

Abstract
B-cell-to-T-cell signaling can shape helper T (Th) cell responses. During infection with Leishmania major, Th response is critical in determining the outcome of disease. Resistance depends on the generation of a protective Th1 response, while susceptibility is mediated by the generation of a Th2 response. In this study, we determined whether B cells are required for the development of polarized Th1 and Th2 responses during infection with L. major. Mice lacking B cells due to disruption of the immunoglobulin M locus (μMT) were infected with L. major, and disease progression and Th cell development were assessed. On the genetically resistant C57BL background, both wild-type and μMT mice controlled the infection and mounted a Th1 response. On the genetically susceptible BALB/c background, both wild-type and μMT mice were susceptible to infection and generated Th2 responses. Thus, during L. major infection, neither direct antigen presentation or costimulation by B cells nor antibody-mediated effector functions are essential for the development of polarized Th responses.
Repetitive stimulation of naive CD4+ helper T (Th) cells in vitro leads to the development of two major types of mature Th cells, which are defined by the type of cytokines they secrete. Th1 cells secrete gamma interferon (IFN-γ) and tumor necrosis factor beta, while Th2 cells secrete interleukin 4 (IL-4), IL-5, and IL-13 (1, 28, 29). During chronic immune responses in vivo, polarized Th responses develop and promote different effector functions. Th1 cells aid phagocytic immune responses, while Th2 cells aid nonphagocytic immune responses. The type of Th response that predominates can be a critical determinant in the outcome of many infections (1, 29).
Many factors which can bias Th differentiation have been identified. Foremost among these are cytokines (1, 29). IL-12 present during the priming of a naive Th cell effectively induces a Th1 response (16), while IL-4 induces Th2 differentiation (24, 46). In addition to cytokines, other factors, such as costimulatory molecules and the strength of T-cell receptor stimulation, can influence Th cell differentiation (4, 6, 14, 30, 47). All of these factors can vary depending on which type of antigen-presenting cell (APC) activates a Th cell. It has, therefore, been postulated that the type of APC may affect Th differentiation.
By presenting antigen directly to Th cells, B cells can potentially influence the Th response. Interaction of B7 and CD40 on B cells with CD28 and CD40 ligand, respectively, on T cells may be required for the acquisition of helper function (25, 49). Additionally, antibodies secreted by B cells alter the epitopes presented to Th cells (2) and activate other cells which may influence the Th response (3). To examine the importance of B cells, mice have been rendered B cell deficient either by antibody depletion (9), by T-cell reconstitution of SCID mice (38, 45), or by genetic disruption of the immunoglobulin (Ig) locus (5, 19). In the absence of B cells, immunization with protein antigen yields Th cells which are less able to provide help for B cells and produce less IL-4 (26). In addition, targeting of antigen to B cells augments T-cell help for other B cells (44). This suggests a model in which APCs augment their own function by promoting the growth of particular Th subsets. Such a model is also supported by in vitro studies demonstrating that Th1 clones are optimally activated by adherent cells, whereas Th2 clones are optimally stimulated by B cells (8). However, the influence of the APC on the Th response during an infection has been less studied. In this report, we characterize the role of B cells during infection with Leishmania major.
The outcome of infection with L. major depends on the type of Th response generated (32). IFN-γ produced by Th1 cells induces infected macrophages to kill the intracellular parasites. Most mouse strains generate a Th1 response to L. major and are therefore resistant. BALB/c mice, however, mount a Th2 response, which is unable to restrain the growth of the parasite (32). If B cells are required for the generation of Th2 responses during L. major infection, then BALB/c mice lacking B cells might be expected to mount a protective Th1 response by default. In fact, anti-IgM-treated BALB/c mice are resistant to L. major (40), and BALB/c X-linked immunodeficient (Xid) mice, which lack the B1 subset of B cells, display enhanced resistance to L. major (13). We have examined the Th response of mice harboring a targeted disruption of the IgM locus (μMT) during infection with L. major. We found that μMT mice from a resistant background were capable of controlling L. major. In contrast to predictions made from other experimental approaches, we found that μMT mice on the BALB/c background were susceptible to L. major infection and developed a Th2 response. Thus, during L. major infection, B cells are not required for polarized Th subset development.
MATERIALS AND METHODS
Mice.
Experiments were performed with μMT mice on the C57BL background (C57.μMT) (19) or μMT mice generated by intercrossing mice which had been backcrossed five times to the BALB/c background. Flow cytometry was used to confirm the absence of B220+ cells in the peripheral blood of μMT mice. Littermate μMT+ or commercially obtained (Jackson Laboratories, Bar Harbor, Maine) wild-type mice were used as controls. Mice were housed in a specific-pathogen-free environment prior to infection and were maintained on trimethoprim-sulfamethoxazole-treated water. All work was performed in accordance with The University of Chicago guidelines for animal care and use.
L. major infection.
L. major (WHOM/IR/−/173) was passaged in BALB/c mice and maintained in vitro for no longer than 2 months prior to infection. Each hind footpad was injected with 106 metacyclic promastigotes, and footpad diameter was measured weekly with a metric caliper. Animals were sacrificed 4 to 10 weeks after infection and individually analyzed. Quantitative parasite cultures of feet and spleens were performed by homogenizing tissue in 3 ml of complete M199 media (20% fetal calf serum, 2 mM l-glutamine, 100 U of penicillin/ml, 100 μg of streptomycin/ml). Aliquots were serially diluted across 96-well plates. Wells were examined for the presence of motile parasites after 2 weeks of culture at 26°C.
Cytokine analysis.
Single-cell suspensions were prepared from draining popliteal lymph nodes of L. major-infected mice. Antigen-specific cytokine secretion was determined by culturing 5 × 105 cells with soluble Leishmania antigen (100 μg/ml) and 2 × 106 irradiated (2,000 rad) major histocompatibility complex (MHC)-matched splenocytes in 96-well round-bottom plates. Where indicated, draining lymph node cells were depleted of B cells prior to analysis by incubation with anti-B220 monoclonal antibody (MAb) and magnetic beads (PerSeptive Biosystems, Framingham, Mass.). Supernatants from the restimulation cultures were removed at 48 h and analyzed for IL-4 and IFN-γ production by an enzyme-linked immunosorbent assay (ELISA) using commercially obtained antibody pairs (PharMingen, San Diego, Calif.). For direct ex vivo analysis of cytokine transcripts, RNA was extracted from B-cell-depleted lymph node samples with TRIzol Reagent (Gibco, Gaithersburg, Md.). RNA was reverse transcribed by using random hexamer primers (Pharmacia, Piscataway, N.J.), and semi-quantitative PCR was performed as previously described (33). Briefly, cDNA was amplified in the presence of a polycompetitor construct that contains addition mutations of authentic cDNA. When these cDNAs are resolved on agarose gels, the higher-molecular-weight bands provide an internal standard for the relative amounts of the lower-molecular-weight experimental cDNAs. Concentrations of cDNAs were adjusted by using the housekeeping gene hypoxanthine-guanine phosphoribosyltransferase (HPRT) prior to analysis of lymphokine gene transcription.
RESULTS
B cells are not required for control of L. major infection.
To assess the role of B cells during infection with L. major, we first determined whether B cells are required for control of infection. μMT mice from the genetically resistant C57BL background were infected with L. major in the hind footpads, and footpad diameter was measured to assess disease progression. Wild-type C57BL and BALB/c mice served as resistant and susceptible controls, respectively. C57.μMT mice, like wild-type C57BL mice, developed far less footpad swelling than BALB/c mice (Fig. 1). Resistance appeared to be long-lived, as mice infected for as long as 24 weeks did not exhibit any enhancement of swelling at later time points (data not shown). Some μMT mice (2 of 15) did develop substantial footpad swelling, reaching footpad diameters greater than 3.5 mm; however, all lesions eventually resolved, and similar lesions were also observed in 1 of 10 wild-type C57BL mice. In contrast, all of the 22 wild-type BALB/c mice developed lesions which did not resolve. Similar results were also obtained with μMT mice bred to the resistant C3H and DBA backgrounds (data not shown). Consistent with the healer phenotype, PCR analysis of the draining lymph nodes from C57.μMT mice revealed a Th1 cytokine profile (data not shown). These results support the previous finding that SCID mice reconstituted only with T cells control L. major as well as mice given both B and T cells (50). Thus, B cells are not required for resistance to L. major.
FIG. 1.

B cells are not required for resistance to L. major. Wild-type and μMT mice from the C57BL background were infected with L. major in the hind footpads, and average footpad diameters over time are depicted. Data for infected mice from the genetically susceptible BALB/c background are shown for comparison. Error bars, standard deviations. Results are representative of six experiments, in which a total of 10 wild-type and 15 μMT mice were analyzed.
B cells are not required for generation of Th2 responses to L. major.
To determine whether B cells were required for the generation of Th2 responses to L. major, C57.μMT mice were backcrossed five times to the genetically susceptible BALB/c background and intercrossed to produce BALB/c B-cell-deficient mice (BALB.μMT). These mice were infected with L. major, and footpad diameter was measured over time. Extensive footpad lesions were apparent in both BALB/c and BALB.μMT mice (Fig. 2A). In total, 28 of 30 BALB/c mice and 27 of 29 BALB.μMT mice exhibited footpad diameters greater than 3.5 mm, compared to 2 of 18 wild-type C57BL mice. Furthermore, none of the lesions from BALB/c or BALB/μMT mice resolved. In some experiments, however, the rates of lesion development for BALB/c and BALB.μMT mice differed slightly. In four of nine experiments, lesions developed at similar rates, (Fig. 2A), but in the remaining experiments, BALB.μMT mice developed lesions approximately 1 week later than wild-type mice. Despite this, parasite loads in feet and spleens were similar for BALB/c and BALB.μMT mice and were greater than those observed in C57BL mice (Fig. 2B). These results demonstrate that B cells are not required for disease susceptibility in BALB/c mice.
FIG. 2.
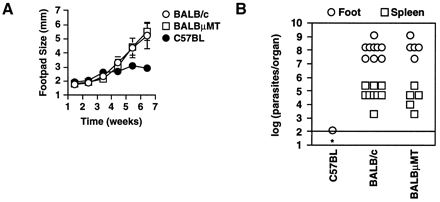
B cells are not required for susceptibility to L. major. (A) Wild-type and μMT mice from the BALB/c background were infected with L. major in the hind footpads, and average footpad diameters over time are depicted. Infected mice from the genetically resistant C57BL background are shown for comparison. Error bars, standard deviations. Results are representative of nine experiments, in of which a total of 30 wild-type and 29 μMT mice were analyzed. (B) Cultures of limiting dilutions were used to determine the parasite loads of mice infected with L. major for 8 weeks. Each symbol depicts the parasite load in a foot or spleen of an individual mouse. The line represents the limit of detection of this assay. ∗, no parasites were detected in the spleen of the C57BL mouse. Results are representative of seven experiments using 23 BALB/c and 24 BALB.μMT mice analyzed between 4 and 10 weeks after infection.
Because C57.μMT mice exhibit normal control of infection with L. major, it was unlikely that BALB.μMT mice were susceptible because of the lack of B cells. It seemed more likely that BALB.μMT mice, like wild-type BALB/c mice, had generated a nonprotective Th2 response. To test this, mice were sacrificed at the onset of lesion development, and cytokine production was measured in cultures of draining lymph node cells stimulated with L. major antigens. Although the time required for the onset of lesion development varied between infections, this did not alter the pattern of cytokines observed. When no exogenous APCs were added, cultures from BALB.μMT mice produced no detectable cytokine (data not shown). When irradiated splenocytes were included as a source of APCs, cells from BALB.μMT mice displayed a cytokine profile similar to that of wild-type BALB/c cells (Fig. 3A). They produced high levels of IL-4 and low levels of IFN-γ, characteristic of a polarized Th2 response. In contrast, cells from resistant C57BL mice produced low levels of IL-4 and high levels of IFN-γ, characteristic of a Th1 response. We obtained similar results regardless of whether unfractionated or B-cell-depleted lymph node cells were stimulated with irradiated APCs and antigen (data not shown). Furthermore, all cytokine production was dependent on Th cells, as it could be completely blocked by the addition of anti-class II MAb to cultures (data not shown). While some cultures from BALB.μMT mice yielded lower levels of IL-4 than wild-type BALB/c cultures, we never observed a switch to a Th1-dominated response. Furthermore, the slightly reduced levels of IL-4 did not correlate with the delayed onset of lesion development observed in some infections. This is consistent with results demonstrating significant Th2 polarization but slightly decreased IL-4 production after infection with Schistosoma mansoni (18). Thus, the modest influence of B cells on Th2 development is insufficient to alter the clinical course of L. major infection.
FIG. 3.
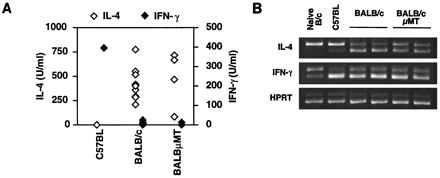
BALB.μMT mice mount a Th2 response to L. major. (A) Unfractionated draining lymph node cells were harvested from mice 8 weeks after infection with L. major. Cells were cultured with L. major antigens and irradiated, MHC-matched splenocytes. After 2 days, IL-4 and IFN-γ in supernatants were measured by an ELISA. Each symbol represents the amount of cytokine detected for an individual mouse. Results are representative of two experiments, using a total of 12 BALB/c and 7 BALB.μMT mice. (B) Draining lymph nodes taken from mice 4 weeks after infection with L. major were depleted of B cells, and cytokine mRNA was measured by competitive RT-PCR. The upper band in each panel corresponds to the amplification product of the competitor DNA, which serves as an internal control for the amplification of endogenous cDNA (lower band). Amplification for the housekeeping gene, HPRT, was used to ensure equivalent loading for all samples. Results are representative of two experiments, using a total of five BALB/c and eight BALB.μMT mice.
Because APC variation can affect the Th response in vitro (8, 12, 26, 44), we also measured cytokines directly ex vivo. B- cell-depleted draining lymph node cells from infected C57BL, BALB/c, and BALB.μMT mice were assayed for cytokine transcripts by competitive reverse transcription-PCR (RT-PCR). In agreement with the results from cells restimulated in vitro, BALB.μMT cells expressed slightly less IL-4 mRNA than B- cell-depleted lymph node cells from BALB/c mice; however, IL-4 expression by both groups was significantly higher than that by C57BL cells (Fig. 3B). Both BALB/c and BALB.μMT mice displayed a Th2 pattern of cytokine expression, with more IL-4 and less IFN-γ mRNA than C57BL mice. Uninfected B- cell-depleted lymph node cells from wild-type BALB/c mice examined for comparison produced lower levels of cytokines than infected lymph nodes. Unlike results obtained by in vitro restimulation, in which exogenous APCs were required for cytokine production, these results demonstrate that sufficient activation occurs in vivo to induce a polarized pattern of cytokine transcription. Thus, B cells serve a unique function during the recall response in vitro which is not essential for the elicitation of cytokines in vivo. Together, these results show that BALB.μMT mice are susceptible to L. major and that susceptibility correlates with a Th2 response.
DISCUSSION
Because they express cytokines, MHC class II, costimulatory molecules, and immunoglobulin, B cells can influence Th cell development through direct interactions or by altering the function of other accessory cells. In this study, we examined the influence of B cells on Th1 and Th2 development during the immune response to L. major. We find that B cells are not required for resistance or Th1 development after challenge with L. major. Using μMT mice bred to the BALB/c background, we also demonstrate that the absence of B cells does not prevent mice from succumbing to the infection. The susceptibility of BALB.μMT mice does not likely reflect a requirement for B cells in controlling L. major infection, since μMT mice on the C57BL background control infection normally. Furthermore, assessment of the cytokine profile by two methods reveals that susceptibility correlates with the generation of a Th2 response. Thus, the genetic polymorphism(s) underlying the distinct disease outcomes of BALB/c and C57BL mice does not require B cells for its manifestation. These results are consistent with previous reports showing efficient Th2 development in the absence of B cells (7, 10, 20, 39).
Our results differ from earlier findings that B cells contribute significantly to susceptibility to L. major (13, 40). The disparate results may be due to the methods used to obtain B-cell-deficient mice. Both BALB/c mice that have been rendered B cell deficient by repeated treatment with anti-IgM and BALB/c Xid mice, which lack the B1 subset of B cells, display enhanced resistance to L. major. Both types of mice, in contrast to μMT mice, have a unique array of circulating antibodies which may alter the response of Fc receptor-bearing cells. In addition, anti-IgM-treated mice may develop T-cell responses to the injected antibody, which in turn may bias the Th response to L. major. Another study, however, demonstrates that BALB/c mice treated with anti-IgM MAb are susceptible to L. major, suggesting that the outcome of infection may depend on the source of antibody used for B-cell depletion (48). To explain the phenotype of Xid mice, it can be reasoned that the selective loss of B1 cells, compared to the loss of both B1 and B2 cells in μMT mice, results in impaired Th2 function. Alternatively, the Xid mutation may result in unrecognized effects on other cell types. Finally, it remains possible that μMT mice have developed compensatory mechanisms which allow for Th2 differentiation in the complete absence of B cells.
Much of the controversy surrounding the role of B cells as APCs stems from variation in the methods used to study Th function. Initial studies examining mice immunized with protein demonstrate decreased proliferative responses of unfractionated lymph node cells during in vitro restimulation (17, 35, 37). Because lymph nodes were unfractionated, results may reflect a role for activated B cells during the in vitro response and not during the in vivo priming. To eliminate this problem, purified Th cells from antigen-primed mice have been stimulated in vitro with antigen and exogenous APCs. When the proliferative response is measured, some groups find that Th cells from B-cell-deficient mice proliferate less than Th cells from control mice (11, 21, 25, 36), while others find Th cell proliferation to be normal (7, 26, 31, 45). Even when proliferation is normal, however, Th cells from protein-immunized B- cell-deficient mice still manifest suboptimal IL-4 production and helper activity (26). Thus, in response to in vivo protein immunization, Th2 responses may be more sensitive to the absence of B cells than proliferative responses.
To determine whether B cells bias the Th response during disease induction, a number of different disease models have been studied. During infection with Trypanosoma cruzi, Xid mice show enhanced resistance to disease accompanied by a decrease in IL-10 production (27). In a mouse model of malaria, μMT mice appear to be selectively deficient in the development of IL-4-producing T cells (22). During experimental autoimmune encephalomyelitis (EAE), targeting of antigen presentation to B cells enhances Th2 responses and lessens pathology (41), while the absence of B cells weakens Th2 responses and augments pathology (51). In contrast to these disease models, during allergic airway sensitization (10, 20) or after injection of schistosome eggs (7), the Th response is only modestly affected. This may result from an ability of certain stimuli to override the influence of B cells by actively directing Th1 or Th2 differentiation, while “neutral” stimuli may be more sensitive to the loss of B cells. Consistent with such a model, Nippostrongylus brasiliensis or S. mansoni, pathogens which normally induce a highly polarized Th2 response, also incite Th2 differentiation in the absence of B cells (7, 39). Also, addition of recombinant IL-4 during in vitro differentiation drives efficient Th2 development regardless of which APC is used (15, 42). By inducing IL-4 very soon after infection (23, 34), L. major may induce Th2 responses in the absence of B cells. Alternatively, suppression of IL-12 secretion by L. major may strongly favor the establishment of a Th2 response (34). Thus, the ability to prime a Th2 response may depend on how a stimulus activates the Th2 pathway.
The role of B cells may depend on the site of the immune response. For example, while lymph node Th cells require B cells for efficient priming to protein antigens, Th cells from the spleen do not (17). The distinct cellular and cytokine microenvironment in the spleen (and perhaps in other sites) may obviate the need for B cells. Such anatomical specialization may explain why B cells are not needed to generate allergic reactions after administration of aerosolized antigen (10, 20). The location of the immune response may also determine the prevailing APC type. For example, lymph nodes may rely on activated B cells as the primary APCs, while the spleen may utilize other APCs. Similarly, because L. major resides inside macrophages (32), antigen presentation by B cells may be relatively insignificant. This may explain why the absence of B cells has little effect on Th cell priming or disease outcome. Thus, during an immune response, the site of T-cell activation or the predominant APC type may determine whether B cells are required for efficient immunity.
The duration of the immune response may also determine whether B cells play a significant role in immunity. Antigen presented by B cells can stimulate previously activated Th cells but cannot effectively stimulate naive Th cells (38). It has been suggested that other APCs initiate the immune response in vivo, and B cells serve to shape the late response (37, 38). In support of this concept, the early responses to EAE induction (51) and Plasmodium chabaudi chabaudi infection (22) are normal in the absence of B cells, but the late responses are characterized by impaired Th2 function. Over time, B cells may provide a proliferative advantage for Th2 cells (8) or may be required for maintaining anergy in Th1 cells (43). The results reported here and those of experiments examining responses to S. mansoni (18) demonstrate, however, that in the absence of B cells, chronic Th2 responses can persist for several weeks. Thus, B cells may be required only for switching of an already established Th1 response to a Th2 response.
A practical goal of immune-based therapy is to minimize pathology by influencing the Th response. To this end, targeting antigen to specific APCs has been shown to have some therapeutic value (41). Our results demonstrate that B cells are not required for the polarized clinical outcomes of L. major infection. Therefore, the ability to bias Th responses may be limited by the particular characteristics of the immune response.
ACKNOWLEDGMENTS
This work was supported by NIH grant AI-42370. S.L.R. is a recipient of a Burroughs Wellcome Fund New Investigator Award in Molecular Parasitology.
We are grateful to F. Finkelman for providing mice and to J. Bird, J. Sider, and N. Reilly for technical assistance.
REFERENCES
- 1.Abbas A K, Murphy K M, Sher A. Functional diversity of helper T lymphocytes. Nature. 1996;383:787–793. doi: 10.1038/383787a0. [DOI] [PubMed] [Google Scholar]
- 2.Amigorena S, Bonnerot C. Role of B-cell and Fc receptors in the selection of T-cell epitopes. Curr Opin Immunol. 1998;10:88–92. doi: 10.1016/s0952-7915(98)80037-x. [DOI] [PubMed] [Google Scholar]
- 3.Ben-Sasson S Z, Le Gros G, Conrad D H, Finkelman F D, Paul W E. Cross-linking Fc receptors stimulate splenic non-B, non-T cells to secrete interleukin 4 and other lymphokines. Proc Natl Acad Sci USA. 1990;87:1421–1425. doi: 10.1073/pnas.87.4.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bluestone J A. New perspectives of CD28-B7-mediated T cell costimulation. Immunity. 1995;2:555–559. doi: 10.1016/1074-7613(95)90000-4. [DOI] [PubMed] [Google Scholar]
- 5.Chen J, Trounstine M, Alt F W, Young F, Kurahara C, Loring J F, Huszar D. Immunoglobulin gene rearrangement in B cell deficient mice generated by targeted deletion of the JH locus. Int Immunol. 1993;5:647–656. doi: 10.1093/intimm/5.6.647. [DOI] [PubMed] [Google Scholar]
- 6.Constant S, Pfeiffer C, Woodard A, Pasqualini T, Bottomly K. Extent of T cell receptor ligation can determine the functional differentiation of naive CD4+ T cells. J Exp Med. 1995;182:1591–1596. doi: 10.1084/jem.182.5.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Epstein M M, Di Rosa F, Jankovic D, Sher A, Matzinger P. Successful T cell priming in B cell-deficient mice. J Exp Med. 1995;182:915–922. doi: 10.1084/jem.182.4.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gajewski T F, Pinnas M, Wong T, Fitch F W. Murine Th1 and Th2 clones proliferate optimally in response to distinct antigen-presenting cell populations. J Immunol. 1991;146:1750–1758. [PubMed] [Google Scholar]
- 9.Gordon J. The B lymphocyte-deprived mouse as a tool in immunobiology. Immunol Methods. 1979;25:227–238. doi: 10.1016/0022-1759(79)90110-8. [DOI] [PubMed] [Google Scholar]
- 10.Hamelmann E, Vella A T, Oshiba A, Kappler J W, Marrack P, Gelfand E W. Allergic airway sensitization induces T cell activation but not airway hyperresponsiveness in B cell-deficient mice. Proc Natl Acad Sci USA. 1997;94:1350–1355. doi: 10.1073/pnas.94.4.1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.HayGlass K T, Naides S J, Scott C F, Jr, Benacerraf B, Sy M S. T cell development in B cell-deficient mice. IV. The role of B cells as antigen-presenting cells in vivo. J Immunol. 1986;136:823–829. [PubMed] [Google Scholar]
- 12.Hernandez H J, Wang Y, Stadecker M J. In infection with Schistosoma mansoni, B cells are required for T helper type 2 cell responses but not for granuloma formation. J Immunol. 1997;158:4832–4837. [PubMed] [Google Scholar]
- 13.Hoerauf A, Solbach W, Lohoff M, Rollinghoff M. The Xid defect determines an improved clinical course of murine leishmaniasis in susceptible mice. Int Immunol. 1994;6:1117–1124. doi: 10.1093/intimm/6.8.1117. [DOI] [PubMed] [Google Scholar]
- 14.Hosken N A, Shibuya K, Heath A W, Murphy K M, O’Garra A. The effect of antigen dose on CD4+ T helper cell phenotype development in a T cell receptor-alpha beta-transgenic model. J Exp Med. 1995;182:1579–1584. doi: 10.1084/jem.182.5.1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsieh C S, Heimberger A B, Gold J S, O’Garra A, Murphy K M. Differential regulation of T helper phenotype development by interleukins 4 and 10 in an alpha beta T-cell-receptor transgenic system. Proc Natl Acad Sci USA. 1992;89:6065–6069. doi: 10.1073/pnas.89.13.6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hsieh C S, Macatonia S E, Tripp C S, Wolf S F, O’Garra A, Murphy K M. Development of TH1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science. 1993;260:547–549. doi: 10.1126/science.8097338. [DOI] [PubMed] [Google Scholar]
- 17.Janeway C A, Jr, Ron J, Katz M E. The B cell is the initiating antigen-presenting cell in peripheral lymph nodes. J Immunol. 1987;138:1051–1055. [PubMed] [Google Scholar]
- 18.Jankovic D, Cheever A W, Kullberg M C, Wynn T A, Yap G, Caspar P, Lewis F A, Clynes R, Ravetch J V, Sher A. CD4+ T cell-mediated granulomatous pathology in schistosomiasis is downregulated by a B cell-dependent mechanism requiring Fc receptor signaling. J Exp Med. 1998;187:619–629. doi: 10.1084/jem.187.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kitamura D, Roes J, Kuhn R, Rajewsky K. A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature. 1991;350:423–426. doi: 10.1038/350423a0. [DOI] [PubMed] [Google Scholar]
- 20.Korsgren M, Erjefält J S, Korsgren O, Sundler F, Persson C G. Allergic eosinophil-rich inflammation develops in lungs and airways of B cell-deficient mice. J Exp Med. 1997;185:885–892. doi: 10.1084/jem.185.5.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kurt-Jones E A, Liano D, HayGlass K A, Benacerraf B, Sy M S, Abbas A K. The role of antigen-presenting B cells in T cell priming in vivo. Studies of B cell-deficient mice. J Immunol. 1988;140:3773–3778. [PubMed] [Google Scholar]
- 22.Langhorne J, Cross C, Seixas E, Li C, von der Weid T. A role for B cells in the development of T cell helper function in a malaria infection in mice. Proc Natl Acad Sci USA. 1998;95:1730–1734. doi: 10.1073/pnas.95.4.1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Launois P, Ohteki T, Swihart K, MacDonald H R, Louis J A. In susceptible mice, Leishmania major induce very rapid interleukin-4 production by CD4+ T cells which are NK1.1−. Eur J Immunol. 1995;25:3298–3307. doi: 10.1002/eji.1830251215. [DOI] [PubMed] [Google Scholar]
- 24.Le Gros G, Ben-Sasson S Z, Seder R, Finkelman F D, Paul W E. Generation of interleukin 4 (IL-4)-producing cells in vivo and in vitro: IL-2 and IL-4 are required for in vitro generation of IL-4-producing cells. J Exp Med. 1990;172:921–929. doi: 10.1084/jem.172.3.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu Y, Wu Y, Ramarathinam L, Guo Y, Huszar D, Trounstine M, Zhao M. Gene-targeted B-deficient mice reveal a critical role for B cells in the CD4 T cell response. Int Immunol. 1995;7:1353–1362. doi: 10.1093/intimm/7.8.1353. [DOI] [PubMed] [Google Scholar]
- 26.Macaulay A E, DeKruyff R H, Umetsu D T. Antigen-primed T cells from B cell-deficient JHD mice fail to provide B cell help. J Immunol. 1998;160:1694–1700. [PubMed] [Google Scholar]
- 27.Minoprio P, el Cheikh M C, Murphy E, Hontebeyrie-Joskowicz M, Coffman R, Coutinho A, O’Garra A. Xid-associated resistance to experimental Chagas’ disease is IFN-gamma dependent. J Immunol. 1993;151:4200–4208. [PubMed] [Google Scholar]
- 28.Mosmann T R, Coffman R L. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. 1989;7:145–173. doi: 10.1146/annurev.iy.07.040189.001045. [DOI] [PubMed] [Google Scholar]
- 29.O’Garra A. Cytokines induce the development of functionally heterogeneous T helper cell subsets. Immunity. 1998;8:275–283. doi: 10.1016/s1074-7613(00)80533-6. [DOI] [PubMed] [Google Scholar]
- 30.Pfeiffer C, Stein J, Southwood S, Ketelaar H, Sette A, Bottomly K. Altered peptide ligands can control CD4 T lymphocyte differentiation in vivo. J Exp Med. 1995;181:1569–1574. doi: 10.1084/jem.181.4.1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Phillips J A, Romball C G, Hobbs M V, Ernst D N, Shultz L, Weigle W O. CD4+ T cell activation and tolerance induction in B cell knockout mice. J Exp Med. 1996;183:1339–1344. doi: 10.1084/jem.183.4.1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reiner S L, Locksley R M. The regulation of immunity to Leishmania major. Annu Rev Immunol. 1995;13:151–177. doi: 10.1146/annurev.iy.13.040195.001055. [DOI] [PubMed] [Google Scholar]
- 33.Reiner S L, Zheng S, Corry D B, Locksley R M. Constructing polycompetitor cDNAs for quantitative PCR. J Immunol Methods. 1993;165:37–46. doi: 10.1016/0022-1759(93)90104-f. [DOI] [PubMed] [Google Scholar]
- 34.Reiner S L, Zheng S, Wang Z E, Stowring L, Locksley R M. Leishmania promastigotes evade interleukin 12 (IL-12) induction by macrophages and stimulate a broad range of cytokines from CD4+ T cells during initiation of infection. J Exp Med. 1994;179:447–456. doi: 10.1084/jem.179.2.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ron Y, De Baetselier P, Gordon J, Feldman M, Segal S. Defective induction of antigen-reactive proliferating T cells in B cell-deprived mice. Eur J Immunol. 1981;11:964–968. doi: 10.1002/eji.1830111203. [DOI] [PubMed] [Google Scholar]
- 36.Ron Y, De Baetselier P, Tzehoval E, Gordon J, Feldman M, Segal S. Defective induction of antigen-reactive proliferating T cells in B cell-deprived mice. II. Anti-mu treatment affects the initiation and recruitment of T cells. Eur J Immunol. 1983;13:167–171. doi: 10.1002/eji.1830130214. [DOI] [PubMed] [Google Scholar]
- 37.Ron Y, Sprent J. T cell priming in vivo: a major role for B cells in presenting antigen to T cells in lymph nodes. J Immunol. 1987;138:2848–2856. [PubMed] [Google Scholar]
- 38.Ronchese F, Hausmann B. B lymphocytes in vivo fail to prime naive T cells but can stimulate antigen-experienced T lymphocytes. J Exp Med. 1993;177:679–690. doi: 10.1084/jem.177.3.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ronchese F, Hausmann B, Le Gros G. Interferon-gamma- and interleukin-4-producing T cells can be primed on dendritic cells in vivo and do not require the presence of B cells. Eur J Immunol. 1994;24:1148–1154. doi: 10.1002/eji.1830240521. [DOI] [PubMed] [Google Scholar]
- 40.Sacks D L, Scott P A, Asofsky R, Sher F A. Cutaneous leishmaniasis in anti-IgM-treated mice: enhanced resistance due to functional depletion of a B cell-dependent T cell involved in the suppressor pathway. J Immunol. 1984;132:2072–2077. [PubMed] [Google Scholar]
- 41.Saoudi A, Simmonds S, Huitinga I, Mason D. Prevention of experimental allergic encephalomyelitis in rats by targeting autoantigen to B cells: evidence that the protective mechanism depends on changes in the cytokine response and migratory properties of the autoantigen-specific T cells. J Exp Med. 1995;182:335–344. doi: 10.1084/jem.182.2.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Seder R A, Paul W E, Davis M M, Fazekas de St. Groth B. The presence of interleukin 4 during in vitro priming determines the lymphokine-producing potential of CD4+ T cells from T cell receptor transgenic mice. J Exp Med. 1992;176:1091–1098. doi: 10.1084/jem.176.4.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stark Aroeira L, Williams O, Borlado L R, Carrera A C, Martinez C. Evidence for B cell participation in the in vitro and in vivo maintenance of in vivo staphylococcal enterotoxin B-induced T cell anergy. Int Immunol. 1997;9:65–72. doi: 10.1093/intimm/9.1.65. [DOI] [PubMed] [Google Scholar]
- 44.Stockinger B, Zal T, Zal A, Gray D. B cells solicit their own help from T cells. J Exp Med. 1996;183:891–899. doi: 10.1084/jem.183.3.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sunshine G H, Jimmo B L, Ianelli C, Jarvis L. Strong priming of T cells adoptively transferred into scid mice. J Exp Med. 1991;174:1653–1656. doi: 10.1084/jem.174.6.1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Swain S L, Weinberg A D, English M, Huston G. IL-4 directs the development of Th2-like helper effectors. J Immunol. 1990;145:3796–3806. [PubMed] [Google Scholar]
- 47.Thompson C B. Distinct roles for the costimulatory ligands B7-1 and B7-2 in T helper cell differentiation? Cell. 1995;81:979–982. doi: 10.1016/s0092-8674(05)80001-7. [DOI] [PubMed] [Google Scholar]
- 48.Titus R G, Muller I, Kimsey P, Cerny A, Behin R, Zinkernagel R M, Louis J A. Exacerbation of experimental murine cutaneous leishmaniasis with CD4+Leishmania major-specific T cell lines or clones which secrete interferon-gamma and mediate parasite-specific delayed-type hypersensitivity. Eur J Immunol. 1991;21:559–567. doi: 10.1002/eji.1830210305. [DOI] [PubMed] [Google Scholar]
- 49.van Essen D, Kikutani H, Gray D. CD40 ligand-transduced costimulation of T cells in the development of helper function. Nature. 1995;378:620–623. doi: 10.1038/378620a0. [DOI] [PubMed] [Google Scholar]
- 50.Varkila K, Chatelain R, Leal L M, Coffman R L. Reconstitution of C.B-17 scid mice with BALB/c T cells initiates a T helper type-1 response and renders them capable of healing Leishmania major infection. Eur J Immunol. 1993;23:262–268. doi: 10.1002/eji.1830230141. [DOI] [PubMed] [Google Scholar]
- 51.Wolf S D, Dittel B N, Hardardottir F, Janeway C A., Jr Experimental autoimmune encephalomyelitis induction in genetically B cell-deficient mice. J Exp Med. 1996;184:2271–2278. doi: 10.1084/jem.184.6.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]