

Abstract
The immune responses to Helicobacter pylori infection play important roles in gastroduodenal diseases. The contribution of gamma interferon (IFN-γ) to the immune responses, especially to the induction of gastric inflammation and to protection from H. pylori infection, was investigated with IFN-γ gene knockout (IFN-γ−/−) mice. We first examined the colonizing abilities of eight H. pylori strains with a short-term infection test in order to select H. pylori strains which could colonize the mouse stomach. Only three strains (ATCC 43504, CPY2052, and HPK127) colonized C57BL/6 wild-type mice, although all of the strains except for ATCC 51110 could colonize IFN-γ−/− mice. The number of H. pylori organisms colonizing the stomach in wild-type mice was lower than that in IFN-γ−/− mice. Oral immunization with the CPY2052 sonicate and cholera toxin protected against infection with strain CPY2052 in both types of mouse. These findings suggested that IFN-γ may play a protective role in H. pylori infection, although the degree of its protective ability was estimated to be low. In contrast, in a long-term infection test done to examine the contribution of IFN-γ to gastric inflammation, CPY2052-infected wild-type mice developed a severe infiltration of mononuclear cells in the lamina propria and erosions in the gastric epithelium 15 months after infection, whereas CPY2052-infected IFN-γ−/− mice showed no inflammatory symptoms. This result clearly demonstrated that IFN-γ plays an important role in the induction of gastric inflammation caused by H. pylori infection.
It is now generally accepted that Helicobacter pylori infection is a major cause of chronic gastritis and peptic ulcers. It has been suggested that the histological damage due to the ammonia produced by H. pylori urease (19), the vacuolization of gastric epithelium caused by the cytotoxin derived from H. pylori (8), and the mucosal injury caused by the host immune responses (1, 10, 15, 16) are important in the pathogenesis of these gastroduodenal diseases. Although it has been suspected that the activation of immune cells is a pivotal factor and that cytokines contribute to their activation, the mechanisms have not been clearly defined.
We previously investigated the cytokine expression patterns of human gastric mucosal biopsy specimens by the reverse transcription-PCR method (20, 21). Since the results showed that the expression of interleukin-8 (IL-8) mRNA was significantly higher in H. pylori-positive gastritis than in H. pylori-negative controls and that there was a close correlation between the expression of IL-8 mRNA and the severity of gastritis, we suggested that IL-8 could play an important role(s) in mucosal inflammation.
In addition, gamma interferon (IFN-γ) and IL-1β mRNAs were detected in the vast majority of the specimens examined in those studies, whereas IL-2, IL-3, IL-4, IL-5, IL-9, IFN-β, and tumor necrosis factor alpha mRNAs were not detected in any specimens and IL-6 mRNA was detected in only a few specimens. IFN-γ is thought to be important in immune responses, because it induces the expression of the class II major histocompatibility complex of antigen-presenting cells and activates macrophages and natural killer cells.
In an experiment with IFN-γ gene knockout (IFN-γ−/−) and wild-type mice, Dalton et al. showed that IFN-γ enhanced the expression of the class II major histocompatibility complex on the surface of peritoneal macrophages during Mycobacterium bovis BCG infection and that IFN-γ contributed to protection from BCG infection (2). Tagawa et al. reported that the development of concanavalin A-induced hepatitis was reduced significantly in IFN-γ gene knockout mice compared with that in wild-type mice (18). Thus, IFN-γ appears to be involved in protection from bacterial infection and in induction of inflammation. Although the expression of IFN-γ mRNA was not specific to H. pylori-associated gastritis, the finding that the expression of IFN-γ mRNA always took place in gastric mucosa (20) may indicate that IFN-γ plays an important role(s) in the induction of immune responses in gastroduodenal mucosa.
For investigations of the role of IFN-γ in H. pylori infection, in vivo studies with IFN-γ gene knockout mice are an effective means. Although colonization by H. pylori of the mouse stomach had previously been considered difficult, Marchetti et al. (12) and Lee et al. (11) recently identified H. pylori strains with colonizing ability in the mouse stomach. Marchetti et al. (12) showed, by examining the colonizing ability of both VacA+ and VacA− H. pylori strains in CD1 mice, that only VacA+ H. pylori strains could provoke the infiltration of inflammatory cells and proposed the VacA+ H. pylori SPM326-infected CD1 mouse model. Lee et al. (11) isolated a strain of H. pylori with high colonizing ability, the Sydney strain, by screening fresh clinical H. pylori isolates with BALB/c and SJL mice and suggested that the Sydney strain could become a standard strain of H. pylori for use in mouse models. Marchetti et al. (12) and Lee et al. (11) noted that their H. pylori-infected mouse models could be used for the study of pathogenesis, the screening of novel therapeutic agents, and the development of vaccines. However, in order to confirm the contribution of IFN-γ to H. pylori-induced gastric inflammation, it is necessary to develop new mouse models which can conclusively define the contribution of this cytokine. In the present study, we designed an H. pylori-infected IFN-γ−/− mouse model and investigated the role of IFN-γ in protection from H. pylori infection and in the development of gastric inflammation.
MATERIALS AND METHODS
Animals.
IFN-γ−/− and wild-type C57BL/6 mice were used in this study. All of the mice were specific-pathogen-free 6-week-old males. The wild-type C57BL/6 mice were purchased from Charles River Japan (Atsugi, Japan). IFN-γ−/− mice were generated by a targeted mutation of the IFN-γ gene as described previously (18). Briefly, a nonfunctional IFN-γ gene was created by inserting the β-galactosidase gene and the neomycin resistance gene at the translation initiation site in the first exon of the IFN-γ gene. The targeting vector containing the disrupted IFN-γ gene was transfected into A3-1 embryonic stem cells. G418-resistant embryonic stem cells were selected and analyzed by PCR and Southern blot hybridization to examine targeted disruption of the IFN-γ gene. These cells were injected into blastocysts from superovulating C57BL/6 females, and then the embryos were transferred to the uteri of pseudopregnant ICR recipients. The male chimeras were crossed with C57BL/6 females, and then embryonic stem cell-derived pups were backcrossed to C57BL/6 mice. The genotypes of the IFN-γ locus were checked by Southern blot hybridization. Homozygous IFN-γ−/− mice were produced by intercrossing heterozygous mice.
The inactivation of the IFN-γ gene function of offspring mice was verified by measuring IFN-γ mRNA in spleen cells from offspring mice stimulated with phorbol myristate acetate and ionomycin and IFN-γ protein in cultures of spleen cells from offspring mice treated in the same manner. IFN-γ mRNA was detected by Northern blot hybridization analysis. IFN-γ protein was measured by an enzyme-linked immunosorbent assay (ELISA) (18).
The mice were all maintained in a barrier animal facility under pathogen-free conditions at Kyoto Prefectural University of Medicine. All equipment and supplies, including cages, water bottles, wooden chips, and food pellets, were sterilized. Under these conditions, both IFN-γ−/− mice and wild-type mice grew normally and were healthy.
Bacteria.
The following H. pylori strains were used in this study. H. pylori ATCC 43504 and ATCC 51110 were obtained from the American Type Culture Collection (Rockville, Md.). H. pylori CPY2052, CPY3401, and HPK127 were kind gifts from M. Karita (Houfu Onsen Hospital, Houfu, Japan). H. pylori KP41a, KP48a, and KP64b were isolated from human gastric biopsy specimens at Kyoto Prefectural University of Medicine. All eight strains were gram-negative spiral rods and were positive for the H. pylori cagA and vacA genes.
Preparation of bacterial inocula.
Lyophilized ATCC 43504 and ATCC 51110 were suspended in phosphate-buffered saline (PBS) (pH 7.4), streaked on Helicobacter-selective brain heart infusion (BHI) agar plates containing 7% (vol/vol) horse blood, vancomycin (10 μg/ml), polymyxin B (2.5 U/ml), trimethoprim (5 μg/ml), and amphotericin B (2 μg/ml) (HSBHI agar plate) (Eiken Chemical Co., Tokyo, Japan), and incubated for 5 days at 37°C with an Aeropack System charged with a gas mixture consisting of 80% N2, 15% CO2, and 5% O2 (Mitsubishi Gas Chemical Co., Tokyo, Japan). One milliliter of PBS was added to each incubated plate, and then the plate was rinsed. Two hundred microliters of the rinse solution was added to 5 ml of BHI broth containing 4% (vol/vol) fetal bovine serum (FBS) in a 20-ml test tube and cultivated for 24 h at 37°C under a gas mixture consisting of 80% N2, 15% CO2, and 5% O2 on a reciprocal shaker (200 rpm). One milliliter of FBS and 1 ml of glycerol were added to this culture, and it was stored at −80°C (stock culture).
Frozen CPY2052, CPY3401, HPK127, KP41a, KP48a, and KP64b in BHI broth containing 15% (vol/vol) FBS and 15% (vol/vol) glycerol were thawed, streaked on HSBHI agar plates, and treated in the same manner as described above. Two hundred microliters of the stock culture was added to 5 ml of BHI broth containing 4% (vol/vol) FBS in a 20-ml test tube and cultivated for about 24 h at 37°C under the gas mixture on a reciprocal shaker (200 rpm). At this time the bacterial growth had reached the late logarithmic phase. After we confirmed that the bacterial concentration of this culture was 2.0 × 108 to 3.0 × 108 CFU/ml by measuring the optical density at 550 nm with a spectrophotometer (UV-120-01; Shimadzu Co., Kyoto, Japan), we used this culture as the inoculum preparation for the mouse infection experiments.
Antigen preparation for oral immunization.
The inoculum preparation of H. pylori CPY2052 described above was centrifuged at 5,000 × g for 10 min. The pellet was washed three times with 20 ml of PBS (pH 7.4) by centrifugation, weighed as wet matter, and suspended in PBS to obtain a 10% precipitate concentration (wet weight of precipitate/volume). The suspension was sonicated twice for 10 min each time with an interval of 10 min by use of an ultrasonic disrupter (Biorupter UCD 130; Tosho Denki Co., Yokohama, Japan) immersed in a melting ice bath. The protein content of the bacterial sonicate was determined by the Lowry method, adjusted to 10 mg of protein/ml with PBS, and used as the antigen for oral immunization and for an ELISA. The antigen preparations were stored at −80°C in aliquots until use.
Experimental protocol for H. pylori infection.
All mice used in this study were challenged only once at the beginning of the experiment, after 8 h of fasting, with 0.5 ml of the inoculum preparation of H. pylori or PBS (pH 7.4) (as an uninfected control) by use of an oral stainless steel catheter. The mice were then maintained on the fast for 4 h and housed as described above. The colonization of gastric mucosa was assessed at specified times. Each experimental group comprised 6 to 10 mice.
Experimental protocol for oral immunization.
Wild-type and IFN-γ−/− mice were each divided into three groups and administered one of the following three kinds of antigen solution: group 1, 0.2 ml of PBS (pH 7.4) alone; group 2, 0.2 ml of PBS containing 100 μl of the CPY2052 sonicate; and group 3, 0.2 ml of PBS containing 100 μl of the CPY2052 sonicate plus 10 μg of cholera toxin (Sigma Chemical Co., St. Louis, Mo.). Antigen administration was carried out at days 0, 7, and 14, after 8 h of fasting, by use of an oral stainless steel catheter. At day 21, all groups were challenged with 0.5 ml of the inoculum preparation of CPY2052 after 8 h of fasting. After the administration of the antigen solution or the inoculum preparation, fasting was continued for 4 h. Colonization of the gastric mucosa and the level of H. pylori-specific serum immunoglobulin G (IgG) in each group were assessed at day 42. Each experimental group comprised 6 to 10 mice.
Microbiological evaluation.
Mice were sacrificed at specified times, and their stomachs were resected. The resected stomachs were divided longitudinally into two halves and weighed. One half of each resected stomach was added to 1 ml of PBS and homogenized with a tissue grinder (Pellet Pestle Mixer; Kimble/Kontes). A 50-μl portion of the homogenized stomach was plated on an HSBHI agar plate and incubated for 5 days at 37°C with an Aeropack System as described above. The colonies on the plate were counted and expressed as CFU per gram of tissue.
For determination of whether the organisms of the colonies were H. pylori, a PCR was carried out with DNA prepared from the colonies. Five of the colonies on the plate were scratched and suspended in 1 ml of PBS. The suspension was centrifuged (10,000 × g for 5 min), and the precipitate was washed with 1 ml of PBS by centrifugation. The precipitate was resuspended in 50 μl of distilled water and boiled for 10 min. The supernatant obtained by centrifugation (10,000 × g for 5 min) was stored at −20°C until use as a PCR template (bacterial DNA preparation).
Four oligonucleotide primers were designed based on the published sequences of the H. pylori ureB and cagA genes and synthesized with a DNA synthesizer (Gene Assembler Plus; Pharmacia Biotech, Uppsala, Sweden) as described previously (21). The primers for the ureB gene were 5′-TGGGATTAGCGAGTATGT-3′ (sense) and 5′-CCCATTTGACTCAATG-3′ (antisense), and the primers for the cagA gene were 5′-GATAACAGGCAAGCTTTTGAGG-3′ (sense) and 5′-CTGCAAAAGATTGTTTGGCAGA-3′ (antisense).
Thirty microliters of the bacterial DNA preparation was denatured by heating at 95°C for 10 min and cooled on ice for 5 min. Five microliters of this denatured DNA preparation was used in the PCR. Fifty microliters of the reaction mixture consisted of 10 mM Tris-HCl (pH 8.8), 0.1% Triton X-100, 50 mM KCl, 1.5 mM MgCl2, 20 μM 2′-deoxynucleotide 5′-phosphates (Pharmacia Biotech), 20 nM each primer, 1.0 U of Taq DNA polymerase (Boehringer GmbH, Mannheim, Germany), and 5 μl of the denatured DNA preparation. Amplification was performed with an automated thermal cycler (TP-3000; Takara Shuzo Co., Kyoto, Japan) for 35 cycles, each of which consisted of 1 min at 95°C for denaturation, 1 min at 50°C for annealing, and 1 min at 72°C for extension. The final cycle included an extension step for 7 min at 72°C to ensure full extension of the product. Ten microliters of each PCR product was analyzed by electrophoresis on a 1.5% agarose gel containing ethidium bromide, and the bands were examined under UV light.
Histological evaluation.
The other half of each resected stomach was used for the histological evaluation. Longitudinal sections from the gastroesophageal junction to the gastroduodenal junction were fixed in neutral buffered 10% formalin, embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin and eosin. The degrees of inflammation were assessed by microscopic observation without knowledge of the experimental groups and was expressed as follows: none, normal appearance of scattered mononuclear cells on the lamina propria (the same degree as in uninfected control mice); mild, moderate infiltration of mononuclear cells in the lamina propria and the submucosa and no erosions in the epithelium; moderate, moderate infiltration of mononuclear cells in the lamina propria and the submucosa and erosions in some parts of the epithelium; and severe, severe infiltration of mononuclear cells in the lamina propria and the submucosa and erosions in many parts of the epithelium.
ELISA for H. pylori-specific serum IgG.
Blood was obtained from mice before killing, and serum was collected. Each well of a 96-well microtiter plate was filled with 100 μl of 0.1 M carbonate buffer (pH 9.6) containing 0.1 μl of the antigen preparation of H. pylori CPY2052 described above, incubated overnight at 4°C, washed five times with PBS (pH 7.4) containing 0.05% (vol/vol) Tween 20 (PBS-Tween 20), and blocked with 100 μl of PBS-Tween 20 containing 2.5% (wt/vol) nonfat dry milk for 1 h at 37°C. The plate was then washed five times with PBS-Tween 20. One hundred microliters of serum samples diluted 1:10 with PBS-Tween 20 was added to each well. The plate was incubated for 1 h at 37°C and rinsed once with PBS-Tween 20. One hundred microliters of alkaline phosphatase-conjugated goat anti-mouse IgG (Cappel, Durham, N.C.) diluted 1:1,000 with PBS-Tween 20 was added to each well, and the plate was incubated for 1 h at 37°C. After the plate was rinsed again with PBS-Tween 20, 100 μl of 3.8 mM p-nitrophenyl phosphate as a substrate was added to each well and incubated for 30 min at room temperature. The optical density at 405 nm was measured with a microplate reader (Titertek Multiscan). Negative control serum was obtained from 13-week-old wild-type mice and included in every assay.
Spectrophotometric urease assay.
Urease activity in live bacteria was measured spectrophotometrically by modifying the method of Fauchère and Blaser (7). H. pylori strains were cultivated for 30 h in BHI broth containing 4% (vol/vol) FBS as described above and harvested by centrifugation at 5,000 × g for 10 min. The precipitate was washed three times with PBS (pH 6.5) by centrifugation and suspended in PBS. The bacterial concentration of the suspension was adjusted to 8 × 108 CFU/ml by measuring the optical density at 550 nm with a Shimadzu UV-120-01 spectrophotometer. Fifty microliters of the bacterial suspension was added to each well of a 96-well microtiter plate, and the reaction was started with the addition of 50 μl of 300 mM urea in PBS containing 0.5% (wt/vol) phenol red. The reaction was carried out at 37°C, and the optical density at 570 nm was measured at 20 and 50 min after the start of the reaction with a microplate reader (Titertek Multiscan). Urease activity was evaluated by calculating the increment in the optical density and was expressed as follows: −, below 0.01; ±, from 0.01 to 0.03; +, from 0.03 to 0.30; and ++, above 0.30.
Statistics.
Colonization and serum IgG values were expressed as means ± standard errors. Sample means were compared by the Mann-Whitney U test. Infection rates were compared by the two-tailed Fisher exact probability test. A P value of <0.05 was considered significant.
RESULTS
Screening of H. pylori strains useful for mouse experimentation.
To identify the H. pylori strains with colonizing ability in the mouse stomach and to determine whether IFN-γ is involved in protection from H. pylori infection, we first examined the colonizing abilities of eight human clinical H. pylori isolates by using C57BL/6 wild-type mice and IFN-γ−/− mice 4 weeks after infection (Table 1). Three strains (ATCC 43504, CPY2052, and HPK127) colonized both wild-type and IFN-γ−/− mice, and the magnitudes of infection were considerably high. However, the colonization values were somewhat lower in the wild-type mice than in the IFN-γ−/− mice (Table 1). Strain ATCC 51110 did not colonize either mouse strain at all, and strain KP48a colonized IFN-γ−/− mice but not wild-type mice. The other three strains (KP41a, KP64b, and CPY3401) colonized IFN-γ−/− mice and partially colonized wild-type mice.
TABLE 1.
Infection rate, colonization, and urease activity of eight H. pylori strains in wild-type and IFN-γ−/− mice 4 weeks after infection
| H. pylori strain | Infection rate in the following micea:
|
Colonization in the following miceb:
|
Urease activityd | ||
|---|---|---|---|---|---|
| Wild type | IFN-γ−/− | Wild type | IFN-γ−/− | ||
| ATCC 43504 | 10/10 | 10/10 | 4.0 ± 0.1 | 4.9 ± 0.1c | + |
| ATCC 51110 | 0/6 | 0/6 | 0 | 0 | − |
| CPY2052 | 10/10 | 7/7 | 3.8 ± 0.1 | 4.8 ± 0.2c | + |
| CPY3401 | 1/8 | 9/9 | 3.4 | 3.7 ± 0.3 | ++ |
| HPK127 | 7/7 | 8/8 | 3.5 ± 0.1 | 4.3 ± 0.2c | + |
| KP41a | 5/6 | 6/6 | 4.2 ± 0.3 | 4.6 ± 0.2 | + |
| KP64b | 3/6 | 6/6 | 3.6 ± 0.1 | 4.3 ± 0.4 | + |
| KP48a | 0/6 | 6/6 | 0 | 4.1 ± 0.2 | + |
Expressed as number of infected mice/number of tested mice.
Expressed as log CFU per gram of tissue.
Significantly different (P < 0.05) from the value for wild-type mice.
Degrees of urease activity were assessed qualitatively (see the text).
As one of the factors regulating colonizing ability, we also evaluated the urease activities of the eight H. pylori strains (Table 1). All strains except for ATCC 51110 showed urease activity.
In addition to the colonizing and urease activities, we examined the histology of the gastric mucosa (Fig. 1). All strains except for CPY2052 resulted in no symptoms of gastric inflammation in both wild-type and IFN-γ−/− mice. The gastric tissues from the wild-type mice infected with CPY2052 showed a moderate infiltration of mononuclear cells in the submucosa and the lamina propria, whereas the same tissues from the IFN-γ−/− mice infected with CPY2052 did not show any infiltration. These findings indicate that only strain CPY2052 can cause the infiltration of inflammatory cells in the presence of IFN-γ.
FIG. 1.
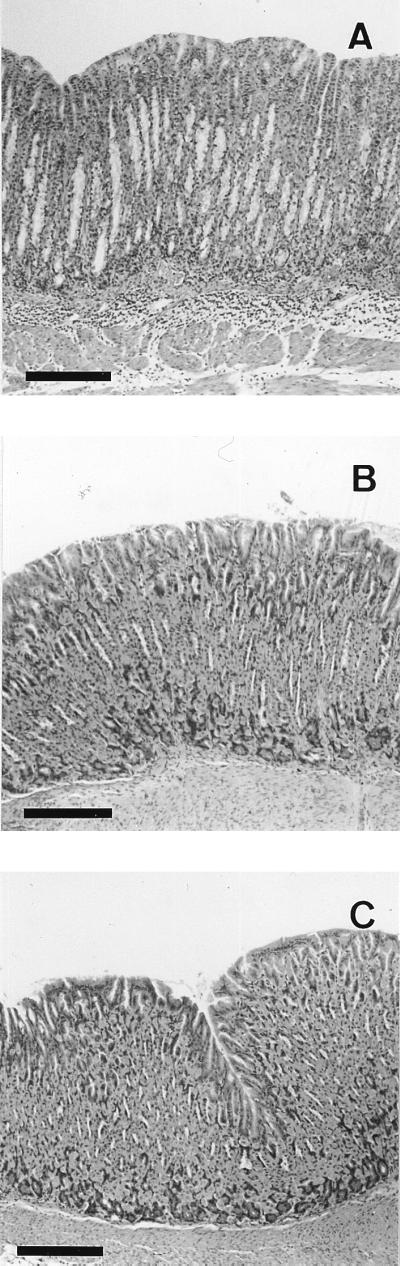
Photomicrographs of gastric tissue from a wild-type mouse (A) and an IFN-γ−/− mouse (B) colonized with CPY2052 and an uninfected control wild-type mouse (C) 4 weeks after infection. Each photomicrograph is representative of a group of at least six mice. Bar, 200 μm.
We were thus able to identify two types of H. pylori strains with colonizing ability in both wild-type and IFN-γ−/− mice, one of which has inflammatory ability (CPY2052) and the other of which does not have inflammatory ability (ATCC 43504 and HPK127).
Long-term infection test for confirming gastric inflammation.
To examine the inflammatory features of the CPY2052-infected wild-type mouse model, we carried out a long-term infection test to compare this model with the CPY2052-infected IFN-γ−/− mouse model (Table 2 and Fig. 2).
TABLE 2.
Colonization and degree of histopathological inflammation in wild-type and IFN-γ−/− mice after long-term infection with H. pylori CPY2052
| Mice | H. pylori strain | Measurement | Value at the indicated time after infection:
|
||
|---|---|---|---|---|---|
| 4 wk | 8 wk | 15 mo | |||
| Wild type | CPY2052 | Colonizationa | 3.8 ± 0.1 | 3.8 ± 0.2 | 5.1 ± 0.5 |
| Infection rateb | 10/10 | 6/6 | 6/6 | ||
| Inflammationc | Mild | Moderate | Severe | ||
| Inflammation rated | 6/10 | 6/6 | 6/6 | ||
| None | Colonization | 0 | 0 | 0 | |
| Infection rate | 0/6 | 0/6 | 0/6 | ||
| Inflammation | None | None | None | ||
| Inflammation rate | 0/6 | 0/6 | 0/6 | ||
| IFN-γ−/− | CPY2052 | Colonization | 4.8 ± 0.2 | 4.5 ± 0.2 | 5.1 ± 0.3 |
| Infection rate | 7/7 | 6/6 | 6/6 | ||
| Inflammation | None | None | None | ||
| Inflammation rate | 0/7 | 0/6 | 0/6 | ||
Colonization was expressed as log CFU per gram of tissue.
Infection rate was expressed as number of infected mice/number of tested mice.
Degree of inflammation was assessed by microscopic observation (see the text).
Inflammation rate was expressed as number of mice with gastric inflammation/number of tested mice.
FIG. 2.

Photomicrographs of gastric tissue from a wild-type mouse colonized with CPY2052 (A, B, and C) and an IFN-γ−/− mouse colonized with CPY2052 (D, E, and F) 15 months after infection. Panels A and D are representative of each group. Panels B and C are close-ups of panel A, and panels E and F are close-ups of panel D (A and D, mucosa and submucosa; B and E, epithelium and lamina propria; C and F, lamina propria and submucosa). Bars, 200 μm for A and D and 50 μm for B, C, E, and F. Infection rates and colonization values are shown in Table 2.
In wild-type mice infected with CPY2052, a moderate infiltration of mononuclear cells in the submucosa and the lamina propria was observed 8 weeks after infection. Erosions were also recognized in some parts of the epithelium. Fifteen months after infection, erosions of the epithelium became more evident and a severe infiltration of mononuclear cells or lymphoid follicles appeared in the submucosa and the lamina propria. However, no vacuoles were observed. The infiltration of mononuclear cells was observed in the body and the cardia but not in the antrum. These histopathological features were thought to indicate the development of chronic gastritis caused by H. pylori infection. In contrast, in CPY2052-infected IFN-γ−/− mice, no inflammatory symptoms were observed, even 15 months after infection.
Protection from H. pylori infection by oral immunization.
To determine whether IFN-γ is involved in protection from H. pylori infection by vaccination, we carried out an oral immunization protection test by using the CPY2052-infected wild-type mouse model and the CPY2052-infected IFN-γ−/− mouse model (Table 3). The CPY2052 bacterial sonicate was given orally to mice with and without cholera toxin, used as an adjuvant. In a control study, PBS was given instead of the antigen. Mice were then exposed to strain CPY2052 as described in Materials and Methods. All mice that received PBS were infected. In mice treated with the bacterial sonicate alone, protection from H. pylori infection was incomplete in both mouse models. In contrast, complete protection was achieved in wild-type mice immunized with the bacterial sonicate and cholera toxin, and almost complete protection was obtained even in IFN-γ−/− mice immunized in the same manner.
TABLE 3.
Protection from H. pylori CPY2052 by oral immunization
| Groupa | Infection rate in the following miceb:
|
Colonization in the following micec:
|
||
|---|---|---|---|---|
| Wild type | IFN-γ−/− | Wild type | IFN-γ−/− | |
| PBS | 10/10 | 6/6 | 4.1 ± 0.2 | 4.7 ± 0.2 |
| HP | 7/10 | 6/8 | 4.2 ± 0.2 | 4.7 ± 0.3 |
| HP + CT | 0/9 | 1/8 | 0 | 3.0 |
HP, H. pylori sonicate; CT, cholera toxin.
Expressed as number of infected mice/number of tested mice.
Expressed as log CFU per gram of tissue.
To identify the effects of IFN-γ on the activation of humoral immune responses in the presence of oral immunization, we performed an additional experiment. The levels of H. pylori-specific serum IgG in both mouse models were determined as one of the indices of the activation of humoral immune responses (Fig. 3). The bacterial sonicate alone induced no increase in the levels of H. pylori-specific serum IgG, whereas the addition of the adjuvant significantly increased the levels of H. pylori-specific serum IgG in both models. However, there was no significant difference in the levels of H. pylori-specific serum IgG between the wild-type mice and the IFN-γ−/− mice.
FIG. 3.

H. pylori-specific serum IgG values in wild-type and IFN-γ−/− mice challenged with H. pylori CPY2052 after oral immunization. Mice were immunized with H. pylori sonicate plus cholera toxin (▪), H. pylori sonicate (▨), or PBS (□). IgG values were assessed by measuring the optical density at 405 nm. Results are expressed as mean ± standard error of the mean for data obtained from 6 to 10 mice per group. a, significantly different (P < 0.05) from values for the H. pylori sonicate group and the PBS group.
DISCUSSION
It appears that there are differences in infective ability among H. pylori strains, since Lee et al. obtained only one H. pylori strain that could infect mice from among biopsy specimens from 23 patients (11). It is not yet known what factors regulate the infective ability of H. pylori. Eaton et al. studied the urease-dependent colonization of gnotobiotic piglets by H. pylori and reported that urease activity was essential for the colonization of gnotobiotic piglets by H. pylori (3, 4). It has therefore been suspected that the urease activity of 8 H. pylori strains has a strong influence on the infective ability of the bacteria. In the present study, strain ATCC 51110 (which has no urease activity) was not able to colonize, while strains ATCC 43504, CPY2052, and HPK127 (which have urease activity) colonized both wild-type and IFN-γ−/− mice. However, strain KP48a, which has urease activity, was not able to colonize wild-type mice. These results indicate that urease is a necessary but not sufficient factor for the colonization of mice by H. pylori. Other bacterial features, such as motility (5, 6) and adherence to epithelial surfaces (9, 14, 17), may influence infective ability.
There have been few reports regarding the protective effect of IFN-γ against Helicobacter infection. Mohammadi et al. suggested that a protective role of IFN-γ against H. felis infection was unlikely, because the neutralization of IFN-γ by anti-IFN-γ antibody treatment had no effect on the magnitude of infection in their H. felis-infected mouse model (13).
On the contrary, in our study, the H. pylori strain (KP48a) without the ability to colonize wild-type mice was able to colonize IFN-γ−/− mice, clearly demonstrating the positive contribution of IFN-γ to protection from H. pylori infection. However, based on our finding that H. pylori ATCC 43504, CPY2052, and HPK127, all with strong infective ability, could colonize wild-type mice despite the expression of IFN-γ, we consider that the protection induced by IFN-γ is not so strong. The present results showed that IFN-γ was not essential for protection by oral immunization, because oral immunization with an H. pylori sonicate and cholera toxin was similarly effective in IFN-γ−/− mice and wild-type mice. The protection mechanisms induced by oral immunization are thought to be different from that based on IFN-γ. After oral immunization with the H. pylori sonicate and cholera toxin, the H. pylori-specific serum IgG levels increased significantly. However, the levels in the immunized wild-type mice were almost the same as those in the immunized IFN-γ−/− mice. These results suggested that the protective effect induced by oral immunization is based on humoral immunity. IFN-γ might exert a protective effect via host defense systems, including cellular immunity and the activation of macrophages and natural killer cells, rather than humoral immunity.
One of the most interesting current issues is how colonization by a noninvasive bacterium, H. pylori, causes gastritis. The vacuolization of epithelial cells by cytotoxin is suspected to be one of the causes. However, because no vacuoles were observed in the epithelium even 15 months after infection in the CPY2052-infected wild-type mouse model, although inflammatory responses were confirmed, it is not likely that vacuolization caused gastritis in this model.
We consider that IFN-γ is involved in the activation of mononuclear cells, because the infiltration of mononuclear cells was observed in CPY2052-infected wild-type mice but not CPY2052-infected IFN-γ−/− mice. Mohammadi et al. reported that anti-IFN-γ antibody treatment of mice caused a significant reduction in in vitro IFN-γ production by splenic and gastric lymphocytes from mice infected with H. felis and that this reduction in IFN-γ production was accompanied by a reduction in the gastric inflammation score (13); these results suggested that IFN-γ was involved in the continuation of gastric inflammation. The present study demonstrated the positive contribution of IFN-γ to gastric inflammation caused by H. pylori infection.
Ye et al. reported that gastric epithelial cells constitutively expressed the class II major histocompatibility complex and that the constitutive expression of the class II major histocompatibility complex on gastric epithelial cells increased markedly during infection with H. pylori. They also suggested that IFN-γ might enhance the antigen-presenting cell function of gastric epithelial cells through the up-regulation of the class II major histocompatibility complex (22). The role of IFN-γ indicated in the present study might involve intensification of the antigen-presenting cell function of gastric epithelial cells.
Based on the present results, we conclude that IFN-γ may play an important role in inflammation rather than protection from H. pylori infection, although IFN-γ is involved in both protection from H. pylori infection and the inflammation induced by this infection. This study also shows that different H. pylori strains can cause various levels of infection or inflammation in the presence or absence of IFN-γ.
ACKNOWLEDGMENTS
We thank K. Kashima, Kyoto Prefectural University of Medicine, for kind advice and helpful discussions and T. Nishino, Kyoto Pharmaceutical University, for technical advice. We are also grateful to M. Karita for generously donating the H. pylori strains.
REFERENCES
- 1.Bayerdörffer E, Lehn N, Hatz R, Mannes G A, Oertel H, Sauerbruch T, Stolte M. Difference in expression of Helicobacter pylori gastritis in antrum and body. Gastroenterology. 1992;102:1575–1582. doi: 10.1016/0016-5085(92)91716-h. [DOI] [PubMed] [Google Scholar]
- 2.Dalton D K, Pitts-Meek S, Keshav S, Figari I S, Bradley A, Stewart T A. Multiple defects of immune cell function in mice with disrupted interferon-γ genes. Science. 1993;259:1739–1742. doi: 10.1126/science.8456300. [DOI] [PubMed] [Google Scholar]
- 3.Eaton K A, Brooks C L, Morgan D R, Krakowka S. Essential role of urease in pathogenesis of gastritis induced by Helicobacter pylori in gnotobiotic piglets. Infect Immun. 1991;59:2470–2475. doi: 10.1128/iai.59.7.2470-2475.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eaton K A, Krakowka S. Effect of gastric pH on urease-dependent colonization of gnotobiotic piglets by Helicobacter pylori. Infect Immun. 1994;62:3604–3607. doi: 10.1128/iai.62.9.3604-3607.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eaton K A, Morgan D R, Krakowka S. Motility as a factor in the colonisation of gnotobiotic piglets by Helicobacter pylori. J Med Microbiol. 1992;37:123–127. doi: 10.1099/00222615-37-2-123. [DOI] [PubMed] [Google Scholar]
- 6.Eaton K A, Suerbaum S, Josenhans C, Krakowka S. Colonization of gnotobiotic piglets by Helicobacter pylori deficient in two flagellin genes. Infect Immun. 1996;64:2445–2448. doi: 10.1128/iai.64.7.2445-2448.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fauchère J-L, Blaser M J. Adherence of Helicobacter pylori cells and their surface components to HeLa cell membranes. Microb Pathog. 1990;9:427–439. doi: 10.1016/0882-4010(90)90061-t. [DOI] [PubMed] [Google Scholar]
- 8.Ghiara P, Marchetti M, Blaser M J, Tummuru M K R, Cover T L, Segal E D, Tompkins L S, Rappuoli R. Role of the Helicobacter pylori virulence factors vacuolating cytotoxin, CagA, and urease in a mouse model of disease. Infect Immun. 1995;63:4154–4160. doi: 10.1128/iai.63.10.4154-4160.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goodwin C S, Armstrong J A, Marshall B J. Campylobacter pyloridis, gastritis and peptic ulceration. J Clin Pathol. 1986;39:353–365. doi: 10.1136/jcp.39.4.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kozol R, Domanowsky A, Jaszewsky R, Czanko R, McCurdy B, Prasad M, Fromm B, Calzada R. Neutrophil chemotaxis in gastric mucosa, a signal-to-response comparison. Dig Dis Sci. 1991;36:1277–1280. doi: 10.1007/BF01307522. [DOI] [PubMed] [Google Scholar]
- 11.Lee A, O’Rourke J, Corazon de Ungria M, Robertson B, Daskalopoulos G, Dixon M F. A standardized mouse model of Helicobacter pylori infection: introducing the Sydney strain. Gastroenterology. 1997;112:1386–1397. doi: 10.1016/s0016-5085(97)70155-0. [DOI] [PubMed] [Google Scholar]
- 12.Marchetti M, Aricò B, Burroni D, Figura N, Rappuoli R, Ghiara P. Development of a mouse model of Helicobacter pylori infection that mimics human disease. Science. 1995;267:1655–1658. doi: 10.1126/science.7886456. [DOI] [PubMed] [Google Scholar]
- 13.Mohammadi M, Czinn S, Redline R, Nedrud J. Helicobacter-specific cell-mediated immune responses display a predominant Th1 phenotype and promote a delayed-type hypersensitivity response in the stomachs of mice. J Immunol. 1996;156:4729–4738. [PubMed] [Google Scholar]
- 14.Neman-Simha V, Mégraud F. In vitro model for Campylobacter pylori adherence properties. Infect Immun. 1988;56:3329–3333. doi: 10.1128/iai.56.12.3329-3333.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nielsen H, Andersen L P. Chemotactic activity of Helicobacter pylori sonicate for human polymorphonuclear leucocytes and monocytes. Gut. 1992;33:738–742. doi: 10.1136/gut.33.6.738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nielsen H, Andersen L P. Activation of human phagocyte oxidative metabolism by Helicobacter pylori. Gastroenterology. 1992;103:1747–1753. doi: 10.1016/0016-5085(92)91430-c. [DOI] [PubMed] [Google Scholar]
- 17.Segal E D, Falkow S, Tompkins L S. Helicobacter pylori attachment to gastric cells induces cytoskeletal rearrangements and tyrosine phosphorylation of host cell proteins. Proc Natl Acad Sci USA. 1996;93:1259–1264. doi: 10.1073/pnas.93.3.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tagawa Y, Sekikawa K, Iwakura Y. Suppression of concanavalin A-induced hepatitis in IFN-γ−/− mice, but not in TNF-α−/− mice: role for IFN-γ in activating apoptosis of hepatocytes. J Immunol. 1997;159:1418–1428. [PubMed] [Google Scholar]
- 19.Tujii M, Kawano S, Tsuji S, Fusamoto H, Kamada T, Sato N. Mechanism of gastric mucosal damage induced by ammonia. Gastroenterology. 1992;102:1881–1888. doi: 10.1016/0016-5085(92)90309-m. [DOI] [PubMed] [Google Scholar]
- 20.Yamaoka Y, Kita M, Kodama T, Sawai N, Kashima K, Imanishi J. Expression of cytokine mRNA in gastric mucosa with Helicobacter pylori infection. Scand J Gastroenterol. 1995;30:1153–1159. doi: 10.3109/00365529509101624. [DOI] [PubMed] [Google Scholar]
- 21.Yamaoka Y, Kita M, Kodama T, Sawai N, Imanishi J. Helicobacter pylori cagA gene and expression of cytokine messenger RNA in gastric mucosa. Gastroenterology. 1996;110:1744–1752. doi: 10.1053/gast.1996.v110.pm8964399. [DOI] [PubMed] [Google Scholar]
- 22.Ye G, Gunasena H, Reyes V E. The role of the gastric epithelium in local T cell activation. In: Ernst P B, Michetti P, Smith P D, editors. The immunobiology of H. pylori: from pathogenesis to prevention. Philadelphia, Pa: Lippincott-Raven Publishers; 1997. pp. 153–165. [Google Scholar]