

Abstract
Escherichia coli heat-labile enterotoxin (LT), an oligomeric protein with one A subunit (LTA) and five B subunits, exerts its effects via the ADP-ribosylation of Gsα, a guanine nucleotide-binding (G) protein that activates adenylyl cyclase. LTA also ADP-ribosylates simple guanidino compounds (e.g., arginine) and catalyzes its own auto-ADP-ribosylation. All LTA-catalyzed reactions are enhanced by ADP-ribosylation factors (ARFs), 20-kDa guanine nucleotide-binding proteins. Replacement of arginine-7 (R7K), valine-53 (V53D), serine-63 (S63K), valine 97 (V97K), or tyrosine-104 (Y104K) in LTA resulted in fully assembled but nontoxic proteins. S63K, V53D, and R7K are catalytic-site mutations, whereas V97K and Y104K are amino acid replacements adjacent to and outside of the catalytic site, respectively. The effects of mutagenesis were quantified by measuring ADP-ribosyltransferase activity (i.e., auto-ADP-ribosylation and ADP-ribosylagmatine synthesis) and interaction with ARF (i.e., inhibition of ARF-stimulated cholera toxin ADP-ribosyltransferase activity and effects of ARF on mutant auto-ADP-ribosylation). All mutants were inactive in the ADP-ribosyltransferase assay; however, auto-ADP-ribosylation in the presence of recombinant human ARF6 was detected, albeit much less than that of native LT (Y104K > V53D > V97K > R7K, S63K). Based on the lack of inhibition by free ADP-ribose, the observed auto-ADP-ribosylation activity was enzymatic and not due to the nonenzymatic addition of free ADP-ribose. V53D, S63K, and R7K were more effective than Y104K or V97K in blocking ARF stimulation of cholera toxin ADP-ribosyltransferase. Based on these data, it appears that ARF-binding and catalytic sites are not identical and that a region outside the NAD cleft may participate in the LTA-ARF interaction.
CT and Escherichia coli LT, which are involved in the pathogenesis of cholera and traveler’s diarrhea, respectively, are oligomeric proteins consisting of an A subunit (CTA or LTA) associated with five B subunits (CTB or LTB) (23). CTA, secreted into the medium by Vibrio cholerae, is proteolytically cleaved to yield A1 (CTA1) and A2 (CTA2) protein fragments linked by a single disulfide, whereas LTA is isolated as a single chain (10, 18, 21). Activation of the proteolytically nicked CTA requires reduction of the disulfide bond, while activation of LTA requires reduction and proteolysis (18, 21). CTA1 and LTA1, which are released from CTA2 and LTA2, respectively, by reduction, are ADP-ribosyltransferases that modify Gsα, a guanine nucleotide-binding protein that activates the adenylyl cyclase catalytic unit and regulates ion flux (2, 23).
The toxin ADP-ribosyltransferase activity is stimulated, in the presence of GTP (or analogues), by ARFs, a multigene family of 20-kDa guanine nucleotide-binding proteins (12, 24, 32, 33). These proteins, ubiquitous in eukaryotic cells, are believed to participate in vesicular trafficking (5, 24, 29). Based on kinetic analyses, it appears that ARFs are allosteric effectors of CTA1; ARF lowers the Km for both substrates and increases the Vmax (25). Both LTA1 and CTA1 are activated by ARF, which preferentially stimulates the reduced and proteolytically nicked species (13, 22, 25). Other bacterial toxins (e.g., C3 ADP-ribosyltransferase) have not been shown to be activated by ARF (31).
To identify the residues responsible for catalytic activity and to produce potential vaccine candidates, amino acid replacements have been introduced into LTA1 and CTA (8, 9, 14, 15, 26, 27, 34). LTA1 mutants were classified into three groups based on the ability of the LTA and LTB to assemble into a stable oligomeric holotoxin and the extent to which ARF-stimulated and basal catalytic activities were affected (26). Prior studies documented the importance of glutamate-112 in LTA1, which corresponds to glutamates in pertussis, diphtheria, and Pseudomonas toxins and C3 exoenzyme that have been cross-linked to the nicotinamide moiety of NAD and are believed to be involved in NAD binding and catalysis (1, 3, 4, 6, 11). Although LT(E112K) had no catalytic activity, it did, following reduction and trypsinization, block the ability of recombinant human ARF6 to stimulate CT, suggesting that it did have an intact ARF-binding site and could complete with active CT for ARF binding (22). It was concluded that reduction and trypsinization of an inactive mutant LTA, which are required to generate catalytically active wild-type LT, led to formation of an ARF-binding site. Further, in LT(E112K), loss of catalytic activity occurred with retention of an intact ARF-binding site (22).
In view of these studies, mutants containing other amino acid replacements in the LTA1 protein were used to define the residues involved in ARF binding and catalysis (7, 26). The LT mutants examined were fully assembled but nontoxic molecules; they contained mutation of arginine-7 to lysine (R7K), valine-53 to aspartate (V53D), serine-63 to lysine (S63K), valine-97 to lysine (V97K), or tyrosine-104 to lysine (Y104K). All mutant proteins were nontoxic to Y1 adrenal cells and inactive in an ADP-ribosylation assay with polyarginine as the substrate (except V53D, which was not tested) (26). Based on the wild-type LT crystallographic structure, R7, V53, and S63 are located in the active site whereas V97 and Y104 are adjacent to and distant from the site, respectively (19, 26, 30). The crystal structures of three of the mutants S63, R7, and V97, appear to be essentially the same as that of wild-type LT (20, 35, 36). By using a sensitive assay, we quantified catalytic activity and evaluated ARF interaction with the mutant toxins.
MATERIALS AND METHODS
Abbreviations used in this paper.
CT, cholera toxin; CTA, cholera toxin A subunit; CTA1, alkylated cholera toxin A1 protein; LT, E. coli heat-labile enterotoxin; LTA, LTA subunits; LTA1, LTA1 protein; G protein, guanine nucleotide-binding protein; LTB, LTB subunit; SDS, sodium dodecyl sulfate; PAGE, polyacrylamide gel electrophoresis; DTT, dithiothreitol; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; ARF, ADP-ribosylation factor; rhARF6, recombinant human ARF6; TCA, trichloroacetic acid; MOPS, [3-(N-morpholino)propane]sulfonic acid.
Materials.
CTA, CT, and anti-choleragenoid (CTB) antibody were purchased from List Biological Laboratories; [adenine-U-14C]NAD (274 mCi/mmol) and [carbonyl-14C]NAD (54 mCi/mmol) were purchased from Amersham; [adenylate-32P]NAD (10 to 50 Ci/mmol) was purchased from Dupont-New England Nuclear; NAD, GTP, ovalbumin, sodium cholate, dimyristoylphosphatidylcholine, thymidine, ADP-ribose, trypsin, soybean trypsin inhibitor, and agmatine were purchased from Sigma; high-molecular-weight standards were purchased from Pharmacia Biotech; DTT was purchased from Schwartz Mann Biotech; low- and high-molecular-weight prestained standards were purchased from Gibco BRL; sodium nitroprusside and MOPS were purchased from Fischer; GAPDH was purchased from Boehringer Mannheim; and horseradish peroxidase-conjugated rabbit immunoglobulin G was purchased from Promega. rhARF6 was prepared by published procedures (28).
NAD:agmatine ADP-ribosyltransferase assay.
The sensitivity of the standard NAD:agmatine ADP-ribosyltransferase assay was enhanced by increasing the specific activity of the NAD and reducing the NAD concentration. The modified assay mixture (total volume, 0.3 ml) contained 50 mM potassium phosphate (pH 7.5), 5 μM [adenine-U-14C]NAD (80,000 cpm), 10 mM agmatine, 20 mM dithiothreitol, 10 mM MgCl2, 100 μM GTP, ovalbumin (0.1 mg/ml), 3 mM dimyristoylphosphatidylcholine, and 0.2% cholate with or without rhARF6 (1 μg). After addition of 1.0 μg of CTA (activated by incubation for 10 min with 70 mM glycine [pH 8.0]–30 mM DTT at 30°C), the samples were incubated for 90 min at 30°C. Duplicate samples (0.1 ml) were applied to columns (4 by 0.5 cm) of AG1-X2 (Bio-Rad) and washed five times with 1 ml of water; the washes were collected for radioassay in a liquid scintillation counter (28).
Auto-ADP-ribosylation assay.
The auto-ADP-ribosylation assay (28) was modified to enhance sensitivity. The assay mixture (total volume, 100 μl), contained 50 mM potassium phosphate (pH 7.5), 5 mM MgCl2, 20 mM thymidine, 100 μM GTP, 0.1 μM [32P]NAD (2 μCi), 0.003% SDS with the indicated amount of toxin, rhARF6 (2 μg), and 0.1 mM ADP-ribose or high-molecular-weight standards (10 μg) as indicated. After 1 h at 30°C, the reaction was stopped by addition of 0.5 ml of cold 20% TCA plus 5 μg of bovine serum albumin as carrier; the mix was stored overnight at 4°C. The precipitate was collected after centrifugation (12,500 × g for 15 min) and dissolved in 2× SDS-PAGE sample buffer (0.125 M Tris-HCl [pH 6.8], 4% SDS, 10% glycerol, 4% β-mercaptoethanol, 0.02% bromophenol blue). The proteins were separated by SDS-PAGE in 14% polyacrylamide gels, transferred to nitrocellulose, and exposed to XAR film (Kodak) or quantified with a PhosphorImager (Molecular Dynamics). In some experiments, nitrocellulose membranes were incubated with antibodies to ARF, CTB, and LTA as described in the figure legends.
RESULTS
All five LT mutants formed stable holotoxins (15, 26). Trypsinization and reduction of each generated a 23-kDa protein, consistent with the formation of LTA1 (Fig. 1). Similar treatment of wild-type LT resulted in a significant increase in ADP-ribosyltransferase activity, whereas after trypsinization and reduction, all LT mutants were inactive in the standard NAD:agmatine ADP-ribosyltransferase assay (data not shown). To determine whether the apparently inactive mutants had an intact ARF-binding site, their ability to interfere with ARF stimulation of CTA ADP-ribosyltransferase activity was examined. The catalytic-site mutants V53D, S63K, and R7K inhibited ARF-stimulated and, to a lesser extent, basal CTA-catalyzed ADP-ribosylagmatine formation in a concentration-dependent manner (Fig. 2). This preferential inhibition of ARF-stimulated activity is consistent with an effect on ARF binding and the conclusion that the mutants have an intact ARF-binding site and can compete with CTA for ARF in the ADP-ribosyltransferase assay. In all three instances, inhibition of the ARF-dependent stimulation of CTA ADP-ribosylation was dependent on DTT. Presumably, reduction by DTT leads to the formation of the mutant A1 protein (Table 1).
FIG. 1.
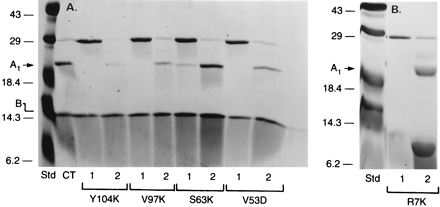
Production of LTA1 from mutants by trypsinization. (A) Y104K (13 μg), V97K (15 μg), S63K (15 μg), or V53D (15 μg) was incubated in 50 mM glycine (pH 8.0)–50 mM DTT with (lane 2) or without (lane 1) 2 μg of trypsin (final volume, 0.1 ml) for 30 min at 30°C before the addition of 0.1 ml of twice-concentrated Laemmli SDS-sample buffer and boiling for 3 min. The proteins were separated by SDS-PAGE in 16% polyacrylamide gels. (B) R7K (30 μg) was incubated in 50 mM glycine (pH 8.0)–20 mM DTT plus trypsin (2 μg) (final volume, 0.1 ml) for 30 min at 30°C before the addition of 20% TCA (0.5 ml). R7K (5 μg, lane 1) and trypsinized R7K (30 μg, lane 2) were subjected to SDS-PAGE, as in panel A, and gels were stained with Coomassie blue. The experiments were repeated at least three times. Std, standard.
FIG. 2.
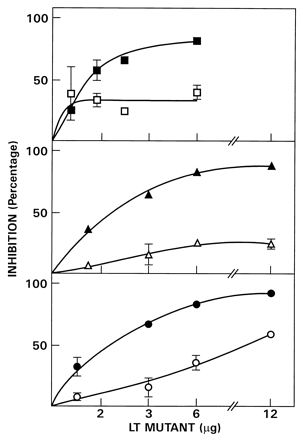
Inhibition of CTA ADP-ribosyltransferase activity by V53D, S63K, and R7K. The LT mutants V53D (30 μg) (▴ and ▵), S63K (30 μg) (■ and □) and R7K (15 μg) (• and ○), were incubated in 50 mM glycine (pH 8.0)–50 mM DTT plus trypsin (2 μg) for 30 min at 30°C, and trypsin inhibitor (10 μg) was added before of the indicated amounts were assayed with (▴, ■, •) or without (▵, □, ○) rhARF6 (1 μg). The percent inhibition of the increase in CTA activity produced by 1 μg of rhARF6 (which was an average of 7.4-fold) is recorded. The CTA NAD:agmatine ADP-ribosyltransferase assay is described in Materials and Methods. Data are the means of values from four assays. Absence of error bars on data points indicates an error too small to record.
TABLE 1.
Effect of reduction of mutant LT on inhibition of CTA1-catalyzed ADP-ribosylagmatine formationa
| Mutant LT | CTA1 ADP-ribosyltransferase activity (pmol · min−1) withb:
|
|||
|---|---|---|---|---|
| 20 mM DTT
|
No DTT
|
|||
| rhARF6 | No ARF | rhARF6 | No ARF | |
| None | 1.30 ± 0.44 | 0.11 ± 0.10 | 1.55 ± 0.04 | 0.11 ± 0.01 |
| S63K | 0.23 ± 0.01 | 0.10 ± 0.01 | 1.98 ± 0.05 | 0.16 ± 0.00 |
| V53D | 0.37 ± 0.07 | 0.15 ± 0.01 | 1.75 ± 0.06 | 0.15 ± 0.02 |
| R7K | 0.35 ± 0.02 | 0.10 ± 0.00 | 2.13 ± 0.21 | 0.20 ± 0.03 |
| Y104K | 1.62 ± 0.13 | 0.10 ± 0.02 | 2.16 ± 0.08 | 0.17 ± 0.01 |
| V97K | 0.92 ± 0.05 | 0.10 ± 0.01 | 2.14 ± 0.11 | 0.19 ± 0.02 |
Reduced and alkylated CTA1 was prepared as described previously (25). Mutant LT S63K, V97K, and V53D (each 30 μg), Y104K (15 μg), and R7K (18 μg) were incubated with trypsin (2 μg) in 50 mM glycine (pH 8.0) at 30°C for 30, and trypsin inhibitor (10 μg) was added before the assay for inhibition of CTA1 (2.0 μg)-catalyzed ADP-ribosylagmatine formation with or without rhARF6 (1 μg) and/or 20 mM DTT in the modified assay mixture. For each mutant, a 12-μg portion was assayed. The experiment was repeated twice.
Data are the averages of four assays (±standard deviation) from one experiment.
In contrast to the catalytic-site mutants, proteins with mutations adjacent to or distant from the site (V97K and Y104K, respectively) only poorly inhibited both ARF-stimulated and basal CTA-catalyzed ADP-ribosylagmatine formation. V97K inhibited both ARF-stimulated and basal activity to the same extent, consistent with the view that the loss of ARF-stimulated activity was due to inhibition of CTA itself (Fig. 3). Y104K had little or no effect at any concentration, suggesting that these mutants lack a functionally active ARF-binding site.
FIG. 3.
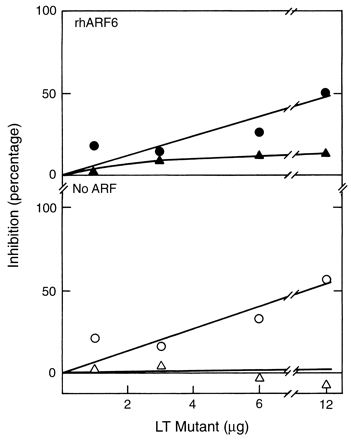
Inhibition of CTA ADP-ribosyltransferase activity by Y104K and V97K. LT mutants Y104K (13 μg) (▴ and ▵), and V97K (22 μg) (• and ○), were incubated as described in the legend to Fig. 2 before being assayed with (▴ and •) or without (▵ and ○) rhARF6 (1 μg).
The presence of an ARF-binding site, as indicated in the competition assay, raised the possibility that the mutants would to some extent, achieve an active structure after reduction and trypsinization. To determine whether residual ADP-ribosyltransferase activity could be detected, the ability of ARF to stimulate auto-ADP-ribosylation by LTA was determined. This assay was considerably more sensitive than quantifying ADP-ribosylagmatine formation. The catalytic-site mutants V53D, S63K, and R7K exhibited auto-ADP-ribosylation, with the activities of S63K and R7K being considerably lower than that of V53D (Fig. 4A). With V53D, ADP-ribosylation of rhARF6, LTA, LTA1 and LTB, as well as of other proteins, was observed; the reaction was enhanced by DTT and ARF. ADP-ribosylation of intact LTA was to some extent ARF independent. The low (relative to V53D) auto-ADP-ribosyltransferase activities of R7K and S63K were, however, enhanced by ARF.
FIG. 4.
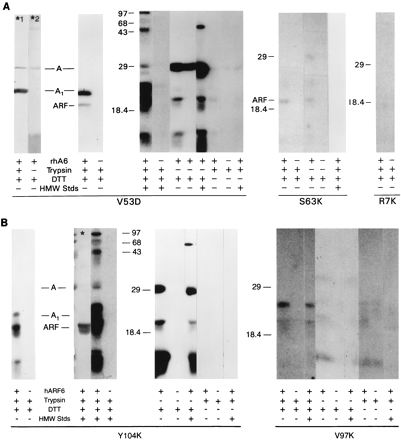
Effects of ARF, trypsin, and DTT on ADP-ribosyltransferase activities of LTA mutants. (A) V53D (30 μg), S63K, (30 μg), and R7K (15 μg) were incubated in 50 mM glycine (pH 8.0), with or without trypsin (2 μg) and/or 50 mM DTT as indicated (final volume, 0.1 ml) for 30 min at 30°C, and trypsin inhibitor (10 μg) was added to trypsinized samples. ADP-ribosylation reactions were performed as described in Materials and Methods with R7K (5.5 μg), S63K (10 μg), or V53D (10 μg), rhARF6 (2 μg), and high-molecular-weight standards (HMW Stds; 10 μg) as indicated. Proteins were separated by SDS-PAGE in 14% polyacrylamide gels, transferred to nitrocellulose, analyzed with a PhosphorImager, and exposed to X-ray film for 3 days (V53D), 7 days (S63K), 10.5 days (S63K, standards), or 14 days (R7K). The autoradiograms for S63K and R7K are computer enhanced. Two of the nitrocellulose lanes were incubated with either LTA monoclonal antibody 4E9 (a gift of R. Holmes, Uniformed Services University of the Health Sciences, Bethesda, Md.) (*1) or polyclonal antibodies against CTB (List Biological Laboratories) (*2) and then with horseradish peroxidase-conjugated second antibody. Anti-CTB antibodies which react with LTB were used to localize LTB on the immunoblots. Anti-CTB antibodies appeared to have some anti-CTA and anti-LTA activity (*2). The high-molecular-weight standard proteins were phosphorylase b (94,000), bovine serum albumin (67,000), ovalbumin (43,000), carbonic anhydrase (30,000), soybean trypsin inhibitor (20,100), and α-lactalbumin (14,400). The experiment was performed at least four times with each mutant. (B) Y104K (13 μg) and V97K (38 μg) were incubated as described for panel A before assay of ADP-ribosyltransferase activities. The asterisk indicates an immunoblot which was reacted with rhARF6 polyclonal antibodies followed by horseradish peroxidase-conjugated second antibody. For all Y104K lanes (4.4 μg), the film was exposed for 1 day, except for the duplicate of the lane with the asterisk, which was exposed for 6 days. For assay mixtures containing V97K (13.7 μg), the film was exposed for 12 days. Experiments were performed at least four times for each mutant. In both panels, lanes marked with asterisks indicate immunoblots; all other lanes represent autoradiograms.
Similar studies also revealed the presence of residual activity in the non-catalytic-site mutants. With Y104K, ARF and the A and B subunits and the A1 protein were labeled in an ARF- and DTT-dependent and trypsin-independent manner (Fig. 4B). The lack of a trypsin requirement may have resulted from the presence of mutant A1 in the preparation, although a mutant A1 band was not visible with protein staining (Fig. 1) or by auto-ADP-ribosylation (Fig. 4B). Trypsin-independent auto-ADP-ribosylation activity was observed previously with holotoxin (13). V97K ADP-ribosylated ARF and its own subunits in an ARF-dependent manner, although the ADP-ribosylation of these and other proteins was considerably lower than that observed with Y104K.
To determine whether the residual ADP-ribosylation reflected the addition of ADP-ribose to the proteins or the covalent attachment of NAD as was observed for the nitroprusside-stimulated incorporation of [32P]NAD into GAPDH (16), V53D and Y104K were activated with trypsin and incubated with ARF, DTT, and either [adenine-14C]NAD or [nicotinamide-14C]NAD (Fig. 5). V53D and Y104K incorporated radiolabel into protein in the presence of [adenine-14C]NAD, but not [nicotinamide-14C]NAD, consistent with an ADP-ribosylation reaction. As expected, GAPDH, but not V53D or Y104K, was labeled in the presence of [nicotinamide-14C]NAD, consistent with the nitroprusside-dependent covalent attachment of NAD rather than ADP-ribose.
FIG. 5.
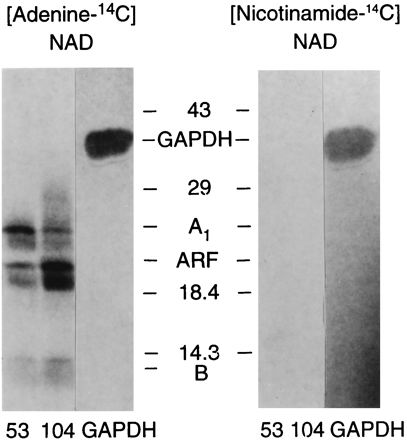
Labelling of V53D and Y104K in the presence of [adenine-14C]NAD or [nicotinamide-14C]NAD. GAPDH (40 μg) was incubated with 0.185 mM [adenine-14C]NAD or [nicotinamide-14C]NAD–1 mM sodium nitroprusside–1 mM DTT–50 mM MOPS (pH 6.9)–1 mM EDTA for 1 h at 37°C (total volume, 0.1 ml); the reaction was stopped by TCA precipitation (21). V53D (30 μg) and Y104K (13 μg) were incubated with trypsin and DTT as described in the legend to Fig. 2; V53D (13.6 μg) and Y104K (5.9 μg) were then assayed in 50 mM potassium phosphate (pH 7.5)–5 mM MgCl2–100 μM GTP–0.003% SDS with rhARF6 (2 μg) and either 0.1 mM [adenine-14C]NAD or [nicotinamide-14C]NAD (each 1.1 × 106 cpm) for 1 h at 30°C (total volume, 0.1 ml). After precipitation with 20% TCA (500 μl), the proteins were dissolved in twice-concentrated sample buffer, separated in 14% gels, transferred to nitrocellulose, and exposed to X-ray film for 7.5 weeks.
To determine whether the ADP-ribosylation reaction involved an enzymatic, as opposed to nonenzymatic, addition of ADP-ribose, the reaction was carried out in the presence of an excess of free ADP-ribose (Fig. 6). Auto-ADP-ribosylation by both mutants was unaffected by the addition of free ADP-ribose, consistent with the conclusion that the ADP-ribosylation did not result from the initial formation of free ADP-ribose. Thus, the reaction appears to be an enzymatic, rather than nonenzymatic, addition of ADP-ribose to protein.
FIG. 6.
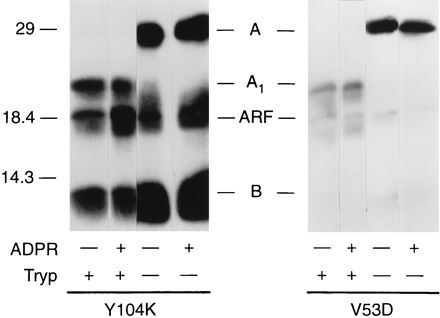
Effect of ADP-ribose on ADP-ribosyltransferase activities of Y104K and V53D. Y104K (13 μg) and V53D (30 μg) were incubated with DTT with or without trypsin as described in the legend to Fig. 2. Samples of Y104K (4.3 μg) or V53D (10 μg) were tested for ADP-ribosyltransferase (ADPR) activity with rhARF6 (2 μg) and 0.1 mM ADP-ribose (see Materials and Methods). Proteins were separated by SDS-PAGE in 14% polyacrylamide gels, transferred to nitrocellulose, and exposed to X-ray film for 1 day. The positions of molecular weight standards are on the left; the positions of LTA (A), LTA1 (A1), rhARF6 (ARF), and LTB (B) are in the center. The experiment was repeated three times in duplicate for Y104K and once in duplicate for V53D.
DISCUSSION
The studies reported here were designed to determine the effects of mutagenesis of catalytic-site and non-catalytic-site amino acids on the interaction of LTA with ARF. Two assays were found to be useful for this purpose, toxin-catalyzed auto-ADP-ribosylation and inhibition by the LT mutants of ARF stimulation of CTA activity. Toxin-catalyzed auto-ADP-ribosylation appeared to be a significantly more sensitive test of catalytic activity than was the ADP-ribosylation of polyarginine or simple guanidino compounds such as agmatine. However, while this assay demonstrates that the mutant can interact with NAD, it does not imply that the mutants are still able to transfer ADP-ribose to G proteins. Since activity was dependent on the presence of ARF-GTP, the loss of an ARF allosteric site might result in the erroneous impression that the mutant lacked catalytic activity when, in fact, it was the allosteric site that was defective. Since ARF may be the principal determinant of activity of some of these or other mutants, an ARF-binding assay, such as the one used in the present study, may be critical to understanding the structure-function relationships in mutant toxins. Specificity in the binding assay required that the mutant toxin be both reduced and trypsinized, conditions similar to those needed to generate a catalytically active molecule. Thus, as noted previously with LT(E112K) (22), the latent form of toxin lacks both transferase and ARF-binding activity. Further characterization of the ARF-binding site and effects of ARF on auto-ADP-ribosylation may yield quantitatively different results, since binding apparently may occur in the absence of an effect on catalytic activity or, if a mutant retained significant auto-ADP-ribosylation activity, catalytic activity might be observed in the presence of a highly compromised ARF-binding site.
An objective of the present study was to develop assays useful in testing vaccine candidates. Nonconservative substitutions were chosen because it is more likely that they will have the most significant effects on activity and, in the larger picture, might have more utility in vaccine development. It is the most aberrant mutant that usually presents the most problems in determining whether the various binding sites have been destroyed. In this study, by using highly sensitive assays, we demonstrated that ARF sites can be detected functionally with these vaccine candidates, either through inhibition of ARF-stimulation of CTA activity or by ARF stimulation of auto-ADP-ribosyltransferase activity.
Of concern in the auto-ADP-ribosylation assay is the possibility that the observed activity does not, in fact, reflect the transferase reaction. Since the toxin is an NAD-binding protein, the radiolabelling might conceivably result from the presence of bound NAD. Alternatively, instead of a concerted SN2-like ADP-ribosyltransferase reaction, perhaps the apparent ADP-ribosylation results from nonenzymatic, covalent association of free ADP-ribose with the protein, a reaction that has been observed in other systems (17). Both of these alternative possibilities appear to have been excluded in the present study. First, association of NAD rather than ADP-ribose with the protein should be reflected by the presence of the nicotinamide label as well as the adenylate-32P, as was observed with the nitroprusside-stimulated attachment of NAD to GAPDH (16), a reaction originally postulated to be ADP-ribosylation. In the latter instance, nicotinamide-14C is bound to the protein, whereas, in the ARF-stimulated auto-ADP-ribosylation, adenylate-32P, but not nicotinamide-14C, was found in the product. To address the issue of nonenzymatic ADP-ribosylation, free ADP-ribose was added to the assay mixture in an attempt to dilute any [adenylate-32P]ADP-ribose released from [32P]NAD. Free ADP-ribose did not significantly inhibit auto-ADP-ribosylation, consistent with the conclusion that an SN2-like reaction, with a direct nucleophilic attack by the protein on NAD, was responsible for the ADP-ribosylation.
It is of interest that the catalytic-site mutants, S63K, V53D, and R7K, inhibited basal CT ADP-ribosyltransferase activity as well as activation by ARF. These data are consistent with the possibility that toxin-toxin interactions affect activity and that complexes containing mutant do not express full activity. Perhaps the presence of the mutant protein promoted some association into a less active species, independent of the effects on the action of ARF. In all cases, however, effects on ARF-stimulated activity were significantly greater than those on basal activity, suggesting two independent mechanisms of inhibition of CTA1, one related to its basal activity and the other related to the ARF-stimulated activity.
The fact that the mutant proteins apparently formed intact holotoxins is crucial to the interpretation of the data, since a loss of activity could stem from nonspecific denaturation as well as from site-specific inactivation. Loss of transferase activity and ARF binding could result from the site-specific effect as well as from generalized pertubation of structure. In this regard, it is noteworthy that the V97K mutant, which appeared to be the least stable in vitro (26), exhibited significantly diminished catalytic and ARF-binding activities. The crystallographic structure of the V97K mutant did not show significant differences from wild-type LT (20).
V97K and Y104K, the non-catalytic-site toxin mutants, appeared not to compete with CTA1 for ARF binding, although the auto-ADP-ribosylation activity of Y104K was stimulated by ARF, suggesting that an ARF-binding site was present but perhaps had a lower affinity than did the wild-type LT. Clearly, the effects of the five LT mutations on ARF binding and catalytic activity were readily distinguished. Mutant Y104K, which weakly inhibited ARF binding to CTA1, had detectable ARF-dependent catalytic activity, whereas S63K and R7K competed for ARF binding but had weaker catalytic activity. V97K had both decreased catalytic activity and ARF binding. These data are consistent with the conclusion that the ARF-binding and catalytic sites are not identical and that a region outside the NAD cleft may participate in the LTA-ARF interaction.
ACKNOWLEDGMENTS
We thank Walter A. Patton for a generous gift of purified rhARF6, Randall Holmes for monoclonal antibody 4E9, and Carol Kosh for expert secretarial assistance.
REFERENCES
- 1.Barbieri J T, Mende-Mueller L M, Rappuoli R, Collier R J. Photolabeling of Glu-129 of the S-1 subunit of pertussis toxin with NAD. Infect Immun. 1989;57:3549–3554. doi: 10.1128/iai.57.11.3549-3554.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Birnbaumer L, Mattera R, Yatani A, Codina J, VanDongen A M J, Brown A M. Recent advances in the understanding of multiple roles of G proteins in coupling of receptors to ionic channels and other effectors. In: Moss J, Vaughan M, editors. ADP-ribosylating toxins and G proteins: insights into signal transduction. Washington, D.C: American Society for Microbiology; 1990. pp. 225–266. [Google Scholar]
- 3.Carroll S F, Collier R J. NAD binding site of diphtheria toxin: identification of a residue within the nicotinamide subsite by photochemical modification with NAD. Proc Natl Acad Sci USA. 1984;81:3307–3311. doi: 10.1073/pnas.81.11.3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carroll S F, Collier R J. Active site of Pseudomonas aeruginosa exotoxin A. Glutamic acid 553 is photolabeled by NAD and shows functional homology with glutamic acid 148 of diphtheria toxin. J Biol Chem. 1987;262:8707–8711. [PubMed] [Google Scholar]
- 5.Cosson P, Letourneur F. Coatomer (COPI)-coated vesicles: role in intracellular transport and protein sorting. Curr Opin Cell Biol. 1997;9:484–487. doi: 10.1016/s0955-0674(97)80023-3. [DOI] [PubMed] [Google Scholar]
- 6.Domenighini M, Magagnoli C, Pizza M, Rappuoli R. Common features of the NAD-binding and catalytic site of ADP-ribosylating toxins. Mol Microbiol. 1994;14:41–50. doi: 10.1111/j.1365-2958.1994.tb01265.x. [DOI] [PubMed] [Google Scholar]
- 7.Douce G, Turcotte C, Cropley I, Roberts M, Pizza M, Domenghini M, Rappuoli R, Dougan G. Mutants of Escherichia coli heat-labile toxin lacking ADP-ribosyltransferase activity act as nontoxic, mucosal adjuvants. Proc Natl Acad Sci USA. 1995;92:1644–1648. doi: 10.1073/pnas.92.5.1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fontana M R, Manetti R, Giannelli V, Magagnoli C, Marchini A, Olivieri R, Domenighini M, Rappuoli R, Pizza M. Construction of nontoxic derivatives of cholera toxin and characterization of the immunological response against the A subunit. Infect Immun. 1995;63:2356–2360. doi: 10.1128/iai.63.6.2356-2360.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giannelli V, Fontana M R, Giuliani M M, Guangcai D, Rappuoli R, Pizza M. Protease susceptibility and toxicity of heat-labile enterotoxins with a mutation in the active site or in the protease-sensitive loop. Infect Immun. 1997;63:331–334. doi: 10.1128/iai.65.1.331-334.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gill D M, Rappaport R S. Origin of the enzymatically actin A1 fragment of cholera toxin. J Infect Dis. 1979;139:674–680. doi: 10.1093/infdis/139.6.674. [DOI] [PubMed] [Google Scholar]
- 11.Jung M, Just I, Damme J, Vandekerckhove J, Aktories K. NAD-binding site of the C3-like ADP-ribosyltransferase from Clostridium limosum. J Biol Chem. 1993;268:23215–23218. [PubMed] [Google Scholar]
- 12.Kahn R A, Gilman A G. Purification of a protein cofactor required for ADP-ribosylation of the stimulatory regulatory component of adenylate cyclase by cholera toxin. J Biol Chem. 1984;259:6228–6234. [PubMed] [Google Scholar]
- 13.Lee C-M, Chang P P, Tsai S-C, Adamik S-C R, Price S R, Kunz B C, Moss J, Twiddy E M, Holmes R K. Activation of Escherichia coli heat-labile enterotoxins by native and recombinant adenosine diphosphate-ribosylation factors, 20-kD guanine nucleotide-binding proteins. J Clin Invest. 1991;87:1780–1786. doi: 10.1172/JCI115197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lobet Y, Cluff C W, Cieplak W., Jr Effect of site-directed mutagenic alterations on ADP-ribosyltransferase activity of the A subunit of Escherichia coli heat-labile enterotoxin. Infect Immun. 1991;59:2870–2879. doi: 10.1128/iai.59.9.2870-2879.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Magagnoli C, Manetti R, Fontana M R, Giannelli V, Giuliani M M, Rappuoli R, Pizza M. Mutations in the A subunit affect yield, stability, and protease sensitivity of nontoxic derivatives of heat-labile enterotoxin. Infect Immun. 1996;64:5434–5438. doi: 10.1128/iai.64.12.5434-5438.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McDonald L J, Moss J. Stimulation by nitric oxide of an NAD linkage to glyceraldehyde-3-phosphate dehydrogenase. Proc Natl Acad Sci USA. 1993;90:6238–6241. doi: 10.1073/pnas.90.13.6238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McDonald L J, Moss J. Nitric oxide-independent, thiol-associated ADP-ribosylation inactivates aldehyde dehydrogenase. J Biol Chem. 1993;268:17878–17882. [PubMed] [Google Scholar]
- 18.Mekalanos J J, Collier R J, Romig W R. Enzymic activity of cholera toxin. II. Relationships to proteolytic processing, disulfide bond reduction, and subunit composition. J Biol Chem. 1979;254:5855–5861. [PubMed] [Google Scholar]
- 19.Merritt E A, Pronk S E, Sixma T K, Kalk K H, Van Zanten B A M, Hol W G J. Structure of partially-activated E. coli heat-labile enterotoxin (LT) at 2.6 Å resolution. FEBS Lett. 1994;337:88–92. doi: 10.1016/0014-5793(94)80635-7. [DOI] [PubMed] [Google Scholar]
- 20.Merrit E A, Sarfaty S, Pizza M, Domenighini M, Rappuoli R, Hol W G J. Mutation of a buried residue causes loss of activity but no conformational change in the heat-labile enterotoxin of Escherichia coli. Nat Struct Biol. 1995;2:269–272. doi: 10.1038/nsb0495-269. [DOI] [PubMed] [Google Scholar]
- 21.Moss J, Osborne Jr J C, Fishman P H, Nakaya S, Robertson D C. Escherichia coli heat-labile enterotoxin: ganglioside specificity and ADP-ribosyltransferase activity. J Biol Chem. 1981;256:12861–12865. [PubMed] [Google Scholar]
- 22.Moss J, Stanley S J, Vaughan M, Tsuji T. Interaction of ADP-ribosylation factor with Escherichia coli enterotoxin that contains an inactivating lysine 112 substitution. J Biol Chem. 1993;268:6383–6387. [PubMed] [Google Scholar]
- 23.Moss J, Vaughan M. ADP-ribosylation of guanyl nucleotide-binding regulatory proteins by bacterial toxins. Adv Enzymol Relat Areas Mol Biol. 1988;61:303–379. doi: 10.1002/9780470123072.ch6. [DOI] [PubMed] [Google Scholar]
- 24.Moss J, Vaughan M. Structure and function of ARF proteins: activators of cholera toxin and critical components of intracellular vesicular transport processes. J Biol Chem. 1995;270:12327–12330. doi: 10.1074/jbc.270.21.12327. [DOI] [PubMed] [Google Scholar]
- 25.Noda M, Tsai S-C, Adamik R, Moss J, Vaughan M. Mechanism of cholera toxin activation by a guanine nucleotide-dependent 19 kDa protein. Biochim Biophys Acta. 1990;1034:195–199. doi: 10.1016/0304-4165(90)90076-9. [DOI] [PubMed] [Google Scholar]
- 26.Pizza M, Domenighini M, Hol W, Giannelli V, Fontana M R, Giuliani M M, Magagnoli C, Peppoloni S, Manetti R, Rappuoli R. Probing the structure-activity relationship of Escherichia coli LT-A by site-directed mutagenesis. Mol Microbiol. 1994;14:51–60. doi: 10.1111/j.1365-2958.1994.tb01266.x. [DOI] [PubMed] [Google Scholar]
- 27.Pizza M, Fontana M R, Giuliani M M, Domenighini M, Magagnoli C, Giannelli V, Nucci D, Hol W, Manetti R, Rappuoli R. A genetically detoxified derivative of heat-labile Escherichia coli enterotoxin induces neutralizing antibodies against the A subunit. J Exp Med. 1994;180:2147–2153. doi: 10.1084/jem.180.6.2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Price S R, Welsh C F, Haun R S, Stanley S J, Moss J, Vaughan M. Effects of phospholipid and GTP on recombinant ADP-ribosylation factors (ARFs). Molecular basis for differences in requirements for activity of mammalian ARFs. J Biol Chem. 1992;267:17766–17772. [PubMed] [Google Scholar]
- 29.Rothman J E. Mechanisms of intracellular protein transport. Nature. 1994;372:55–63. doi: 10.1038/372055a0. [DOI] [PubMed] [Google Scholar]
- 30.Sixma T K, Pronk S E, Kalk K H, Wartna E S, vanZanten B A M, Witholt B, Hol W G J. Crystal structure of a cholera toxin-related heat-labile enterotoxin from E. coli. Nature. 1991;351:371–377. doi: 10.1038/351371a0. [DOI] [PubMed] [Google Scholar]
- 31.Tsai S-C, Adamik R, Moss J, Aktories K. Separation of the 24 kDa substrate for Botulinum C3 ADP-ribosyltransferase and the cholera toxin ADP-ribosylation factor. Biochem Biophys Res Commun. 1988;152:957–961. doi: 10.1016/s0006-291x(88)80376-0. [DOI] [PubMed] [Google Scholar]
- 32.Tsai S-C, Noda M, Adamik R, Chang P P, Chen H-C, Moss J, Vaughan M. Stimulation of choleragen enzymatic activities by GTP and two soluble proteins purified from bovine brain. J Biol Chem. 1988;263:1768–1772. [PubMed] [Google Scholar]
- 33.Tsuchiya M, Price S R, Tsai S-C, Moss J, Vaughan M. Molecular identification of ADP-ribosylation factor mRNAs and their expression in mammalian cells. J Biol Chem. 1991;266:2772–2777. [PubMed] [Google Scholar]
- 34.Tsuji T, Inoue T, Miyama A, Okamoto K, Honda T, Miwatani T. A single amino acid substitution in the A subunit of Escherichia coli enterotoxin results in a loss of its toxic activity. J Biol Chem. 1990;265:22520–22525. [PubMed] [Google Scholar]
- 35.Van den Akker F, Merritt E A, Pizza M G, Domenighini M, Rappuoli R, Hol W G J. The Arg7Lys mutant of heat-labile enterotoxin exhibits great flexibility of active site loop 47-56 of the A subunit. Biochemistry. 1995;34:10996–11004. doi: 10.1021/bi00035a005. [DOI] [PubMed] [Google Scholar]
- 36.Van den Akker F, Pizza M, Rappuoli R, Hol W G. Crystal structure of a non-toxic mutant of heat-labile enterotoxin, which is a potent mucosal adjuvant. Protein Sci. 1997;6:2650–2654. doi: 10.1002/pro.5560061220. [DOI] [PMC free article] [PubMed] [Google Scholar]