

Abstract
Disseminated cryptococcosis is accompanied by cryptococcal polysaccharides in the serum and the lack of cellular infiltrates in infected tissues. Cryptococcal polysaccharides given intravenously to mice inhibit the influx of T lymphocytes into the sites of cell-mediated immune response. The focus here was to determine whether cryptococcal polysaccharides modulate the expression of molecules, such as L-selectin, that are important in extravasation of T cells. Cryptococcal glucuronoxylomannan (GXM), but not galactoxylomannan or mannoprotein, was found to cause loss of L-selectin from freshly isolated human T cells of both CD4 and CD8 subsets and from Jurkat cells. With the signaling-pathway inhibitors staurosporine (which inhibits protein kinase C) and herbimycin A (which inhibits protein tyrosine kinases), we showed that GXM or the cryptococcal culture filtrate antigen CneF directly induces L-selectin loss from CD4+ and CD8+ T cells via a herbimycin A-sensitive pathway(s) presumably involving one or more protein tyrosine kinases but not via a pathway involving protein kinase C. Loss of L-selectin from the T cells before the T cells have a chance to bind to L-selectin ligands on endothelial cells would be expected to prevent T-cell migration into inflamed tissues and/or lymph organs.
Cryptococcus neoformans is an encapsulated yeast-like organism that infects both immunologically competent and immunocompromised individuals. Cryptococcosis ranks in the top four infectious diseases causing death among patients with AIDS (2, 31). Systemic cryptococcosis is characterized by high titers of cryptococcal polysaccharides in serum and minimal cellular infiltration into the infected tissues of patients (11, 16, 33). By using a mouse model, we have found that an intravenous injection of the cryptococcal polysaccharides, to simulate the antigenemia seen in cryptococcosis patients, inhibits leukocyte (neutrophil, monocyte, and T lymphocyte) accumulation at the site of a cell-mediated immune reaction (13). The inhibition of leukocyte infiltration by intravascular cryptococcal polysaccharides occurs irrespective of whether the delayed-type hypersensitivity (DTH) response is to C. neoformans antigen or Mycobacterium tuberculosis antigen (13). Our findings suggest that the minimal cellular infiltrates observed in infected tissues of patients with cryptococcosis may be due, in part, to the circulating cryptococcal polysaccharides. The fact that high titers of cryptococcal polysaccharides in the sera of patients with cryptococcosis can diminish leukocyte migration in response to stimuli other than C. neoformans may be an important confounding factor in AIDS patients with cryptococcosis.
The mechanisms underlying the inhibitory effects of circulating cryptococcal polysaccharides on leukocyte influx are not clear. It is well known that leukocyte attachment to endothelial cells is an important step for leukocyte extravasation. Leukocyte-endothelial cell interactions are mediated by two different sets of receptor-ligand interactions (7, 35). One set of receptors involves the selectin family and their ligands and the other involves the integrins and their ligands. It is possible that circulating cryptococcal polysaccharides may down-regulate leukocyte surface expression of functionally important selectins or integrin molecules and/or may block the receptor-ligand interaction by binding to the receptor or the ligand. Any of these interactions of cryptococcal polysaccharides would result in the inhibition of leukocyte attachment to endothelial cells, which in turn would inhibit leukocyte migration into inflammatory sites.
L-selectin is a molecule constitutively expressed on the surface of most leukocytes, including human T cells (22). It initiates leukocyte attachment to venular endothelium during lymphocyte recirculation through the lymph nodes and during leukocyte recruitment into sites of inflammation (22). The density of L-selectin on the cell surface is a major determinant of binding activity and entry into tissues. A unique feature of L-selectin is that it is shed from the surfaces of leukocytes after activation of the cells or after cross-linking of L-selectin (20, 30). Changes in surface L-selectin expression have been found to have profound effects on the migration and location of T cells in vivo (1, 10). In an earlier study, we found that cryptococcal polysaccharides induce L-selectin loss from the surfaces of human neutrophils (12). Consequently, we were interested in whether cryptococcal polysaccharides would also down-regulate L-selectin expression on the surfaces of human T cells.
The concentrated supernatant (CneF) from a 5-day culture of C. neoformans contains two polysaccharides, glucuronoxylomannan (GXM) and galactoxylomannan (GalXM), and mannoprotein (MP) (9). GXM is a high-molecular-weight polysaccharide which is the predominant component of CneF (9, 28). GXM is also readily detected in sera of patients with disseminated cryptococcosis (11, 16, 33). The two minor constituents of CneF, GalXM and MP, are most likely also present in body fluids from patients with disseminated cryptococcosis (32). In our previous studies, we found that GXM, but not GalXM or MP, directly triggers human neutrophils to shed surface L-selectin (12).
The first objective of this study was to determine whether GXM could directly induce L-selectin shedding from human T cells. After finding that GXM did induce a reduction in L-selectin on the surfaces of human peripheral blood T cells, our second objective was to study the T-cell surface receptors and the possible intracellular signaling molecules involved in GXM-induced L-selectin shedding.
MATERIALS AND METHODS
Maintenance of endotoxin-free conditions.
To prevent lipopolysaccharide (LPS) or endotoxin from influencing the experimental results, all experiments were performed under conditions which minimize endotoxin contamination, as defined previously (12).
MAbs.
The following purified and fluorochrome-conjugated monoclonal antibodies (MAbs) were purchased from Becton Dickinson Immunocytometry System (Mountain View, Calif.): anti-CD28 (mouse immunoglobulin G1 [IgG1]), anti-CD2 (mouse IgG2a), fluorescein isothiocyanate (FITC)-conjugated anti-CD4 (anti-Leu-3a, a mouse IgG1), anti-CD45RA (mouse IgG1), phycoerythrin (PE)-conjugated anti-L-selectin MAb (anti-Leu-8, a mouse IgG2a), and FITC-conjugated mouse irrelevant IgG1 and PE-conjugated mouse irrelevant IgG2a for isotype controls. FITC-conjugated goat F(ab′)2 anti-mouse IgG, used as a secondary reagent to recognize mouse IgG, was purchased from Caltag Laboratories (South San Francisco, Calif.). Cy-chrome-conjugated anti-CD8 (mouse IgG1) and a similarly labeled irrelevant IgG1 control antibody were purchased from PharMingen (San Diego, Calif.). Anti-CD3 (mouse IgG2a), anti-CD4 (mouse IgG2b), anti-CD11a (mouse IgG1), and anti-CD18 (mouse IgG1) were purified on a G-bind column from supernatants from ATCC hybridoma cell lines CRL8001, CRL8002, HB202, and HB203, respectively. The mouse myeloma proteins MOPC21 (IgG1), MOPC195 (IgG2b), and UPC10 (IgG2a) were obtained from Organon Teknika-Cappel (Malvern, Pa.) and were used as isotype control antibodies.
Chemical reagents.
Herbimycin A was obtained from Calbiochem (La Jolla, Calif.). Phorbol myristate acetate (PMA) and staurosporine were purchased from Sigma Chemical Co. (St. Louis, Mo.). All reagents were dissolved in dimethyl sulfoxide and stored at −20°C. The final concentration of dimethyl sulfoxide used in our experiments was 0.1% or less and did not affect surface L-selectin expression on the T cells.
Preparation of CneF, GXM, GalXM, and MP.
CneF was prepared as previously reported by culturing C. neoformans isolate 184A in a defined medium (5). CneF was analyzed by anodic polyacrylamide slab gel electrophoresis, staining of the gels with periodic acid-Schiff’s (PAS) stain, and development of blots from the gels with concanavalin A (ConA). In PAS-stained gels, three bands were observed, and in the blot developed with ConA, a broad diffuse band was observed. These results are consistent with the bands expected for GXM, GalXM, and MP (28). GXM, GalXM, and MP were isolated as previously described (14). All fractions were subjected to electrophoresis and ConA blotting to determine the efficiency of the fractionation, and then they were lyophilized and reconstituted in endotoxin-free, sterile physiologic saline solution to 2 mg (dry weight) per ml.
Cell lines.
The Jurkat (clone E6-1; ATCC TIB 152), J.RT3-T3.5 (ATCC TIB 153), and J.CaM1.6 (ATCC CRL 2063) T-cell lines were maintained in complete medium (CM) consisting of RPMI 1640 with 100 U of penicillin/ml, 100 μg of streptomycin/ml, and 10% fetal bovine serum (FBS) (CM plus 10% FBS). The surface phenotype of Jurkat cells is CD3+ CD4+ CD28+ CD11a+ CD18+ CD2+ LFA3+ CD45RA+ Leu-8 (L-selectin)+ HLA-DR−. The J.RT3-T3.5 cells were derived from the E6-1 clone of the Jurkat cell line (40), and they lack the surface T-cell receptor (TCR)-CD3 complex due to the absence of production of the β chain of the TCR (29, 40). The J.CaM1.6 line was also derived from the E6-1 clone of the Jurkat line. J.CaM1.6 expresses a structurally normal TCR-CD3 complex but is defective in Lck kinase, a protein tyrosine kinase (PTK) of T cells (36). The lack of CD3 expression on J.RT3-T3.5 cells and similar expression of CD3 on Jurkat and J.CaM1.6 cells were confirmed at the time of performing each experiment. In addition to confirming the phenotypes of the Jurkat, J.RT3-T3.5, and J.CaM1.6 cells, we also found that Jurkat and J.CaM1.6 cells expressed CD28 on their surfaces but the J.RT3-T3.5 cells did not. Furthermore, the amounts of CD3 and CD28 expressed on Jurkat and J.CaM1.6 cells were similar. The three cell lines were routinely passaged 2 days prior to use.
Human T-cell purification.
Human T cells were obtained by the method previously described (27). Briefly, buffy coats were obtained from the blood of healthy volunteers at the Sylvan E. Goldman Blood Institute (Oklahoma City, Okla.). The peripheral blood mononuclear cells (PBMC) were isolated by centrifugation on Ficoll-Hypaque (Pharmacia Fine Chemicals, Piscataway, N.J.) at 400 × g for 30 min at room temperature. The PBMC either were used for studies of L-selectin on CD4+ and CD8+ cells or were further separated for other studies. For further fractionation, PBMC were suspended in CM plus 10% FBS and incubated in plastic tissue culture flasks for 1 h at 37°C. PBMC that did not adhere to the plastic were further depleted of adherent monocytes and B cells by incubation on nylon wool columns for 1 h at 37°C. The peripheral blood lymphocytes that did not adhere to the nylon-wool were fractionated by centrifugation on three-step discontinuous Percoll (Pharmacia Inc.) gradients to remove NK cells. The T-cell-enriched fraction was suspended in CM without 10% FBS at a concentration of 107 cells per ml. The T-cell-enriched fractions were composed of >92% CD3+ cells, a phenotype characteristic of T cells, and the cells in these fractions were >95% viable based on trypan blue dye exclusion.
Stimulation of T cells with various reagents.
Freshly isolated human peripheral blood T cells or Jurkat, J.RT3-T3.5, or J.CaM1.6 cells (106) were incubated in 0.5-ml polypropylene microcentrifuge tubes for the designated time (1, 2, or 3 h) at 37°C either in 100 μl of RPMI 1640 medium alone or 100 μl of PMA (100 ng/ml), LPS (500, 250, 100, 50, 25, or 10 ng/ml), CneF (carbohydrate concentration, 1 mg/ml as determined by phenol sulfuric acid assay), GXM (1, 0.5, or 0.25 mg/ml), GalXM (1, 0.5, or 0.25 mg/ml), or MP (1, 0.5, or 0.25 mg/ml) diluted in RPMI 1640. After culturing the T cells for the designated time with one of the reagents, the T cells were immunostained for surface receptor expression and subjected to flow cytometric analysis. The viability of the T cells was determined by trypan blue exclusion and the MTT (a tetrazolium dye) colorimetric assay after the T cells were incubated separately with one of the reagents for 2 or 3 h at 37°C. There was no significant loss in viability following culture in any of the reagents compared to incubation of the T cells in RPMI 1640 (>90% viable). After the Jurkat cells were incubated with PMA, GXM, GalXM, or MP for 2 h at 37°C, the supernatants were collected and tested for the presence of soluble L-selectin with an L-selectin enzyme-linked immunosorbent assay (ELISA) kit (Bender MedSystems, Vienna, Austria). The assay was performed according to the kit insert. This ELISA detects soluble L-selectin in concentrations ranging from 0.4 to 25 ng per ml.
Pretreatment of cells with various antibodies.
Jurkat T cells (106) were preincubated for 30 min at 4°C either in 50 μl of phosphate-buffered saline (PBS) supplemented with 0.1% bovine serum albumin (BSA) or in 50 μl of anti-CD3, anti-CD4, anti-CD28, anti-CD11a, anti-CD18, or an isotype-matched mouse myeloma protein (MOPC21, MOPC195, or UPC10, each at a 20-μg/ml final concentration). The concentration of each MAb used in this study exceeded the saturating concentration as determined by flow cytometry. After incubating the Jurkat cells with the MAb, 50 μl of serum-free RPMI 1640 medium or 50 μl of RPMI 1640 medium containing 1 mg of GXM/ml was added before incubating the mixture for 2 h at 37°C. To compare the effects of anti-CD3 and anti-CD28 MAbs on L-selectin expression on Jurkat cells, J.RT3-T3.5 T cells, and freshly isolated human T cells, each cell population (106) was preincubated for 30 min at 4°C either in 50 μl of PBS supplemented with 0.1% BSA or with 50 μl of anti-CD3, anti-CD28, or one of the isotype-matched mouse myeloma proteins UPC10 or MOPC21 (each at a 20-μg/ml final concentration). Following the MAb treatment, 50 μl of serum-free RPMI 1640 medium was added to the cells and the mixtures were incubated for 2 h at 37°C before the level of L-selectin expression on the cells was detected.
Effects of inhibitors on modulation of L-selectin expression.
Jurkat T cells or freshly isolated PBMC (106) were preincubated in 50 μl of CM alone, in 50 μl of staurosporine (62.5 ng/ml, final concentration), or in 50 μl of herbimycin A (5 or 2.5 μg/ml, final concentration) for 30 min at 37°C. Fifty microliters of PMA (100 ng/ml), GXM (1 mg/ml), anti-CD3 MAb (20 μg/ml), or an isotype-matched MAb, UPC-10 (20 μg/ml), diluted in CM was added to the cells, and the cell mixtures were incubated for 2 h at 37°C.
Immunofluorescence staining for flow cytometric analysis.
The medium used throughout the immunofluorescent staining procedures was PBS supplemented with 0.1% BSA and 0.1% sodium azide. For direct immunofluorescent analysis of L-selectin on the surface of freshly isolated human T cells, Jurkat T cells, J.RT3-T3.5 T cells, or J.CaM1.6 T cells, 106 cells were incubated for 30 min at 4°C in 50 μl of medium containing 0.2 μg of PE-conjugated mouse anti-Leu-8 MAb or containing 0.2 μg of PE-conjugated mouse irrelevant IgG2a. After three washes in medium, the cells were fixed in 1% (wt/vol) paraformaldehyde in PBS and stored at 4°C in the dark until they were analyzed. For indirect labeling of cells with anti-CD28, -CD2, -CD45RA, or -CD18, 106 Jurkat or J.RT3-T3.5 cells were suspended in 50 μl of medium with 1 μg of goat IgG (Sigma Chemical Co.) for preblocking and were incubated for 30 min at 4°C. After being washed twice in medium, the cells were suspended in 50 μl of medium containing 1 μg of mouse anti-human CD28 (IgG1) MAb, 0.25 μg of mouse anti-human CD2 (IgG2a) MAb, 0.5 μg of mouse anti-human CD45RA (IgG1), 1 μg of mouse anti-human CD18 (IgG1), 0.5 μg or 1 μg of MOPC (IgG1) protein, or 0.25 μg of UPC10 (IgG2a) and were incubated for 30 min at 4°C. After three washes in medium, the cells were suspended in medium containing 1 μg of FITC-conjugated goat F(ab′)2 anti-mouse IgG and were incubated for 30 min at 4°C. After another three washes in medium, the cells were fixed and stored as indicated above.
For three-color flow cytometric analysis, human PBMC were collected from the Ficoll-Hypaque gradient, washed twice with PBS, and resuspended in RPMI 1640. The viability of the PBMC was found to be greater than 98% by trypan blue dye exclusion. PBMC (106) were incubated for 30 min at 4°C in 50 μl of medium containing 0.2 μg each of PE-conjugated mouse anti-Leu-8 MAb, FITC-conjugated mouse anti-CD4 MAb, and Cy-chrome-conjugated mouse anti-CD8 MAb. For the isotype controls, PBMC were incubated with 0.2 μg each of PE-conjugated mouse IgG2a, FITC-conjugated mouse IgG1, and Cy-chrome-conjugated mouse IgG1.
The immunofluorescently stained cells were analyzed with a FACStarPLUS (Becton Dickinson & Co., Mountain View, Calif.) flow cytometer gated to exclude cells that stained with the isotype-matched control antibody (12).
Statistical analysis.
The mean, standard error of the mean (SEM), and paired or unpaired Student’s t test results were used to analyze the data. When two groups were being compared, a P value of ≤0.05 was considered to indicate a significant difference between the groups.
RESULTS
GXM, but not GalXM or MP, induces surface L-selectin loss from human peripheral blood T cells.
It has been reported that L-selectin is constitutively expressed on the surface of the majority of peripheral blood T cells from healthy individuals (37). With PE-conjugated anti-Leu-8 MAb in a direct immunofluorescence assay, we also found that the majority (71% ± 6%; mean results from four healthy individuals) of freshly isolated human blood T cells expressed L-selectin. Culturing blood T cells in RPMI 1640 medium at 37°C for 1 to 3 h did not significantly affect L-selectin expression. Furthermore, when 10 to 500 ng of LPS/ml was added to the medium and the cells were incubated at 37°C for 2 h, L-selectin on the surfaces of the T cells did not diminish (data not shown). In agreement with findings by other investigators (34), culturing T cells with PMA for 1 h resulted in a dramatic decrease in the amount of cell surface L-selectin. Eighty percent of the cells were L-selectin negative, and the remaining 20% of the cells had very low densities of L-selectin on their surfaces (approximately half of the density found on unstimulated T cells) (Fig. 1). On average, after 1 h of PMA exposure, the density of L-selectin on the total T-cell population was reduced 85%, and culturing the T cells with PMA for another hour resulted in only slightly more reduction in L-selectin (Fig. 1). GXM-induced L-selectin loss from human blood T cells was less than that induced by PMA (Fig. 1). GXM-induced loss, like PMA-induced loss of L-selectin from T cells, increased slightly between 1 and 2 h, with near-maximal L-selectin loss by 2 h after induction (Fig. 1). On average, after 2 h of exposure to GXM at 1 mg/ml, 50% of the T cells were L-selectin negative, with the remaining 50% expressing a level of L-selectin slightly lower than or similar to that observed on unstimulated T cells. The density of L-selectin on the total T-cell population was reduced 50% when GXM at 1 mg/ml was used as the stimulant (Fig. 1). GXM at 0.5 mg/ml induced a 24% reduction in L-selectin from the T cells’ surfaces, whereas GXM at 0.25 mg/ml did not significantly affect L-selectin expression on T cells. In contrast, neither GalXM nor MP induced L-selectin loss from human blood T cells (Fig. 1).
FIG. 1.
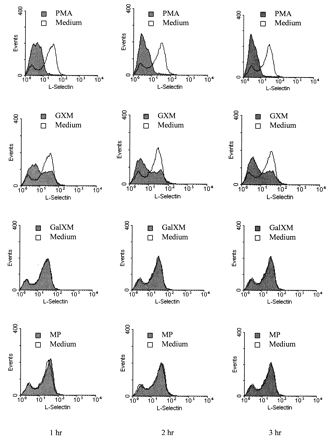
GXM, but not GalXM or MP, induces surface L-selectin loss from freshly isolated human peripheral blood T cells. T lymphocytes (106) were incubated in RPMI 1640 medium alone or with PMA (100 ng/ml), GXM, GalXM, or MP (1 mg/ml) at 37°C for the designated time prior to immunostaining and flow cytometric analysis for L-selectin. y axis, number of cells; x axis, log fluorescence. The shaded areas represent fluorescence patterns after reagent-treated lymphocytes were stained with anti-Leu-8; the unshaded areas represent fluorescence patterns after medium-treated lymphocytes were stained with anti-Leu-8. The data are from one representative experiment of three experiments done with cells obtained from three different donors.
Both CD4+ and CD8+ T cells have reduced surface L-selectin after treatment with cryptococcal antigen.
Considering that GXM induced complete loss of L-selectin from approximately 50% of the freshly isolated human T cells, we were interested in determining if L-selectin was preferentially lost from CD4+ compared to CD8+ T cells. Using three-color flow cytometric analysis for CD4, CD8, and L-selectin on PBMC either untreated (incubated in medium) or treated with CneF (1 mg/ml), which is approximately 88% GXM (28), we observed that CneF induced both CD4+ and CD8+ T cells to lose L-selectin (Fig. 2). Loss of L-selectin from CD4+ and CD8+ cells after a 2-h treatment with CneF was assessed on PBMC from 10 different individuals; the mean percent L-selectin-positive CD4+ and CD8+ cells and the mean fluorescence intensity after L-selectin staining of PBMC incubated in medium or in CneF are shown in Fig. 2C. The two subsets of T cells were equally susceptible to cryptococcal antigen-induced L-selectin loss, as indicated by the fact that after CneF treatment 15.2% (the mean of 10 samples) of the CD4+ cells and 22.9% (the mean of 10 samples) of the CD8+ cells totally lost L-selectin. Furthermore, CneF-treated CD4+ and CD8+ T cells that did not completely lose L-selectin were similar with regard to the reduction in mean fluorescence intensity of L-selectin after incubation in CneF (Fig. 2C).
FIG. 2.
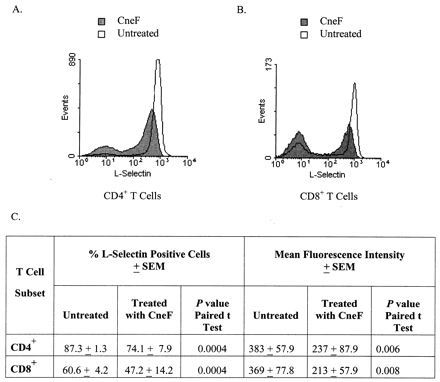
Both CD4+ and CD8+ T cells lose L-selectin in response to cryptococcal antigen. (A and B) Profiles of L-selectin-stained CD4+ and CD8+ cells, respectively, from one representative individual of the 10 individuals whose PBMC were analyzed after a 2-h incubation in medium alone (open areas). and after a 2-h treatment with CneF (shaded areas). (C) Mean percentage of L-selectin positive cells ± SEM and the mean fluorescence intensity ± SEM of L-selectin staining in the CD4+ and CD8+ cell populations which did not completely lose L-selectin after a 2-h treatment with medium alone (untreated) and after a 2-h treatment with CneF. P values from a paired t test for the comparisons of the CneF-treated cells with the untreated cells are also shown.
GXM, but not GalXM or MP, induces surface L-selectin loss from Jurkat T lymphoblastic cells.
Jurkat is a human CD4+-T-cell line that expresses CD3 along with a number of other T-cell surface receptors which are typical of freshly isolated CD4+ human peripheral blood T cells. For instance, Jurkat cells have been shown to resemble normal T cells in many respects (17, 23). In particular, we found that more than 95% of the Jurkat cells constitutively expressed high levels of L-selectin on their surfaces, and as with freshly isolated human peripheral blood CD4+ T cells, Jurkat cells exposed to PMA for 2 h lost on average 84% of their surface L-selectin (Fig. 3A). Culturing Jurkat cells with GXM for 2 h resulted in loss of L-selectin ranging from 37 to 60%, with a mean ± SEM of 47% ± 4%. The variation in percent L-selectin loss induced by GXM in different experiments was most likely due to the use of different lots of GXM. Neither GalXM nor MP induced L-selectin loss from Jurkat cells (Fig. 3A). Based on these data, we established the fact that Jurkat cells were like freshly isolated T cells with respect to loss of L-selectin after stimulation with PMA, GXM, GalXM, or MP.
FIG. 3.
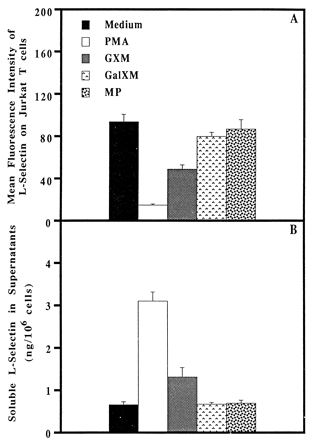
GXM, but not GalXM or MP, induces surface L-selectin loss from a CD4+ T-cell line, Jurkat. (A) Effects of PMA, GXM, GalXM, and MP on surface L-selectin expression on Jurkat cells. L-selectin on Jurkat cells was measured by direct immunofluorescence staining with flow cytometric analysis after the cells were incubated with the designated reagents at 37°C for 2 h. The values represent the mean ± SEM obtained from six individual experiments. (B) Quantification of soluble L-selectin levels in supernatants of Jurkat T cells after incubation with serum-free RPMI 1640 medium, PMA, GXM, GalXM, or MP at 37°C for 2 h. The supernatants were examined for soluble L-selectin with a commercial ELISA kit. The values represent the means ± SEMs obtained from quadruplicate determinations for each sample. The data are from one representative experiment of two. The concentration of GXM, GalXM, or MP used for treatment was 1 mg (dry weight)/ml.
GXM-induced L-selectin loss was due to shedding of L-selectin.
It has been reported that loss of L-selectin from the surfaces of T cells induced by PMA is due to proteolytic cleavage of the extracellular region of L-selectin from the cell surface rather than to internalization of L-selectin (34). To determine if GXM-induced L-selectin loss from T cells was also due to shedding, levels of soluble L-selectin in supernatants from Jurkat cells stimulated with GXM were measured and compared to levels of soluble L-selectin in supernatants from Jurkat cells cultured in medium or stimulated with PMA, GalXM, or MP. The supernatants from Jurkat cells cultured in medium at 37°C for 2 h contained detectable concentrations of soluble L-selectin (Fig. 3B), suggesting that cell surface proteolysis may be a part of the normal turnover of L-selectin. Stimulation of Jurkat cells with PMA or GXM induced increases in soluble L-selectin in the supernatants (Fig. 3B) corresponding to the loss of surface L-selectin induced by the respective agent (Fig. 3A). In contrast, incubation of Jurkat cells with GalXM or MP which did not alter the expression of L-selectin on the cell surface (Fig. 3A) did not result in an increase in soluble L-selectin in the supernatants (Fig. 3B).
Antibodies to CD3, CD4, CD28, CD11a, or CD18 do not block GXM-induced L-selectin loss from T cells.
To determine whether GXM might bind to one or more known T-cell surface receptors and induce L-selectin loss via that receptor(s), a panel of MAbs reactive with designated receptors on human T cells was tested for the ability to block GXM-induced L-selectin loss from Jurkat cells. Candidate receptors were chosen primarily on the basis of previous studies suggesting the involvement of the receptors in T-cell activation (25). As shown in Table 1, GXM induced 53% loss of L-selectin from the surfaces of Jurkat cells. None of the irrelevant isotype control MAbs nor antibodies to CD11a, CD18, or CD4 significantly affected surface L-selectin expression on Jurkat cells. Furthermore, neither irrelevant isotype control MAbs nor antibodies to CD11a, CD18, or CD4 blocked GXM-induced L-selectin loss from Jurkat cells (Table 1). In contrast, anti-CD3 and anti-CD28 MAbs independently were able to trigger loss of L-selectin from Jurkat cells (Table 1). Anti-CD3 MAb induced a greater reduction in L-selectin than did anti-CD28 MAb. The level of L-selectin loss in response to GXM was intermediate to those induced by anti-CD3 and anti-CD28 (Table 1). Anti-CD28 did not block GXM-induced L-selectin loss. In fact, when Jurkat cells were treated with anti-CD28 MAb and GXM, more L-selectin was lost from the Jurkat cells than was lost with either reagent alone (Table 1). Anti-CD3 MAb stimulated a greater loss of L-selectin than was stimulated by GXM. When Jurkat cells were treated with anti-CD3 and GXM together, the loss of L-selectin was no greater than when Jurkat cells were stimulated with anti-CD3 alone (Table 1). In other words, anti-CD3 MAb and GXM did not have additive effects on Jurkat cells with respect to loss of L-selectin. Several explanations can be given for this finding. First, GXM and anti-CD3 could be engaging the same receptor at different sites. Second, anti-CD3 could be blocking GXM binding to its ligand but at the same time signaling the cells to shed L-selectin. Third, anti-CD3 could be stimulating the maximum amount of L-selectin loss possible and therefore the effects of GXM would be lost. In the third case, GXM could be binding to a different receptor than CD3. To assess whether anti-CD3 and GXM were binding to the same site, we performed binding assays and did flow cytometric analysis. We found that GXM did not affect anti-CD3 MAb binding to Jurkat cells and anti-CD3 MAb did not block binding of radioisotope-labeled CneF, which is 88% GXM, to Jurkat cells (data not shown). Consequently, we excluded the second possibility.
TABLE 1.
Effects of various MAbs against T-cell surface receptors on GXM-induced L-selectin loss from Jurkat T cellsa
| Incubated MAb | Isotype | Mean fluorescence intensityb
|
|
|---|---|---|---|
| Medium | GXM | ||
| None | 115 | 54 (53) | |
| MOPC21 | IgG1 | 120 | 63 (45) |
| Anti-CD28 | IgG1 | 75 (35) | 18 (84) |
| Anti-CD11a | IgG1 | 120 | 56 (51) |
| Anti-CD18 | IgG1 | 120 | 52 (55) |
| UPC10 | IgG2a | 120 | 58 (50) |
| Anti-CD3 | IgG2a | 31 (73) | 25 (78) |
| MOPC195 | IgG2b | 120 | 60 (48) |
| Anti-CD4 | IgG2b | 120 | 56 (51) |
The Jurkat cells were preincubated at 4°C for 30 min with irrelevant isotype control MAb MOPC21, MOPC195, or UPC10 or with one of various MAbs to a T-cell surface receptor. After incubation, serum-free RPMI 1640 medium or GXM (1 mg/ml) was added and the T cells were incubated for an additional 2 h at 37°C. Expression of surface L-selectin was measured by immunofluorescence staining with flow cytometric analysis. The data are from one experiment representive of the two experiments performed.
Values in parentheses indicate the percent reduction in intensity of L-selectin on Jurkat cells after stimulation as compared to that of Jurkat cells after incubation in medium.
Effects of staurosporine, a PKC inhibitor, on GXM-induced L-selectin loss.
It has been reported that the density of surface L-selectin on lymphocytes is regulated by signaling through C-type protein kinases (23). Selective pharmacological activation of this pathway with phorbol esters, such as PMA, results in L-selectin shedding from lymphocytes within minutes after stimulation, and L-selectin does not reappear on the surfaces of the lymphocytes for several hours (23). To determine whether GXM-induced L-selectin loss was via activation of signaling mediated by protein kinase C (PKC), a PKC inhibitor, staurosporine, which has been found to block PMA-induced L-selectin loss from the lymph node cells of mice, was used in this study (20). In preliminary experiments, we found that staurosporine at 125 ng/ml or more blocked PMA-induced L-selectin loss from Jurkat cells but also was toxic to Jurkat cells after a 2.5-h incubation. Through further studies, we found that staurosporine over a limited range of concentrations, which included the concentration (62.5 ng/ml) used in the studies shown in Fig. 4, did not significantly affect the viability or the surface expression of L-selectin on Jurkat cells but did completely block PMA-induced L-selectin loss. In the presence of staurosporine (62.5 ng/ml), the loss of L-selectin induced by anti-CD3 MAb was reduced; however, staurosporine did not affect the level of GXM-induced L-selectin loss. These results suggest that anti-CD3 MAb stimulates L-selectin loss via a pathway which includes PKC, whereas the pathway through which GXM signals L-selectin loss is not dependent on PKC.
FIG. 4.
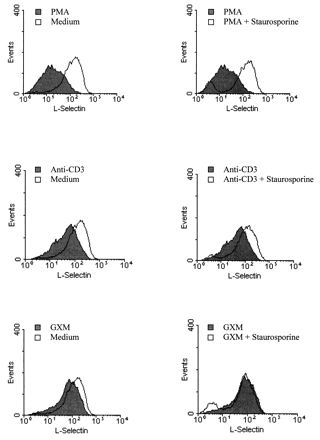
Effects of a PKC inhibitor, staurosporine (62.5 ng/ml), on PMA-, anti-CD3-, and GXM-induced L-selectin loss from Jurkat T cells. Jurkat cells were preincubated with RPMI 1640 medium or staurosporine at 37°C for 30 min, followed by stimulation with PMA (100 ng/ml), anti-CD3 (20 μg/ml), or GXM (1 mg/ml) at 37°C for 2 h. The Jurkat cells were then stained with anti-Leu-8 MAb to determine the level of L-selectin expression. The data are from one representative experiment of three.
Effects of herbimycin A, a PTK inhibitor, on GXM-induced L-selectin loss.
It is well documented that TCR-CD3 complex-driven T-cell proliferation involves a group of PTKs (39). Therefore, it is possible that engagement of CD3 by anti-CD3 MAb to induce L-selectin loss is also directed via a pathway involving PTK. Likewise, it is possible that PTK might be a participant in the pathway controlling GXM-induced L-selectin loss. To address these possibilities, a PTK inhibitor, herbimycin A, was added at two different concentrations to Jurkat cells stimulated with PMA, anti-CD3, or GXM. Also included in these experiments were controls in which Jurkat cells were incubated with each of the two concentrations of herbimycin A. Neither concentration of herbimycin A alone induced measurable changes in L-selectin on the surfaces of Jurkat cells (data not shown), and neither concentration affected PMA-induced loss of L-selectin (Fig. 5). These data demonstrate that herbimycin A does not induce L-selectin loss and that herbimycin A does not block signaling pathways in which PTK is not a component. When herbimycin A was incubated for 30 min with Jurkat cells before the Jurkat cells were stimulated with anti-CD3 MAb or GXM for an additional 2 h, the loss of L-selectin from the surfaces of the Jurkat cells was partially blocked (Fig. 5). When the time for which Jurkat cells were incubated with herbimycin A was increased from 30 min to 4 h, anti-CD3-induced L-selectin loss was inhibited to a greater extent; however, increasing the preincubation time with herbimycin A did not alter the GXM-induced L-selectin loss from the Jurkat cells (data not shown).
FIG. 5.
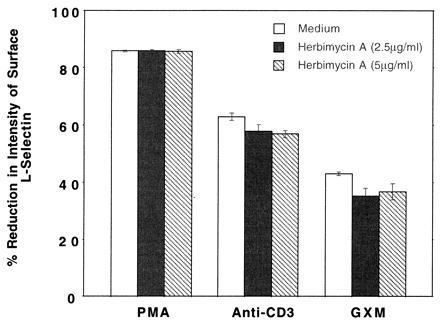
Effects of a PTK inhibitor, herbimycin A, on PMA-, anti-CD3-, and GXM-induced L-selectin loss from Jurkat T cells. The Jurkat cells were preincubated with RPMI 1640 medium or various concentrations of herbimycin A at 37°C for 30 min, followed by stimulation with PMA (100 ng/ml), anti-CD3 (20 μg/ml), or GXM (1 mg/ml) for 2 h. The cells were then stained with anti-Leu-8 MAb to determine the level of L-selectin expression. The percent reduction in L-selectin was determined by comparing the stimulated cells with the unstimulated control cells. The data are pooled from three experiments.
To confirm the effects of herbimycin A on GXM-induced L-selectin loss in T cells and to assess whether PTK was in the signaling pathway of CneF-induced L-selectin loss from freshly isolated human CD4+ and CD8+ T cells, we repeated the herbimycin A experiments, replacing Jurkat cells with freshly isolated T cells. Herbimycin A at both concentrations used (2.5 and 5.0 μg/ml) partially but significantly blocked loss of L-selectin induced by anti-CD3 from CD4+ T cells (Fig. 6A). The higher concentration of herbimycin A significantly reduced the level of L-selectin loss from CD4+ cells incubated in CneF. In contrast to CD4+ cells, neither concentration of herbimycin A affected L-selectin loss from CD8+ cells stimulated with anti-CD3 MAb; however, the 5.0-μg/ml concentration of herbimycin A did significantly reduce loss of L-selectin from CD8+ T cells stimulated with CneF (Fig. 6B). The large error bars in the figure result from the large variance in L-selectin loss from one individual to another; however, in all cases where we obtained significant differences we noted that herbimycin A inhibited to some degree the loss of L-selectin and that inhibition was significant at a 95%-or-greater confidence level.
FIG. 6.
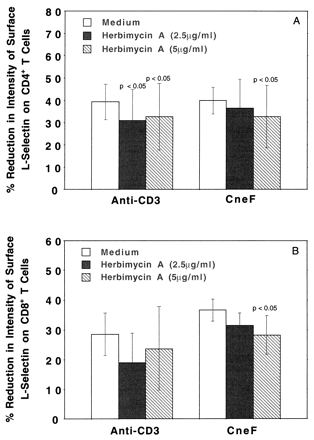
Effects of a PTK inhibitor, herbimycin A, on anti-CD3- and CneF-induced L-selectin loss from freshly isolated human CD4+ and CD8+ T cells. Freshly isolated PBMC from four different individuals were preincubated with herbimycin A for 30 min and then incubated with anti-CD3 or CneF for an additional 2 h before being immunostained for CD4, CD8, and L-selectin. Three-color flow cytometric analysis was performed on the immunolabeled cells. The mean percent reductions in surface L-selectin after treatment of CD4+ cells (A) or CD8+ cells (B) with anti-CD3 or CneF are shown. The error bars indicate the SEMs, and the P values for comparisons with no herbimycin A (medium) are shown above the bars where a significant difference was observed in a paired t test.
Assessment of surface L-selectin after stimulation of Jurkat, J.RT3-T3.5, and J.CaM1.6 T cells with GXM or CneF.
Having found that Jurkat cells treated with anti-CD3 did not shed additional L-selectin when stimulated with GXM (Table 1), we were interested in whether GXM could induce loss of L-selectin in a Jurkat mutant cell line, J.RT3-T3.5, which is defective in expression of the TCR-CD3 complex (29, 40), and in J.CaM1.6, which has normal TCR-CD3 complex expression but defective Lck kinase (36). Before determining if the TCR-CD3 complex influenced GXM-induced L-selectin loss from T cells, it was necessary to demonstrate that the expression of L-selectin was equivalent on Jurkat, J.RT3-T3.5, and J.CaM1.6 cells. The surface densities of L-selectin on Jurkat, J.RT3-T3.5, and J.CaM1.6 cells were comparable. The loss of L-selectin in Jurkat, J.RT3-T3.5, and J.CaM1.6 cells after treatment with PMA or CneF was compared in six additional experiments. PMA-induced losses of L-selectin, which are dependent on PKC in the signaling pathway, were similar in the Jurkat and J.RT3-T3.5 cell lines, as one might predict (Table 2); however, PMA-induced loss of L-selectin in the J.CaM1.6 cell line was significantly higher than that in the Jurkat or J.RT3-T3.5 cell line (P < 0.008) (data not shown). There was no significant difference between the Jurkat and J.CaM1.6 cell lines with regard to L-selectin loss in response to CneF. In contrast, CneF induced a significantly higher level of L-selectin loss from Jurkat cells (51.8% ± 7.1% reduction) and J.CaM1.6 cells (56.5% ± 8.3% reduction) than from J.RT3-T3.5 cells (33.5% ± 6.0% reduction) (P < 0.03 in a paired t test). Consistent with our earlier results, neither GalXM nor MP had an effect on L-selectin expression on either the Jurkat or J.RT3-T3.5 cell line (Table 2).
TABLE 2.
Expression of various T-cell surface molecules on two T-cell lines after incubation with various reagentsa
| Reagent | Mean fluorescence intensity
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Anti-CD28
|
Anti-CD45RA
|
Anti-CD2
|
Anti-CD18
|
Anti-L-selectin
|
||||||
| Ju | JR | Ju | JR | Ju | JR | Ju | JR | Ju | JR | |
| Medium | 15 | 0 | 28 | 19 | 43 | 43 | 21 | 20 | 100 | 93 |
| PMA | 13 | 0 | 29 | 21 | 51 | 51 | 24 | 19 | 17 | 13 |
| GXM | 16 | 0 | 28 | 21 | 47 | 51 | 21 | 20 | 41 | 70 |
| GalXM | 16 | 0 | 28 | 21 | 44 | 45 | 21 | 22 | 87 | 87 |
| MP | 15 | 0 | 28 | 21 | 44 | 44 | 21 | 20 | 97 | 87 |
Ju, Jurkat; JR, J.RT3-T3.5. The T cells (106) were incubated for 2 h at 37°C with 100 ng of PMA/ml or 1 mg of GXM, GalXM, or MP/ml in serum-free RPMI 1640 medium. CD28, CD45RA, CD2, CD18, or L-selectin expression on the two T-cell lines was determined by immunofluorescence staining with the designated antibody and subjecting the cells to flow cytometric analysis. The data are from one experiment representative of the two experiments performed.
To our surprise, we also found that CD28 was lacking on J.RT3-T3.5 cells (Table 2). At first glance, it may seem that the absence of both CD28 and CD3 on the surfaces of J.RT3-T3.5 cells might complicate the interpretation of data on GXM-induced loss of L-selectin from this mutant line. However, the data in Table 1 demonstrate that anti-CD28 was unable to block GXM-induced loss of L-selectin from Jurkat cells which display CD28 on their surfaces. In fact, anti-CD28 MAb had an additive effect along with GXM on the loss of L-selectin from the surfaces of Jurkat cells, indicating that GXM most likely was not signaling through CD28 to induce L-selectin loss. Consequently, it is highly unlikely that the lack of CD28 expression on J.RT3-T3.5 cells would affect GXM-induced L-selectin loss, but rather any effects on L-selectin loss after stimulation with GXM would be more likely to be associated with the lack of the TCR-CD3 complex.
Several controls were included in our studies to demonstrate that the Jurkat and J.RT3-T3.5 cells were behaving as expected and were similar in their behavior to freshly isolated human T cells. We observed that PMA stimulated 78% loss of L-selectin from Jurkat cells, 87% loss from J.RT3-T3.5 cells, and 88% loss from freshly isolated human T cells, whereas anti-CD3 stimulated 70% loss of L-selectin from Jurkat cells and 61% loss from freshly isolated human T cells but did not affect the L-selectin levels on J.RT3-T3.5 cells that lacked CD3. Similarly, anti-CD28 stimulated 28% loss of L-selectin from Jurkat cells and 16% loss from freshly isolated human T cells but did not affect surface L-selectin on J.RT3-T3.5 cells which did not have CD28.
Effects of GXM, GalXM, or MP on expression of CD28, CD45RA, CD2, and CD18 on Jurkat and J.RT3-T3.5 cells.
To determine whether GXM affects the expression of other T-cell surface receptors, such as CD28, CD45RA, CD2, or CD18, the densities of these receptors were measured on the surfaces of both cell lines after incubation with either medium or GXM for 2 h. Candidate receptors were chosen for this study primarily on the basis of reports that these receptors were up- or down-regulated during T-cell activation (18, 37). As shown in Table 2, GXM did not modulate the level of CD28, CD45RA, or CD18 but slightly up-regulated CD2 and significantly down-regulated L-selectin expression. Similar results were obtained when PMA was used as a stimulant for Jurkat and J.RT3-T3.5 cells. Neither GalXM nor MP influenced the expression of any of the surface receptors analyzed.
DISCUSSION
L-selectin (also termed Leu-8, TQ1, LECAM-1, and LAM-1) is a well-characterized adhesion molecule expressed on circulating leukocytes, including lymphocytes, neutrophils, monocytes, and eosinophils (22, 37). A unique feature of L-selectin is that it is shed from leukocyte surfaces after cellular activation (34). Proteolytic cleavage of the extracellular region of L-selectin results in the formation of soluble L-selectin, which has been proposed to have regulatory functions (34). PMA, calcium ionophore, anti-CD3, and mitogenic lectins have been found to trigger L-selectin shedding from the surfaces of T cells within minutes to several hours after stimulation (6, 21, 24). However, exposure of blood lymphocytes to biologically active concentrations of LPS or several different cytokines, including interleukin-1 (IL-1), IL-3, IL-4, IL-5, IL-6, gamma interferon, granulocyte-macrophage colony stimulating factor, and tumor necrosis factor fails to significantly alter cell surface L-selectin expression over a 24-h period (37). In the present study, we confirmed that LPS has no effect on L-selectin expression on T lymphocytes whereas PMA or anti-CD3 MAb triggers T cells to lose L-selectin. Furthermore, we demonstrated that a well-characterized polysaccharide, termed GXM, from the yeast-like organism C. neoformans also modulates L-selectin expression on T cells.
In contrast to PMA-induced loss of L-selectin from T cells, GXM-induced loss of L-selectin from human peripheral blood T cells displays a distinct pattern. After T cells were treated with PMA, 80% of the cells completely shed L-selectin, with the remaining 20% expressing half the level of L-selectin expressed on unstimulated T cells. These data imply that almost all T cells are affected by PMA. Because all T cells have PKC, the target of PMA, our finding that PMA causes L-selectin loss from all T cells is not unexpected. In contrast to PMA, GXM stimulates only about 50% of T cells to completely shed L-selectin, whereas the other 50% of T cells either do not shed L-selectin or shed only a small amount. These findings suggested to us that possibly a subset of T cells, such as CD4+ or CD8+ T cells, were shedding L-selectin in response to GXM. When this question was addressed with three-color flow cytometric analysis of freshly isolated human PBMC, we found that CD4+ and CD8+ T cells have similar patterns of L-selectin loss in response to CneF, which is 88% GXM (9, 28). Between 15 and 23% of the CD4+ and CD8+ T cells completely shed L-selectin, whereas the remaining cells lost none or only a part of their surface L-selectin. Although it is clear that CD4+ and CD8+ T cells shed L-selectin in similar patterns after stimulation with CneF, at the present time it is still uncertain whether the populations of CD4+ and CD8+ T cells that completely shed L-selectin represent specific subsets distinct from the cells that only partially shed or did not shed L-selectin in response to GXM.
Surface L-selectin on normal T cells is implicated in lymphocyte homing and the extravasation of cells into areas of inflammation (22). L-selectin initiates the trafficking of lymphocytes to the lymph nodes by binding to unique carbohydrates on at least two mucin-like ligands, GlyCAM-1 and CD34 (3, 24). If lymphocytes in the blood stream have reduced L-selectin, then induction of the cell-mediated immune response may be greatly reduced (38). L-selectin may also play an important role in the recruitment of naive T cells to sites of inflammation in vivo (8, 38). In this regard, the expression of T-cell-mediated immunity, such as DTH responses, and protection against infection may depend in part on L-selectin-mediated T-cell recruitment. However, it is important to mention that the idea of L-selectin functioning in this latter capacity has not met with universal acceptance. If L-selectin is induced to be completely shed from the surfaces of specific subsets of T lymphocytes by intravascular GXM, then those subsets would not move from the bloodstream to lymph nodes and possibly would not move to the site of acute inflammation or a DTH reaction. In addition, partial reduction in L-selectin on the T cells’ surfaces could also affect the migration of the affected T cells by reducing their rolling on the endothelial cell surface and thus could diminish the subsequent migration of the T cells into the tissues.
GXM-induced shedding of L-selectin from T cells may play an important role in the pathogenesis of cryptococcosis. First, GXM is a major capsular polysaccharide of C. neoformans and can readily be detected in sera of patients with disseminated cryptococcosis (9). Cryptococcal polysaccharide (mainly GXM) titers in sera from patients with cryptococcosis who do not have AIDS typically range from 1:64 to 1:1,028 (11). In AIDS patients with cryptococcosis, titers of cryptococcal polysaccharides (mainly GXM) in both spinal fluid and serum generally are higher than titers in cryptococcosis patients who do not have AIDS. There are reports of cryptococcal antigen titers exceeding 1:100,000 (equivalent to approximately 1 mg of cryptococcal polysaccharides per ml) (15). With therapy, cryptococcal polysaccharide levels in the spinal fluid of patients with AIDS show decreases at predictable rates (15). However, serum cryptococcal polysaccharide levels do not usually decrease greatly (15). Consequently, in some AIDS patients with disseminated cryptococcosis, there is a sufficient amount of cryptococcal polysaccharide in serum to induce the shedding of L-selectin from lymphocytes. In vivo, concentrations of cryptococcal polysaccharides may be higher in regional blood vessels where the polysaccharide is entering the bloodstream than is detected in serum samples from the patients with cryptococcosis. Leukocytes encountering the high concentrations of cryptococcal polysaccharide in regional vessels might shed L-selectin to a greater extent than would be predicted from the serum cryptococcal antigen levels. Also, prolonged exposure to cryptococcal polysaccharide in vivo may cause extensive shedding of L-selectin. Preliminary data from our studies have demonstrated that all leukocyte types (including T lymphocytes) from four AIDS patients with disseminated cryptococcosis display significantly lower amounts of surface L-selectin than do leukocytes from healthy individuals (unpublished data). Furthermore, simulation of antigenemia in mice by injecting CneF intravenously results in limited leukocyte influx into inflammatory sites (13). Second, it has been demonstrated that T-cell-mediated immunity is essential for clearance of C. neoformans from the infected host (26). As mentioned above, both the induction and the expression of T-cell-mediated immunity appear to depend on L-selectin-mediated T-cell homing and recruitment into the reaction sites (35). Therefore, it is reasonable to predict that circulating GXM, by inducing the shedding of L-selectin from the surfaces of blood T cells, modulates anticryptococcal T-cell-mediated immunity. In fact, in our previous studies, we have shown that an intravenous injection of GXM into mice inhibits the accumulation of leukocytes, including T cells, at sites of anticryptococcal DTH response (13).
One objective of this study was to determine how GXM induces surface L-selectin shedding from T cells. It has been reported that the loss of leukocyte L-selectin can be induced either by cellular activators (an activation-dependent mechanism) (20) or by cross-linking of L-selectin with a chemical cross-linker or specific MAbs (an activation-independent mechanism) (30). In this study, we found that GXM-induced L-selectin loss from Jurkat cells and freshly isolated human CD4+ and CD8+ T cells is blocked in part by a PTK inhibitor, herbimycin A, indicating that GXM induces L-selectin shedding from T cells mainly by an activation-dependent mechanism. Furthermore, it has previously been reported by others that the loss of L-selectin expression after treatment of blood lymphocytes with PMA was accompanied by an increase in the density of CD2 expressed on the cell surface (37). In this study, we found that treatment of Jurkat cells with PMA for 2 h induced a reduction in L-selectin, with a slight increase in CD2 expression. Similar results were obtained when Jurkat cells were treated with GXM. The ability of GXM to induce a slight increase in CD2 expression also suggests that GXM activates T cells.
Activation-dependent down-regulation of L-selectin expression on T cells is a well-known phenomenon (22). However, the signaling pathway that regulates L-selectin expression on T cells is poorly understood (22). PKC has been reported to participate in down-regulation of L-selectin expression on T cells (23). In agreement with these findings (23), in this study, we found that a PKC activator, PMA, can reduce L-selectin expression by about 80 to 85% on both freshly isolated human peripheral T cells and Jurkat cells. Furthermore, a PKC inhibitor, staurosporine, blocks PMA-induced L-selectin loss.
In addition to showing that anti-CD3 MAb can induce L-selectin loss, we also found that engagement of CD28 by specific anti-CD28 MAb induced L-selectin loss from T cells, implying that activation of T cells via CD28 induces L-selectin loss. The support for this statement comes from the following observations. First, in the same experiment, both isotype-matched irrelevant control MAb (IgG1) and anti-CD11a (IgG1) and anti-CD18 (IgG1) MAbs did not affect L-selectin expression; however, anti-CD28 (IgG1) MAb induced a reduction in surface L-selectin. Second, the anti-CD28 MAb-induced L-selectin loss correlated directly with the density of surface CD28 on the T cells before stimulation. For example, Jurkat cells and freshly isolated human T cells express CD28 and show a significant loss of L-selectin when treated with anti-CD28 MAb whereas J.RT3-T3.5 cells do not express detectable levels of surface CD28 and do not lose surface L-selectin when stimulated with anti-CD28 MAb. To our knowledge, these data are the first demonstration of anti-CD28 MAb inducing L-selectin loss from T cells. These results are not surprising when one considers that CD28 is a well-characterized costimulation receptor for T-cell activation and proliferation (4). Unfortunately, in this study, we did not determine the signaling pathway for anti-CD28 MAb-induced L-selectin loss. However, since anti-CD28 MAb and GXM had additive effects in inducing L-selectin loss from Jurkat cells, it is reasonable to predict that anti-CD28-induced L-selectin loss is mediated by a different signaling pathway than that used by GXM.
To dissect GXM signaling in the induction of L-selectin loss, we assessed the abilities of PTK and PKC inhibitors to interfere with induction of L-selectin shedding by GXM. In designing and performing these experiments, we were cautious about several parameters. First, we chose a panel of well-characterized and extensively used inhibitors of components in signaling pathways. Second, we examined the effects of each inhibitor alone at different concentrations on cell viability and on L-selectin expression. Inhibitors that altered cell viability and/or affected L-selectin expression were not used in the experiments. Finally, in each experiment, we included at least one positive control to check whether the inhibitor worked under the conditions we were using. We compared data obtained with the designated inhibitors added to Jurkat cells stimulated with anti-CD3 or PMA to results obtained with the inhibitors added to Jurkat cells stimulated with GXM. Our results suggest that both anti-CD3- and GXM-induced loss of L-selectin are signaled at least in part through a pathway involving a PTK. Herbimycin A has been reported to inhibit the T-cell PTKs Lck and Fyn, so it is likely that Lck and/or Fyn are in the GXM-induced signaling pathway for loss of L-selectin from T cells (19). The possibility of a need for Lck to be functional in the CneF-induced signaling pathway for L-selectin loss was eliminated by the fact that CneF (88% GXM) induced J.CaM1.6 cells, which lack Lck expression (36), to shed L-selectin to the same degree as the CneF-induced shedding of L-selectin seen in Jurkat cells, which have surface receptor expression similar to that of J.CaM1.6 cells and which have normal Lck function.
In an attempt to identify the GXM receptor(s) which is involved in GXM-induced L-selectin loss, a panel of MAbs reactive with receptors on human T cells was tested for the ability to inhibit GXM-induced L-selectin loss. We did not find a MAb that blocked GXM-induced L-selectin loss. However, the results must be interpreted with great caution, as the MAb may not block the relevant epitope on the receptor to which GXM binds. Consequently, at the present time, we still do not know which molecule(s) on the T-cell surface can bind GXM and mediate GXM-induced L-selectin loss.
In summary, in our earlier study, we showed that GXM, but not GalXM or MP, can induce surface L-selectin shedding from human neutrophils (12). In the present study, we found that GXM displayed similar modulating effects on L-selectin expression on both human peripheral blood CD4+ and CD8+ T cells and the Jurkat CD4+ T-cell lines. Peripheral blood neutrophils lacking L-selectin are not expected to migrate into inflammatory tissues (1), whereas naive T cells lacking L-selectin are not expected to migrate into peripheral lymph nodes and/or inflammatory tissues (22). In the presence of high levels of cryptococcal polysaccharides, especially GXM, as is found in sera of patients with disseminated cryptococcosis, we would expect to see a profound effect on the migration and location of leukocytes. Indeed, there is very limited cellular infiltration into infected tissues in disseminated cryptococcosis (16). The mechanisms responsible for GXM-induced L-selectin loss from T cells were partly characterized. GXM-induced L-selectin loss from T cells is mainly due to an activation-dependent mechanism(s). More than one signal transduction pathway appears to be operating in GXM-induced L-selectin loss. GXM-induced L-selectin loss does not require activation of PKC. One or more unidentified PTKs which do not include Lck appear to be in the signal transduction pathway induced by GXM that results in L-selectin loss from human T cells. These findings have general implications for studies involving characterization of the molecular basis for modulation of L-selectin on T cells as well as implications for effects of circulating GXM on T-cell extravasation.
ACKNOWLEDGMENTS
The assistance of Jim Henthorn in the flow cytometric analyses is greatly appreciated. Flow cytometric analysis was performed at the Flow Cytometry and Cell Sorting Core Facility of the Oklahoma Center for Molecular Medicine, Oklahoma City, Okla. The authors also thank Anqiang Zhou for his help with the studies of freshly isolated human T cells.
This work was supported by Public Health Service grant AI-18895 from the National Institute of Allergy and Infectious Diseases.
REFERENCES
- 1.Arbones M L, Ord D C, Ley K, Ratech H, Maynard-Curry C, Otten G, Capon D J, Tedder T F. Lymphocyte homing and leukocyte rolling and migration are impaired in L-selectin (CD62L) deficient mice. Immunity. 1994;1:247–260. doi: 10.1016/1074-7613(94)90076-0. [DOI] [PubMed] [Google Scholar]
- 2.Armstrong D. Life-threatening opportunistic fungal infection in patients with the acquired immunodeficiency syndrome. Ann N Y Acad Sci. 1988;544:443–450. doi: 10.1111/j.1749-6632.1988.tb40442.x. [DOI] [PubMed] [Google Scholar]
- 3.Baumheter S, Singer M S, Henzel W, Hemmerich S, Renz M, Rosen S D, Lasky L A. Binding of L-selectin to the vascular sialomucin CD34. Science. 1993;262:443–446. doi: 10.1126/science.7692600. [DOI] [PubMed] [Google Scholar]
- 4.Bryan R G, Li Y, Lai J H, Van M, Rice N R, Rich R R, Tan T H. Effect of CD28 signal transduction on c-Rel in human peripheral blood T cells. Mol Cell Biol. 1994;14:7933–7942. doi: 10.1128/mcb.14.12.7933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buchanan K L, Murphy J W. Characterization of cellular infiltrates and cytokine production during the expression phase of the anticryptococcal delayed-type hypersensitivity response. Infect Immun. 1993;61:2854–2865. doi: 10.1128/iai.61.7.2854-2865.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buhrer C, Berlin C, Jablonski-Westrich D, Holzmann B, Thiele H G, Hamann A. Lymphocyte activation and regulation of three adhesion molecules with supposed function in homing: LECAM-1 (MEL-14 antigen), LPAM-1/2 (alpha 4-integrin) and CD44 (Pgp-1) Scand J Immunol. 1992;35:107–120. doi: 10.1111/j.1365-3083.1992.tb02839.x. [DOI] [PubMed] [Google Scholar]
- 7.Butcher E C. Leukocyte-endothelial cell recognition: three (or more) steps to specificity and diversity. Cell. 1991;76:1033–1036. doi: 10.1016/0092-8674(91)90279-8. [DOI] [PubMed] [Google Scholar]
- 8.Carlos T M, Harlan J M. Leukocyte-endothelial adhesion molecules. Blood. 1994;84:2068–2101. [PubMed] [Google Scholar]
- 9.Cherniak R, Sundstrom J B. Polysaccharide antigens of the capsule of Cryptococcus neoformans. Infect Immun. 1994;62:1507–1512. doi: 10.1128/iai.62.5.1507-1512.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dawson J, Sedgwick A D, Edwards J C W, Lees P. The monoclonal antibody MEL-14 can block lymphocyte migration into a site of chronic inflammation. Eur J Immunol. 1992;22:1647–1650. doi: 10.1002/eji.1830220646. [DOI] [PubMed] [Google Scholar]
- 11.Diamond R D, Bennett J E. Prognostic factors in cryptococcal meningitis. A study in 111 cases. Ann Intern Med. 1974;80:176–181. doi: 10.7326/0003-4819-80-2-176. [DOI] [PubMed] [Google Scholar]
- 12.Dong Z M, Murphy J W. Cryptococcal polysaccharides induce L-selectin shedding and tumor necrosis factor receptor loss from the surface of human neutrophils. J Clin Investig. 1996;97:689–698. doi: 10.1172/JCI118466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dong Z M, Murphy J W. Intravascular cryptococcal culture filtrate (CneF) and its major component, glucuronoxylomannan, are potent inhibitors of leukocyte accumulation. Infect Immun. 1995;63:770–778. doi: 10.1128/iai.63.3.770-778.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dong Z M, Murphy J W. Mobility of human neutrophils in response to Cryptococcus neoformans cells, culture filtrate antigen, and individual components of the antigen. Infect Immun. 1993;61:5067–5077. doi: 10.1128/iai.61.12.5067-5077.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eng R H K, Bishburg E, Smith S M, Kapila R. Cryptococcal infections in patients with acquired immune deficiency syndrome. Am J Med. 1986;81:19–23. doi: 10.1016/0002-9343(86)90176-2. [DOI] [PubMed] [Google Scholar]
- 16.Farmer S G, Komoromski R A. Histologic response to capsule-deficient Cryptococcus neoformans. Arch Pathol. 1973;96:383–387. [PubMed] [Google Scholar]
- 17.Imboden J B, Weiss A, Stobo J D. The antigen receptor on a human T cell line initiates activation by increasing cytoplasmic free calcium. J Immunol. 1985;134:663–665. [PubMed] [Google Scholar]
- 18.Jenkins M K. The ups and downs of T cell costimulation. Immunity. 1994;1:443–446. doi: 10.1016/1074-7613(94)90086-8. [DOI] [PubMed] [Google Scholar]
- 19.June C H, Fletcher M C, Ledbetter J A, Schieven G L, Siegel J N, Phillips A F, Samelson L E. Inhibition of tyrosine phosphorylation prevents T-cell receptor-mediated signal transduction. Proc Natl Acad Sci USA. 1990;87:7722–7726. doi: 10.1073/pnas.87.19.7722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jung T M, Dailey M O. Rapid modulation of homing receptors (gp90MEL-14) induced by activators of protein kinase C. J Immunol. 1990;144:3130–3136. [PubMed] [Google Scholar]
- 21.Jung T M, Gallatin W M, Weissman I L, Dailey M O. Down-regulation of homing receptors after T cell activation. J Immunol. 1988;141:4110–4117. [PubMed] [Google Scholar]
- 22.Jutila M A. Function and regulation of leukocyte homing receptors. J Leukoc Biol. 1993;55:133–140. doi: 10.1002/jlb.55.1.133. [DOI] [PubMed] [Google Scholar]
- 23.Kaldjian E P, Stoolman L M. Regulation of L-selectin mRNA in Jurkat cells. J Immunol. 1995;154:4351–4362. [PubMed] [Google Scholar]
- 24.Lasky L A, Singer M S, Dowbenko D, Imai Y, Henzel W J, Grimley C, Fennie C, Gillett N, Watson S R, Rosen S D. An endothelial ligand for L-selectin is a novel mucin-like molecule. Cell. 1992;69:927–938. doi: 10.1016/0092-8674(92)90612-g. [DOI] [PubMed] [Google Scholar]
- 25.Mondino A, Jenkins M K. Surface proteins involved in T cell costimulation. J Leukoc Biol. 1994;55:805–815. doi: 10.1002/jlb.55.6.805. [DOI] [PubMed] [Google Scholar]
- 26.Murphy J W. Cell-mediated immunity. In: Howard D H, Miller J D, editors. The mycota. VI. Human and animal relationships. Berlin, Germany: Springer-Verlag; 1996. pp. 67–97. [Google Scholar]
- 27.Murphy J W, Hidore M R, Wong S C. Direct interactions of human lymphocytes with the yeast-like organism, Cryptococcus neoformans. J Clin Investig. 1993;91:1553–1566. doi: 10.1172/JCI116361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murphy J W, Mosley R L, Cherniak R, Reyes G H, Kozel T R, Reiss E. Serological, electrophoretic, and biological properties of Cryptococcus neoformans antigens. Infect Immun. 1988;56:424–431. doi: 10.1128/iai.56.2.424-431.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ohashi P S, Mak T W, Van den Elsen P, Yanagi Y, Yoshikai Y, Calman A F, Terhorst C, Stobo J D, Weiss A. Reconstitution of an active surface T3/T-cell antigen receptor by DNA transfer. Nature. 1985;316:606–609. doi: 10.1038/316606a0. [DOI] [PubMed] [Google Scholar]
- 30.Palecanda A, Walcheck B, Bishop D K, Jutila M A. Rapid activation-independent shedding of leukocyte L-selectin induced by cross-linking of the surface antigen. Eur J Immunol. 1992;22:1279–1286. doi: 10.1002/eji.1830220524. [DOI] [PubMed] [Google Scholar]
- 31.Powderly W G. Therapy for cryptococcal meningitis in patients with AIDS. Clin Infect Dis. 1992;14:S54–S59. doi: 10.1093/clinids/14.supplement_1.s54. [DOI] [PubMed] [Google Scholar]
- 32.Reiss E, Cherniak R, Eby E, Kaufman L. Enzyme immunoassay detection of IgM to galactoxylomannan of Cryptococcus neoformans. Diagn Immunol. 1984;2:109–115. [PubMed] [Google Scholar]
- 33.Rippon J W. Cryptococcosis. In: Rippon J W, editor. Medical mycology. 3rd ed. Philadelphia, Pa: W. B. Saunders Company; 1988. pp. 582–609. [Google Scholar]
- 34.Schleiffenbaum B, Spertini O, Tedder T F. Soluble L-selectin is present in human plasma at high levels and retains functional activity. J Cell Biol. 1992;119:229–238. doi: 10.1083/jcb.119.1.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Springer T A. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 36.Straus D B, Weiss A. Genetic evidence for the involvement of the lck tyrosine kinase in signal transduction through the T cell antigen receptor. Cell. 1992;70:585–593. doi: 10.1016/0092-8674(92)90428-f. [DOI] [PubMed] [Google Scholar]
- 37.Tedder T F, Penta A C, Levine H B, Freedman A S. Expression of the human leukocyte adhesion molecule, LAM1. J Immunol. 1990;144:532–540. [PubMed] [Google Scholar]
- 38.Tedder T F, Steeber D A, Pizcueta P. L-selectin-deficient mice have impaired leukocyte recruitment into inflammatory sites. J Exp Med. 1995;181:2259–2264. doi: 10.1084/jem.181.6.2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weiss A, Littman D R. Signal transduction by lymphocyte antigen receptors. Cell. 1994;76:236–274. doi: 10.1016/0092-8674(94)90334-4. [DOI] [PubMed] [Google Scholar]
- 40.Weiss A, Stobo J D. Requirement for the coexpression of T3 and the T cell antigen receptor on a malignant human T cell line. J Exp Med. 1984;160:1284–1299. doi: 10.1084/jem.160.5.1284. [DOI] [PMC free article] [PubMed] [Google Scholar]