

Abstract
Lipoprotein(a) (Lp(a)) represents a unique subclass of circulating lipoprotein particles and consists of an apolipoprotein(a) (apo(a)) molecule covalently bound to apolipoprotein B-100. The metabolism of Lp(a) particles is distinct from that of low-density lipoprotein (LDL) cholesterol, and currently approved lipid-lowering drugs do not provide substantial reductions in Lp(a), a causal risk factor for cardiovascular disease. Somatic genome editing has the potential to be a one-time therapy for individuals with extremely high Lp(a). We generated an LPA transgenic mouse model expressing apo(a) of physiologically relevant size. Adeno-associated virus (AAV) vector delivery of CRISPR-Cas9 was used to disrupt the LPA transgene in the liver. AAV-CRISPR nearly completely eliminated apo(a) from the circulation within a week. We performed genome-wide off-target assays to determine the specificity of CRISPR-Cas9 editing within the context of the human genome. Interestingly, we identified intrachromosomal rearrangements within the LPA cDNA in the transgenic mice as well as in the LPA gene in HEK293T cells, due to the repetitive sequences within LPA itself and neighboring pseudogenes. This proof-of-concept study establishes the feasibility of using CRISPR-Cas9 to disrupt LPA in vivo, and highlights the importance of examining the diverse consequences of CRISPR cutting within repetitive loci and in the genome globally.
Keywords: lipoprotein(a), LPA, apolipoprotein(a), liver, gene therapy, adeno-associated virus, somatic genome editing, CRISPR-Cas9, AAV-CRISPR, atherosclerosis
Graphical abstract
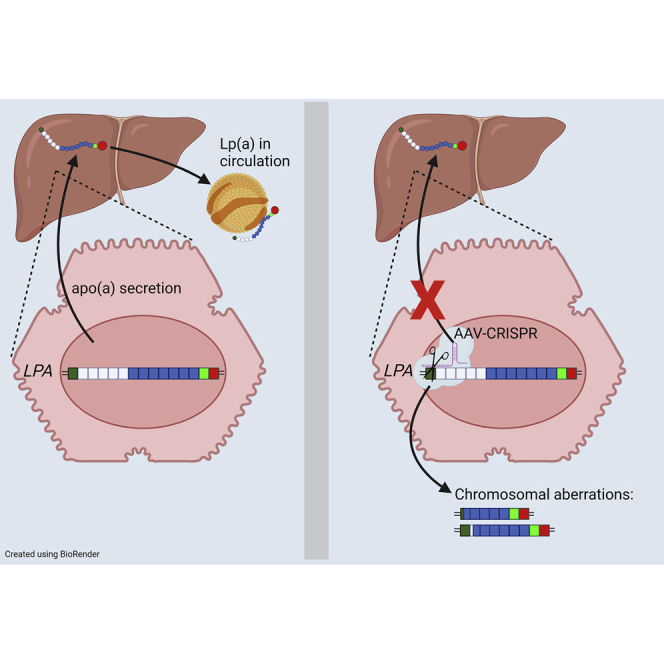
Doerfler and Park et al. show that editing of the LPA gene is a promising therapeutic approach to lower lipoprotein(a), a circulating lipid particle strongly associated with heart disease. AAV delivery of CRISPR-Cas9 nearly completely removed apolipoprotein(a) levels in a transgenic mouse model with a single treatment.
Introduction
Lipoprotein(a) (Lp(a)) is a lipoprotein particle that is well established as an independent and causal risk factor for atherosclerotic cardiovascular disease (ASCVD) and calcific aortic valve disease (CAVD).1, 2, 3, 4, 5, 6, 7, 8, 9 Lp(a) is composed of a low-density lipoprotein (LDL)-like particle with apolipoprotein(a) (apo(a)) covalently linked to apolipoprotein B-100 (apoB-100).10 The apo(a) protein itself is encoded by the LPA gene and is highly similar to plasminogen (PLG), a zymogen that plays a key role in fibrinolysis.11 Both apo(a) and plasminogen contain a series of kringle (K) domains, which are triple loop structures stabilized by three internal disulfide bonds. The apo(a) protein consists of 10 kringle type IV (KIV) domains, denoted as KIV1–KIV10, followed by KV and protease-like domains. Lp(a) concentrations are strongly heritable and vary widely in humans based on the number of KIV2 domains (ranging from two to greater than 40) that correspond to allele size of the LPA gene. Smaller isoforms of LPA encode apo(a) proteins that are more efficiently secreted from the liver, resulting in higher Lp(a) levels in the circulation.12 Individuals with plasma Lp(a) levels over 30–50 mg/dL, or about one in five individuals in the United States,13 are at increased risk for CAVD,4,14 making it an important target for drug development.
Current lipid-lowering therapies, including PCSK9 inhibitors, do not target Lp(a) specifically or adequately lower plasma Lp(a) concentrations. RNA interference and antisense technologies targeting the LPA transcript show tremendous promise for reducing Lp(a) levels in high-risk patients.15 Currently, there is an ongoing phase III clinical trial to assess whether antisense-mediated knockdown of apo(a) production by the liver can reduce the risk of major adverse cardiovascular events in patients with established cardiovascular disease (CVD)) and elevated Lp(a) levels (Lp(a) HORIZON, NCT04043552).16 There are also small interfering RNA (siRNA) molecules in development that can effectively lower Lp(a), with a recent phase I study (NCT03626662) showing 71%–97% reductions after a single dose.17 However, there is a strong case to be made for genome editing to achieve Lp(a) reduction. First, the LPA gene is exclusively expressed in the liver, an organ that is amenable to targeting with both viral vectors and non-viral delivery systems. Second, although the biological function of apo(a) remains unknown, there appear to be no adverse phenotypes associated with homozygous loss of this gene in humans.16,18,19 Third, oligonucleotides and siRNAs require repeated injections, whereas a single treatment with genome-editing enzymes could permanently eliminate apo(a) production, providing the greatest degree of durable CVD risk reduction without a need for additional dosing.
Clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9 (CRISPR-Cas9) is the most widely used system for in vivo gene editing, with many potential applications for human therapeutics.20,21 Off-target effects remain a primary concern in the use of CRISPR-Cas9 for gene therapy as they can result in impaired cellular viability or tumorigenesis. Therefore, rigorous selection of single guide RNA (sgRNA) is required to reduce off-target effects and maximize editing efficiency. In addition, highly efficient delivery is a critical factor to maximize editing efficiency. Adeno-associated virus (AAV) has a high tropism for the liver and is a promising delivery vehicle for cardiovascular targets in the liver. AAV delivery of CRISPR-Cas9 has already been used successfully to edit Pcsk9, Apob, and Ldlr.22, 23, 24, 25 At the present time, preclinical gene editing to disrupt LPA has not been reported.
In this study, we utilized a gRNA specific for the first kringle in apo(a), and used AAV for delivery of the gRNA and Cas9. We measured the effect of editing on plasma apo(a) concentrations, and we documented on- and off-target effects on a genome-wide basis. This work demonstrates that liver-directed genome editing with CRISPR-Cas9 can effectively lower apo(a) levels, and highlights the importance of surveillance for unintended editing events within highly repetitive genes and globally within the genome.
Results
Generation of transgenic mice expressing apo(a)
The LPA gene is found only in humans and a subset of primates, although a similar protein exists in hedgehogs that arose independently through a gene duplication event.26 Therefore, we developed transgenic mice expressing a small version of human apo(a) by pronuclear injection of a linearized LPA cDNA driven by the liver-specific promoter from APOE (Figure 1). The LPA transgene was bred onto an Ldlr-null background to more closely reflect a human lipoprotein distribution with high levels of LDL. We found using droplet digital PCR (ddPCR) with transgene-specific primers that LPA+/0Ldlr−/− mice have three LPA transgene copies per diploid genome (c/dg) (Figure S1). Sanger sequencing identified the junction containing contiguous sequence between an intergenic site on chr3:120164677 in the mouse genome (mm10) (Figure S2A) and the 5′ end of the LPA construct, with two types of the tandem junctions of the 3′ and 5′ ends of the LPA construct (Figures S2B and S2C). Therefore, the transgenic mice have three tandem repeats of the LPA construct in direct orientation integrated into the chr3:120164677 (Figure S2D). The original transgene encoded a 14-kringle form of apo(a), containing all the invariant KIV types and five copies of KIV2. However, Sanger sequencing of the integrated transgene revealed the presence of an in-frame 342-bp deletion of most of KIV8 and a small amount of KIV7; the junction point was at the homologous site in the respective kringles and thus an intact hybrid of KIV7 and KIV8 was formed. Since no other alternative sequences were observed, the deletion occurred prior to tandem multiplication of the transgene insertion.
Figure 1.

Generation of transgenic mice expressing apo(a)
(A) Schematic representation of the LPA transgene used to generate transgenic mice expressing human LPA. LPA+/0 mice were then crossed with mice lacking the LDL receptor (Ldlr−/−). (B) Relative LPA mRNA abundance in different LPA+/0 mouse tissues as measured by qPCR (n = 10). Different letters denote data that are significantly different by ANOVA with post hoc Tukey test, p < 0.05. Values are mean SD. (C) Western blot for apo(a) in mice either lacking (0/0) or expressing (+/0) the LPA transgene, run under non-reduced or reduced conditions. M, male; F, female; C, control human plasma. ∗Aggregated material that did not enter the resolving gel. (D) Cholesterol and apo(a) distribution among lipoprotein fractions in pooled plasma (n = 4 per group).
Characterization of Lp(a) transgenic mice
The LPA transgene was maintained in the hemizygous or null state for all experimental animals (LPA+/0, LPA+/0Ldlr−/−, or LPA0/0Ldlr−/−) (Figure 1A). LPA transgene mRNA abundance in the liver, kidney, spleen, heart, small intestine, brain, and quadriceps was assessed by real-time RT-qPCR relative to Gapdh (Figure 1B). The highest levels of LPA expression were detected in liver and brain tissue, consistent with the APOE promoter elements driving the transgene. We performed a western blot to determine if apo(a) is present in the plasma and covalently bound with murine apoB-100 (Figure 1C). Human plasma expressing a form of Lp(a) that has 16 KIV domains (16K) was used as a control (C, lanes 1 and 10). Plasma from male and female mice lacking (0/0) or expressing (+/0) the human LPA transgene were resolved under reduced and non-reduced conditions. We observed that the apo(a) protein is detectable in plasma from both male and female mice but does not form a covalent disulfide bond with murine apoB-100 (Figure 1C) because the expected band of slightly higher mobility than the human Lp(a) control is absent. Apo(a) distribution on lipoproteins was assessed in lipoprotein fractions separated by size-exclusion chromatography from male mice that were fasted for 5 h. Cholesterol was distributed as expected within the profile for Ldlr−/− mice on a chow diet, and did not differ with respect to LPA genotype. Interestingly, apo(a) was found only in the lipoprotein fractions corresponding to very-low-density lipoprotein (VLDL)- and intermediate-density lipoprotein (IDL)/LDL-sized particles, with none detectable in high-density lipoprotein (HDL) or the lipoprotein free fractions (Figure 1D). Despite the inability to form a covalent disulfide bond with murine apoB-100, apo(a) still resides on apoB-100-containing particles in this mouse model, presumably through non-covalent interactions.
LPA gene editing rapidly and efficiently lowers circulating apo(a) protein levels
We generated an AAV vector co-expressing a sgRNA targeting LPA exon 2 in the KIV1-encoding region of the LPA cDNA transgene. Given the highly repetitive nature of KIV domains in the LPA gene, and the need to achieve complete removal of apo(a) protein, we aimed to target as close as possible to the translation start site in the coding sequence. Exon 1 encodes only the first 49 bp of the signal peptide, and was not targetable by SaCas9. Multiple gRNAs were designed for exon 2 based on the presence of a canonical NNGRRT protospacer adjacent motif (PAM). The optimal gRNA for this region was found to reside in antisense orientation from +147 to +124 relative to the ATG start codon, within the first KIV domain. The designed 23-nucleotide sgRNA (GCAGGTCCTTCCTGTGACAGTGG, PAM-TGGAGT) had no exact-match off-targets identified in the LPA transgene. Expression of the sgRNA was driven by a U6 promoter, along with SaCas9 expressed under the control of the liver-specific hybrid liver promoter (HLP). Male and female LPA+/0Ldlr−/− mice were injected intraperitoneally at 8 weeks of age with 1 × 1012 genome copies (GC) of AAV expressing either GFP as a control (AAV-GFP) or sgRNA and Cas9 (AAV-CRISPR). Plasma and body weights were collected at weekly intervals until the terminal time point at 12 weeks of age when livers were harvested (Figure 2A). There were no significant differences in body weights following treatment in either male or female mice throughout the study (Figure 2B). Liver to body weight ratios were modestly but significantly higher in male LPA+/0Ldlr−/− mice that received AAV-CRISPR, while female ratios were unchanged between groups (Figure 2C). Cholesterol levels also remained unchanged between groups in male and female mice (Figure 2D). Liver histology for proliferation (Ki-67) and cell death (TUNEL) was quantified, and no significant changes were observed in mice that received AAV-CRISPR (Figure S3). Alanine transaminase (ALT) and aspartate transaminase (AST) levels were not significantly altered by AAV treatment, and remained in a normal range throughout the study (Figure S4). Finally, vector genome analysis by qPCR revealed approximately one to nine genome copies per diploid genome equivalent (dge) in mice that received AAV-GFP, while mice that received AAV-CRISPR ranged from one to 39 AAV genome copies per diploid genome (Figure S5). Although not significant, there is a trend toward lower AAV GC/dge in female mice, in accordance with previous studies that have shown reduced transduction in livers of female mice receiving AAV.27 Robust expression of GFP and SaCas9 protein was confirmed by western blotting in mice receiving the respective AAV vectors (Figures 2E–2H). Plasma apo(a) protein expression was assessed by western blot, revealing undetectable levels in male and female LPA+/0Ldlr−/− mice that received AAV-CRISPR at 4 weeks post injection (Figure 2I). An ELISA specific for apo(a) revealed essentially complete elimination of the protein from plasma as early as 1 week post injection, which was sustained for the duration of the study (Figure 2J). We also observed that apo(a) levels are approximately 2-fold higher in male transgenic mice relative to females at baseline.
Figure 2.

Knockdown of apo(a) in LPA+/0Ldlr−/− mice by AAV-CRISPR
(A) Male and female LPA+/0Ldlr−/− mice were injected at 8 weeks of age with 1 × 1012 genome copies of AAV expressing either GFP as a control (AAV-GFP) or Cas9 and an sgRNA targeting LPA exon 2 in the KIV1 domain of the LPA transgene (AAV-CRISPR). Plasma and body weights were collected weekly until 12 weeks of age, at which point plasma and liver tissue were harvested. (B) Weekly body weights in male and female mice. (C) Liver to body weight ratios and (D) fasted plasma cholesterol levels in male and female mice given either AAV-GFP or AAV-CRISPR. (E) Western blot for GFP in male and female LPA+/0Ldlr−/− mice given AAV-GFP or AAV-CRISPR. (F) Quantification for GFP western blot. All groups normalized to male mice given AAV-GFP vector. (G) Western blot for SaCas9 in male and female LPA+/0Ldlr−/− mice given AAV-GFP or AAV-CRISPR. (H) Quantification for SaCas9 western blot. All groups normalized to male mice given AAV-GFP vector. (I) Western blot for apo(a) in male and female LPA+/0Ldlr−/− mice given AAV-GFP or AAV-CRISPR. (J) Time course for apo(a) expression in male and female LPA+/0Ldlr−/− mice given AAV-GFP or AAV-CRISPR measured by ELISA (n ≥ 6). Statistical analysis was done using Mann-Whitney test on non-normally distributed data with ∗p < 0.05 (GFP western blot males, apo(a) ELISA males and females). Statistical analysis was done using a Welch’s t test on normally distributed data with ∗p < 0.05 (body weight male and females at each time point, liver:body weight ratio males and females, cholesterol males and females, GFP western blot females, SaCas9 western blot males and females). Values are mean SD.
Assessment of gene editing in mice receiving AAV-CRISPR
We first measured editing at the on-target site in KIV1, and assessed insertions and deletions (indels) by T7 endonuclease I (T7E1) assay and inference of CRISPR edits (ICE; data not shown) (Figures S6A and S6B). Both assays showed no measurable indels in all treated animals compared with control, which was surprising given the near-complete degree of plasma apo(a) protein knockdown. Although the LPA sgRNA has only one exact match in the LPA transgene, which is at the on-target site in KIV1, many potential off-target sites within the LPA gene on other KIV domains share high sequence homology. We hypothesized that efficient simultaneous cleavages at on-target and off-target sites within the LPA transgene could cause large deletions, leaving only a small amount of KIV1 unrearranged. To test this hypothesis, we checked the frequency of KIV1 unrearranged on-target sites by ddPCR (Figures 3A and S7). The unrearranged KIV1 region normalized by LPA construct copy number was 36.8% ± 4.2% and 36.7% ± 1.8% in edited male and female animals, respectively. This is compared with 98.8% ± 3.7% and 101.1% ± 2.9% in control male and female animals, respectively, indicating frequent rearrangement at on-target double-strand breaks (DSBs) (Figure 3A).
Figure 3.

Chromosomal rearrangements in LPA+/0Ldlr−/− mouse livers injected with AAV-CRISPR
(A) On-target unrearranged KIV1 normalized by LPA construct copy number, indicating frequent rearrangement at on-target DSB in AAV-treated mice. Statistical analysis was done using Welch’s t test with ∗p < 0.05. Values are mean SD. (B–E). For CAST-seq, a bait primer binding upstream of the on-target site on KIV1 was used to identify chromosomal aberrations derived from on-target and off-target activities of CRISPR. CAST-seq reads aligned to the 13K LPA are represented by bars in Integrated Genomics Viewer (IGV) plots. The green triangle marks the position of the on-target site in KIV1. (B and C) In AAV-GFP sample, 96% of reads align to KIV1 with a minimal background at other KIV domains indicating intact KIV1 without chromosomal rearrangement. (D and E) Frequent chromosomal deletions and inversions between CRISPR-induced on- and off-target DSBs within the LPA transgene and small indels at the breakpoint junctions in AAV-CRISPR sample, all contributing to LPA disruption. The percentage of aligned CAST-seq read at each KIV domain in descending order. Fifteen percent of CAST-seq reads aligned at KIV2 domains were displayed in gray due to low alignment scores as those reads are equally likely to map to five KIV2 domains; 9.92% of reads aligned at KIV5 are shown in blue or red, indicating deletion and inversion of sequences between KIV1 and KIV5, respectively. CAST-seq also revealed deletions between the on-target site and off-target sites in other KIV domains. (F) Agarose gel image showing a full-length PCR amplicon between KIV1 and KIV5 (3,211 bp) in AAV-GFP-treated mice, and truncated amplicons in all AAV-CRISPR-treated mice.
For a comprehensive understanding of the chromosomal aberrations within the LPA transgene, we performed chromosomal aberrations analysis by single targeted linker-mediated PCR sequencing (CAST-seq) in control (Figures 3B and 3C) and edited (Figures 3D and 3E) female mice.28 A bait primer binding upstream (PAM-proximal side) of the on-target site on KIV1 was used to identify chromosomal aberrations derived from on-target and off-target activities of CRISPR by mapping the chromosomal sequences fused to PAM-proximal arm of the on-target DSB. Alignment of CAST-seq reads to the LPA transgene revealed the frequent chromosomal deletions and inversions between CRISPR-induced on- and off-target DSBs within the LPA transgene and small indels at the breakpoint junctions in edited mouse, all contributing to LPA disruption (Figures 3D and 3E). The most retrieved CAST-seq reads included large deletions between the on-target site in KIV1 and off-target sites in KIV2(1-5) (OT10, OT12, OT13, and OT18 in Table S1) domains. The second most frequent event involved large deletion and inversion with an off-target site on KIV5 (OT2 in Table S1). CAST-seq also revealed large deletions between the on-target site and off-target sites in KIV10 (OT6), KIV6 (OT4), KIV9 (OT3), and KIV3 (OT9) (Figures 3D and 3E; Table S1).
To validate the CAST-seq-identified local chromosomal rearrangements between the on-target site in KIV1 and potential off-target site on KIV5, we used a forward primer specific to the on-target site PAM upstream region and a reverse primer specific to KIV5 off-target site PAM downstream region (Figures S8A and S8B). We detected a full-length amplicon (3,211 bp) in control animals and truncated amplicons in all AAV-CRISPR-treated animals, indicating frequent local chromosomal rearrangement occurring between KIV1 and KIV5 (Figure 3E). Truncated amplicons of 1,174 and 490 bp are the product of a 2.7-kb large deletion created by on- and off-target DSBs re-joining (Figures S8A and S8B) or a 2-kb large deletion between two off-target DSBs at KIV2(1) and KIV4 (OT15 in Table S1; Figures S8C and S8D).
To further investigate these complex rearrangements, we performed PCR amplification of full-length LPA cDNA (5,219 bp) followed by Pacific Biosciences (PacBio) Single-Molecule, Real-Time (SMRT) sequencing.29 High-fidelity (HiFi) reads from AAV-GFP and AAV-CRISPR samples were aligned to the 13K LPA cDNA reference sequence (Figures S9A and S9B), and diverse insertions and deletions were analyzed (Figure S9C). In the AAV-GFP control sample, 70% of reads had expected full-length 13K cDNA sequence and remaining reads had unexpected homologous insertions or deletions, possibly due to the homologous recombination in the germline of founder mice30 or limitation of long-range PCR across the repetitive regions (Figures S9A, S9C, and S10).31 In the AAV-CRISPR-treated sample, 22% of reads had the expected full-length 13K cDNA sequence and an impressive 72% of reads showed large deletions ranging from 1,710 to 4,079 bp in increments of 342 bp, corresponding to the individual KIV domain size (Figures S9B and S9C). Deletions in the AAV-CRISPR sample closely matched CRISPR DSBs at on- and off-target sites, further confirming the ligation of these sequences identified through CAST-seq (Figure S9B). We also observed alleles containing multiple deletions at on- and off-target cut sites and between separate off-target sites (Figure S9B). Collectively, these data indicate that off-target cutting within the LPA transgene causes chromosomal deletions and rearrangements, explaining the robust degree of apo(a) knockdown despite a lack of small indels at the on-target site.
Identification of off-target sites within the context of the human genome
To investigate the efficiency and safety of our LPA targeting gene therapy in the context of the human genome, we performed in depth on- and off-target analysis in HEK293T cells (Figure 4). We electroporated px601 plasmid encoding LPA sgRNA/SaCas9 or LPA sgRNA/SaCas9 ribonucleoprotein (RNP) into HEK293T at 1–4 μg and 30–90 pmol dose, respectively. T7E1 showed minimal indel activity and no evidence of editing by ICE analysis in HEK293T (data not shown), similar to what we observed in transgenic mice (Figure S6). The ddPCR copy number assay was optimized for the human LPA gene, and we quantified the rearrangement at the on-target site in exon 2. We detected up to 92% on-target rearrangement with a dose of 4 μg of plasmid (Figure 4A) and up to 90% on-target rearrangement with a dose of 90 pmol RNP demonstrating highly active LPA sgRNA in HEK293T cells (Figure 4C). Next, we performed genome-wide unbiased identification of DSBs enabled by sequencing (GUIDE-seq) in HEK293T cells treated with electroporation of CRISPR-Cas9 as plasmid (Figure 4B) or RNP (Figure 4D), along with a blunt double-stranded oligodeoxynucleotides (dsODN). GUIDE-seq relies on capture of a dsODN into DSBs to identify CRISPR-induced off-target activities in the genome globally by sequencing.32,33 Positive linear correlations between GUIDE-seq read counts and indel mutation frequencies have been reported.33
Figure 4.

GUIDE-seq off-target analysis in HEK293T cells treated with LPA sgRNA/SaCas9 plasmid and RNP
(A) HEK293T cells were electroporated using 1–4 μg of px601 plasmid encoding LPA sgRNA/SaCas9, and percentage of unrearranged on-target site was measured by ddPCR showing plasmid dose-dependent decrease in the unrearranged on-target site. (B) GUIDE-seq results from HEK293T cells treated by electroporation of 2.5 μg of plasmid along with 20 pmol of dsTag showing on-target site (On) and 25 off-target sites. The reference LPA on-target site is shown at the top (N, any nucleotide; R, purine). Nucleotide mismatches between on- and off-target sites are highlighted. Off-target sites were labeled according to the descending order of GUIDE-seq read count (see also Table S1). (C) HEK293T cells were electroporated using 30–90 pmol LPA sgRNA/SaCas9 as RNP. RNP dose-dependent decrease in the unrearranged on-target site is shown. (D) GUIDE-seq results from HEK293T cells electroporated with 90 pmol RNP and 20 pmol dsTag showing 13 identified off-target sites. (E) Ratio of GUIDE-seq read counts (on- or off-target read counts/on read count) for the sites identified by both plasmid and RNP-treated samples. Nearly all off-target sites identified by RNP (except OT26) were also found by plasmid, with a higher ratio of GUIDE-seq read counts indicating a higher degree of off-target mutagenesis in plasmid-treated sample. OT, off target.
In the plasmid-treated sample, GUIDE-seq identified 25 off-target sites, which were labeled according to the descending order of read count (OT1–OT25) (Figure 4B and Table S1). Of 25 off-target sites, 11 sites with one to four mismatches (MMs) to the target site are at LPA exons corresponding to KIV2–10, five off-target sites with two to four MMs are at pseudogenes nearby LPA, and nine off-target sites with three to seven MMs and low read counts are at other loci. Due to highly homologous KIV domains, some of the off-target sites (e.g., OT4 and OT5; OT7 and OT8; OT9 and OT10; OT12, OT13, and OT18) have the same target and PAM sequences but occur at different genomic locations. The off-target site with the highest read count, OT1, has one MM on PAM and three PAM-distal MMs and is located at LOC107986665 pseudogene, 256 kbp away from LPA on-target site (Table S1; Figure S11). In the RNP-treated sample, a total of 13 off-target sites were identified, with 12 sites also found in the plasmid-treated sample (Figures 4D and 4E). GUIDE-seq performed on the HEK293T cells receiving CRISPR-Cas9 delivered as a plasmid and RNP identified the overlapping off-target sites at or near LPA with high abundance, but the plasmid-treated sample generated a higher number of identified off-target sites as well as higher ratio of GUIDE-seq read count normalized by that of on-target sites (Figure 4E).
Analysis of chromosomal rearrangements as a result of LPA gene editing within the context of the human genome
Genomic DNA from plasmid- (Figures 5A and 5B) and RNP (Figures 5C and 5D)-treated HEK293T cells were subjected to CAST-seq to identify chromosomal rearrangements involving the on-target DSB in the human genome (hg38). CAST-seq performed using plasmid-treated HEK293T cells identified 14 off-target rearrangements, of which eight are located at LPA, five at nearby pseudogenes, and one at PLG (Figure 5A). CAST-seq performed using RNP-treated HEK293T cells identified nine off-target rearrangements, of which eight were found in the plasmid-treated sample (Figure 5C). CAST-seq showed frequent rearrangements between on- and off-target sites within the LPA, particularly at OT2 on exon 20 and OT3 on exon 28 (Figures 5A and 5C; Table S1). The on- and off-target rearrangement identified by CAST-seq in plasmid- (Figure 5B) and RNP (Figure 5D)-treated samples was in good agreement with off-target DSBs identified by GUIDE-seq, demonstrating efficient chromosomal rearrangement between on- and off-target DSBs. CAST-seq-identified chromosomal rearrangement events were validated by endpoint PCR using the primers designed to amplify across the expected large deletion junction between the PAM upstream side of on-target DSB and the off-target DSBs at LPA (OT2, OT3, OT4, OT6, OT12, OT13, OT18, and OT26) or pseudogene (OT1) (Figure 5E). Next, we adapted a KIV2 copy number variant (CNV) genotyping qPCR assay34 for ddPCR and quantified the KIV2 repeat number in unmodified and edited HEK293T. The primers and probe used for the assay were specific for KIV2 and KIV3 domains. We found that HEK293T contains ∼18 c/dg of KIV2 and KIV3 domains, which decreased to 5 c/dg with 4 μg of plasmid delivery and was also reduced in a dose-dependent manner with RNP treatment (Figure 5F). Since LPA harbors intragenic multiallelic CNV and the number of KIV2 copies varies from one to >40, these results indicate that the number of CRISPR-induced DSBs and extent of chromosomal arrangement would be influenced by KIV2 CNV polymorphism in the individual genome.
Figure 5.

CAST-seq off-target analysis and on-target LPA rearrangements in HEK293T cells treated with LPA sgRNA/SaCas9 plasmid and RNP
(A) CAST-seq was performed on RNP and plasmid-treated HEK293T using the bait primer on the PAM upstream region of the on-target site to identify chromosomal rearrangements between on-target and off-target DSBs. CAST-seq identified on- and off-target chromosomal rearrangement in HEK293T cells electroporated with 2.5 μg of plasmid. The reference LPA on-target site is shown at the top. Mismatched nucleotides and indels (−1/1) are highlighted. (B) Correlation between GUIDE-seq read counts and CAST-seq read counts for the OT sites identified by both assays in plasmid-treated HEK293T. (C) CAST-seq identified on- and off-target chromosomal rearrangement in HEK293T cells electroporated with 90 pmol of RNP. (D) Correlation between GUIDE-seq read counts and CAST-seq read counts for the OT sites identified by both assays in RNP-treated HEK293T. (E) CAST-seq-identified chromosomal rearrangement events were validated by endpoint PCR using the primers designed to amplify across the expected large deletion junction between the PAM upstream side of on-target DSB and the off-target DSBs at selected OT sites. Lane 1, OT2 (404 bp); 2, OT3 (448 bp); 3, OT4 (579 bp); 4, OT6 (530 bp); 5, OT1 (376 bp); and 6, OT12, OT13, OT18, and OT26 (399 bp). PCR products in lane 6 could be from deletion between the on-target site and four different OT sites (OT12, OT13, OT18, and OT26) that share the same target sequence and PCR primer binding sequence. (F) ddPCR quantification of the KIV2 CNV in untreated and plasmid- or RNP-treated HEK293T. The primers and probe used for the assay were specific for KIV2 and KIV3 domains. Untreated HEK293T contains ∼18 c/dg of KIV2 and KIV3 domains, which decreased in a plasmid- and RNP dose-dependent manner.
Discussion
Despite the causal role of Lp(a) in ASCVD and CAVD disease processes, there is currently no US Food and Drug Administration (FDA)-approved therapy that specifically targets Lp(a). Efforts to inhibit Lp(a) production by the liver through antisense and RNA interference approaches are promising, but it remains to be seen if these drugs will decrease cardiovascular events and associated mortality. Here, we present the first evidence that CRISPR-Cas9 can be used to permanently disrupt the LPA gene in vivo, rapidly and efficiently lowering plasma apo(a) levels.
We developed a new transgenic mouse model that expresses physiologically relevant levels of apo(a) in the circulation. Although intended to be liver specific, expression of LPA mRNA was also detected in the brain, which is likely a consequence of the APOE regulatory elements contained in the pLIV promoter. It is unlikely that brain-derived apo(a) could be secreted and enter the systemic circulation, as apoB is not expressed in this tissue, and apoE-containing lipoproteins constitute a pool that does not cross the blood-brain barrier.35,36 This mouse model expresses a small isoform of LPA containing five KIV2 domains, an important advantage for therapeutic testing and disease modeling as shorter isoforms are associated with more efficient secretion from the liver and higher Lp(a) levels in humans.12 Although these studies were not performed in mice expressing human apoB-100, we found by size-exclusion chromatography of mouse plasma that apo(a) was localized in the VLDL and IDL/LDL fractions, with no detectable free apo(a) in plasma or on HDL. Note that KIV7 and KIV8 have been identified as key for non-covalent lysine-dependent interactions between apo(a) and apoB-100.37,38 Most of KIV8 is deleted in our transgene, but, since KIV6 contains an identical lysine-binding site to KIV7 and KIV8, it appears that the KIV6-KIV7 sequence sufficiently compensates in non-covalent binding to mouse apoB-100. Future studies designed to assess the impact of LPA gene editing on atherosclerosis in mouse models should also include the human gene encoding apoB-100 to afford the production of bona fide covalent Lp(a) particles.
In humans, endogenous apo(a) is produced exclusively by the liver, making it an excellent target for therapeutic disruption with AAV-delivered CRISPR-Cas9. In order to achieve the greatest degree of knockout and specificity, we chose a sgRNA targeting exon 2 of the LPA transgene in the KIV1 domain. AAV-CRISPR delivery to mice resulted in very efficient removal of apo(a) protein from the circulation, as demonstrated by western blotting and ELISA. This nearly complete loss of apo(a) (over 99% in male and over 96% in female mice) was stable for the 4-week observation period. Although sex differences in AAV gene delivery to the liver have been reported,23 with males being more efficiently transduced, we obtained a comparable extent of apo(a) removal in female mice. This is likely due to the dose used, which is approximately 5- to 10-fold higher than that required for complete hepatocyte transduction in male mice. Nonetheless, this is a clinically relevant dose of AAV8 (4 × 1013 GC/kg) that has been used in humans without major adverse events.39 Evidence of safety was also supported by the lack of changes to overall body weight, ALT, AST, plasma cholesterol, as well as normal liver histology.
We were initially surprised to find no editing at the on-target site in the AAV-CRISPR mice by T7E1 or ICE analysis, given that we observed complete protein knockdown. Further analysis showed that many off-target edits occurred within the LPA transgene itself. Through ddPCR analysis, we determined that approximately 64% of LPA transgene alleles had a rearranged KIV1-encoding domain. CAST-seq and PacBio long-read sequencing analysis revealed that off-target indels in other KIV domains in LPA and deletions between off-target sites also account for further loss of apo(a) expression. Considering that 20%–40% of the cells within the liver are non-parenchymal cells40 that do not express LPA41 but still contribute DNA to the pool, it stands to reason that we achieved editing in the vast majority of hepatocytes within the liver. Therefore, the primary effect of CRISPR editing in our model was segmental deletions of the LPA transgene mediated by on- and off-target cutting within similar KIV domains. AAV-CRISPR reduced plasma apo(a) to nearly undetectable levels mediated by cumulative consequences of LPA disruptive mutations.
Given the unanticipated editing events in the transgenic mouse model, we wanted to carefully assess this targeting strategy on the full LPA gene within the context of the human genome using human embryonic kidney (HEK293T) cells. Again, we saw no editing in the cells by T7E1 or ICE analysis, but a remarkable 92% on-target rearrangement by ddPCR. Using a combination of CAST-seq and GUIDE-seq analysis, we determined that there were a large number of chromosomal rearrangement events between on- and off-target DSBs within LPA itself. The difference in the off-target effects observed between using Cas9 expressing plasmid versus RNP is likely due to RNP’s short active span.42 A higher degree of off-target mutagenesis could be expected with AAV-CRISPR treatment due to robust expression of Cas9 at 4 weeks post injection (Figures 2G and 2H).
Human LPA harbors intragenic multiallelic CNV, and the number of KIV2 copies varies from two to >40.18 As such, the number of CRISPR-induced DSBs and extent of chromosomal arrangement would vary based on KIV2 CNV polymorphism in the individual genome. The majority of identified off-target activity appears to be localized to LPA exons or pseudogenes near the LPA locus. GUIDE-seq did not detect off-target sites within the plasminogen (PLG) gene, nor were any potential sites in PLG identified in silico by COSMID43 (data not shown). Nonetheless, CAST-seq in plasmid-treated samples identified deletion between LPA exon 2 and PLG intron 9, although with low read counts (Figure 5A), and we detected a 255-kb deletion between on-target and OT1 (Figure 5E), resulting in the removal of the entire PLG gene located between LPA and LOC107986665 (Figure S11). The frequency of this deletion event is expected to be low in comparison with other edits. However, since plasminogen is a critical enzyme in fibrinolysis, LPA gene-editing approaches should be carefully assessed to avoid off-target editing or loss of PLG.
In summary, we report the first demonstration of successful in vivo editing of LPA, a major emerging target in CVD risk reduction. A one-time treatment with a gene-editing nuclease could provide permanent removal of Lp(a) for patients at the greatest risk. We also identified challenges in targeting such a polymorphic and repetitive gene with the CRISPR-Cas9 system. Despite the heterogeneity of editing events, we achieved very efficient apo(a) protein removal from the circulation, establishing valuable proof of concept for a new class of Lp(a) therapeutics.
Materials and methods
The data that support the findings of this study are available from the corresponding author upon request. The next-generation sequencing (NGS) data are accessible at NCBI’s Sequence Read Archive available at: https://www.ncbi.nlm.nih.gov/sra.
Plasmid design and cloning
The 1162-pAAV-HLP-EmGFP-SpA plasmid, which encodes an emerald GFP driven by a liver-specific promoter was previously reported and is publicly available through Addgene (Watertown, MA).44 An sgRNA targeting exon 2 of LPA, encoding the KIV1 domain, was designed based on the presence of an NNGRRT PAM for Staphylococcus aureus Cas9 (SaCas9). Cloning of the sgRNA (GCAGGTCCTTCCTGTGACAGTGG) was accomplished by annealing oligonucleotides (Sigma-Aldrich, St. Louis, MO) and ligating into the BbsI site of 1313-pAAV-U6-SA-BbsI-MluI-gRNA-HLP-SACas9-HA-OLLAS-spA (Addgene, Watertown, MA)44 to obtain 1718-pAAV-U6-SA-AMD-LPa-gRNA3-HLP-SACas9-HA-OLLAS-spA.45 The total size of the recombinant AAV genome is 4,662 bp, including the inverted terminal repeats (ITRs). Complete plasmid sequences are available upon request.
AAV production
Recombinant AAV8 vectors were generated as previously described,46 with several modifications.24 Plasmids required for AAV packaging, adenoviral helper plasmid pAdDeltaF6 (PL-F- PVADF6), and AAV8 packaging plasmid pAAV2/8 (PL-T-PV0007) were obtained from the University of Pennsylvania Vector Core. Cell pellets were harvested and purified using a single cesium chloride density gradient centrifugation. Fractions containing AAV were pooled and then dialyzed against PBS using a 100-kDa Spectra-Por Float-A-Lyzer G2 dialysis device (Fisher Scientific, Hampton, NH) to remove the cesium chloride. Purified AAV (now referred to as AAV-GFP and AAV-CRISPR, respectively) were concentrated using a Sartorius Vivaspin Turbo 4 Ultrafiltration Unit and stored at −80°C until use. AAV titers were calculated after DNase digestion using qPCR relative to a standard curve of the transgene plasmid. Primers used for titer are included in Table S2.
Animals
The LPA transgene was constructed in the pLIV.7 vector.47 The vector contains ∼3 kb of the 5′ flanking region of the human APOE gene containing the promotor, exon 1, intron 1 and part of exon 2 of APOE, a 254-bp fragment from the 3′-flanking region of APOE containing the polyadenylation signal sequence, and a liver element consisting of a 1.7-kb fragment from the hepatic control region of the APOE/APOC1 gene locus. An apo(a) cDNA encoding a 14-kringle form of apo(a) (i.e., with all KIV domains as well as the KV and protease domains present and five copies of KIV2) was excised from the corresponding pRK5 mammalian expression plasmid48 by digestion with SalI and inserted into pLIV.7 digested with XhoI. The 14K cDNA also harbors the rs3798220 SNP encoding an Ile-Met substitution in the protease-like domain.49 A linear fragment encompassing the regulatory regions from pLIV.7 and the 14K cDNA were excised by digestion with SalI and SpeI. Transgenic mice were generated by pronuclear injection of the fragment into a C57BL/6 strain at the London Regional Transgenic and Gene Targeting Facility. Founder mice were screened for the transgene and presence of apo(a) in plasma by western blot (described below), and a strain of transgenic mice was established by subsequent breeding of a founder mouse with C57BL/6 mice. The transgene was then bred into Ldlr−/− mice from The Jackson Laboratory (B6.129S7-Ldlrtm1Her/J, stock number 002207, Bar Harbor, ME) to more closely mimic a human lipoprotein profile. The transgene was maintained in the hemizygous or null state (LPA+/0 Ldlr−/− or LPA0/0 Ldlr−/−). The PCR-based amplification of full-length cDNA and PacBio single-molecule long-read sequencing confirmed the 13K cDNA with 5 KIV2 domains and a KIV7-KIV8 hybrid within the LPA+/0LDLR−/− mice. This is still a physiologically relevant form of apo(a) as it contains a small number of KIV2 repeats, and it appears to non-covalently bind to murine apoB-100 likely because KIV6 contains an identical lysine-binding site to KIV7 and KIV8. Mice were housed in a pathogen-free animal facility with a daylight cycle from 0700 to 1900 h. Animals were allowed free access to food and water. Mice were maintained on a standard laboratory diet (irradiated PicoLab Select Rodent 50 IF/6F, LabDiet product code 5V5R, St. Louis, MO). Both male and female mice were used in all experiments unless otherwise stated. Mice were injected intraperitoneally at 8 weeks of age with 1 × 1012 genome copies of AAV expressing either the control (AAV-GFP) or sgRNA and CRISPR-Cas9 (AAV-CRISPR). Fasting for lipid measurements was performed for 5 h, and plasma was obtained by retro-orbital bleeding with heparinized Natelson collecting tubes under the influence of isofluorane anesthesia. Plasma was collected weekly until 12 weeks of age, at which point animals were euthanized to harvest livers for analysis. All procedures were performed according to the regulations and with the prior approval of the Institutional Animal Care and Use Committees of the Baylor College of Medicine (protocol AN-6243) and The University of Western Ontario (protocols 2016-087 and 2020-104).
Genotyping
The LPA transgene was detected by PCR using the primers 5′-CCCCTGTGGTCCGGCAGTGCTACC-3′ and 5′-GGGATGGCAGACAAGCTGGC-3′, which amplify a 955-bp fragment encompassing KIV10, KV, and part of the protease-like domain. The following β-globin control primer pair was used in the same reaction: 5′-CCAATCTGCTCACACAGGATAGAGAGGGCAGG-3′ and 5′-CCTTGAGGCTGCCAAGTGATTCAGGCCATCG-3′. The wild-type Ldlr allele was detected by PCR using the primers 5′-CCATATGCATCCCCAGTCTT-3′ and 5′-GCGATGGATACACTCACTGC-3′, and the null Ldlr allele was detected using primer pair 5′-AATCCATCTTGTTCAATGGCCGATC-3′ and 5′-CCATATGCATCCCCAGTCTT-3′. Cycling conditions were as follows: 5 min at 95°C followed by 35 cycles of 30 s at 95°C, 1 min at 60°C, and 1 min at 72°C, then 3 min at 72°C, and holding at 4°C.
RNA analysis
Mice were fasted for 6 h and then anesthetized using Avertin (0.015–0.017 mL/g body weight; intraperitoneal injection). Hearts and livers were perfused with 10 mL of PBS containing 100 U/mL heparin. The liver, kidney, spleen, heart, small intestine, brain, and quadriceps muscles were removed, weighed, snap-frozen in liquid nitrogen, and stored at −80°C. RNA was extracted from all tissues using TriZol reagent (Invitrogen, Waltham, MA) as per manufacturer’s instructions, and 3 μg of RNA was reverse-transcribed to cDNA using the iScript Advanced cDNA Synthesis kit for RT-qPCR (Bio-Rad, Hercules, CA). Tissue LPA mRNA levels were determined by real-time qPCR using the SsoAdvanced Universal SYBR Green Supermix kit (Bio-Rad, Hercules, CA) and calculated using the ΔΔCT method. The primers used were as follows: LPA 5′-GGCCTCCTTCTGAACAAGAC-3′ and 5′-GAAGAGGATGCACAGAGAGGG-3′; Gapdh 5′-GCGACTTCAACAGCAACTCC-3′ and 5′-TAGCCGTATTCATGGTCATACC-3′.
Western blotting
Mouse plasma (4 μL) was added to 16 μL of 4× SDS-PAGE sample buffer in the presence or absence of 4 μL of 1 M DTT. Samples were then boiled for 7 min, briefly centrifuged, and loaded onto 4%–20% polyacrylamide gradient mini-gels (Bio-Rad, Hercules, CA). After electrophoresis at 120 V for 90 min, proteins were electroblotted (100 V, 90 min) onto polyvinylidene fluoride (PVDF) membranes (Millipore IPFL00010). Membranes were then blocked in 5% (v/v) nonfat dry milk in 20 mM Tris pH 8.0, 150 mM NaCl, 0.1% (v/v) Tween 20 for 1 h, then incubated with a primary antibody against apo(a) in blocking buffer overnight. The primary antibody was either the monoclonal antibody A550 (1:5,000) or a rabbit monoclonal antibody (1:3,000) purchased from Abcam. After washing three times in 20 mM Tris pH 8.0, 150 mM NaCl, 0.1% (v/v) Tween 20, membranes were incubated with horseradish peroxidase-conjugated secondary antibody (sheep anti-mouse immunoglobulin [Ig] G or goat anti-rabbit IgG, as appropriate) (1:5,000) in blocking buffer for 1 h. After three washes, immunoreactive bands were detected using SuperSignal West Pico PLUS chemiluminescent substrate (Thermo Fisher, Waltham, MA) on a Chemi-Doc imager (Bio-Rad, Hercules, CA). Liver tissue was homogenized in ∼10 volumes of radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris pH 8.0, 1 mM EDTA, 1% Triton X-100, 0.1% sodium dodecyl sulfate, 0.5% sodium deoxycholate, 150 mM sodium chloride, and protease inhibitors [Roche 11836153001]) using a Bead Blaster 24 (Benchmark D2400). Protein concentrations were determined using BCA assay (Thermo-Pierce #23227). Liver lysates (38 μg for HA-Tag and 20 μg for GFP) were diluted in 4× LDS buffer (Life Technologies, Ref. NP0007) supplemented with 5% beta-mercaptoethanol and separated by SDS-PAGE using 4%–12% gradient gels (Life Technologies NP0322BOX) at 150 V for 1.5 h. Proteins were transferred to PVDF membranes for either 1.5 h (GFP blot) or 4 h (HA-Tag blot), followed by blocking for 2 h at room temperature in a 2:1 solution of Intercept (PBS) Blocking Buffer (Li-Cor, 927-70001) and PBS with 0.05% Tween 20 (PBS-T). Primary antibodies to the HA-Tag (1:1,000, rabbit, C29F4, 3724, Cell Signaling), GFP (1:3,000, rabbit, A-11122, Thermo Fisher Scientific), and beta tubulin (1:500, mouse, University of Iowa Developmental Studies Hybridoma Bank E7) were diluted in 1% BSA in PBS-T and membranes were incubated overnight at 4 C. Then, blots were washed three times (10 min each) with PBS-T. Goat anti-rabbit 680-nm and anti-mouse 800-nm antibodies (1:15,000, 611-144-002-0.5, and 610-145-002- 0.5, Rockland) were incubated at room temperature for 1.5 h, washed three times (10 min each) with PBS-T, and imaged using an Odyssey Classic Imager (Li-Cor).
Plasma analysis and fast protein liquid chromatography
Plasma lipoprotein separation was performed by the Mouse Metabolic Core (MMC) at Baylor College of Medicine. Plasma pooled from four mice per group (250 μL total) was fractionated by fast protein liquid chromatography (FPLC) using two Superose-6 columns connected in series (Pharmacia FPLC System, Amersham Pharmacia Biotech, Amersham, United Kingdom) as previously described.51, 52, 53 Total plasma cholesterol in lipoprotein fractions was measured using the Cholesterol E Kit (Wako Pure Chemical Industries, Mountain View, CA).
Histology
Mouse livers were fixed overnight in 10% formalin, washed in 70% ethanol for 48 h, and stored in 70% ethanol. Paraffin embedding, sectioning, and antibody staining were performed by the Texas Digestive Diseases Morphology core as previously described.54 Slides were imaged at ×10 magnification on a Nikon Ci-L bright-field microscope at the Integrated Microscopy Core (Baylor College of Medicine). Ki67-and TUNEL-positive cell quantification was performed by manual count, and nuclei were quantified by ImageJ (https://imagej.nih.gov).
ALT and AST assays
Plasma ALT and AST were measured using the Teco Diagnostics ALT (SGPT) Liquid Reagent (A524-150) and Teco Diagnostics AST (SGOT) Liquid Reagent (A559-150), respectively. Plasma was diluted 1:10 and 10 μL of plasma dilution was mixed with 100 μL of reagent mix (five volumes of reagent 1 with one volume of reagent 2, prewarmed at 37 C). NADH absorbance at 340 nm was read at time 0 and every 2–4 min up to 28 min at 37°C using a Tecan Infinite M200 PRO plater reader. ALT and AST levels in plasma from mice with known liver damage (assay positive control) were around 200 and 300 U/L, respectively (data not shown).
Vector genome analysis
DNA was isolated from liver samples using the Qiagen DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany). Serial dilution of a known standard was used as control. The qPCR was completed with 250 ng of DNA using primers against either emGFP 5′- GCATCGACTTCAAGGAGGAC-3′ and 5′- TGCACGCTGCCGTCCTCGATG-3′ or SaCas9 5′-GTACTACGAGGAAACCGGGAAC-3′ and 5′-GTTGTTGTAGAAGGAGGCGATAAAC-3′.
Apo(a) ELISA
Apo(a) was measured using the Mercodia Lp(a) ELISA kit (Mercodia, Winston-Salem, NC) with some modifications to the sample preparation. For sample preparation, 5 μL of sample and 5 μL of pre-treatment solution were mixed and incubated for 1 h at room temperature. Then 1 mL of sample buffer was added to the sample and pre-treatment solution after 1 h and mixed. After the test procedure was completed, the absorbance was then read at 450 nm on an Infinite M200 PRO (Tecan, Morrisville, NC). The U/L measurements were then converted to mg/dL by multiplying by 0.1254 according to the manufacturer’s instructions. LPA0/0 Ldlr−/− pooled controls were used to subtract background absorbance measurements from all samples in the time-course experiments.
Transgene copy number analysis by ddPCR
Fifty micrograms of liver tissue was perfused with PBS and genomic DNA was extracted using the DNeasy Blood and Tissue kit (Qiagen, Hilden, Germany). Concentrations were determined with the Qubit 4 Fluorometer and dsDNA broad-range assay kit (Thermo Fisher, Waltham, MA). Multiple EvaGreen PCR reactions were run simultaneously to quantify the copy number of target and reference loci. EvaGreen-based reaction mixes were prepared with 20 ng of genomic DNA template, 1× QX200 ddPCR EvaGreen Supermix (Bio-Rad, Hercules, CA), 100 nM target or reference primers, and 10 U of EcoRI-HF restriction enzyme in each 20 μL of reaction mix. The copy number of target loci was normalized by the copy number of reference loci to quantify the target loci copy number in mock and treated samples. For the KIV2 CNV assay, multiplex probe-based reaction mixes were prepared with 20 ng of genomic DNA template, 1× ddPCR Supermix for Probes (Bio-Rad, Hercules, CA), 900 nM each KIV2 and RPP30 primers, 250 nM each KIV2 (FAM) and RPP30 (VIC) probes, and 10 U EcoRI-HF restriction enzyme in each 20 μL of reaction mix. The copy number of KIV2 was normalized by RPP30 to quantify the KIV2 CNV in mock and treated samples. PCR was performed according to the manufacturer’s cycling protocol with optimized annealing temperature. Primer and probe sequences are provided in Table S3.
Adaptor ligation-mediated PCR
Genomic DNA from the LPA+/0 Ldlr−/− mouse was fragmented with either the restriction enzyme EcoRI-HF (NEB, Ipswich, MA) or BamHI-HF (NEB, Ipswich, MA). Then, each digested genome was treated with the NEBNext Ultra II EndRepair/dA-Tailing Module (NEB, Ipswich, MA), followed by ligation of the CAST-seq adaptor to the fragments via the NEBNext Ultra II Ligation Module (NEB, Ipswich, MA), providing a bidirectional binding site on every fragment for a universal primer complementary to the adaptor sequence. Both a reverse primer complementary to the 5′ beginning of the LPA construct and a forward primer complementary to the 3′ end of the construct were designed such that endpoint PCR could be performed by pairing each one of these two primers with the universal primer and using the ligated fragmented genomic DNA as template. PCR amplicons were gel extracted and analyzed by Sanger sequencing to identify the LPA transgene construct knockin location in the mouse genome (Figure S2A). A similar assay was performed using enzymatic fragmentation following the manufacturer’s instructions (NEBNext Ultra II FS DNA Library Prep) and sequenced on NGS to identify tandem junctions (Figures S2B and S2C). The primer sequences used for the LPA transgene knockin identification are provided in Table S3.
CAST-seq
The CAST-seq assay was optimized for LPA sgRNA, including decoy primers and a bait primer for the PAM upstream side of DSB, following the protocol described previously.28 Decoy primers that substantially blocked on-target amplification across the LPA target site were selected. Then 500 ng of genomic DNA from AAV-CRISPR- and AAV-GFP-treated male and female mice were subjected to enzymatic fragmentation, end repair, A-tailing, and CAST-seq adapter ligation according to the manufacturer’s instructions (NEBNext Ultra II FS DNA Library Prep). The first PCR was performed using the primers complementary to the linker sequence (linker primer) and a bait primer. Decoy primers were introduced to ensure that only when the binding sites of the decoy primers were lost because of large deletion or translocation did second and third PCR steps amplify the full-length product for NGS. The second PCR utilized nested primers to reduce the non-specific amplification. The third PCR introduced the barcoded Illumina adaptor for sequencing (NEB, NEBNext Multiplex Oligos for Illumina). The libraries were loaded into an Illumina MiSeq Reagent Kit V2-500 cycle (Illumina, San Diego, CA) according to the manufacturer’s instructions. The LPA transgene construct sequence was used to map NGS reads on Integrated Genome Viewer (IGV). For CAST-seq analysis in HEK293T, sequences were processed using the CAST-seq pipeline publicly available on GitHub (https://github.com/AG-Boerries/CAST-Seq).
GUIDE-seq
Samples were prepared for GUIDE-seq analysis, largely following the protocol described in Tsai et al. 2015 and Nobles et al. 2019.32, 33 Briefly, a 2.5-μg px601 plasmid expressing Cas9 and LPA sgRNA and 90-pmol SaCas9/LPA sgRNA RNP along with a 20-pmol iGUIDE dsODN tag were nucleofected into HEK293T using SF buffer and CM-130 program. Genomic DNA was extracted 4 days post electroporation using DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany). Then 500 ng of genomic DNA were subjected to enzymatic fragmentation, end repair, A-tailing, and Y-adapter ligation according to the manufacturer’s instructions (NEBNext Ultra II FS DNA Library Prep). The first round of discovery PCR was done for sense and antisense strands individually using a primer specific to the dsODN tag (GSP1) and a P5 primer. The second round of discovery PCR was done individually for sense and antisense strands with nested primers for the dsODN (GSP2) and P5 primer. Samples were purified and concentrated using 0.7× beads (GE, Sera-Mag Select). Final library quantification and QC were done using Qubit dsDNA HS Assay Kit and KAPA Library Quantification Kit. Libraries were mixed in equal molar concentrations, loaded into a 300-cycle version 2 Illumina kit, and run on a MiSeq (Illumina, San Diego, CA). Sequences were processed using the iGUIDE pipeline publicly available on GitHub (https://github.com/cnobles/iGUIDE). GRCh38/hg38 was used as a reference genome.
T7E1 assay
PCR reactions were carried out with primers surrounding the expected cut site of the LPA gRNA using Q5 Hot Start High-Fidelity DNA Polymerase (NEB, Ipswich, MA). The purified PCR products of the unedited control and experimental samples were denatured and reannealed to produce heteroduplex DNA. The reannealed products were digested with T7E1 (NEB, Ipswich, MA) according to the manufacturer’s protocol. The results were analyzed by agarose gel electrophoresis. The primer sequences used for the T7E1 assay are provided in Table S3.
ICE analysis
PCR reactions were carried out with primers surrounding the expected cutsite of the LPA gRNA using Q5 Hot Start High-Fidelity DNA Polymerase (NEB, Ipswich, MA). The purified PCR products of the unedited control and experimental samples were processed by Sanger sequencing. The resulting sequence trace files (.ab1) were then uploaded into the ICE Web tool (available at https://ice.synthego.com/#/) for indels quantification. The primer sequences used for the ICE assay are provided in Table S3.
Library preparation for PacBio sequencing
The long-range PCR was performed using genomic DNA (gDNA) from AAV-GFP- or AAV-CRISPR-treated mouse. PCR reaction contained 100 ng of gDNA, 200 nM of primers in 100-μL reaction (LongAmp Hot Start Taq 2X Master Mix, NEB). The PCR program consisted of initial denaturation (2 min at 94°C) and 30 cycles of denaturation (30 s at 94°C), annealing (30 s at 60°C), and extension (6 min at 65°C). Five-hundred nanograms of the long-range PCR amplicons were used for PacBio library preparation, which consists of DNA damage repair, end repair/A-tail, SMRTbell adaptor ligation (SMRTbell Express Template Prep Kit 2.0), nuclease treatment (SMRTbell Enzyme Clean Up Kit), and AMPure bead purification following the standard protocol. The SMRT-bell library was sequenced on a PacBio Sequel II 8M flowcell in circular consensus sequencing (CCS) mode following the standard protocol with 1-h pre-extension and 30-h collection time (Pacific Biosciences). The PacBio subreads were converted to HiFi reads; 48,824 and 56,878 HiFi reads with sequencing accuracy above 99.9% in AAV-GFP- and AAV-CRISPR-treated samples, respectively, were used for analysis.
Statistics
Animal numbers were estimated based on previous experience with each specific assay and expected effect size. No pre-randomization was performed, and researchers were not blind to genotype. Animals were sex- and age-matched for all experiments as detailed in the figure legends. Data were tested for normal distribution using the Shapiro-Wilk normality tests. Differences between two groups were assessed with either Welch’s t test for normally distributed data or Mann-Whitney test for non-normally distributed data as detailed in figure legends. All data are shown as the mean ± standard deviation (SD). Comparisons involving two groups was done using Welch’s t test with ∗p < 0.05. For comparisons involving three or more groups, an ANOVA with post hoc Tukey test was applied and ∗p < 0.05. GraphPad Prism 7 was used for statistical analyses.
Data availability
The data that support the findings of this study are available from the corresponding author upon request.
Acknowledgments
We would like to thank Dr. Pradip Kumar Saha and the Mouse Metabolic Core at Baylor College of Medicine for performing the fast performance liquid chromatography. This work was supported by American Heart Association fellowships (19PRE34380467 to A.M.D. and 19POST34430092 to M.D.G.), National Institutes of Health (R01HL152314 and UG3HL151545 to G.B.; U42OD026645, R01HL132840 and R01DK124477 to W.R.L.), NIH T32 training grant (T32HL07676 to A.M.D.), Canadian Institutes of Health Research project grant (PG-45375 to M.L.K. and M.B.B.), Heart and Stroke Foundation of Canada grant in aid (G-17-0018740 to M.L.K. and M.B.B.), and Natural Sciences and Engineering Research Council of Canada of Canada discovery grant (RGPIN-2015-05006 to M.L.K.). Imaging for this project was supported by the Integrated Microscopy Core at Baylor College of Medicine and the Center for Advanced Microscopy and Image Informatics (CAMII) with funding from NIH (DK56338, CA125123, ES030285) and CPRIT (RP150578, RP170719). This work was also supported by the Texas Digestive Diseases Morphology Core (P30DK56338).
Author contributions
A.M.D., S.H.P., J.M.A., G.B., M.B.B., M.L.K., and W.R.L. conceived the project and designed the studies. A.M.D. and S.H.P. performed and analyzed most of the experiments. A.E.H. produced the viral vectors. A.M.D., A.L., M.D.G., and M.C. conducted in vivo experiments. J.M.A., M.B.B., and M.L.K. generated the transgenic mice. J.M.A. performed RNA and western blot analysis. L.S. conducted experiments to assess gene-editing frequencies. A.Y. completed the adaptor ligation-mediated PCR. M.D.G. performed AST, ALT, and western blot experiments. A.M.D., S.H.P., M.B.B., M.L.K., and W.R.L. wrote the manuscript, which was revised and approved by all authors.
Declaration of interests
M.L.K. has held a research grant from Pfizer; is a member of advisory boards for Amya Therapeutics and Novartis; has received speaker’s honoraria/consulting fees from Amgen, Regeneron, and Eli Lilly; and holds/has held research contracts with Sanofi, Ionis, Eli Lilly, and Abcentra. M.B.B. has held a research contract with Ionis. S.M.M. has received consulting fees from Roche, Denka, and Novartis and research support from Amgen through Medpace.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2022.10.009.
Supplemental information
References
- 1.Berg K. A new serum type in man- the Lp system. Acta Pathol. Microbiol. Scand. 1963;59:369–382. doi: 10.1111/j.1699-0463.1963.tb01808.x. [DOI] [PubMed] [Google Scholar]
- 2.Berg K., Dahlén G., Frick M.H. Lp(a) lipoprotein and pre-β1-lipoprotein in patients with coronary heart disease. Clin. Genet. 1974;6:230–235. doi: 10.1111/j.1399-0004.1974.tb00657.x. [DOI] [PubMed] [Google Scholar]
- 3.Djurovic S., Berg K. Epidemiology of Lp(a) lipoprotein: its role in atherosclerotic/thrombotic disease. Clin. Genet. 1997;52:281–292. doi: 10.1111/j.1399-0004.1997.tb04345.x. [DOI] [PubMed] [Google Scholar]
- 4.Emerging Risk Factors Collaboration. Erqou S., Kaptoge S., Perry P.L., Di Angelantonio E., Thompson A., White I.R., Marcovina S.M., Collins R., Thompson S.G., Danesh J. Lipoprotein(a) concentration and the risk of coronary heart disease, Stroke, and nonvascular mortality the emerging risk factors collaboration ∗ Europe PMC funders group. JAMA. 2009;302:412–423. doi: 10.1001/jama.2009.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Erqou S., Thompson A., Di Angelantonio E., Saleheen D., Kaptoge S., Marcovina S., Danesh J. Apolipoprotein(a) isoforms and the risk of vascular disease. Systematic review of 40 studies involving 58, 000 participants. J. Am. Coll. Cardiol. 2010;55:2160–2167. doi: 10.1016/j.jacc.2009.10.080. [DOI] [PubMed] [Google Scholar]
- 6.Clarke R., Peden J.F., Hopewell J.C., Kyriakou T., Goel A., Heath S.C., Parish S., Barlera S., Franzosi M.G., Rust S., et al. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N. Engl. J. Med. 2009;361:2518–2528. doi: 10.1056/NEJMoa0902604. [DOI] [PubMed] [Google Scholar]
- 7.Kamstrup P.R., Tybjærg-hansen A., Steffensen R., Nordestgaard B.G. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301:2331–2339. doi: 10.1001/jama.2009.801. [DOI] [PubMed] [Google Scholar]
- 8.Tsimikas S., Fazio S., Ferdinand K.C., Ginsberg H.N., Koschinsky M.L., Marcovina S.M., Moriarty P.M., Rader D.J., Remaley A.T., Reyes-Soffer G., et al. NHLBI working group recommendations to reduce lipoprotein(a)-mediated risk of cardiovascular disease and aortic stenosis. J. Am. Coll. Cardiol. 2018;71:177–192. doi: 10.1016/j.jacc.2017.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kamstrup P.R., Tybjærg-Hansen A., Nordestgaard B.G. Elevated lipoprotein(a) and risk of aortic valve stenosis in the general population. J. Am. Coll. Cardiol. 2014;63:470–477. doi: 10.1016/j.jacc.2013.09.038. [DOI] [PubMed] [Google Scholar]
- 10.Koschinsky M.L., Marcovina S.M. Structure-function relationships in apolipoprotein(a): insights into lipoprotein(a) assembly and pathogenicity. Curr. Opin. Lipidol. 2004;15:167–174. doi: 10.1097/00041433-200404000-00009. [DOI] [PubMed] [Google Scholar]
- 11.McLean J.W., Tomlinson J.E., Kuang W.J., Eaton D.L., Chen E.Y., Fless G.M., Scanu A.M., Lawn R.M. cDNA sequence of human apolipoprotein(a) is homologous to plasminogen. Nature. 1987;330:132–137. doi: 10.1038/330132a0. [DOI] [PubMed] [Google Scholar]
- 12.Rader D.J., Cain W., Ikewaki K., Talley G., Zech L.A., Usher D., Brewer H.B. The inverse association of plasma lipoprotein(a) concentrations with apolipoprotein(a) isoform size is not due to differences in Lp(a) catabolism but to differences in production rate. J. Clin. Invest. 1994;93:2758–2763. doi: 10.1172/JCI117292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thanassoulis G. Screening for high lipoprotein(a) the time is now. Circulation. 2019;139:1493–1496. doi: 10.1161/CIRCULATIONAHA.119.038989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nordestgaard B.G., Langlois M.R., Langsted A., Chapman M.J., Aakre K.M., Baum H., Borén J., Bruckert E., Catapano A., Cobbaert C., et al. Quantifying atherogenic lipoproteins for lipid-lowering strategies: consensus-based recommendations from EAS and EFLM. Atherosclerosis. 2020;294:46–61. doi: 10.1016/j.atherosclerosis.2019.12.005. [DOI] [PubMed] [Google Scholar]
- 15.Reyes-Soffer G., Ginsberg H.N., Berglund L., Duell P.B., Heffron S.P., Kamstrup P.R., Lloyd-Jones D.M., Marcovina S.M., Yeang C., Koschinsky M.L., American Heart Association Council on Arteriosclerosis, Thrombosis and Vascular Biology; Council on Cardiovascular Radiology and Intervention; and Council on Peripheral Vascular Disease Lipoprotein(a): a genetically determined, causal, and prevalent risk factor for atherosclerotic cardiovascular disease: a scientific statement from the American heart association. Arterioscler. Thromb. Vasc. Biol. 2022;42:E48–E60. doi: 10.1161/ATV.0000000000000147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsimikas S., Karwatowska-Prokopczuk E., Gouni-Berthold I., Tardif J.-C., Baum S.J., Steinhagen-Thiessen E., Shapiro M.D., Stroes E.S., Moriarty P.M., Nordestgaard B.G., et al. Lipoprotein(a) reduction in persons with cardiovascular disease. N. Engl. J. Med. 2020;382:244–255. doi: 10.1056/NEJMoa1905239. [DOI] [PubMed] [Google Scholar]
- 17.Koren M.J., Moriarty P.M., Baum S.J., Neutel J., Hernandez-Illas M., Weintraub H.S., Florio M., Kassahun H., Melquist S., Varrieur T., et al. Preclinical development and phase 1 trial of a novel siRNA targeting lipoprotein(a) Nat. Med. 2022;28:96–103. doi: 10.1038/s41591-021-01634-w. [DOI] [PubMed] [Google Scholar]
- 18.Schmidt K., Noureen A., Kronenberg F., Utermann G. Structure, function, and genetics of lipoprotein (a) J. Lipid Res. 2016;57:1339–1359. doi: 10.1194/jlr.R067314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Emdin C.A., Khera A.V., Natarajan P., Klarin D., Won H.-H., Peloso G.M., Stitziel N.O., Nomura A., Zekavat S.M., Bick A.G., et al. Phenotypic characterization of genetically lowered human lipoprotein(a) levels. J. Am. Coll. Cardiol. 2016;68:2761–2772. doi: 10.1016/j.jacc.2016.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adli M. The CRISPR tool kit for genome editing and beyond. Nat. Commun. 2018;9:1911. doi: 10.1038/s41467-018-04252-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doudna J.A., Charpentier E. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- 22.Ran F.A., Cong L., Yan W.X., Scott D.A., Gootenberg J.S., Kriz A.J., Zetsche B., Shalem O., Wu X., Makarova K.S., et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–191. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jarrett K.E., Lee C.M., Yeh Y.H., Hsu R.H., Gupta R., Zhang M., Rodriguez P.J., Lee C.S., Gillard B.K., Bissig K.D., et al. Somatic genome editing with CRISPR/Cas9 generates and corrects a metabolic disease. Sci. Rep. 2017;7:44624. doi: 10.1038/srep44624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jarrett K.E., Lee C., De Giorgi M., Hurley A., Gillard B.K., Doerfler A.M., Li A., Pownall H.J., Bao G., Lagor W.R. Somatic editing of Ldlr with adeno-associated viral-CRISPR is an efficient tool for atherosclerosis research. Arterioscler. Thromb. Vasc. Biol. 2018;38:1997–2006. doi: 10.1161/ATVBAHA.118.311221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chadwick A.C., Evitt N.H., Lv W., Musunuru K. Reduced blood lipid levels with in vivo CRISPR-Cas9 base editing of ANGPTL3. Circulation. 2018;137:975–977. doi: 10.1161/CIRCULATIONAHA.117.031335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boffelli D., Cheng J.F., Rubin E.M. Convergent evolution in primates and an insectivore. Genomics. 2004;83:19–23. doi: 10.1016/s0888-7543(03)00148-4. [DOI] [PubMed] [Google Scholar]
- 27.Davidoff A.M., Ng C.Y.C., Zhou J., Spence Y., Nathwani A.C. Sex significantly influences transduction of murine liver by recombinant adeno-associated viral vectors through an androgen-dependent pathway. Blood. 2003;102:480–488. doi: 10.1182/blood-2002-09-2889. [DOI] [PubMed] [Google Scholar]
- 28.Turchiano G., Andrieux G., Klermund J., Blattner G., Pennucci V., el Gaz M., Monaco G., Poddar S., Mussolino C., Cornu T.I., et al. Quantitative evaluation of chromosomal rearrangements in gene-edited human stem cells by CAST-Seq. Cell Stem Cell. 2021;28:1136–1147.e5. doi: 10.1016/j.stem.2021.02.002. [DOI] [PubMed] [Google Scholar]
- 29.Amarasinghe S.L., Su S., Dong X., Zappia L., Ritchie M.E., Gouil Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020;21:30. doi: 10.1186/s13059-020-1935-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Würtele H., Gusew N., Lussier R., Chartrand P. Characterization of in vivo recombination activities in the mouse embryo. Mol. Genet. Genomics. 2005;273:252–263. doi: 10.1007/s00438-005-1112-2. [DOI] [PubMed] [Google Scholar]
- 31.Hommelsheim C.M., Frantzeskakis L., Huang M., Ülker B. PCR amplification of repetitive DNA: a limitation to genome editing technologies and many other applications. Sci. Rep. 2014;4:5052. doi: 10.1038/srep05052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nobles C.L., Reddy S., Salas-Mckee J., Liu X., June C.H., Melenhorst J.J., Davis M.M., Zhao Y., Bushman F.D. IGUIDE: an improved pipeline for analyzing CRISPR cleavage specificity. Genome Biol. 2019;20:14. doi: 10.1186/s13059-019-1625-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsai S.Q., Zheng Z., Nguyen N.T., Liebers M., Topkar V.V., Thapar V., Wyvekens N., Khayter C., Iafrate A.J., Le L.P., et al. GUIDEseq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015;33:187–197. doi: 10.1038/nbt.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lanktree M.B., Rajakumar C., Brunt J.H., Koschinsky M.L., Connelly P.W., Hegele R.A. Determination of lipoprotein(a) kringle repeat number from genomic DNA: copy number variation genotyping using qPCR. J. Lipid Res. 2009;50:768–772. doi: 10.1194/jlr.D800050-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mahley R.W. Central nervous system lipoproteins: ApoE and regulation of cholesterol metabolism. Arterioscler. Thromb. Vasc. Biol. 2016;36:1305–1315. doi: 10.1161/ATVBAHA.116.307023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu M., Kuhel D.G., Shen L., Hui D.Y., Woods S.C. Apolipoprotein E does not cross the blood-cerebrospinal fluid barrier, as revealed by an improved technique for sampling CSF from mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012;303:R903–R908. doi: 10.1152/ajpregu.00219.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Becker L., McLeod R.S., Marcovina S.M., Yao Z., Koschinsky M.L. Identification of a critical lysine residue in apolipoprotein B-100 that mediates noncovalent interaction with apolipoprotein(a) J. Biol. Chem. 2001;276:36155–36162. doi: 10.1074/jbc.M104789200. [DOI] [PubMed] [Google Scholar]
- 38.Becker L., Cook P.M., Wright T.G., Koschinsky M.L. Quantitative evaluation of the contribution of weak lysine-binding sites present within apolipoprotein(a) kringle IV types 6-8 to lipoprotein(a) assembly. J. Biol. Chem. 2004;279:2679–2688. doi: 10.1074/jbc.M309414200. [DOI] [PubMed] [Google Scholar]
- 39.Pasi K.J., Rangarajan S., Mitchell N., Lester W., Symington E., Madan B., Laffan M., Russell C.B., Li M., Pierce G.F., et al. Multiyear follow-up of AAV5-hFVIII-SQ gene therapy for Hemophilia A. N. Engl. J. Med. 2020;382:29–40. doi: 10.1056/NEJMoa1908490. [DOI] [PubMed] [Google Scholar]
- 40.Racanelli V., Rehermann B. The liver as an immunological organ. Hepatology. 2006;43:S54–S62. doi: 10.1002/hep.21060. [DOI] [PubMed] [Google Scholar]
- 41.Brancale J., Vilarinho S. A single cell gene expression atlas of 28 human livers. J. Hepatol. 2021;75:219–220. doi: 10.1016/j.jhep.2021.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vakulskas C.A., Behlke M.A. Evaluation and reduction of crispr off-target cleavage events. Nucleic Acid Ther. 2019;29:167–174. doi: 10.1089/nat.2019.0790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cradick T.J., Qiu P., Lee C.M., Fine E.J., Bao G. COSMID: a web-based tool for identifying and validating CRISPR/Cas off-target sites. Mol. Ther. Nucleic Acids. 2014;3:e214. doi: 10.1038/mtna.2014.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li A., Lee C.M., Hurley A.E., Jarrett K.E., De Giorgi M., Lu W., Balderrama K.S., Doerfler A.M., Deshmukh H., Ray A., et al. A self-deleting AAV-CRISPR system for in vivo genome editing. Mol. Ther. Methods Clin. Dev. 2019:111–122. doi: 10.1016/j.omtm.2018.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.De Giorgi M., Jarrett K.E., de Aguiar Vallim T.Q., Lagor W.R. In: Atherosclerosis: Methods and Protocols. Ramji D., editor. Springer US; 2022. In vivo gene editing in lipid and atherosclerosis research; pp. 673–713. [DOI] [PubMed] [Google Scholar]
- 46.Lagor W.R., Johnston J.C., Lock M., Vandenberghe L.H., Rader D.J. Adeno-associated viruses as liver-directed gene delivery vehicles: focus on lipoprotein metabolism. Methods Mol. Biol. 2013;1027:273–307. doi: 10.1007/978-1-60327-369-5_13. [DOI] [PubMed] [Google Scholar]
- 47.Fan J., Wang J., Bensadouni A., Lauer S.J., Dang Q., Mahley R.W., Ii J.M.T. Overexpression of hepatic lipase in transgenic rabbits leads to a marked reduction of plasma high density lipoproteins and intermediate density lipoproteins. Proc. Natl. Acad. Sci. USA. 1994:8724–8728. doi: 10.1073/pnas.91.18.8724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gabel B., Yao Z., McLeod R.S., Young S.G., Koschinsky M.L. Carboxyl-terminal truncation of apolipoproteinB-100 inhibits lipoprotein(a) particle formation. FEBS Lett. 1994;350:77–81. doi: 10.1016/0014-5793(94)00737-3. [DOI] [PubMed] [Google Scholar]
- 49.Scipione C.A., McAiney J.T., Simard D.J., Bazzi Z.A., Gemin M., Romagnuolo R., Macrae F.L., Ariëns R.A., Hegele R.A., Auld J., et al. Characterization of the I4399M variant of apolipoprotein(a): implications for altered prothrombotic properties of lipoprotein(a) J. Thromb. Haemost. 2017;15:1834–1844. doi: 10.1111/jth.13759. [DOI] [PubMed] [Google Scholar]
- 50.Marcovina S.M., Albers J.J., Gabel B., Koschinsky M.L., Gaur V.P. Effect of the number of apolipoprotein(a) kringle 4 domains on immunochemical measurements of lipoprotein(a) Clin. Chem. 1995;41:246–255. [PubMed] [Google Scholar]
- 51.Chang B.H.J., Liao W., Li L., Nakamuta M., Mack D., Chan L. Liver-specific inactivation of the abetalipoproteinemia gene completely abrogates very low density lipoprotein/low density lipoprotein production in a viable conditional knockout mouse. J. Biol. Chem. 1999;274:6051–6055. doi: 10.1074/jbc.274.10.6051. [DOI] [PubMed] [Google Scholar]
- 52.Son S.H., Goo Y.H., Choi M., Saha P.K., Oka K., Chan L.C.B., Paul A. Enhanced atheroprotection and lesion remodelling by targeting the foam cell and increasing plasma cholesterol acceptors. Cardiovasc. Res. 2016;109:294–304. doi: 10.1093/cvr/cvv241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bissig-Choisat B., Wang L., Legras X., Saha P.K., Chen L., Bell P., Pankowicz F.P., Hill M.C., Barzi M., Leyton C.K., et al. Development and rescue of human familial hypercholesterolaemia in a xenograft mouse model. Nat. Commun. 2015;6:7339. doi: 10.1038/ncomms8339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.De Giorgi M., Jarrett K.E., Burton J.C., Doerfler A.M., Hurley A., Li A., Hsu R.H., Furgurson M., Patel K.R., Han J., et al. Depletion of essential isoprenoids and ER stress induction following acute liver-specific deletion of HMG-CoA Reductase. J. Lipid Res. 2020;61:1675–1686. doi: 10.1194/jlr.RA120001006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon request.