

Abstract
We have investigated the in vivo growth kinetics of a Salmonella typhimurium strain (P11D10) carrying a mutation in ssaJ, a Salmonella pathogenicity island 2 (SPI2) gene encoding a component of a type III secretion system required for systemic growth in mice. Similar numbers of mutant and wild-type cells were recovered from the spleens and livers of BALB/c mice up to 8 h after inoculation by the intraperitoneal route. Thereafter, the numbers of wild-type cells continued to increase logarithmically in these organs, whereas those of P11D10 remained relatively static for several days before being cleared. Gentamicin protection experiments on spleen cell suspensions recovered from infected mice showed that viable intracellular wild-type bacteria accumulated over time but that intracellular P11D10 cells did not. Infection experiments were also performed with wild-type and P11D10 cells carrying the temperature-sensitive plasmid pHSG422 to distinguish between bacterial growth rates and killing in vivo. At 16 h postinoculation there were 10-fold more wild-type cells than mutant cells in the spleens of infected mice, but the numbers of cells of both strains carrying the nonreplicating plasmid were very similar, showing that there was little difference in the degree of killing sustained by the two strains and that the SPI2 secretion system must be required for bacterial replication, rather than survival, in vivo. The SPI2 mutant phenotype in mice is similar to that of strains carrying mutations in the Salmonella virulence plasmid spv genes. To determine if these two sets of genes interact together, a double mutant strain carrying SPI2 and spv mutations was constructed and compared with strains carrying single mutations in terms of virulence attenuation. These experiments failed to provide any evidence showing that the SPI2 and spv gene products interact together as part of the same virulence mechanism.
Infection of mice by Salmonella typhimurium produces a systemic disease similar to human typhoid (6, 9). This process involves a large number of bacterial virulence genes, many of which are required for invading, surviving, and replicating within host cells (18). Recent work has shown that a significant proportion of S. typhimurium virulence genes are located in distinct chromosomal regions called pathogenicity islands (18, 22). Pathogenicity islands are flanked by repeat sequences, insertion elements, or tRNA genes and can be unstable. Their G+C content is usually different from that of the remainder of the chromosome, suggesting that they were acquired by horizontal transmission (3).
SPI1 (38) and SPI2 (41, 45) are two pathogenicity islands, located at 63 and 30 centisomes, respectively, on the Salmonella chromosome, containing genes that encode two distinct type III secretion systems. Type III secretion systems are present in a number of bacterial pathogens of animals and plants (28, 30). The secretion apparatus is encoded by at least 17 different genes, and homologues of several of these are present in each bacterial species (28). The secreted effector proteins studied to date have different functions, causing bacterial uptake into host cells, as in the case of Shigella flexneri (36), or prevention of phagocytosis, as in the case of Yersinia sp. (10). In addition to the secretion apparatus and effector proteins, type III secretion systems usually include proteins that translocate the effector proteins through the plasma membranes of eukaryotic host cells, chaperones that associate with the effector proteins in the bacterial cytosol, and various regulatory proteins that control the expression of the secretion system genes or the secretion of effector and translocator proteins (10, 28, 30).
The S. typhimurium SPI1 secretion system mediates bacterial invasion of host intestinal epithelial cells but is apparently dispensible for subsequent stages of pathogenesis (16, 17, 29). The SPI2 secretion system plays a crucial role in systemic pathogenesis because mutant strains are highly attenuated in virulence when administered by the oral, intraperitoneal, or intravenous route (24, 45).
S. typhimurium is believed to replicate within macrophages during growth in the spleen and liver (15, 31, 33, 44). Some S. typhimurium strains carrying mutations in SPI2 secretion system genes fail to accumulate inside cultured macrophages, suggesting that the secretion system is involved in this process (8, 27, 41). Since SPI2 secretion system genes are also induced inside macrophages and other host cell types (47), it has been proposed that the secretion apparatus might be assembled inside host cells and translocate bacterial proteins across the vacuolar membrane containing bacteria into the cytosol (41, 47).
To learn more about the function of the SPI2 secretion system in vivo, we investigated the growth kinetics of a SPI2 mutant strain in different body sites during the course of infection. Using a nonreplicating plasmid to measure the relative rates of bacterial growth and killing, we show that the SPI2 type III secretion system does not contribute significantly to the ability of S. typhimurium to survive in vivo but that it is required for the rapid bacterial growth that occurs in the spleen and liver a few hours postinoculation. Although there are similarities in the behavior of the SPI2 mutant strain and strains carrying mutations in the spv genes of the virulence plasmid, mixed-infection experiments with single- and double-mutant strains clearly show that the virulence functions of these two sets of genes are different from each other.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The bacterial strains used in this study are listed in Table 1. The 12023s derivative strain P11D10 (ssaJ::mTn5) has been described previously (45) and was chosen for these studies because it has a similar growth rate to 12023 and is, in contrast to some SPI2 mutants, resistant to killing by complement (11). Bacteria were grown in Luria-Bertani (LB) medium supplemented with ampicillin (50 μg/ml), kanamycin (50 μg/ml), streptomycin (1 mg/ml), nalidixic acid (50 μg/ml), or chloramphenicol (50 μg/ml for plasmid containing strains and 8 μg/ml for chromosomal integrants) as appropriate.
TABLE 1.
Bacterial strains used in this study
| Strain | Description | Source or reference |
|---|---|---|
| S. typhimurium | ||
| 12023s | Wild type | 49 |
| 12023Nalr | Nalidixic acid-resistant spontaneous mutant | This study |
| 12023Smr | Streptomycin-resistant spontaneous mutant | This study |
| SL1344 | Wild type | NCTC, Colindale, United Kingdoma |
| SLD10 | ssaJ::mTn5 in SL1344 | This study |
| P11D10 | ssaJ::mTn5 in 12023s | 45 |
| P4E3 | purD::mTn5 in 12023s | 24 |
| P5D10 | spvA::mTn5 in 12023s | 24 |
| HH104 | sseC::aphT in 12023s | 27 |
| HH109 | ssaV::aphT in 12023s | 11 |
| HH110 | ssaV::Cmr in 12023s | This study |
| HH111 | ssaV::Cmr, purD::mTn5 in 12023s | This study |
| HH112 | ssaV::Cmr, spvA::mTn5 in 12023s | This study |
| HH114 | ssaV::Cmr, sseC::aphT in 12023s | This study |
| E. coli S17-1λpir | λpir phage lysogen | 37 |
NCTC, National Collection of Type Cultures.
Construction of a nonpolar ssaV mutation.
Plasmid p7-2 is a 7.5-kb PstI fragment containing the ssaV gene from λ clone 7 (45) subcloned into pUC18. Plasmid p7-2 was used as a template for PCR with primers ssaVL1 (5′-TAGGCCTGATGCGCAGTTTATGCTG-3′) and ssaVR1 (5′-TAGGCCTCACGACTTCAGCAAGTTC-3′). The 2.6-kb PCR product containing the entire ssaV gene was ligated to pCRII-TOPO (Invitrogen) to generate plasmid pID800. The SmaI fragment of pSL1 (34) containing a chloramphenicol resistance cassette without promoter or terminator sequences was ligated into the SmaI site of ssaV in pID800. The resulting plasmid, pID801, was used as a template for PCR with primers ssaVL2 (5′-TACGACTCTAGAGATGCGCAGTTTATGCTG-3′) and ssaVR2 (5′-TTACGATCTAGACACGACTTCAGCAAGTTC-3′). The PCR product, which contained terminal XbaI sites, was digested with XbaI and ligated to the suicide vector pKAS32 (46) which had been cut with XbaI to give plasmid pID802. Plasmid pID802 was transferred by conjugation from E. coli S17-1λpir to S. typhimurium 12023Nalr. Exconjugants were selected for nalidixic acid and ampicillin resistance to obtain a merodiploid strain. The integrated plasmid was transferred by P22 transduction to 12023Smr, and transductants were selected by ampicillin resistance. Transductants were plated onto green plates (35) without any selection to permit spontaneous segregation of the merodiploid by homologous recombination. Resolution of the merodiploid was selected for by resistance to streptomycin, and the Smr colonies were screened for resistance to chloramphenicol and sensitivity to ampicillin. The resulting strain was designated HH110, and the nonpolar mutation was confirmed by Southern hybridization analysis. Double-mutant strains were constructed by P22 transduction of the ssaV::Cm mutation into the required single-mutant strain backgrounds.
Mouse infection studies.
Female BALB/c mice (20 to 25 g) were used for all infection studies, except for 50% lethal dose (LD50) studies, where C3H/HeN mice (20 to 25 g) were also used, and were inoculated intraperitoneally (i.p.) with a 0.2-ml volume of bacterial cells suspended in physiological saline. To prepare the inocula, bacteria were grown overnight at 37°C in 10 ml of LB broth in a 20-ml container (Sterilin) with shaking (150 rpm) and were then used to inoculate fresh medium (1:100) and were grown under the same conditions for 2 to 3 h until an optical density at 550 nm of 0.35 to 0.6 was reached. Bacterial cultures were then diluted in physiological saline, and the CFU were enumerated by plating a dilution series of the inoculum. For mixed infections, wild-type and mutant strains were grown separately and then mixed prior to inoculation. The number of viable bacteria and the proportion of both strains were checked by plating a dilution series of the inoculum onto LB plates with or without the appropriate antibiotic. Mice were sacrificed at appropriate time points postinoculation by carbon dioxide inhalation. An intraperitoneal lavage was performed before removal of mouse organs by injecting 10 ml of ice-cold saline into the peritoneal cavity, followed by gentle massage and removal of the peritoneal exudate suspension by needle and syringe. To determine the bacterial load in the spleen, liver, and mesenteric lymph nodes, the organs were removed and placed in 1 to 3 ml of saline, and a homogeneous suspension was made by using glass or plastic homogenizers. Eukaryotic cells were then harvested by centrifugation at 1,500 × g and resuspended in 0.01% sodium deoxycholate at room temperature for 15 min to lyse the eukaryotic cells. Viable bacterial CFU were determined by plating aliquots of a dilution series of the lysate onto LB agar. For mixed infections, wild-type and mutant strains were distinguished by plating a dilution series onto LB agar alone and LB agar containing kanamycin.
Gentamicin protection assays.
The percentage of intracellular bacteria in mouse spleens was determined by using a gentamicin protection assay. The spleens of salmonella-infected mice were excised and placed individually in 3 ml of ice-cold RPMI in a petri dish. Spleens were teased apart with 18-guage needles bent at right angles, followed by gentle repeat pipetting to form a homogeneous suspension. The suspension was transferred to a 15-ml plastic tube and placed on ice for 5 min to allow any debris to settle before transfer to a fresh tube and adjustment of the volume to 3 ml with ice-cold RPMI. A 0.5-ml aliquot was then removed for determination of the total bacterial load in the spleen. The eukaryotic cells in the remaining suspension were harvested by centrifugation at 800 × g for 10 mins at 4°C, resuspended in 1 ml RPMI containing 50 μg/ml gentamicin and placed at 37°C to kill any extracellular bacteria. After 1 h of incubation, gentamicin was removed from the suspension by harvesting the eukaryotic cells as described above and resuspension in 1 ml of saline before transfer to a microfuge tube. Bacteria and eukaryotic cells in both the total and the gentamicin-treated aliquots were then harvested by centrifugation at 15,000 × g for 2 min in a microfuge, resuspension in 1 ml of 0.01% sodium deoxycholate, and treatment at room temperature for 15 min with vortexing to lyse the eukaryotic cells. Total and gentamicin-resistant bacterial CFU were determined by plating serial dilutions of the lysate onto LB plates. The numbers of extracellular bacteria were calculated by subtracting the number of gentamicin-protected bacteria from the total.
Measurement of Salmonella replication and killing within the mouse spleen.
The temperature-sensitive plasmid pHSG422 (23) was used to measure Salmonella growth and killing within the mouse spleen as described by Benjamin et al. (2). Strains 12023 and P11D10 were transformed with plasmid pHSG422, and the plasmid was maintained at 30°C during growth in LB by selection for ampicillin resistance. Both strains containing pHSG422 were found to lose the plasmid at similar rates upon transfer to 37°C in the absence of antibiotic selection, and plasmid carriage did not affect virulence as determined by the LD50 (data not shown). Incubation at 37°C with shaking at 150 rpm for 2.25 to 2.5 h in the absence of antibiotic selection was required to obtain a plasmid copy number of one or none per cell as described by Benjamin et al. (2), with 60 to 90% of the bacterial cells harboring a plasmid (data not shown). After injection of mice, the proportion of bacteria carrying the plasmid was determined by treating spleen cell suspensions with gentamicin as described above and then plating a dilution series to LB medium containing appropriate antibiotics to enumerate the total number of bacteria and the number carrying pHSG422.
Statistical analysis.
The measurements of plasmid pHSG422 carriage were analyzed by Student t test. Competitive index data were analyzed by using the Kruskall-Wallis one-way analysis of variance and the Mann-Whitney U test for statistical significance. Probabilities (P) of 0.05 or less were considered significant.
RESULTS
In vivo growth kinetics of an SPI2 mutant strain.
Strain P11D10 (ssaJ::mTn5) was used to investigate the growth kinetics of S. typhimurium cells lacking a functional SPI2 type III secretion system. This strain contains a transposon insertion in the ssaJ gene, which is thought to encode a structural component of the SPI2 type III secretion apparatus and is the terminal gene of an operon (26). The transposon insertion in ssaJ was also transferred from P11D10 by P22 phage transduction to strain SL1344 to generate strain SLD10, and the LD50s (i.p.) of ssaJ mutants were determined in both strain backgrounds with BALB/c (itys) mice, and with C3H/HeN (ityr) mice for strain P11D10 (Table 2). The LD50 of both bacterial strains carrying the ssaJ mutation with BALB/c mice is similar to other SPI2 mutants that have been tested (24, 41, 45, and our own unpublished data). In C3H/HeN mice, P11D10 was also attenuated in virulence, but the degree of attenuation compared with 12023 was less than that observed with BALB/c mice (Table 2).
TABLE 2.
LD50 values of S. typhimurium strainsa
| Bacterial strain | Mutation | Mouse strain | LD50 (CFU) |
|---|---|---|---|
| 12023 | BALB/c | <10 | |
| SL1344 | BALB/c | <13 | |
| 12023 | C3H/HeN | 4.20 × 103 | |
| P11D10 | ssaJ::mTn5 | BALB/c | 1.35 × 106 |
| P11D10 | ssaJ::mTn5 | C3H/HeN | 6.27 × 105 |
| SLD10 | ssaJ::mTn5 | BALB/c | 9.27 × 105 |
Groups of five female BALB/c or C3H/HeN mice were inoculated i.p. with 1:10 dilutions of bacteria suspended in physiological saline. The LD50 values were calculated after 28 days by the method of Reed and Muench (42).
To determine the body site and time at which a SPI2 mutant strain first exhibits a difference in total cell numbers compared with the wild-type strain, mice were inoculated i.p. with a mixture of approximately 5 × 104 CFU of strain 12023 and 5 × 104 CFU of strain P11D10. At 4, 8, 16, and 24 h postinoculation, groups of 5 mice were killed, a peritoneal lavage was performed, and the spleen, liver, and mesenteric lymph nodes (MLN) were excised and homogenized. Host cells were lysed by treatment with sodium deoxycholate to release intracellular bacteria, and the total CFU were determined. At these time points, very few bacteria were recovered from venous blood.
At 4 and 8 h postinoculation the numbers of P11D10 were similar to or greater than those for strain 12023 in the peritoneal lavage, MLN, spleen, and liver (Fig. 1). By 16 h the percentage of mutant bacteria recovered from the spleen and liver had fallen to approximately 14 and 17%, respectively, and by 24 h to 9 and 12%, respectively. At all time points the proportion of mutant bacteria was lowest in the spleens compared with other body sites (Fig. 1). From 24 h onwards, the percentage of wild-type bacteria continued to increase and caused 100% mortality by 4 days postinoculation.
FIG. 1.
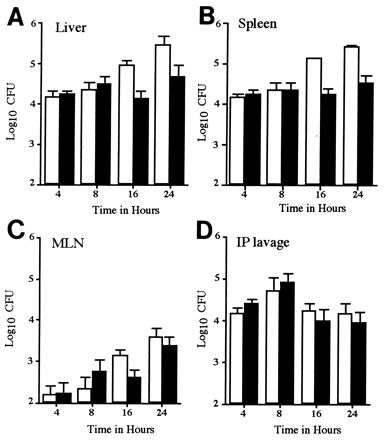
In vivo growth kinetics of a SPI2 mutant strain in a mixed infection. Mice were inoculated i.p. with 105 CFU of a mixed inoculum of 12023 and P11D10 strains. The numbers of 12023 (open bars) and P11D10 (filled bars) bacterial CFU in the liver (A), spleen (B), MLN (C), and peritoneal lavage (D) were determined at various time points postinoculation. Data shown are the means ± standard deviation (SD) for groups of 5 mice and are normalized to a 50:50 ratio in the inoculum.
To examine the kinetics of P11D10 infection over a longer period of time, mice were inoculated i.p. with 105 CFU of strain 12023 or P11D10. Groups of 5 mice were sacrificed at different time points, their livers and spleens were excised, a cell suspension was prepared, and the numbers of bacterial CFU were enumerated. Mice infected with strain 12023 all died by 3 to 4 days postinoculation, at which time the bacterial load exceeded 108 CFU in both the spleen and the liver, whereas mice inoculated with P11D10 all appeared to be healthy 4 weeks after inoculation (Fig. 2). Approximately one-fifth of the initial inoculum of both strains was recovered from both the spleen and the liver at 4 h postinoculation. An increase in cell numbers of both strains then occurred in both the liver and the spleen over the next 4 h. There was no significant increase in the numbers of mutant cells in the spleen or liver over the next 68 h (Fig. 2) and the following 7 days (data not shown). Mutant CFU decreased over the next 3 weeks (Fig. 2), but mice still contained low numbers (approximately 100 CFU/spleen) of P11D10 at up to 12 weeks postinoculation. By 16 weeks postinoculation three of five mice had cleared the infection from both the spleen and the liver.
FIG. 2.
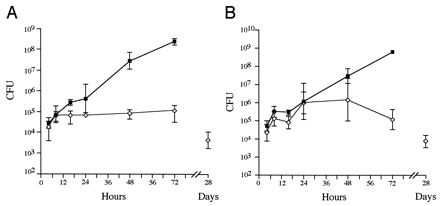
In vivo growth kinetics in single-strain infections. The bacterial loads in the spleen (A) and the liver (B) were determined over time for single infections with 12023 (squares) and P11D10 (diamonds). Data are expressed as the means ± 1 SD for groups of 5 mice.
In P11D10-inoculated mice, hepatic abcessess occurred between 2 and 4 weeks postinoculation. The spleens of these mice enlarged to approximately five times their normal weight between days 5 and 14 (data not shown). This splenomegaly resolved between 8 and 12 weeks postinoculation, shortly before bacterial clearance.
These results show that strains 12023 and P11D10 are transported from the peritoneal cavity to the spleen and liver in approximately equivalent numbers, suggesting that the mutant cells are capable of limited growth in these organs. The absence of the SPI2 secretion system effectively prevents further accumulation of bacterial cells in these organs, leading to their eventual clearance.
Failure of an SPI2 mutant strain to accumulate within spleen cells.
As several SPI2 mutant strains fail to accumulate within cultured host cells (8, 27, 41), a gentamicin protection assay was performed on spleen cell suspensions recovered from infected mice to determine whether this phenotypic defect could also be observed in vivo.
Since the numbers of bacteria recovered at the same time from the spleens of different animals vary considerably (Fig. 2), these experiments were done by using mixed infections. Figure 3 confirms the results shown in Fig. 1: over a period of 16 h there was an approximately 10-fold increase in the total numbers of 12023 cells recovered from spleens, whereas the overall numbers of P11D10 cells remained relatively static. The majority of cells of both strains were sensitive to gentamicin. These cells likely represent the products of bacterium-induced host cell lysis in vivo (7, 33, 39, 44), host cells ruptured during spleen cell preparation, and/or extracellular bacterial replication in vivo. Within the gentamicin-protected (intracellular) population, there was a significant net increase in the numbers of 12023 cells over the 16-h time period, but this was not observed for P11D10 cells. These data are unlikely to have arisen from the relatively large inocula used, because single infections by 12023 and P11D10 with inocula of approximately 1,000 CFU showed that the gentamicin-protected wild-type cells underwent a 14-fold increase over a 24-h period, whereas the P11D10 cells increased by 3.5-fold over a similar length of time. This failure to detect an increase in intracellular CFU of mutant cells in vivo is consistent with the intracellular growth defect observed in cell culture.
FIG. 3.
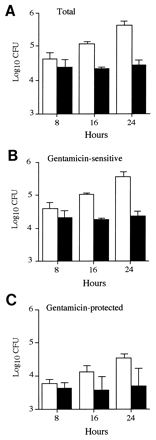
Gentamicin protection assays of spleen cell suspensions after mixed infections. The total numbers of P11D10 (solid bars) and wild-type (open bars) bacteria within the spleen (A), as well as the number of these that were gentamicin-sensitive (B) or protected (C), were determined over time in mixed infections. Data are expressed as the mean ± 1 SD for groups of 5 mice.
Mutations in the SPI2 secretion system affect bacterial growth rate in the spleen.
The failure of P11D10 cells to accumulate in the spleen could be due to a slower overall growth rate, an increased sensitivity to killing by the host, or a combination of both. To distinguish between these possibilities, we used the temperature-sensitive plasmid pHSG422 (23), which cannot divide at 37°C or above and therefore becomes diluted in a bacterial population during growth in the mouse. This plasmid has previously been used to measure Salmonella growth rates and killing in vivo (2, 14, 19, 20). Since the plasmid antibiotic resistance markers precluded analysis by mixed infection, mice were inoculated separately with 12023 or P11D10 containing pHSG422 at a copy number of approximately one per cell. Spleen cell suspensions from infected mice were treated with gentamicin, and total numbers of gentamicin-sensitive and gentamicin-protected bacteria were determined, as well as the proportion containing pHSG422. Table 3 shows that the total numbers of 12023 bacteria were significantly greater than the numbers of mutant bacteria at 16 h in the spleen (see also Fig. 2) but that the numbers of 12023 and mutant bacteria carrying pHSG422 were approximately the same, indicating that there was no difference in the degree of killing sustained by the two bacterial strains. The calculated proportional distance provides a measure of the dilution of pHSG422 within a bacterial population and shows that gentamicin-sensitive 12023 bacteria had undergone a greater number of replications than the gentamicin-resistant population. This can also be seen by the approximate number of generations calculated by comparison of the pHSG422 carriage rates in the inoculum and in bacteria recovered from mice. In mice infected with P11D10 the proportional distance was similar in both gentamicin-sensitive and gentamicin-protected populations and was significantly less than that calculated for strain 12023, indicating a lower bacterial replication rate in vivo. From these experiments it can be concluded that the difference in numbers of 12023 and P11D10 bacteria at 16 h is due to a slower growth rate of the mutant strain rather than a greater susceptibility to killing by the host.
TABLE 3.
Bacteria carrying plasmid pHSG422 in gentamicin-treated spleen cell suspensions at 16 h postinoculationa
| Strain | Total CFU (log10) | CFU/pHSG422 (log10) | Proportional distance (log10) | Approx. no. of generations |
|---|---|---|---|---|
| 12023 | ||||
| Gentamicin protected | 4.54 ± 0.09 | 3.21 ± 0.12* | 1.23 ± 0.33‡ | 4 |
| Gentamicin sensitive | 5.40 ± 0.12 | 3.55 ± 0.20* | 1.84 ± 0.18‡ | 5–6 |
| Total | 5.46 ± 0.11 | 3.74 ± 0.15** | 1.73 ± 0.14 | 5–6 |
| P11D10 | ||||
| Gentamicin protected | 4.18 ± 0.21 | 3.09 ± 0.14* | 1.09 ± 0.10† | 3 |
| Gentamicin sensitive | 4.66 ± 0.23 | 3.63 ± 0.12* | 1.05 ± 0.15† | 3 |
| Total | 4.79 ± 0.22 | 3.73 ± 0.11** | 1.06 ± 0.13 | 3 |
Each figure is the mean of 10 mice ± 1 SD. The proportional distance was calculated as the log10 total CFU − log10 plasmid bearing CFU for each mouse and the calculated mean of 10 mice. P values are for the null hypothesis that means are not statistically different and are as follows: ∗, P > 0.05; ∗∗, P > 0.5; †, P > 0.25; ‡, P < 0.001.
The function of the SPI2 secretion system is independent from that of the virulence plasmid spv genes.
Previous studies have shown that mutations in the spv genes located on the virulence plasmid also affect S. typhimurium growth rates within the mouse spleen (19). To determine if the SPI2 secretion system genes and the spv genes are functionally related, we constructed an ssaV::Cmr, spvA::mTn5 double-mutant strain and compared its level of virulence attenuation with strains carrying single mutations by measuring their competitive indices in mixed infections.
A Cmr marked ssaV mutant strain (HH110) was first constructed as described in the Materials and Methods and then combined by transduction with mutations carrying Kmr markers in other genes. Mixed infections performed with an ssaV::aphT (Kmr) mutant strain (HH109) and strain HH110 showed that the two strains were indistinguishable in terms of virulence attenuation (Table 4). Similarly, when the ssaV::Cmr mutation was combined with a mutation in sseC (encoding a putative translocator of the type III secretion system, which leads to a typical degree of attenuation for a SPI2 mutant [27]), there was no additive effect on virulence attenuation (Table 4). This shows that the products of ssaV and sseC are involved in the same virulence function. By contrast, when the ssaV::Cmr mutation was combined with a functionally unrelated purD::mTn5 mutation, the competitive index (CI) of the resulting mutant strain (HH111) against HH110 was significantly lower and similar to the CI of the purD strain against the wild-type strain. A significantly lower CI was also obtained with HH112 (ssaV::Cmr, spvA::mTn5) against HH110, and this was similar to that of P5D10 (spvA::mTn5) against the wild-type strain. These results show that when either purD or spvA mutations are combined with an ssaV mutation the effects on virulence are entirely additive and that therefore the functions of the genes are unrelated.
TABLE 4.
CI valuesa for mixed infections
| Strain | Genotype | CI |
|---|---|---|
| Mixed infection with wild-type strain | ||
| HH110 | ssaV::Cmr | 0.058 |
| P4E3 | purD::mTn5 | 0.005 |
| P5D10 | spvA::mTn5 | 0.068 |
| Mixed infection with HH110 strain | ||
| HH109 | ssaV::aphT | 1.14 |
| HH111 | ssaV::Cmr, purD::mTn5 | 0.008b |
| HH112 | ssaV::Cmr, spvA::mTn5 | 0.052b |
| HH104 | ssaV::Cmr, sseC::aphT | 1.28c |
The CI was calculated as the output ratio of mutant to wild-type bacteria divided by the input ratio of mutant to wild-type bacteria. In mixed infections with two mutant strains, strain HH110 represents the “wild-type” strain for the calculation of CI. The CI shown is the median of the CIs obtained from three mice for the mixed infections with HH110 and the mean of the CIs obtained from two mice for mixed infections with the wild-type strain. Mice were inoculated i.p. with approximately 105 CFU of a mixed inoculum. Mouse spleens were harvested after 48 h for enumeration of bacterial CFU. The different strains used were differentiated on the basis of antibiotic sensitivity.
Significantly different (P < 0.05) to the CI obtained for mixed infections with strains HH109 and HH110.
The CI was not significantly different (P > 0.5) to the CI obtained for mixed infections with strains HH109 and HH110.
DISCUSSION
The SPI2 type III secretion system genes were identified by the failure of corresponding mutant strains to be recovered from the spleens of infected BALB/c mice following i.p. inoculation of a mixture of different S. typhimurium mutant strains (24). The itys allele renders the BALB/c mouse highly susceptible to infection by S. typhimurium, but we have shown here that the secretion system is also involved in virulence during infection of ityr mice, and other work has shown that corresponding genes in Salmonella cholerasuis are required for pathogenesis in mice and pigs (12). As SPI2 is present in all S. enterica strains examined to date (25, 40), it seems likely to be a key virulence determinant in systemic Salmonella infection.
To establish the time and body site where the phenotype of SPI2 mutants first becomes apparent, we investigated the in vivo growth kinetics of a SPI2 mutant strain both in the presence of a wild-type strain and following single strain inoculations. Mice were inoculated by the i.p. route since it is known that SPI2 mutations can affect the SPI1 type III secretion system (11, 26), whose function is important for, but apparently restricted to, bacterial translocation across the gut epithelium (17). Therefore, differences in bacterial numbers recovered from mice orally inoculated with wild-type and SPI2 mutants (8) might reflect effects on SPI1 function. We found similar numbers of wild-type and mutant cells in the mesenteric lymph nodes and peritoneal cavity during the first 24 h, indicating that a functional secretion system is not required for bacterial survival in these body sites following i.p. inoculation. By contrast, although the initial numbers of mutant and wild-type cells were similar in both the liver and the spleen, mutant cells failed to accumulate in high numbers in these organs, the defect first being apparent in the spleen at between 8 and 16 h postinoculation in both single and mixed infections. The numbers of pHSG422-containing wild-type and mutant bacteria in the spleen at 16 h were also similar, which shows that mutant bacteria are transported from the peritoneal cavity to the spleen in similar numbers. Whereas the numbers of wild-type cells continued to increase logarithmically, the net load of mutant bacteria remained relatively static for several days before eventual clearance, showing that the secretion system is required for accumulation of S. typhimurium in the spleen and liver.
By treating spleen cell suspensions recovered from infected mice with gentamicin, it was possible to observe intracellular accumulation of viable wild-type bacteria over time. The fact that no such increase was observed in the SPI2 mutant strain is consistent with the in vitro intracellular growth defects observed with mutant SPI2 strains (8, 27, 41), although it does not rule out a role for SPI2 in extracellular bacterial growth if S. typhimurium undergoes significant extracellular replication in the mouse spleen.
Experiments with the temperature-sensitive plasmid pHSG422 showed that the SPI2 secretion system does not contribute significantly to the ability of S. typhimurium to survive in vivo. At 16 h postinoculation there were approximately 10-fold more wild-type than mutant cells in the spleens of infected mice, but the numbers of wild-type and mutant bacteria carrying the marker plasmid were very similar. This shows that there is little difference in the degree of cell killing sustained by the two bacterial strains and that the SPI2 secretion system must be required for bacterial replication rather than for surviving a host immune response that would otherwise kill replication-proficient cells.
The phenotype of the SPI2 mutant strain is reminiscent of the phenotype of plasmid-cured Salmonella strains and strains carrying mutations in the virulence plasmid spv genes. First, both types of mutants are affected in their ability to cause systemic infection in mice (21, 24, and the present study). Second, these mutants cannot be transcomplemented in vivo by coinfection with virulent cells (24). Third, both classes of mutants have been reported to be defective for intracellular growth (27, 32, 41). Fourth, both spv and SPI2 genes are transcribed within host cells (43, 47, 48). Fifth, both mutants are affected in growth rate rather than in an increased susceptibility to host killing mechanisms (19 and the present study).
Despite these similarities, the comparison of virulence attenuation between single- and double-mutant strains failed to provide any evidence showing that they interact together as part of the same virulence mechanism. The mTn5 insertion in strain P10D5 is located within spvA and is likely to have polar effects on downstream genes of the operon. However, we have not examined whether SpvD is secreted by the S. typhimurium SPI2 secretion system, as has been proposed (13).
Previous studies with cultured host cells have shown that SPI2 genes are induced intracellularly and are required for intracellular bacterial proliferation (27, 41, 47). As S. typhimurium replicates within intracellular vacuoles (1, 4, 5), the secretion system could direct effector proteins across the vacuolar membrane and influence the fate of the Salmonella vacuole. The absence of a functional secretion system does not appear to increase the susceptibility of bacteria to killing by the host. The secretion system could influence the growth rate of S. typhimurium either by suppression of or resistance to a normal growth inhibition response from the macrophage. Alternatively, the SPI2 secretion system could direct salmonellae to a subcellular niche favorable for bacterial replication. Further investigation of these and other hypotheses will be aided by knowledge of the predicted effector proteins of the secretion system.
ACKNOWLEDGMENTS
We thank Christoph Tang and Michael Hensel for reviewing this manuscript and T. Hashimoto-Gotoh for the gift of plasmid pHSG422. We also thank Scott R. Waterman for helpful suggestions and practical assistance.
This work was supported by a grant from the United Kingdom Medical Research Council to D. W. Holden. C. R. Beuzon was supported by an EMBO Fellowship.
REFERENCES
- 1.Alpuche-Aranda C M, Racoosin E L, Swanson J A, Miller S I. Salmonella stimulate macrophage macropinocytosis and persist within spacious phagosomes. J Exp Med. 1994;179:601–608. doi: 10.1084/jem.179.2.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benjamin W H, Jr, Hall P, Roberts S J, Briles D E. The primary effect of the ity locus is on the rate of growth of Salmonella typhimurium that are relatively protected from killing. J Immunol. 1990;144:3143–3151. [PubMed] [Google Scholar]
- 3.Blum G, Ott M, Lischewski A, Ritter A, Imrich H, Tschape H, Hacker J. Excision of large DNA regions termed pathogenicity islands from tRNA-specific loci in the chromosome of an Escherichia coli wild-type pathogen. Infect Immun. 1994;62:606–614. doi: 10.1128/iai.62.2.606-614.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buchmeier N A, Heffron F. Intracellular survival of wild-type Salmonella typhimurium and macrophage-sensitive mutants in diverse populations of macrophages. Infect Immun. 1989;57:1–7. doi: 10.1128/iai.57.1.1-7.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carrol M E W, Jackett P S, Aber V R, Lowrie D B. Phagolysosome formation, cyclic adenosine 3′:5′-monophosphate and the fate of Salmonella typhimurium within mouse peritoneal macrophages. J Gen Microbiol. 1979;110:421–429. doi: 10.1099/00221287-110-2-421. [DOI] [PubMed] [Google Scholar]
- 6.Carter P B, Collins F M. The route of enteric infection in normal mice. J Exp Med. 1974;139:1189–1203. doi: 10.1084/jem.139.5.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen L M, Kaniga K, Galan J E. Salmonella spp. are cytotoxic for cultured macrophages. Mol Microbiol. 1996;21:1101–1115. doi: 10.1046/j.1365-2958.1996.471410.x. [DOI] [PubMed] [Google Scholar]
- 8.Cirrillo D M, Valdivia R H, Monack D M, Falkow S. Macrophage-dependent induction of the Salmonella pathogenicity island 2 type III secretion system and its role in intracellular survival. Mol Microbiol. 1999;30:175–188. doi: 10.1046/j.1365-2958.1998.01048.x. [DOI] [PubMed] [Google Scholar]
- 9.Collins F M. Vaccines and cell mediated immunity. Bacteriol Rev. 1974;38:371–402. doi: 10.1128/br.38.4.371-402.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cornelis G R, Wolf-Watz H. The Yersinia Yop virulon: a bacterial system for subverting eukaryotic cells. Mol Microbiol. 1997;23:861–867. doi: 10.1046/j.1365-2958.1997.2731623.x. [DOI] [PubMed] [Google Scholar]
- 11.Deiwick J, Shea J E, Gleeson C, Holden D W, Hensel M. Mutations in SPI2 genes affecting transcription of SPI1 genes and resistance to antimicrobial agents. J Bacteriol. 1999;180:4775–4780. doi: 10.1128/jb.180.18.4775-4780.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dunyak D S, Teel J F, Kennedy M J, Holden D W. Abstracts of the 97th General Meeting of the American Society for Microbiology 1997. Washington, D.C: American Society for Microbiology; 1997. Identification of Salmonella pathogenicity island 2 (SPI2) genes in Salmonella choleraesuis using signature-tagged mutagenesis, abstr. B-275; p. 76. [Google Scholar]
- 13.El Gedaily A, Paesold G, Krause M. Expression profile and subcellular location of the plasmid-encoded virulence (Spv) proteins in wild-type Salmonella dublin. Infect Immun. 1997;65:3406–3411. doi: 10.1128/iai.65.8.3406-3411.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fallon M T, Benjamin W H, Schoeb T R, Briles D E. Mouse hepatitis virus strain UAB infection enhances resistance to Salmonella typhimurium in mice by inducing suppression of bacterial growth. Infect Immun. 1991;59:852–856. doi: 10.1128/iai.59.3.852-856.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fields P I, Swanson R V, Haidaris C G, Heffron F. Mutants of Salmonella typhimurium that cannot survive within the macrophage are avirulent. Proc Natl Acad Sci USA. 1986;83:5189–5193. doi: 10.1073/pnas.83.14.5189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galan J E. Molecular genetic bases of Salmonella entry into host cells. Mol Microbiol. 1996;20:263–271. doi: 10.1111/j.1365-2958.1996.tb02615.x. [DOI] [PubMed] [Google Scholar]
- 17.Galan J E, Curtiss R., III Cloning and molecular characterization of genes whose products allow Salmonella typhimurium to penetrate tissue culture cells. Proc Natl Acad Sci USA. 1989;86:6383–6387. doi: 10.1073/pnas.86.16.6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Groisman E A, Ochman H. How Salmonella became a pathogen. Trends Microbiol. 1997;5:343–349. doi: 10.1016/S0966-842X(97)01099-8. [DOI] [PubMed] [Google Scholar]
- 19.Gulig P A, Doyle T J. The Salmonella typhimurium virulence plasmid increases the growth rate of salmonellae in mice. Infect Immun. 1993;61:504–511. doi: 10.1128/iai.61.2.504-511.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gulig P A, Doyle T J, Clare-Salzler M J, Maiese R L, Matsui H. Systemic infection of mice by wild-type but not Spv−Salmonella typhimurium is enhanced by neutralization of gamma interferon and tumor necrosis factor alpha. Infect Immun. 1997;65:5191–5197. doi: 10.1128/iai.65.12.5191-5197.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gulig P A, Danbara H, Guiney D G, Lax A J, Norel F, Rhen M. Molecular analysis of spv virulence genes of the Salmonella virulence plasmids. Mol Microbiol. 1993;7:825–830. doi: 10.1111/j.1365-2958.1993.tb01172.x. [DOI] [PubMed] [Google Scholar]
- 22.Hacker J, Blum-Oehler G, Muhldorfer I, Tschape H. Pathogenicity islands of virulent bacteria: structure, function and impact on microbial evolution. Mol Microbiol. 1997;23:1089–1097. doi: 10.1046/j.1365-2958.1997.3101672.x. [DOI] [PubMed] [Google Scholar]
- 23.Hashimoto-Gotoh T, Franklin F C H, Nordheim A, Timmis K N. Specific-purpose plasmid cloning vectors. I. Low copy number, temperature-sensitive, mobilization-defective pSC101-derived containment vectors. Gene. 1981;16:227–235. doi: 10.1016/0378-1119(81)90079-2. [DOI] [PubMed] [Google Scholar]
- 24.Hensel M, Shea J E, Gleeson C, Jones M D, Dalton E, Holden D W. Simultaneous identification of bacterial virulence genes by negative selection. Science. 1995;269:400–403. doi: 10.1126/science.7618105. [DOI] [PubMed] [Google Scholar]
- 25.Hensel M, Shea J E, Bäumler A J, Gleeson C, Blattner F, Holden D W. Analysis of the boundaries of Salmonella pathogenicity island 2 and the corresponding chromosomal region of Escherichia coli K-12. J Bacteriol. 1997;179:1105–1111. doi: 10.1128/jb.179.4.1105-1111.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hensel M, Shea J E, Raupach B, Monack D, Falkow S, Gleeson C, Kubo T, Holden D W. Functional analysis of ssaJ and the ssaK/U operon, thirteen genes encoding components of the type III secretion apparatus of Salmonella pathogenicity island 2. Mol Microbiol. 1997;24:155–167. doi: 10.1046/j.1365-2958.1997.3271699.x. [DOI] [PubMed] [Google Scholar]
- 27.Hensel M, Shea J E, Waterman S R, Mundy R, Nickolaus T, Banks G, Vasquez-Torres A, Gleeson C, Fang F C, Holden D W. Genes encoding putative effector proteins of the type III secretion system of Salmonella pathogenicity island 2 are required for bacterial virulence and proliferation in macrophages. Mol Microbiol. 1999;30:163–174. doi: 10.1046/j.1365-2958.1998.01047.x. [DOI] [PubMed] [Google Scholar]
- 28.Heuck C J. Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol Mol Biol Rev. 1998;62:379–433. doi: 10.1128/mmbr.62.2.379-433.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jones B D, Falkow S. Identification and characterization of a Salmonella typhimurium oxygen-regulated gene required for bacterial internalization. Infect Immun. 1994;62:3745–3752. doi: 10.1128/iai.62.9.3745-3752.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee C. Type III secretion systems: machines to deliver bacterial proteins into eukaryotic cells? Trends Microbiol. 1997;5:148–156. doi: 10.1016/S0966-842X(97)01029-9. [DOI] [PubMed] [Google Scholar]
- 31.Leung K Y, Finlay B B. Intracellular replication is essential for the virulence of Salmonella typhimurium. Proc Natl Acad Sci USA. 1991;88:11470–11474. doi: 10.1073/pnas.88.24.11470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Libby S J, Adams L G, Ficht T A, Allen C, Whitford H A, Buchmeirer N A, Bossie S, Guiney D G. The spv genes on the Salmonella dublin virulence plasmid are required for severe enteritis and systemic infection in the natural host. Infect Immun. 1997;65:1786–1792. doi: 10.1128/iai.65.5.1786-1792.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lindgren S W, Stojiljkovic I, Heffron F. Macrophage killing is an essential virulence mechanism of Salmonella typhimurium. Proc Natl Acad Sci USA. 1996;93:4197–4201. doi: 10.1073/pnas.93.9.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lukomski S R, Hull R A, Hull S I. Identification of the O antigen polymerase (rfc) gene in Escherichia coli O4 by insertional mutagenesis using a nonpolar chloramphenicol resistance cassette. J Bacteriol. 1996;178:240–247. doi: 10.1128/jb.178.1.240-247.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maloy S R, Steward V L, Taylor R K. Genetic analysis of pathogenic bacteria. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1996. [Google Scholar]
- 36.Menard R, Prevost M-C, Gounon P, Sansonetti P, Dehio C. The secreted Ipa complex of Shigella flexneri promotes entry into mammalian cells. Proc Natl Acad Sci USA. 1996;93:4197–4201. doi: 10.1073/pnas.93.3.1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miller V I, Mekalanos J. A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J Bacteriol. 1988;170:2575–2583. doi: 10.1128/jb.170.6.2575-2583.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mills D M, Bajaj V, Lee C A. A 40 kb chromosomal fragment encoding Salmonella typhimurium invasion genes is absent from the corresponding region of the Escherichia coli K-12 chromosome. Mol Microbiol. 1995;15:749–759. doi: 10.1111/j.1365-2958.1995.tb02382.x. [DOI] [PubMed] [Google Scholar]
- 39.Monack D M, Raupach B, Hromockyj A E, Falkow S. Salmonella typhimurium invasion induces apoptosis in infected macrophages. Proc Natl Acad Sci USA. 1996;93:9833–9838. doi: 10.1073/pnas.93.18.9833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ochman H, Groisman E A. Distribution of pathogenicity islands in Salmonella spp. Infect Immun. 1996;64:5410–5412. doi: 10.1128/iai.64.12.5410-5412.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ochman H, Soncini F C, Solomon F, Groisman E L. Identification of a pathogenicity island required for Salmonella survival in host cells. Proc Natl Acad Sci USA. 1996;93:7800–7804. doi: 10.1073/pnas.93.15.7800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reed L J, Muench H. A simple method of estimating fifty percent end points. Am J Hyg. 1938;27:493–497. [Google Scholar]
- 43.Rhen M, Riikonen P, Taira S. Transcriptional regulation of Salmonella enterica virulence plasmid genes in cultured macrophages. Mol Microbiol. 1993;10:45–56. doi: 10.1111/j.1365-2958.1993.tb00902.x. [DOI] [PubMed] [Google Scholar]
- 44.Richter-Dahlfors A, Buchan A M J, Finlay B B. Murine salmonellosis studied by confocal microscopy: Salmonella typhimurium resides intracellularly inside macrophages and exerts a cytotoxic effect on phagocytes in vivo. J Exp Med. 1997;184:569–580. doi: 10.1084/jem.186.4.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shea J E, Hensel M, Gleeson C, Holden D W. Identification of a virulence locus encoding a second type III secretion system in Salmonella typhimurium. Proc Natl Acad Sci USA. 1996;93:2593–2597. doi: 10.1073/pnas.93.6.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Skorupski K, Taylor R K. Positive selection vectors for allelic exchange. Gene. 1996;169:47–52. doi: 10.1016/0378-1119(95)00793-8. [DOI] [PubMed] [Google Scholar]
- 47.Valdivia R H, Falkow S. Fluorescence-based isolation of bacterial genes expressed within host cells. Science. 1997;277:2007–2011. doi: 10.1126/science.277.5334.2007. [DOI] [PubMed] [Google Scholar]
- 48.Wilson J A, Doyle T J, Gulig P A. Exponential phase expression of spvA of the Salmonella typhimurium virulence plasmids: induction in intracellular salts medium and intracellularly in mice and cultured mammalian cells. Microbiology. 1997;143:3827–3839. doi: 10.1099/00221287-143-12-3827. [DOI] [PubMed] [Google Scholar]
- 49.Wray C, Sojka W J. Experimental Salmonella typhimurium infection in calves. Res Vet Sci. 1978;25:139–143. [PubMed] [Google Scholar]