [PubMed] [Google Scholar]
- Hsu Y-P, Hall E, Booher G.et al. Fluorogenic d-amino acids enable real-time monitoring of peptidoglycan biosynthesis and high-throughput transpeptidation assays. Nat Chem. 2019;11:335–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imdahl F, Saliba A-E.. Advances and challenges in single-cell RNA-seq of microbial communities. Curr Opin Microbiol. 2020;57:102–10. [DOI] [PubMed] [Google Scholar]
- Iyer-Biswas S, Wright CS, Henry JT.et al. Scaling laws governing stochastic growth and division of single bacterial cells. Proc Natl Acad Sci. 2014;111:15912–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jõers A, Kaldalu N, Tenson T.. The frequency of persisters in Escherichia coli reflects the kinetics of awakening from dormancy. J Bacteriol. 2010;192:3379–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang C-M, Nyayapathy S, Lee J-Y.et al. Wag31, a homologue of the cell division protein DivIVA, regulates growth, morphology and polar cell wall synthesis in mycobacteria. Microbiology. 2008;154:725–35. [DOI] [PubMed] [Google Scholar]
- Kelly CD, Rahn O.. The growth rate of individual bacterial cells. J Bacteriol. 1932;23:147–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A ul M, Torelli A, Wolf I.et al. AutoCellSeg: robust automatic colony forming unit (CFU)/cell analysis using adaptive image segmentation and easy-to-use post-editing techniques. Sci Rep. 2018;8:7302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandekar S, Liebens V, Fauvart M.et al. The putative de-N-acetylase DnpA contributes to intracellular and biofilm-associated persistence of Pseudomonas aeruginosa exposed to fluoroquinolones. Front Microbiol. 2018;9:1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiviet DJ, Nghe P, Walker N.et al. Stochasticity of metabolism and growth at the single-cell level. Nature. 2014;514:376–9. [DOI] [PubMed] [Google Scholar]
- Kohram M, Vashistha H, Leibler S.et al. Bacterial growth control mechanisms inferred from multivariate statistical analysis of single-cell measurements. Curr Biol. 2021;31:955–964.e4. [DOI] [PubMed] [Google Scholar]
- Kolpen M, Kragh KN, Bjarnsholt T.et al. Denitrification by cystic fibrosis pathogens - Stenotrophomonas maltophilia is dormant in sputum. Int J Med Microbiol. 2015;305:1–10. [DOI] [PubMed] [Google Scholar]
- Kopf SH, Sessions AL, Cowley ES.et al. Trace incorporation of heavy water reveals slow and heterogeneous pathogen growth rates in cystic fibrosis sputum. Proc Natl Acad Sci. 2016;113:E110–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kragh KN, Alhede M, Jensen PØ.et al. Polymorphonuclear leukocytes restrict growth of Pseudomonas aeruginosa in the lungs of cystic fibrosis patients. McCormick BA (ed.). Infect Immun. 2014;82:4477–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuru E, Hughes HV, Brown PJ.et al. In situ probing of newly synthesized peptidoglycan in live bacteria with fluorescent D-amino acids. Angew Chem Int Ed Engl. 2012;51:12519–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuru E, Tekkam S, Hall E.et al. Synthesis of fluorescent D-amino acids and their use for probing peptidoglycan synthesis and bacterial growth in situ. Nat Protoc. 2015;10:33–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahav-Mankovski N, Prasad PK, Oppenheimer-Low N.et al. Decorating bacteria with self-assembled synthetic receptors. Nat Commun. 2020;11:1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leveau JHJ, Lindow SE.. Predictive and interpretive simulation of green fluorescent protein expression in reporter bacteria. J Bacteriol. 2001;183:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin-Reisman I, Gefen O, Fridman O.et al. Automated imaging with ScanLag reveals previously undetectable bacterial growth phenotypes. Nat Methods. 2010;7:737–9. [DOI] [PubMed] [Google Scholar]
- Licht TR, Krogfelt KA, Cohen PS.et al. Role of lipopolysaccharide in colonization of the mouse intestine by Salmonella typhimurium studied by in situ hybridization. Infect Immun. 1996;64:3811–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licht TR, Tolker-Nielsen T, Holmstrom K.et al. Inhibition of Escherichia coli precursor-16S rRNA processing by mouse intestinal contents. Environ Microbiol. 1999;1:23–32. [DOI] [PubMed] [Google Scholar]
- Liechti GW, Kuru E, Hall E. et al. A new metabolic cell-wall labelling method reveals peptidoglycan in Chlamydia trachomatis. Nature. 2014;506:507–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Shah SJ, Wilmes LJ.et al. Quantitative tissue oxygen measurement in multiple organs using 19 F MRI in a rat model. Magn Reson Med. 2011;66:1722–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long Z, Nugent E, Javer A.et al. Microfluidic chemostat for measuring single cell dynamics in bacteria. Lab Chip. 2013;13:947. [DOI] [PubMed] [Google Scholar]
- Luk CH, Valenzuela C, Gil M. et al. Salmonella enters a dormant state within human epithelial cells for persistent infection. Celli J (ed.). PLOS Pathog. 2021;17:e1009550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Ehrhardt DW, Margolin W.. Colocalization of cell division proteins FtsZ and FtsA to cytoskeletal structures in living Escherichia coli cells by using green fluorescent protein. Proc Natl Acad Sci USA. 1996;93:12998–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailloux BJ, Fuller ME.. Determination of in situ bacterial growth rates in aquifers and aquifer sediments. Appl Environ Microbiol. 2003;69:3798–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manina G, Dhar N, McKinney JD.. Stress and host immunity amplify Mycobacterium tuberculosis phenotypic heterogeneity and induce non-growing metabolically active forms. Cell Host Microbe. 2015;17:32–46. [DOI] [PubMed] [Google Scholar]
- Mannik J, Driessen R, Galajda P. et al. Bacterial growth and motility in sub-micron constrictions. Proc Natl Acad Sci. 2009;106:14861–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannik J, Wu F, Hol FJH. et al. Robustness and accuracy of cell division in Escherichia coli in diverse cell shapes. Proc Natl Acad Sci. 2012;109:6957–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuse S, Shan Y, Canas-Duarte SJ.et al. Bacterial persisters are a stochastically formed subpopulation of low-energy cells. Bollenbach T (ed.). PLOS Biol. 2021;19:e3001194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meselson M, Stahl FW.. The replication of DNA in Escherichia coli. Proc Natl Acad Sci. 1958;44:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel J-B, Yeh PJ, Chait R. et al. Drug interactions modulate the potential for evolution of resistance. Proc Natl Acad Sci. 2008;105:14918–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffatt JH, Harper M, Harrison P. et al. Colistin resistance in Acinetobacter baumannii is mediated by complete loss of lipopolysaccharide production. Antimicrob Agents Chemother. 2010;54:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffitt JR, Lee JB, Cluzel P.. The single-cell chemostat: an agarose-based, microfluidic device for high-throughput, single-cell studies of bacteria and bacterial communities. Lab Chip. 2012;12:1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohiuddin SG, Kavousi P, Orman MA.. Flow-cytometry analysis reveals persister resuscitation characteristics. BMC Microbiol. 2020;20:202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moller S, Kristensen CS, Poulsen LK.et al. Bacterial growth on surfaces: automated image analysis for quantification of growth rate-related parameters. Appl Environ Microbiol. 1995;61:741–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouton JM, Helaine S, Holden DW.et al. Elucidating population-wide mycobacterial replication dynamics at the single-cell level. Microbiology. 2016;162:966–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myhrvold C, Kotula JW, Hicks WM.et al. A distributed cell division counter reveals growth dynamics in the gut microbiota. Nat Commun. 2015;6:10039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman TM, Lord ND, Paulsson J.et al. Memory and modularity in cell-fate decision making. Nature. 2013;503:481–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olrichs NK, Aarsman MEG, Verheul J. et al. A novel in vivo cell-wall labeling approach sheds new light on peptidoglycan synthesis in Escherichia coli. ChemBioChem. 2011;12:1124–33. [DOI] [PubMed] [Google Scholar]
- Orman MA, Brynildsen MP.. Dormancy is not necessary or sufficient for bacterial persistence. Antimicrob Agents Chemother. 2013;57:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orman MA, Brynildsen MP.. Inhibition of stationary phase respiration impairs persister formation in E. coli. Nat Commun. 2015;6:7983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ørskov J. Method for the isolation of bacteria in pure culture from single cells and procedure for the direct tracing of bacterial growth on a solid medium. J Bacteriol. 1922;7:537–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel P, O'Hara BJ, Aunins E. et al. Modifying TIMER to generate a slow-folding DsRed derivative for optimal use in quickly-dividing bacteria. Helaine S (ed.). PLOS Pathog. 2021;17:e1009284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Personnic N, Striednig B, Hilbi H.. Quorum sensing controls persistence, resuscitation, and virulence of Legionella subpopulations in biofilms. ISME J. 2021;15:196–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Personnic N, Striednig B, Lezan E. et al. Quorum sensing modulates the formation of virulent Legionella persisters within infected cells. Nat Commun. 2019;10:5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyrusson F, Varet H, Nguyen TK. et al. Intracellular Staphylococcus aureus persisters upon antibiotic exposure. Nat Commun. 2020;11:2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulsen LK, Ballard G, Stahl DA.. Use of rRNA fluorescence in situ hybridization for measuring the activity of single cells in young and established biofilms. Appl Environ Microbiol. 1993;59:1354–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulsen LK, Licht TR, Rang C.et al. Physiological state of Escherichia coli BJ4 growing in the large intestines of streptomycin-treated mice. J Bacteriol. 1995;177:5840–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putrinš M, Kogermann K, Lukk E. et al. Phenotypic heterogeneity enables uropathogenic Escherichia coli to evade killing by antibiotics and serum complement. Infect Immun. 2015;83:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raybourne RB, Bunning VK.. Bacterium-host cell interactions at the cellular level: fluorescent labeling of bacteria and analysis of short-term bacterium-phagocyte interaction by flow cytometry. Infect Immun. 1994;62:665–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichenbach H. Die Absterbeordnung der Bakterien und ihre Bedeutung für Theorie und Praxis der Desinfektion. Zeitschrift für Hygiene und Infektionskrankheiten. 1911;69:171–222. [Google Scholar]
- Remus-Emsermann MN, Leveau JH.. Linking environmental heterogeneity and reproductive success at single-cell resolution. ISME J. 2010;4:215–22. [DOI] [PubMed] [Google Scholar]
- Remus-Emsermann MNP, Kowalchuk GA, Leveau JHJ.. Single-cell versus population-level reproductive success of bacterial immigrants to pre-colonized leaf surfaces. Environ Microbiol Rep. 2013;5:387–92. [DOI] [PubMed] [Google Scholar]
- Rocha EPC, Fralick J, Vediyappan G.et al. A strand-specific model for chromosome segregation in bacteria. Mol Microbiol. 2003;49:895–903. [DOI] [PubMed] [Google Scholar]
- Roostalu J, Jõers A, Luidalepp H.et al. Cell division in Escherichia coli cultures monitored at single cell resolution. BMC Microbiol. 2008;8:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saliba A-E, Li L, Westermann AJ.et al. Single-cell RNA-seq ties macrophage polarization to growth rate of intracellular Salmonella. Nat Microbiol. 2017;2:16206. [DOI] [PubMed] [Google Scholar]
- Santi I, Dhar N, Bousbaine D.et al. Single-cell dynamics of the chromosome replication and cell division cycles in mycobacteria. Nat Commun. 2013;4:2470. [DOI] [PubMed] [Google Scholar]
- Schaechter M, Maaloe O, Kjeldgaard NO.. Dependency on medium and temperature of cell size and chemical composition during balanced grown of Salmonella typhimurium. J Gen Microbiol. 1958;19:592–606. [DOI] [PubMed] [Google Scholar]
- Schulte M, Olschewski K, Hensel M.. Fluorescent protein-based reporters reveal stress response of intracellular Salmonella enterica at level of single bacterial cells. Cell Microbiol. 2021;23:e13293. [DOI] [PubMed] [Google Scholar]
- Shah NA, Laws RJ, Wardman B.et al. Accurate, precise modeling of cell proliferation kinetics from time-lapse imaging and automated image analysis of agar yeast culture arrays. BMC Syst Biol. 2007;1:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Campbell RE, Steinbach PA. et al. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 2004;22:1567–72. [DOI] [PubMed] [Google Scholar]
- Shcherbo D, Merzlyak EM, Chepurnykh TV.et al. Bright far-red fluorescent protein for whole-body imaging. Nat Methods. 2007;4:741–6. [DOI] [PubMed] [Google Scholar]
- Siegrist MS, Whiteside S, Jewett JC.et al. Amino acid chemical reporters reveal peptidoglycan dynamics of an intracellular pathogen. ACS Chem Biol. 2013;8:500–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stapels DAC, Hill PWS, Westermann AJ.. et al. Salmonella persisters undermine host immune defenses during antibiotic treatment. Science. 2018;362:1156–60. [DOI] [PubMed] [Google Scholar]
- Strack RL, Strongin D E, Mets L.et al. Chromophore formation in DsRed occurs by a branched pathway. J Am Chem Soc. 2010;132:8496–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stretton S, Techkarnjanaruk S, Mclennan AM.et al. Use of green fluorescent protein to tag and investigate gene expression in marine bacteria. 1998;64:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturm A, Heinemann M, Arnoldini M. et al. The cost of virulence: retarded growth of Salmonella typhimurium cells expressing type III secretion system 1. Ausubel FM (ed.). PLoS Pathog. 2011;7:e1002143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumar N, Tan S, Aldridge BB.et al. Exploitation of Mycobacterium tuberculosis reporter strains to probe the impact of vaccination at sites of infection. Salgame P (ed.). PLoS Pathog. 2014;10:e1004394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi R, Tamura T, Nakayashiki T.et al. Colony-live — a high-throughput method for measuring microbial colony growth kinetics— reveals diverse growth effects of gene knockouts in Escherichia coli. BMC Microbiol. 2014;14:171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanouchi Y, Pai A, Park H. et al. A noisy linear map underlies oscillations in cell size and gene expression in bacteria. Nature. 2015;523:357–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor GT, Suter EA, Li ZQ. et al. Single-cell growth rates in photoautotrophic populations measured by stable isotope probing and resonance Raman microspectrometry. Front Microbiol. 2017;8:1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira RC, Baêta BA, Ferreira JS. et al. Fluorescent membrane markers elucidate the association of Borrelia burgdorferi with tick cell lines. Braz J Med Biol Res. 2016;49:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terskikh A. “Fluorescent Timer”: Protein That Changes Color with Time. Science. 2000;290:1585–8. [DOI] [PubMed] [Google Scholar]
- Tiyanont K, Doan T, Lazarus MBet al. Imaging peptidoglycan biosynthesis in Bacillus subtilis with fluorescent antibiotics. Proc Natl Acad Sci. 2006;103:11033–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueckert JE, Nebe von-Caron G, Bos AP.et al. Flow cytometric analysis of Lactobacillus plantarum to monitor lag times, cell division and injury. Lett Appl Microbiol. 1997;25:295–9. [DOI] [PubMed] [Google Scholar]
- Ullman G, Wallden M, Marklund EG. et al. High-throughput gene expression analysis at the level of single proteins using a microfluidic turbidostat and automated cell tracking. Philos Trans R Soc B Biol Sci. 2013;368:20120025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ursell TS, Nguyen J, Monds RD. et al. Rod-like bacterial shape is maintained by feedback between cell curvature and cytoskeletal localization. Proc Natl Acad Sci. 2014;111:E1025–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhusha VV, Chudakov DM, Gurskaya NG. et al. Common Pathway for the Red Chromophore Formation in Fluorescent Proteins and Chromoproteins. Chem Biol. 2004;11:845–54. [DOI] [PubMed] [Google Scholar]
- Wakamoto Y, Dhar N, Chait R.et al. Dynamic persistence of antibiotic-stressed mycobacteria. Science. 2013;339:6. [DOI] [PubMed] [Google Scholar]
- Wakamoto Y, Ramsden J, Yasuda K.. Single-cell growth and division dynamics showing epigenetic correlations. Analyst. 2005;130:7. [DOI] [PubMed] [Google Scholar]
- Wallden M, Fange D, Lundius EG.et al. The synchronization of replication and division cycles in individual E. coli Cells. Cell. 2016;166:729–39. [DOI] [PubMed] [Google Scholar]
- Wang P, Robert L, Pelletier J. et al. Robust growth of Escherichia coli. Curr Biol. 2010;20:1099–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber F, Zaliznyak T, Edgcomb VP.et al. Using stable isotope probing and Raman microspectroscopy to measure growth rates of heterotrophic bacteria. Appl Environ Microbiol. 2021;87:e0146021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson GS. The proportion of viable bacteria in young cultures with especial reference to the technique employed in counting. J Bacteriol. 1922;7:405–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong C-F, Yeo JY, Gan SK-E.. APD colony counter app: using watershed algorithm for improved colony counting. Nat Methods Appl Notes. 2016;1–3. [Google Scholar]
- Wong FH-S, Cai Y, Leck H.et al. Determining the development of persisters in extensively drug-resistant Acinetobacter baumannii upon exposure to polymyxin B-based antibiotic combinations using flow cytometry. Antimicrob Agents Chemother. 2019;64:e01712–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia A, Han J, Jin Z.et al. Dual-color fluorescent timer enables detection of growth-arrested pathogenic bacterium. ACS Infect Dis. 2018;4:1666–70. [DOI] [PubMed] [Google Scholar]
- Yang D, Jennings AD, Borrego E.et al. Analysis of factors limiting bacterial growth in PDMS mother machine devices. Front Microbiol. 2018;9:871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Haagensen JAJ, Jelsbak L.et al. In situ growth rates and biofilm development of Pseudomonas aeruginosa populations in chronic lung infections. J Bacteriol. 2008;190:2767–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Liu Y, Yin H.et al. Regrowth-delay body as a bacterial subcellular structure marking multidrug-tolerant persisters. Cell Discov. 2019;5:8. [DOI] [PMC free article] [PubMed] [Google Scholar]