

Abstract
CRISPR/Cas9 technology has greatly accelerated genome engineering research. The CRISPR/Cas9 complex, a bacterial immune response system, is widely adopted for RNA-driven targeted genome editing. The systematic mapping study presented in this paper examines the literature on machine learning (ML) techniques employed in the prediction of CRISPR/Cas9 sgRNA on/off-target cleavage, focusing on improving support in sgRNA design activities and identifying areas currently being researched.
This area of research has greatly expanded recently, and we found it appropriate to work on a Systematic Mapping Study (SMS), an investigation that has proven to be an effective secondary study method. Unlike a classic review, in an SMS, no comparison of methods or results is made, while this task can instead be the subject of a systematic literature review that chooses one theme among those highlighted in this SMS. The study is illustrated in this paper. To the best of the authors' knowledge, no other SMS studies have been published on this topic.
Fifty-seven papers published in the period 2017–2022 (April, 30) were analyzed. This study reveals that the most widely used ML model is the convolutional neural network (CNN), followed by the feedforward neural network (FNN), while the use of other models is marginal. Other interesting information has emerged, such as the wide availability of both open code and platforms dedicated to supporting the activity of researchers or the fact that there is a clear prevalence of public funds that finance research on this topic.
Keywords: CRISPR/Cas9, On/Off-target cleavage, Deep learning, Machine learning, sgRNA
1. Introduction
Genetic engineering in several living organisms is increasingly used for treating specific diseases, creating species with particular genetic characteristics, and many other tasks. These objectives can be achieved with various biotechnological techniques, which are generally quite complex. However, the introduction of CRISPR-based techniques, an acronym for clustered regularly interspaced short palindromic repeats, has facilitated genome editing with specific objectives. The first clues about the CRISPR system were noticed in bacteria and archaea in the late eighties. A group of DNA segments was discovered containing short palindromic repeated sequences separated by DNA fragments (spacers). These spacers appear after cellular infection by viruses, and form an adaptive immune system, because after a viral infection, viral DNA is embedded into the CRISPR site in the form of spacers. This recurrent cluster is associated with a set of genes called CRISPR-Cas genes that transcribe CRISPR-associated system proteins.
In a possible subsequent viral attack, part of the CRISPR locus is transcribed into crRNA (CRISPR RNA), which joins a transactivating RNA (tracrRNA). These sequences bind to a Cas9 protein and guide it to the DNA target site of the virus. The Cas9 protein inactivates the virus by unwinding its DNA and performing a double-stranded cut.
The sequence targeted by Cas9 is closed with a n-base pair sequence (n = 2–6) called PAM (protospacer adjacent motif), which is part of the attacker virus DNA but not of the CRISPR locus; Cas9 will avoid binding to a target sequence if it is not followed by PAM. This way it will prevent cutting the CRISPR locus itself.
Following the study of Jinek et al. [1], Doudna and Charpentier [2] redesigned the union of CRISPR-RNA and transactivating RNA by creating single-RNA sequences called single guide RNAs (sgRNAs). These guides, when linked to Cas9, identify and cut the target DNA specified by the sgRNA. By manipulating the sequence, the Cas9 system can identify and cut any DNA sequence. The CRISPR/Cas9 system thus has become a powerful genome editing technique: its activity is based on a 23-base pair (bp) sequence, of which the last 3 bp make up the PAM sequence, to perform recognition and knockout.
Proper development of a sgRNA is extremely delicate because not all of them are equally effective. The efficiency of sgRNA depends on characteristics such as the target site, the properties of the endonuclease and the profile of the sequence itself. Furthermore, the cell tends to repair the cut DNA, possibly resulting in more or less severe mutations. The prediction of DNA cleavage efficiency together with its effects and mutations (off-target profile or effects) plays a fundamental role in the design of sgRNAs. Furthermore, it would be of great help for researchers to obtain the parameters of a sequence performing genetic modification in silico, saving valuable resources by performing only a limited number of in vitro experiments.
Software systems are therefore of particular interest, including those that use artificial intelligence techniques, to automatically extract and learn the characteristics and determinants of the sequence and predict the cleavage efficiency on the target.
The aim of this study is the analysis of the literature related to the ML techniques employed in the prediction of CRISPR/Cas9 sgRNA on/off-target cleavage by means of a SMS, considered as one of the optimal approaches, since it defines an accurate process for data retrieval and interpretation [3].
This document has the following structure: Section 2 underlines the need for a SMS; Section 3 shows the method implemented in this study to select/classify documents; Section 4 reports the results of the analysis Section 5 discloses and briefly discuss the answers to research questions and finally, Section 6 summarizes conclusions, together with possible future research directions.
2. The reason for a systematic mapping study
Systematic investigations have proven to be a secondary study method over the past two decades, evolving from research based on evidence-based primary studies. Recently, the concept of systematic investigation has been adopted in various fields, such as software engineering [4], [5], [6], [7], education [8], [9], digital clinical diagnosis and human care [10], [11]. The lack of studies that collect and summarize the results of the primary empirical studies was addressed by Pickard as early as 1998 [12]. The proposal of combining the results of primary studies by means of meta-analysis and the idea of research synthesis were addressed by Miller and Hayes, respectively [13], [14]. Basili's work in the field of software engineering [15], is the first attempt to create knowledge by synthesizing primary studies.
Bioinformatics is a methodological approach that supports more than one discipline from medicine to engineering to information technology; research that provides results based on empirical evidence can benefit these disciplines. In the field of bioinformatics, ML techniques employed in the prediction of CRISPR/Cas9 sgRNA on/off-target cleavage focusing on improving support in sgRNA design are attracting the interest of researchers.
As this area of research has expanded recently, the authors found it appropriate to work on an SMS. Research communities could benefit from the results of this study, and to the best of the authors' knowledge, no other SMS messages have been sent on the subject.
Aimed to a wide overview of this research area, this work used the SMS approach described in [3] to collect data and interpret results on research/development alignment, scope of interest, and empirical evidence collected in the literature. The results of this SMS can identify areas suitable for conducting systematic literature reviews (SLR), and areas where a primary study is more appropriate. An SLR is a means of interpreting available and empirically relevant research on a particular research question or phenomenon of interest [3], [6].
The SMS is illustrated, and if risks (or biases) are detected by other investigators in this study, they can vary the SMS process in such a way that the risk can be mitigated, the results strengthened, and the review window or objectives changed/extended.
3. Research method
The research process adopted here follows the guidelines proposed by Kitchenham [3] to perform the SMS.
3.1. Research questions
The SMS presented here uses the population, intervention, output (PIO) paradigm described in [3]: the use of a “comparison” factor is also proposed there, but this factor is not taken into consideration here since documents are partially scanned in the SMS. The components of the PIO paradigm, for the purposes of this study, are defined below:
-
-
Population: researchers, biotechnologists, doctors, institutions, pharmaceutical companies;
-
-
Intervention: machine learning/deep learning techniques to design CRISPR/Cas9 sgRNA and predict target cleavage;
-
-
Output: all of the benefits that lead to better CRISPR/Cas9 sgRNA on-target cleavage, clinical applications, gene editing applications, and investigation of RNA functions.
Taking into consideration what is suggested by Arksey et al. [16] and in accordance with the PIO paradigm defined by the authors, the general questions of the SMS have been structured to make them correspond to the dimensions we intend to investigate in this mapping study. Table 1 lists the research questions (RQ).
Table 1.
Research questions.
| ID | Question | Reason |
|---|---|---|
| RQ1 | What is the temporal and geographical distribution of the research? | Understand how documents develop over time and how they are distributed among countries interested in genome editing, specifically on deep learning/machine learning techniques to predict CRISPR/Cas9 sgRNA target cleavage |
| RQ2 | Which stakeholders do the documents refer to? | Identify specific stakeholders interested in this research |
| RQ3 | What are the ML models of interest? | Discern the themes that stimulate researchers, listing the use of the most used models. Identify possible techniques that are considered to be most effective by researchers. |
| RQ4 | What are the most frequent research objectives? | Understand the objectives to which the researchers aim, in particular with reference to the target on/off. |
| RQ5 | What types of cells are used in the studies? | Identify the datasets used in the documents, to understand the species involved and the availability of the data. |
| RQ6 | What results are presented in the papers supporting the research? | Understand if the solutions developed by the researchers are working and if they are freely offered to research. |
| RQ7 | Financing? | Investigate which lenders are most interested in this type of research, which indirectly also helps to identify any stakeholders. |
3.2. Search protocol
3.2.1. Search string
A well-constructed search string ensures that we automatically extract a good sample of literature papers relevant to our study. To construct the string, it is necessary to identify some terms commonly used in the literature relating to the principles of the abovementioned PIO. These words are collected through the validation process described in Section 3.2.2; the words are the following:
-
-
Population: researcher, doctor, hospital, drug company, medicine, CRISPR/Cas9, sgRNA, on-target, off-target, cleavage, genome engineering, genome editing, knockout efficacy;
-
-
Intervention: machine learning, deep learning;
-
-
Output: prediction of CRISPR/Cas9 sgRNA on-target and off-target cleavage, gRNA design, enhancing CRISPR–Cas9 gRNA efficiency, accurate prediction, identification of sgRNA sites, interactive gRNA design webserver, cloud-based service.
The search strings used in 3.2.2 were constructed with the above words, linked with “OR” or “AND”.
3.2.2. Search strategy, string generation, and validation
A search string should be designed before searching for the overall timeframe elected to ensure that possibly most of the representative studies of the literature on deep/machine learning techniques used to predict CRISPR/Cas9 sgRNA on/off-target cleavage are extracted from automated search. To design the search string, the method recommended by Zhang [17] was followed, taking into consideration the number of significant studies found on the topic under investigation, automatically extracted, and manually extracted significant studies.
A 12-months publication period has been chosen for the validation of the search string [17] runs from May 2021 to April 2022. First, we conducted a manual search based on the snowball technique; this technique of searching for relevant literature guarantees good completeness of the automatic search. References were selected and collected from each eligible paper, and then, focusing on these references we found further relevant studies on the topic of this SMS. Then, the articles that were cited in each paper of interest were also taken into consideration. Specifically, the most significant papers in the period May 2021 – Apr. 2022 were searched from specialized sources, such as the following:
-
·
Nature biotechnology
-
·
Nucleic acid research
-
·
Nature communications
-
·
Bioinformatics
In that period, three relevant papers were discovered [18], [19], [75], and the references included in those papers were analyzed and collected, thus increasing the group of relevant papers on this topic. Then, we made different assemblies of the search strings that contained the most interesting keywords; as chosen by the authors, a search was conducted by using well-known repositories, with the following scheme:
-
·Scopus (https://www.scopus.com/) considering the following filters:
-
oSearch within: Article title, Abstract, Keywords.
-
oYear: 2017–2022.
-
oArticle.
-
oJournal.
-
oEnglish.
-
o
-
·WoS (https://www.webofscience.com) considering the following filters:
-
oSearch within: ALL.
-
oYear: 2017–2022.
-
oArticle.
-
oNOT Document Types: Proceedings Papers.
-
oEnglish.
-
o
A different quantity of papers was extracted, with the aim of verifying the string validation described in [17]. The final search string used is:
-
-
Scopus: ((Machine learning) OR (Deep learning)) AND (CRISPR) AND ((cas9) OR (sgrna))
-
-
WoS: (ALL=(“machine learning” or “Deep learning”)) AND ALL=(“CRISPR”) AND (ALL=(“Cas9″) OR ALL=(”sgrna“))
Despite the numerous words included in the PIO, the search string that gave a satisfactory result is slim. The opinion of the authors is that many authors use many keywords and concepts, even quite different from each other, while addressing the same themes. Therefore, a more comprehensive string made it possible to find as many relevant papers as possible. In any case, for the purpose of this study the goal of the collection is achieved.
With the above strings, the search for papers in the two Scopus and WoS databases, using the same filters indicated, for the entire research period defined by the authors, i.e., January 2017 – April 2022 produced the search results shown in Table 2. The documents extracted from Scopus and WoS were compared to eliminate duplicates, finally obtaining 210 unique documents.
Table 2.
Number of documents found in Scopus/WoS.
| Source | # papers retrieved |
|---|---|
| Scopus | 176 |
| WoS | 94 |
| Unique papers | 210 |
3.2.3. Random assignment of articles to reviewing authors and screening
The role of a reviewer was assumed by the authors who evaluated all papers, primarily to decide on inclusion or exclusion and, for the included papers, analyze the content according to the rules of the SMS. For each of the 210 articles to be guaranteed three revisions, the papers were divided into 3 groups of 105 papers (210 * 3 = 630 = 105 * 6). The eligibility of each study and therefore its inclusion or exclusion was decided through a screening process: as stated above, each paper has undergone three reviews from three reviewers; each reviewer decided whether to include or not the study; and subsequently, the reviewers had a short discussion to reach an agreement and if an agreement was not reached, the acceptance or exclusion of the document was decided by majority.
As indicated above, articles published between January 2017 and April 2022 were taken into consideration. The publication period can be chosen as an arbitrary parameter [3]: we consider a period of approximately 5 years to be adequate for the purpose of this study. The document quality was not considered to be a critical factor, because all documents can provide useful information for the SMS. In any case, we have adopted rather stringent criteria, i.e., inclusion only of papers in journals, only in English, and only in the 'article' category. Furthermore, having used the Scopus and WoS databases, to a certain extent already guarantees the exclusion of documents of dubious quality level. Table 3 reports the quantity of the selected and included publications.
Table 3.
Selected documents.
| Source | Number of publications |
|||
|---|---|---|---|---|
| Retrieved | Excluded | Included | Included (%) | |
| Digital libraries | 210 | 153 | 57 | 27.14 |
From each document included, we then extracted phrases and concepts that could be useful to set up classification schemes and map them in the research questions.
The reviewers extracted words from the title and abstracts. Where the abstract was found to be insufficient, the introduction and possibly also the conclusions were analyzed. An article would have been excluded if a reviewer could not extract any useful words to classify the document and answer the RQs, but this circumstance did not occur. A total of 57 documents were classified [18], [19], [24], [25], [26], [27], [28], [29], [30], [31], [32], [33], [34], [35], [36], [37], [38], [39], [40], [41], [42], [43], [44], [45], [46], [47], [48], [49], [50], [51], [52], [53], [54], [55], [56], [57], [58], [59], [60], [61], [62], [63], [64], [65], [66], [67], [68], [69], [70], [71], [72], [73], [74], [75], [76], [77], [78].
4. Results
This section presents the results obtained during the screening process useful to answer the 7 RQs and briefly argue the results that relate to each “dimension”.
4.1. Time and geographic distribution of publications
Fig. 1 shows the number of documents published annually and how they varied from 2017 to 2022 (April, 30). It is evident that interest in studying the topic discussed in this paper is growing. Given the young age of CRISPR/Cas9 technology combined with machine learning techniques, a positive evolution is expected in the coming years; however, this aspect is also discussed later. Fig. 2 shows the geographical distribution of the documents. In the case of articles with authors from multiple countries, the majority of the authors was taken into consideration; in the case in which there is no evident majority among the nationalities, the document was classified as belonging to the country of the first author. The 57 selected studies were conducted in only 13 countries.
Fig. 1.

Distribution of documents based on years.
Fig. 2.
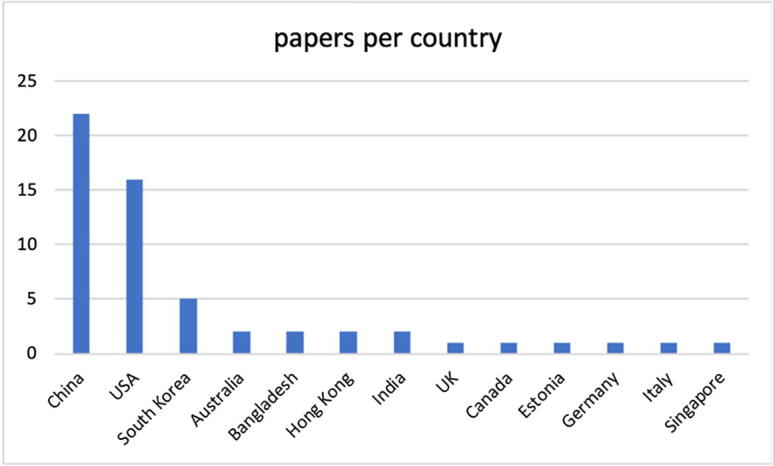
Most productive countries in the SMS topic.
The most active countries on the subject appear to be China and the USA, with 22 and 16 documents, respectively, and many studies classified as “USA” in the research teams include researchers working at other universities, including Chinese. This result could be due to many factors that are difficult to determine, with one of the many factors being China's strong investment in artificial intelligence techniques in recent years as well as a growing interest in biotechnologies and specifically in CRISPR techniques. The latter is a promising technology, and it is almost certain that it will represent the future of biotechnology. The scope of this technique is as relevant to the future of biotechnology as machine learning is to the future of the IT industry, and not only. The most industrialized countries are aware of this circumstance, and have wasted no time in promoting, through a government program, the facilitation of the vast applications of genomics and other biotechnologies and the large-scale development of personalized medical treatments, new drugs, and next-generation biotechnology products and services. Parallel to what is occurring for biotechnologies, China has quickly caught up with the United States in the field of artificial intelligence (e.g., machine learning), which is proving to be an excellent enabling technology. Europe appears to be far behind on the topic of this SMS.
4.2. Stakeholder
In general, the systems described in the documents seek to optimize the design of sgRNAs by maximizing their activity on the target and minimizing their potential off-target mutations. This field of study is focused on a very specific objective that can find extremely interesting applications in a myriad of fields of science. CRISPR/Cas9 is a revolutionary gene-editing technology that can be widely utilized in biology, biotechnology and medicine. The use of machine learning techniques or, more specifically, deep learning techniques could provide useful guidelines for selecting effective target sites and assist users in designing more efficient gRNAs. The analysis of the selected studies therefore does not provide significant specific indications on the objective of each of them, since CRISPR/Cas9 is a basic genetic technique. We can share the opinion expressed by the authors in some documents, for example, the reference to practical uses of this system for clinical applications such as cancer modeling [20], treatment of HIV [21], or other gene editing applications, including the defining mechanisms of neurodegenerative diseases [22]. These are of course only a few examples. For the benefit of the readers, we report that interesting application perspectives for the technique have been outlined since 2014 in [23]. Essentially, identification of single-guide RNA activity is critical for theoretical research, such as investigation of RNA functions, and new applications in the genome editing and synthetic biology fields.
4.3. Machine learning techniques
Fig. 3 and Table 4 show the machine learning techniques most commonly used by the scientific community in the relevant documents found.
Fig. 3.

Most used machine learning models: CNN (convolutional neural networks), FNN (feedforward neural networks), BiLSTM (bidirectional long short-term memory), RNN (recurrent neural network), LRCN (long-term recurrent convolutional networks), GCN (graph convolutional networks).
Table 4.
Hybrid Models: DCDNN (deep convolutional denoising neural network), BGRU (bidirectional gated recurrent unit), SVR (support vector regression).
| Hybrid models | n. |
|---|---|
| BiLSTM + CNN | 2 |
| CNN-XGboost | 1 |
| DCDNN + CNN | 1 |
| CNN + LSTM | 1 |
| CNN + LRCN | 1 |
| CNN + RNN | 1 |
| CNN + BGRU | 1 |
| CNN + SVR | 1 |
| Random Forest + SVM | 1 |
| SVM + XGBoost + Linear regression | 1 |
As shown in Fig. 3, the most commonly used model is the CNN (convolutional neural network). Most documents use this model for learning and inference, and this result is most likely due to the model's ability to automatically extract the input features necessary for training, organized in matrices (e.g., images). In digital images, pixel values are organized in a two-dimensional array of numbers. A small set of parameters, an optimizable feature extractor, is applied at each image zone, which makes CNNs highly efficient for image analysis since a feature can occur anywhere in the image. The process of defining optimal parameters such as kernels is known as training. This group of neural networks is becoming dominant in many computer vision tasks and interesting across a variety of domains, including biology. In many cases, as in [46], an attempt was made to organize data into matrices assimilated to images, thus making it possible to adopt this powerful deep learning model. In 8 studies, FNN networks were used and were often also used as a comparison model. A single document adopts an RNN network that shows the ability to process long and entire sequences such as genetic sequences. Other models and techniques used in the documents are LSTM-bidirectional, an extension of traditional LSTM networks that improve their performance; LRCN (long-term recurrent convolutional network), consisting of an LSTM as an input model and a CNN as an output model; and GCN (graph convolutional network), a neural network architecture used in machine learning on graphs. Other studies adopt hybrid models by combining the basic models listed above. These models, indicated in Table 4, do not rely solely on a single machine learning model (such as a CNN, an FNN or a random forest), but are composite models formed by two or more basic models. It is interesting to note that the majority of the hybrid models identified still use a convolutional neural network. In fact, 9 out of 11 models are composed of a CNN joint with another model, and only two models have in common the use of an SVM (support vector machine).
Fig. 4 shows further ML models, which in the analyzed documents are mainly used for a comparison of the obtained performances with proposed models.
Fig. 4.

Popular machine learning models used mainly for comparison.
5. Research objectives
The objective of the studies analyzed is the result that is given by the solution proposed and discussed. Because it is easy to guess from the terms themselves, the papers that we indicate with the objective 'on-target site prediction' are those that develop models that aim to provide a prediction of the on-target cleavage efficiency, while those indicated as 'off site prediction – target' develop models that aim to provide a prediction of the off-target cleavage efficiency. Some papers study techniques for predicting the cleavage efficiency at both sites. Fig. 5 shows that more than 50 % of the models designed, built and described in the documents aim to predict the on-target cleavage efficiency and approximately 30 % the off-target cleavage efficiency; only 10 documents, describe ML models that provide an efficiency prediction at both sites. On the sidelines, from the review of the documents, it emerged that authors mainly propose the creation of new models or experiments on different sets of data or different arrangement of the features.
Fig. 5.

Objective of the models presented in the studies.
5.1. Cells used in the experiment
In Fig. 6 below, a pie chart is shown that summarizes the cell types that are included in the datasets used for the studies. The majority uses human cells (70 %), while 9 % used datasets containing human and mouse cells; the minority uses datasets that contain cells of the following species: Zebrafish, Ciona, Caenorhabditis elegans, Ascidians, Drosophila, Mouse, or bacterial cell lines and plant cell lines.
Fig. 6.

Cell types used in studies.
5.2. Resources provided by the authors
In Table 5, we can observe that 63 % (36) of the studies provide a link to a GitHub or Code Ocean repository, containing the source code of the models proposed, while 14 % (8) of the research groups offer both the link to a repository containing the software designed and an online service to support researchers in sgRNA design by providing an estimate of the on/off-target cleavage efficiency. In 10 % (6) of the cases, only the public online platform was made available, and few of the studies did not share online functions or source code of the models developed and illustrated. See Appendixes 2 and 3.
Table 5.
Services or software made available.
| Resources provided | Papers |
|---|---|
| just the code | 36 |
| web server | 6 |
| code + webserver | 8 |
| nothing | 7 |
5.3. Trends
Table 6 shows a couple of cases in multiple dimensions to highlight some trends in the selected literature, specifically the temporal distribution of the two models that have been the most trusted by researchers over time. From the graph, it can be seen that in 2019 and 2021, studies were published that were mainly based on CNN experimentation. However, CNN is used throughout the period under observation, and considering the partial figure of 2022, confidence in CNNs appears to be growing. Among the selected documents, FNNs appeared in 2018, and they continue to attract marginal interest. All other models, at least up to the date of our research, appear to be the basis of occasional experiments, and they are understood to be attempts to identify more effective models.
Table 6.
ML models per year.
| Models | 2017 | 2018 | 2019 | 2020 | 2021 | 2022 |
|---|---|---|---|---|---|---|
| CNN | 1 | 2 | 5 | 4 | 9 | 3 |
| FNN | 0 | 3 | 1 | 4 | 1 | 0 |
5.4. Financing
The graph in Fig. 7 is intended to highlight the interests of the lenders. There is no doubt that today, the main interest is in the public sphere. This circumstance is not very surprising, because this area is basic research with a rather uncertain future, and thus, private lenders are still watching, perhaps with extreme caution. The problems that relate to the lack of explainability of machine learning models, the effectiveness of prediction, and the impossibility of operating on the genome with a significant margin of uncertainty are issues that still require a large amount of effort from the scientific community. In essence, this research, albeit fascinating, does not appear to be attractive at the moment for those who want to transform it into business.
Fig. 7.

Financing.
6. Discussion
As already mentioned, the purpose of this work is to analyze how research is developing in the context of the ML techniques used in the prediction of CRISPR/Cas9 sgRNA on/off-target cleavage. Unlike a classic review, in an SMS, no comparison of methods or results is made, and no suggestion is given about the use of one method or technique rather than another. These themes can instead be the subject of a systematic literature review that chooses one theme among those highlighted in this SMS, for example, the comparison of studies using the CNN. This study which can be conducted later. The analysis conducted and the results found allow us to partially answer the RQs listed in Table 1.
(RQ1) Section 4.1, Fig. 1, shows the temporal distribution of documents: it should be noted that from 2017 to 2021, the number of documents increased significantly. Some papers published at the beginning of 2022, may not be available on the date of the search. It can be observed that in 2022 (January-April), 3 articles have been published, which is a value that is comparable to those of previous years, and thus, it is expected that at the end of 2022, the number of documents could align with previous years or possibly increase. It should be considered that other articles could actually be published, at least online, by their respective publishers, but they are not yet on Scopus/WoS due to the delay in the inclusion times of the papers in those repositories, which on average are 8 weeks but can arrive even after several months, when the papers have not yet been effectively assigned an issue. With reference to the geographic distribution of the studies, the two nations that are most likely farther ahead in both artificial intelligence and biotechnology techniques are the USA and China; therefore, the result that we proposed in Fig. 2 confirms this.
(RQ2) We do not think that we can give an actually useful answer about the specific stakeholders interested in the research indicated in the selected papers. In Section 4.2, we have provided our interpretation, and here, we can conclude that identification of single-guide RNA activity is critical for theoretical research, such as investigation of RNA functions, and new applications in the genome editing and synthetic biology fields and that the adoption of machine learning techniques or, more specifically, CNN techniques could provide useful support for selecting effective target sites and assisting biotechnologists in designing efficient gRNAs.
(RQ3) Section 4.3, by means of Fig. 3 and Table 4, highlights that the most used technique is the CNN, which is an interesting solution for the analysis of genomic sequences due to the ability to perform automatic and 'parallel' extraction of features. In addition, 11 documents present projects and realizations of hybrid solutions, but 9 of these hybrid solutions still employ a CNN. To underline the advantage of using a CNN, many authors compare this technique with many others, the first being random forest. A more precise indication can be deduced from Table 4. Furthermore, in Section 4.7 in Table 6, the 2 machine learning models most used in the documents are related to their respective temporal trends.
(RQ4) In Section 4.4 in Fig. 5, it is noted how in most of the studies, models based on ML or hybrid are designed that predict the on-target site.
(RQ5) Section 4.5 in Fig. 6 shows how most of the datasets used in the documents refer to human cells. Only two studies tend to generalize and use mixed datasets, making use of both human and other species cells, in which five research groups employ human and mouse cell lines, and the remainder employ datasets that contain cell lines from a single species, such as bacteria, plants or mouse.
(RQ6) Section 4.6 in Table 5 shows the solutions offered to support researchers; surprisingly, in a positive sense, source code is made available in most documents. Other authors, on the other hand, offer a more sophisticated solution, i.e., a free online platform made available to other research groups or different stakeholders.
(RQ7) Section 4.8 in Fig. 7 shows the distribution on the origin of the funds declared by the authors. Generally, as it should be, there is no mention of the amount of economic effort; in several cases, part of the funds, or the fund offered by 'other' than public financiers, is dedicated to paying scholarships for researchers. Up to date, the presence of typical lenders in the medical/biological/pharmaceutical sector, i.e., industrial companies, specifically pharmaceutical companies, is not significant.
In the Appendix 1 we have introduced a description of the CNN approach, which we believe is useful to enable the reader to immediately understand the issues to be addressed and the benefits that can be obtained. We chose the CNN model because, as reported above, it is the most chosen model and of growing interest to address the research in prediction of CRISPR/Cas9 sgRNA target cleavage. The researcher who is interested will then have to deepen the literature that we have identified in the study and will be able to try his hand at some experiments using the large amount of code and on-line services publicly available.
7. Conclusions
The Cas9 nucleases are proving to be valid tools for genome editing. Their broad applications are slowed by the lack of knowledge of the rules governing guide RNA activity. A key prerequisite for CRISPR/Cas9 success is its ability to distinguish between on-target single guide RNA and off-target homologous sites. Therefore, rigorous design of sgRNAs that maximize their on-target activity and minimize their potential flaws are crucial concerns for this technique. Early in 2015, the importance of eliminating errors was emphasized, and in [79], it was highlighted that the margins of error in the use of CRISPR were still too high to be able to apply the technique (in that case on embryos) safely.
In recent years, numerous documents have been published that aim to create classic or hybrid ML models that aim to predict the on/off-target site with the least possible margin of error. In many cases, such utilities are made available to the research world in the form of free online services or source code.
The aim of this paper was to present a systematic mapping study that summarizes the existing knowledge on the models studied by numerous research groups. From an initial series of 210 scientific articles, 57 were selected, those that are most relevant to this study. These articles allowed the analysis, discussion of the results, and answer to the questions posed in the study.
The results showed that the most used model is CNN. Some research seeks to improve the performance of CNNs by designing hybrid models, which are in most cases composed of CNN with LSTM, biLSTM or RNN. CNNs have achieved excellent results across a variety of domains, recently including biology, while an increasing interest has emerged in biotechnology, specifically for the prediction of sgRNA target cleavage efficacy. Despite it is becoming a widely used technique in a variety of highly complex tasks, such as image classification and object detection, it is not the famous Columbus egg. Being familiar with key concepts and advantages of deep learning as well as its limitations is essential to leverage it in biotechnology research with the goal of improving CRISPR performance and, eventually, new concerns.
The models designed in the classified documents aim for the most part to alternatively estimate on-target or off-target sites. The models were trained with datasets referenced by a specific cell type. This approach makes the algorithms incompatible with other cell types and species; thus, even if they perform well, it is still unclear how effectively these models are able to generalize.
In the opinion of the authors, the primary objectives of the research on this topic remain the reduction of the margin of error and the explainability of the models used. Second, a possible research direction is to design and implement systems that have a greater generalization capacity, to make them easier for researchers to use.
Despite its development and positive applications in medicine and biology, there are several ethical concerns about CRISPR genome editing technology. CRISPR technology has shown technical limitations such as mosaicism effect, highly variable efficiency, and lacking accuracy as reported in experiments on animals and human cell lines. Moreover, while it is greatly simple to edit a genome, the duration and the effects are still unknown especially when the edited genes are transferred to the next generations. Further uncertainty resides in the influence of such modifications on complex biological traits, thus highlighting other potential risks. The SMS we present here could be helpful in improving the efficiency of CRISPR genome editing technology partially addressing the concerns raised by the use of this technology.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix 1. Why CNN for the prediction of CRISPR/Cas9 sgRNA target cleavage
The sgRNAs designed to cut a target DNA are not equally effective, while their efficiency depends on the features like the target site, the properties of the endonuclease, and the design of the sequence. Furthermore, when DNA gets cut, the cell tries to repair it, leading to possible mutations. Predicting efficiency in cutting DNAs and its side effects and mutations is an important task in sgRNA design. To refine sgRNA design task, various efficiency prediction systems have been developed, for example, locating PAM sequence (CasFinder [80]), scoring efficiencies empirically based on sequence key features (CHOPCHOP [81]), or predicting them with training models (sgRNA designer [82], sgRNA scorer [24], [83], SSC [84], CRISPRscan [85]). Prediction systems based on Convolutional Neural Networks are becoming competitive both in predicting on-target and off-target efficiencies. Convolutional Neural Networks have attracted attention in computational biology because they excel at pattern recognition, can detect and process important parts of an image input, can process raw sequences, without manual feature engineering, which can expedite model creation. Here we cite a couple of examples to illustrate some of the techniques that are becoming widespread.
A CNN approach to predict both off-target and on-target efficiencies is described in Chuai [29]. In this system, called DeepCRISPR, the sequence is encoded into a one-hot matrix, composed of 4 rows, one for each nucleobase, and 23 columns, one for each nucleobase in a 23-bp sgRNA sequence. The matrix gets augmented with additional rows corresponding to epigenetic features, to build a generalized model. This matrix then gets passed as image input to a CNN, which is able to use both linear regression and classification to predict efficiencies. In the first case, the predicted value is a real value, while in the second case a class is predicted (0 low efficiency, 1 high efficiency). Fig. A shows an example of one-hot encoding of a sequence.
Fig. A.

One-hot encoding of a sequence.
The system developed by Xue [36], called DeepCas9, uses a CNN too. A sequence up to 30-bp is encoded into a one-hot matrix using the same one-hot encoding scheme used in Fig. A. Also, in [36] the obtained matrix gets passed as input to the CNN that uses linear regression to predict efficiency represented by a real value. The above examples use a similar encoding mechanism to transform each sgRNA sequence into a data format suitable for the CNN. In fact, CNNs take as input a matrix of values, corresponding to the pixel matrix of an image. In this way, it is possible to take advantage of CNN's ability to work with raw data encoded in the form of images.
Several models have been developed so far, to which we refer in the references, with comparable performance, while recently Xiang et Al [19] argued that prediction improvement can be achieved with large high-quality datasets rather than working on the model. For a neural network to acquire the ability to provide a useful result, it must be trained. Network training also uses coded sequences as images: to make the training effective it is necessary to have generously sized datasets and high-performance processing systems. As an example, Xue in [36] used different sgRNA efficiency datasets covering several cell species types, some of these are:
-
•
Chari dataset [83], consisting in 1234 guides targeting Human 293T cells.
-
•
Wang dataset [84], consisting in 2076 guides targeting 221 genes in Human HL-60 cells.
-
•
Doench dataset [82], consisting in 2333 guides targeting CCDC101, MED12, TADA2B, TADA1, HPRT, CUL3, NF1, and NF2 genes from Human A375 cells.
-
•
Hart dataset [87], consisting in 4239 guides targeting 829 genes in Human Hct116 cells.
-
•
Moreno-Mateos dataset [85], consisting in 1020 guides targeting 128 genes in Zebrafish genome.
Furthermore, many datasets were aggregated in [86], creating a dataset of 31,625 sgRNAs. Recently Xiang et Al [19] report on the generation of on-target gRNA activity data for 10,592 gRNAs: integrating these with complementary published data, they train a deep learning model, called CRISPRon, on 23,902 gRNAs.
When large datasets are not available (for example for a specific species) typically the results obtained by using a CNN model are unsatisfactory. Modifying the CNN architecture or changing some of its hyperparameters do not improve model performances. In this case, it can be adopted a data augmentation technique. Augmenting data has proved to be a key step in improving CNN performances. An interesting example is reported in [29].
Many systems have been developed using Python language, because of its simplicity and popularity compared to other programming languages. An example of an integrated development environment used was Pycharm, in combination with VSCode. Also, Git version control system was used, in combination with GitHub. To develop the core of the system, the convolutional neural network behind the prediction task, Tensorflow has been mostly used, including Keras. Keras uses a data structure to represent the way that neural network layers are organized. Other libraries used are Scipy, an open library dedicated to scientific computing, NumPy, a library for scientific calculation that provides many functions for operations between matrices, Scikit-learn, a library for machine learning supporting algorithms and Python default libraries for miscellaneous purposes. Interesting examples of model flow for the CNN models can be found in Supplementary Fig. 13 in [19], in Table 2 in [46], in Fig. 3 in [72], in Fig. 1 in [71], in Fig. 10 in [69] and in Fig. 1 in [70].
Appendix 2. Support platforms for researchers (links)
Last accessed October 15, 2022.
[19] https://rth.dk/resources/crispr/.
[26] https://crista.tau.ac.il/.
[27] https://crispr.ml.
[29] https://www.deepcrispr.net/(*).
[35] https://www.crisprindelphi.design/.
[37] https://bioinfolab.miamioh.edu/CRISPR-DT (*).
[42] https://www.DeepHF.com/.
[45] https://deepcrispr.info/DeepSpCas9.
[49] https://deepcrispr.info/DeepxCas9, https://deepcrispr.info/DeepSpCas9-NG.
[50] https://crisprdb.org/wu-crispr/.
[51] https://bliulab.net/sgRNA-PSM/.
[54] https://big.hanyang.ac.kr:2195/CGD.
[55] https://web.iitd.ac.in/crispcut/off-targets/(*).
[70] https://www.innovebioinfo.com/Sequencing_Analysis/RNAediting/RNA1.php.
(*) website was down on October 15, 2022.
Appendix 3. Open code (links)
Last accessed October 15, 2022.
[18] https://github.com/vli31/CROTON .
[19] https://github.com/RTH-tools/crispron/.
[25] https://github.com/khaled-buet/CRISPRpred.
[27] https://www.microsoft.com/en-us/research/project/crispr.
[28] https://github.com/BauerLab/TUSCAN.
[30] https://github.com/zhangchonglab/sgRNA-cleavage-activity-prediction.git.
[31] https://github.com/yuuuuzhang/dl-CRISPR_offtarget_prediction.
[32] https://github.com/MichaelLinn/off_target_prediction.
[33] https://github.com/penn-hui/OfftargetPredict.
[34] gitlab.com/bauerlab/crispro.
[35] https://www.github.com/gifford-lab/inDelphi-dataprocessinganalysis.
[36] https://github.com/lje00006/DeepCas9.
[38] https://github.com/luslab/crispr-indels.
[40] https://github.com/BauerLab/VARSCOT.
[41] https://www.github.com/czbiohub/Primer3Wrapper.
[42] https://github.com/izhangcd/DeepHF.
[43] https://github.com/biomedBit/DeepSgrnaBacteria.
[44] https://github.com/qiaoliuhub/AttnToCrispr.
[45] https://github.com/MyungjaeSong/Paired-Library, https://github.com/CRISPRJWCHOI/BaseEditing_tool.
[47] https://github.com/TerminatorJ/GNL_Scorer.
[50] https://github.com/wang-lab/sgDesigner.
[52] https://github.com/LQYoLH/CnnCrispr.
[53] https://github.com/Peppags/C_RNNCrispr.
[54] https://github.com/vipinmenon1989/CGD.
[56] https://github.com/Rafid013/CRISPRpredSEQ.
[57] https://deepcrispr.info/DeepSpCas9variants.
[58] https://github.com/tsailabSJ/changeseq, https://github.com/aryeelab/guideseq.
[59] https://github.com/MyungjaeSong/Paired-Library.
[60] https://codeocean.com/capsule/9553651/tree/v1.
[61] https://github.com/jingry/autoBioSeqpy.
[62] https://github.com/wangyi-fudan/SeqCor.
[63] https://github.com/nmt315320/sgRNACNN.git.
[64] https://github.com/gifford-lab/skipguide-analysis.
[66] https://github.com/Peppags/CRISPRont-CRISPRofft.
[67] https://github.com/kundajelab/PREUSS.
[69] https://github.com/BioinfoVirgo/CRISPR-IP.
[70] https://github.com/wjd198605/EditPredict.
[71] https://github.com/MoonLBH/CNN-XG.
[73] https://github.com/JiazhiHuLab/CNN_predict.
[74] https://github.com/South-Walker/AttCRISPR.
[75] https://github.com/cabbi-bio/cropsr.
[76] https://github.com/dDipankar/DeepGuide.
[77] https://github.com/AWHKU/RunMLDE_SpCas9.
[78] https://github.com/xuhi1996.
References
- 1.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012 Aug 17;337(6096):816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014 Nov 28;346(6213):1258096. doi: 10.1126/science.1258096. [DOI] [PubMed]
- 3.B. Kitchenham and S. Chartres, “Guidelines for performing systematic literature reviews in software engineering,” Keele Univ., Newcastle, U.K., and Durham Univ., Durham, U.K., Rep. EBSE-2007-01, 2007. Available online: https://www.researchgate.net/publication/302924724_Guidelines_for_performing_Systematic_Literature_Reviews_in_Software_Engineering (accessed on October 25, 2021).
- 4.Wieringa R., Maiden N., Mead N., Rolland C. Requirements engineering paper classification and evaluation criteria: A proposal and a discussion. Requir Eng. 2006;11:102–107. doi: 10.1007/s00766-005-0021-6. [DOI] [Google Scholar]
- 5.MacDonell S., Shepperd M., Kitchenham B., Mendes E. How Reliable Are Systematic Reviews in Empirical Software Engineering? IEEE Trans Softw Eng. 2010;36:676–687. doi: 10.1109/TSE.2010.28. [DOI] [Google Scholar]
- 6.Kitchenham, B.A.; Dyba, T.; Jorgensen, M. Evidence-based software engineering. In Proceedings of the Proceedings. 26th International Conference on Software Engineering, Edinburgh, UK, 23-28 May 2004; pp. 273-281.
- 7.Sjoberg D.I.K., Dyba T., Jorgensen M. The Future of Empirical Methods in Software Engineering Research. 2007;23–25:358–378. [Google Scholar]
- 8.Scalera M., Gentile E., Plantamura P., Dimauro G. A Systematic Mapping Study in Cloud for Educational Innovation. Appl Sci. 2020;10:4531. doi: 10.3390/app10134531. [DOI] [Google Scholar]
- 9.Baldassarre M.T., Caivano D., Dimauro G., Gentile E., Visaggio G. Cloud Computing for Education: A Systematic Mapping Study. IEEE Trans Educ. Aug. 2018;61(3):234–244. doi: 10.1109/TE.2018.2796558. [DOI] [Google Scholar]
- 10.Dimauro G., Caivano D., Di Pilato P., Dipalma A., Camporeale M.G. A Systematic Mapping Study on Research in Anemia Assessment with Non-Invasive Devices. Appl Sci. 2020;10:4804. doi: 10.3390/app10144804. [DOI] [Google Scholar]
- 11.Nícolas B. Santos, Rodrigo S. Bavaresco, João E.R. Tavares, Gabriel de O. Ramos, Jorge L.V. Barbosa,A systematic mapping study of robotics in human care,Robotics and Autonomous Systems,Volume 144,2021,103833, 10.1016/j.robot.2021.103833.
- 12.Pickard L.M., Kitchenham B.A., Jones P.W. Combining empirical results in software engineering. Inf Softw Technol. 1998;40:811–821. doi: 10.1016/S0950-5849(98)00101-3. [DOI] [Google Scholar]
- 13.Miller, J. Can results from software engineering experiments be safely combined? In Proceedings of the Proceedings Sixth International Software Metrics Symposium (Cat. No.PR00403), Boca Raton, FL, USA, 4–6 November 1999; pp. 152–158.
- 14.W. Hayes, “Research synthesis in software engineering: A case for meta- analysis,” in Proc. 6th IEEE Int. Softw. Metrics Symp., Boca Raton, FL, USA, 1999, pp. 143–151.
- 15.Basili V.R., Shull F., Lanubile F. Building knowledge through families of experiments. IEEE Trans Softw Eng. 1999;25:456–473. doi: 10.1109/32.799939. [DOI] [Google Scholar]
- 16.Arksey H., O’Malley L. Scoping studies: Towards a methodological framework. Int J Soc Res Methodol. 2005;8(1):19–32. [Google Scholar]
- 17.Zhang H., Babar M.A., Tell P. Identifying relevant studies in software engineering. Inf Softw Technol. 2011;53:625–637. doi: 10.1016/j.infsof.2010.12.010. [DOI] [Google Scholar]
- 18.Victoria R Li, Zijun Zhang, Olga G Troyanskaya, CROTON: an automated and variant-aware deep learning framework for predicting CRISPR/Cas9 editing outcomes, Bioinformatics, Volume 37, Issue Supplement_1, July 2021, Pages i342–i348, 10.1093/bioinformatics/btab268. [DOI] [PMC free article] [PubMed]
- 19.Xiang X., Corsi G.I., Anthon C., et al. Enhancing CRISPR-Cas9 gRNA efficiency prediction by data integration and deep learning. Nat Commun. 2021;12:3238. doi: 10.1038/s41467-021-23576-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roper J, Tammela T, Akkad A, Almeqdadi M, Santos SB, Jacks T, Yilmaz ÖH. Colonoscopy-based colorectal cancer modeling in mice with CRISPR-Cas9 genome editing and organoid transplantation. Nat Protoc. 2018 Feb;13(2):217-234. doi: 10.1038/nprot.2017.136. Epub 2018 Jan 4. PMID: 29300388; PMCID: PMC6145089. [DOI] [PMC free article] [PubMed]
- 21.Yin C, Zhang T, Qu X, Zhang Y, Putatunda R, Xiao X, Li F, Xiao W, Zhao H, Dai S, Qin X, Mo X, Young WB, Khalili K, Hu W. In Vivo Excision of HIV-1 Provirus by saCas9 and Multiplex Single-Guide RNAs in Animal Models. Mol Ther. 2017 May 3;25(5):1168-1186. doi: 10.1016/j.ymthe.2017.03.012. Epub 2017 Mar 30. PMID: 28366764; PMCID: PMC5417847. [DOI] [PMC free article] [PubMed]
- 22.Kramer NJ, Haney MS, Morgens DW, Jovičić A, Couthouis J, Li A, Ousey J, Ma R, Bieri G, Tsui CK, Shi Y, Hertz NT, Tessier-Lavigne M, Ichida JK, Bassik MC, Gitler AD. CRISPR-Cas9 screens in human cells and primary neurons identify modifiers of C9ORF72 dipeptide-repeat-protein toxicity. Nat Genet. 2018 Apr;50(4):603-612. doi: 10.1038/s41588-018-0070-7. Epub 2018 Mar 5. PMID: 29507424; PMCID: PMC5893388. [DOI] [PMC free article] [PubMed]
- 23.Hsu P.D., Lander E.S., Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014 Jun 5;157(6):1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chari R., Yeo N.C., Chavez A., George M. Church, sgRNA Scorer 2.0: A Species-Independent Model To Predict CRISPR/Cas9 Activity. ACS Synth Biol. 2017;6(5):902–904. doi: 10.1021/acssynbio.6b00343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rahman M.K., Rahman M.S. CRISPRpred: A flexible and efficient tool for sgRNAs on-target activity prediction in CRISPR/Cas9 systems. PLoS ONE. 2017;12(8):e0181943. doi: 10.1371/journal.pone.0181943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abadi S., Yan W.X., Amar D., Mayrose I. A machine learning approach for predicting CRISPR-Cas9 cleavage efficiencies and patterns underlying its mechanism of action. PLoS Comput Biol. 2017;13(10):e1005807. doi: 10.1371/journal.pcbi.1005807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Listgarten J., Weinstein M., Kleinstiver B.P., et al. Prediction of off-target activities for the end-to-end design of CRISPR guide RNAs. Nat Biomed Eng. 2018;2:38–47. doi: 10.1038/s41551-017-0178-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wilson L.O.W., Reti D., O'Brien A.R., Dunne R.A., Bauer D.C. High Activity Target-Site Identification Using Phenotypic Independent CRISPR-Cas9 Core Functionality. The CRISPR Journal. Apr 2018.182-190. doi: 10.1089/crispr.2017.0021. [DOI] [PubMed] [Google Scholar]
- 29.Chuai G., Ma H., Yan J., et al. DeepCRISPR: optimized CRISPR guide RNA design by deep learning. Genome Biol. 2018;19:80. doi: 10.1186/s13059-018-1459-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guo J., Wang T., Guan C., Liu B., Luo C., Xie Z., et al. Improved sgRNA design in bacteria via genome-wide activity profiling. Nucleic Acids Res. 2018;46(14):7052–7069. doi: 10.1093/nar/gky572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Y., Long Y., Yin R., Kwoh C.K. DL-CRISPR: A Deep Learning Method for Off-Target Activity Prediction in CRISPR/Cas9 With Data Augmentation. IEEE Access. 2020;8:76610–76617. doi: 10.1109/ACCESS.2020.2989454. [DOI] [Google Scholar]
- 32.Lin J., Wong K.-C. Off-target predictions in CRISPR-Cas9 gene editing using deep learning. Bioinformatics. 2018;34(17):i656. doi: 10.1093/bioinformatics/bty554. i663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peng H., Zheng Y.i., Zhao Z., Liu T., Li J. Recognition of CRISPR/Cas9 off-target sites through ensemble learning of uneven mismatch distributions. Bioinformatics. 2018;34(17):i757. doi: 10.1093/bioinformatics/bty558. i765. [DOI] [PubMed] [Google Scholar]
- 34.Schoonenberg V.A.C., Cole M.A., Yao Q., et al. CRISPRO: identification of functional protein coding sequences based on genome editing dense mutagenesis. Genome Biol. 2018;19:169. doi: 10.1186/s13059-018-1563-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shen M.W., Arbab M., Hsu J.Y., et al. Predictable and precise template-free CRISPR editing of pathogenic variants. Nature. 2018;563:646–651. doi: 10.1038/s41586-018-0686-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xue L.i., Tang B., Chen W., Luo J. Prediction of CRISPR sgRNA Activity Using a Deep Convolutional Neural Network. J Chem Inf Model. 2019;59(1):615–624. doi: 10.1021/acs.jcim.8b00368. [DOI] [PubMed] [Google Scholar]
- 37.Zhu H., Liang C. CRISPR-DT: designing gRNAs for the CRISPR-Cpf1 system with improved target efficiency and specificity. Bioinformatics. 2019;35(16):2783–2789. doi: 10.1093/bioinformatics/bty1061. [DOI] [PubMed] [Google Scholar]
- 38.Anob M. Chakrabarti, Tristan Henser-Brownhill, Josep Monserrat, Anna R. Poetsch, Nicholas M. Luscombe, Paola Scaffidi,Target-Specific Precision of CRISPR-Mediated Genome Editing, Molecular Cell,Volume 73, Issue 4,2019,Pages 699-713.e6, 10.1016j.molcel.2018.11.031. [DOI] [PMC free article] [PubMed]
- 39.Shrawgi H. Dilip Singh Sisodia, Convolution neural network model for predicting single guide RNA efficiency in CRISPR/Cas9 system. Chemometrics and Intelligent Laboratory Systems. 2019;189:149–154. doi: 10.1016/j.chemolab.2019.04.008. [DOI] [Google Scholar]
- 40.Wilson L.O.W., Hetzel S., Pockrandt C., et al. VARSCOT: variant-aware detection and scoring enables sensitive and personalized off-target detection for CRISPR-Cas9. BMC Biotechnol. 2019;19:40. doi: 10.1186/s12896-019-0535-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leenay R.T., Aghazadeh A., Hiatt J., et al. Large dataset enables prediction of repair after CRISPR–Cas9 editing in primary T cells. Nat Biotechnol. 2019;37:1034–1037. doi: 10.1038/s41587-019-0203-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang D., Zhang C., Wang B., et al. Optimized CRISPR guide RNA design for two high-fidelity Cas9 variants by deep learning. Nat Commun. 2019;10:4284. doi: 10.1038/s41467-019-12281-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang L., Zhang J. Prediction of sgRNA on-target activity in bacteria by deep learning. BMC Bioinf. 2019;20:517. doi: 10.1186/s12859-019-3151-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu Q., He D., Xie L. Prediction of off-target specificity and cell specific fitness of CRISPR-Cas System using attention boosted deep learning and network-based gene feature. PLoS Comput Biol. 2019;15(10):e1007480. doi: 10.1371/journal.pcbi.1007480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim H.K., Kim Y., Lee S., et al. SpCas9 activity prediction by DeepSpCas9, a deep learning-based model with high generalization performance. Sci Adv. 2019;5:eaax9249. doi: 10.1126/sciadv.aax9249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dimauro G., Colagrande P., Carlucci R., Ventura M., Bevilacqua V., Caivano D. CRISPRLearner: A Deep Learning-Based System to Predict CRISPR/Cas9 sgRNA On-Target Cleavage Efficiency. Electronics. 2019;8:1478. doi: 10.3390/electronics8121478. [DOI] [Google Scholar]
- 47.Wang J., Xiang X.i., Bolund L., Zhang X., Cheng L., Luo Y. GNL-Scorer: a generalized model for predicting CRISPR on-target activity by machine learning and featurization. J Mol Cell Biol. 2020;12(11):909–911. doi: 10.1093/jmcb/mjz116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang G., Dai Z., Dai X. A Novel Hybrid CNN-SVR for CRISPR/Cas9 Guide RNA Activity Prediction. Front Genet. 2020;10:1303. doi: 10.3389/fgene.2019.01303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim H.K., Lee S., Kim Y., et al. High-throughput analysis of the activities of xCas9, SpCas9-NG and SpCas9 at matched and mismatched target sequences in human cells. Nat Biomed Eng. 2020;4:111–124. doi: 10.1038/s41551-019-0505-1. [DOI] [PubMed] [Google Scholar]
- 50.Hiranniramol K., Chen Y., Liu W., Wang X. Generalizable sgRNA design for improved CRISPR/Cas9 editing efficiency. Bioinformatics. 2020;36(9):2684–2689. doi: 10.1093/bioinformatics/btaa041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu B., Luo Z., He J. sgRNA-PSM: Predict sgRNAs On-Target Activity Based on Position-Specific Mismatch. Mol Ther Nucleic Acids. 2020;20:323–330. doi: 10.1016/j.omtn.2020.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu Q., Cheng X., Liu G., et al. Deep learning improves the ability of sgRNA off-target propensity prediction. BMC Bioinf. 2020;21:51. doi: 10.1186/s12859-020-3395-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guishan Zhang, Zhiming Dai, Xianhua Dai, C-RNNCrispr: Prediction of CRISPR/Cas9 sgRNA activity using convolutional and recurrent neural networks, Computational and Structural Biotechnology Journal, Volume 18, 2020, Pages 344-354, ISSN 2001-0370, 10.1016/j.csbj.2020.01.013. [DOI] [PMC free article] [PubMed]
- 54.Vipin Menon A., Sohn J.-i., Nam J.-W. CGD: Comprehensive guide designer for CRISPR-Cas systems, Computational and Structural. Biotechnol J. 2020;18:814–820. doi: 10.1016/j.csbj.2020.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jaspreet Kaur Dhanjal Samvit Dammalapati, Shreya Pal, Durai Sundar, Evaluation of off-targets predicted by sgRNA design tools. Genomics. 2020;112(5):3609–3614. doi: 10.1016/j.ygeno.2020.04.024. [DOI] [PubMed] [Google Scholar]
- 56.Muhammad Rafid A.H., Toufikuzzaman M., Rahman M.S., et al. CRISPRpred(SEQ): a sequence-based method for sgRNA on target activity prediction using traditional machine learning. BMC Bioinf. 2020;21:223. doi: 10.1186/s12859-020-3531-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim N., Kim H.K., Lee S., et al. Prediction of the sequence-specific cleavage activity of Cas9 variants. Nat Biotechnol. 2020;38:1328–1336. doi: 10.1038/s41587-020-0537-9. [DOI] [PubMed] [Google Scholar]
- 58.Lazzarotto C.R., Malinin N.L., Li Y., et al. CHANGE-seq reveals genetic and epigenetic effects on CRISPR–Cas9 genome-wide activity. Nat Biotechnol. 2020;38:1317–1327. doi: 10.1038/s41587-020-0555-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Song M., Kim H.K., Lee S., et al. Sequence-specific prediction of the efficiencies of adenine and cytosine base editors. Nat Biotechnol. 2020;38:1037–1043. doi: 10.1038/s41587-020-0573-5. [DOI] [PubMed] [Google Scholar]
- 60.Lin J., Zhang Z., Zhang S., Chen J., Wong K.C. CRISPR net: A recurrent convolutional network quantifies CRISPR off-target activities with mismatches and Indels. Adv Sci. 2020;1903562 doi: 10.1002/advs.201903562. [DOI] [Google Scholar]
- 61.Jing R., Li Y., Xue L.i., Liu F., Li M., Luo J. autoBioSeqpy: A Deep Learning Tool for the Classification of Biological Sequences. J Chem Inf Model. 2020;60(8):3755–3764. doi: 10.1021/acs.jcim.0c00409. [DOI] [PubMed] [Google Scholar]
- 62.Liu X., Yang Y., Yan Qiu M.d., Reyad-ul-ferdous Q.D., Wang Y.i. SeqCor: correct the effect of guide RNA sequences in clustered regularly interspaced short palindromic repeats/Cas9 screening by machine learning algorithm. Journal of Genetics and Genomics. 2020;47(11):672–680. doi: 10.1016/j.jgg.2020.10.007. [DOI] [PubMed] [Google Scholar]
- 63.Niu M., Lin Y., Zou Q. sgRNACNN: identifying sgRNA on-target activity in four crops using ensembles of convolutional neural networks. Plant Mol Biol. 2021;105:483–495. doi: 10.1007/s11103-020-01102-y. [DOI] [PubMed] [Google Scholar]
- 64.Louie W., Shen M.W., Tahiry Z., Zhang S., Worstell D., Cassa C.A., et al. Machine learning based CRISPR gRNA design for therapeutic exon skipping. PLoS Comput Biol. 2021;17(1):e1008605. doi: 10.1371/journal.pcbi.1008605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Charlier J., Nadon R., Makarenkov V. Accurate deep learning off-target prediction with novel sgRNA-DNA sequence encoding in CRISPR-Cas9 gene editing. Bioinformatics. 2021;37(16):2299–2307. doi: 10.1093/bioinformatics/btab112. [DOI] [PubMed] [Google Scholar]
- 66.Zhang G., Zeng T., Dai Z., Dai X. Prediction of CRISPR/Cas9 single guide RNA cleavage efficiency and specificity by attention-based convolutional neural networks, Computational and Structural. Biotechnol J. 2021;19:1445–1457. doi: 10.1016/j.csbj.2021.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu X., Sun T., Shcherbina A., et al. Learning cis-regulatory principles of ADAR-based RNA editing from CRISPR-mediated mutagenesis. Nat Commun. 2021;12:2165. doi: 10.1038/s41467-021-22489-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vinodkumar P.K., Ozcinar C., Anbarjafari G. Prediction of sgRNA Off-Target Activity in CRISPR/Cas9 Gene Editing Using Graph Convolution Network. Entropy. 2021;23:608. doi: 10.3390/e23050608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang Z.-R., Jiang Z.-R. Effective use of sequence information to predict CRISPR-Cas9 off-target. 2022;Volume 20:650–661. doi: 10.1016/j.csbj.2022.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang J., Ness S., Brown R., Yu H., Oyebamii O., Jiang L., et al. EditPredict: Prediction of RNA editable sites with convolutional neural network. Genomics. November 2021;113(6):3864–3871. doi: 10.1016/j.ygeno.2021.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li B., Ai D., Liu X. CNN-XG: A Hybrid Framework for sgRNA On-Target Prediction. Biomolecules. 2022;12:409. doi: 10.3390/biom12030409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Niu R., Peng J., Zhang Z., Shang X. R-CRISPR: A Deep Learning Network to Predict Off-Target Activities with Mismatch, Insertion and Deletion in CRISPR-Cas9 System. Genes. 1878;2021:12. doi: 10.3390/genes12121878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang W., Yin J., Zhang-Ding Z., Xin C., Liu M., Wang Y., et al. In-depth assessment of the PAM compatibility and editing activities of Cas9 variants. Nucleic Acids Res. 2021;49(15):8785–8795. doi: 10.1093/nar/gkab507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xiao L.M., Wan Y.Q., Jiang Z.R. AttCRISPR: a spacetime interpretable model for prediction of sgRNA on-target activity. BMC Bioinf. 2021;22:589. doi: 10.1186/s12859-021-04509-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Müller Paul H., Istanto D.D., Heldenbrand J., et al. CROPSR: an automated platform for complex genome-wide CRISPR gRNA design and validation. BMC Bioinf. 2022;23:74. doi: 10.1186/s12859-022-04593-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Baisya D., Ramesh A., Schwartz C., et al. Genome-wide functional screens enable the prediction of high activity guides in Yarrowia lipolytica. Nat Commun. 2022;13:922. doi: 10.1038/s41467-022-28540-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Thean D.G.L., Chu H.Y., Fong J.H.C., et al. Machine learning-coupled combinatorial mutagenesis enables resource-efficient engineering of CRISPR-Cas9 genome editor activities. Nat Commun. 2022;13:2219. doi: 10.1038/s41467-022-29874-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fan Yongxian, Xu Haibo, Prediction of Off-Target Effects in CRISPR/Cas9 System by Ensemble Learning, Current Bioinformatics 2021; 16(9). https://dx.doi.org/10.2174/1574893616666210811100938.
- 79.Liang P., Xu Y., Zhang X., Ding C., Huang R., Zhang Z., et al. CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein Cell. 2015 May;6(5):363–372. doi: 10.1007/s13238-015-0153-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Aach J., Mali P., Church G.M. CasFinder: Flexible algorithm for identifying specific Cas9 targets in genomes. bioRxiv. 2014 [Google Scholar]
- 81.Labun K., Montague T.G., Gagnon J.A., Thyme S.B., Valen E. CHOPCHOP v2: A web tool for the next generation of CRISPR genome engineering. Nucleic Acids Res. 2016;44:W272. doi: 10.1093/nar/gkw398. W276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Doench J.G., Fusi N., Sullender M., Hegde M., Vaimberg E.W., Donovan K.F., et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol. 2016;34:184–191. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chari R., Mali P., Moosburner M., Church G.M. Unraveling CRISPR-Cas9 genome engineering parameters via a library-on-library approach. Nat Methods. 2015;12:823–826. doi: 10.1038/nmeth.3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Xu H., Xiao T., Chen C.-H., Li W., Meyer C.A., Wu Q., et al. Sequence determinants of improved CRISPR sgRNA design. Genome Res. 2015;25:1147–1157. doi: 10.1101/gr.191452.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Moreno-Mateos M., Vejnar C., Beaudoin J.D., et al. CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat Methods. 2015;12:982–988. doi: 10.1038/nmeth.3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Haeussler M., Schonig K., Eckert H., Eschstruth A., Mianne J., Renaud J.-B., et al. Evaluation of off- target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016;17:148. doi: 10.1186/s13059-016-1012-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hart T., Chandrashekhar M., Aregger M., Steinhart Z., Brown K.R., MacLeod G., et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell. 2015;163:1515–1526. doi: 10.1016/j.cell.2015.11.015. [DOI] [PubMed] [Google Scholar]