

Abstract
Staphylococcus aureus is considered as one of the most widespread bacterial pathogens and continues to be a prevalent cause of mortality and morbidity across the globe. FmtA is a key factor linked with methicillin resistance in S. aureus. Consequently, new antibacterial compounds are crucial to combat S. aureus resistance. Here, we present the virtual screening of a set of compounds against the available crystal structure of FmtA. The findings indicate that gemifloxacin, paromomycin, streptomycin, and tobramycin were the top-ranked potential drug molecules based on the binding affinity. Furthermore, these drug molecules were analyzed with molecular dynamics simulations, which showed that the identified molecules formed highly stable FmtA–inhibitor(s) complexes. Molecular mechanics Poisson–Boltzmann surface area and quantum mechanics/molecular mechanics calculations suggested that the active site residues (Ser127, Lys130, Tyr211, and Asp213) of FmtA are crucial for the interaction with the inhibitor(s) to form stable protein–inhibitor(s) complexes. Moreover, fluorescence- and isothermal calorimetry-based binding studies showed that all the molecules possess dissociation constant values in the micromolar scale, revealing a strong binding affinity with FmtAΔ80, leading to stable protein–drug(s) complexes. The findings of this study present potential beginning points for the rational development of advanced, safe, and efficacious antibacterial agents targeting FmtA.
1. Introduction
Staphylococcus aureus is a robust Gram-positive bacterium that causes various types of illnesses, including bacteremia, meningitis, endocarditis, pneumonia, skin infections, toxic shock syndrome, and other hospital-acquired infections.1−3 The fatality caused by S. aureus is mainly due to the substantial use of β-lactam antibiotics, which is also responsible for developing its resistant strains.2,4 The mortality of 20 to 40% and the development of methicillin-resistant strains make S. aureus a pathogen of greatest concern.5,6 Moreover, the evolution of methicillin-resistant Staphylococcus aureus (MRSA) strains for developing resistance to vancomycin ensures few treatment strategies that provoke an utmost need to develop novel compounds or antibiotics for treating S. aureus infection.7,8 Earlier, it was reported that S. aureus can be vulnerable to vancomycin and β-lactamases by inhibiting non-essential genes, which are crucial for forming cell wall constituents.9,10 However, in late 1960s, it was found that FmtA is responsible for the development of methicillin resistance in MRSA strains and that the removal of fmtA gene disturbs the homogeneity of methicillin resistance.11,12 The expression of fmtA is enhanced in the presence of cell wall inhibitors like methicillin, oxacillin, fosfomycin, and bacitracin.13,14 In S. aureus, the wall teichoic acids (WTAs) are associated with the peptidoglycan layer or the exterior face of the cytoplasmic membrane.15 It has been documented that FmtA functions as an esterase which removes d-alanine from teichoic acid.11 Furthermore, in the cell wall, teichoic acid is known for developing virulence, biofilm generation, cellular activities, adhesion, metal regulation, and resistance.16,17 Boles et al. stated that the fmtA mutants are devoid of WTAs, and the inactivation of fmtA is associated with the functioning of teichoic acids.18,19
The structure of FmtA is similar to penicillin-binding proteins (PBPs), β-lactamases, and penicillin-recognizing proteins (PRE) like esterases, aminopeptidases, endopeptidases, and amidases.20−22 FmtA bears two of three conserved motifs, that is, the first and second motifs, namely SXXK and Y/SXN, respectively, which are found in β-lactamase and PBPs, whereas the third motif, namely KTG, is lacking. Our earlier reported crystal structure and mutational studies revealed that Ser-Lys-Asp might function as a catalytic triad for catalyzing the wall teichoic acid in FmtA.23 The conserved motifs comprising Ser, Lys, and Tyr are crucial for the functioning of FmtA, that is, Ser is known to function as a nucleophile, Lys works in acylation/deacylation, while Tyr holds the substrate in the course of hydrolysis of WTA. Further, the quantum mechanics/molecular mechanics (QM/MM) study showed that the backbone amides of Ser127 and Gly344 act as oxyanion hole residues to stabilize the tetrahedral intermediates for the catalysis of WTA by FmtA.24
In the current study, we have utilized the available crystal structure of FmtA (PDB ID: 5ZH8) for performing structure-based virtual screening against the drugs of e-LEA3D. The binding affinity of the screened compounds (gemifloxacin, paromomycin, streptomycin, and tobramycin) was cross-confirmed by molecular docking studies utilizing HADDOCK and AutoDockTools. Density functional theory (DFT) calculations were performed to examine the chemical reactivity of compounds. Molecular dynamics simulation and molecular mechanics generalized Born surface area calculations were done to assess the stability of the protein after the binding of the ligands. FmtAΔ80 was cloned, expressed, and purified via a cation exchanger and size-exclusion chromatography. Fluorescence spectroscopy was conducted to study the structural and conformational alterations in FmtAΔ80 after the interaction with the selected compounds. Moreover, isothermal calorimetry (ITC) was utilized to determine the thermodynamic parameters. The sequential work done to screen the potent molecules and their binding interaction against FmtAΔ80 is represented in Figure 1.
Figure 1.

Diagrammatic illustration of the work done to identify the inhibitor molecules and their binding interactions against FmtA.
2. Materials and Methods
2.1. Virtual Screening and Molecular Docking
The crystal structure of FmtA (PDB ID: 5ZH8) was considered as a protein receptor to screen potential compounds.25 The virtual screening of FmtA against 1930 drugs of e-LEA3D: Cheminformatics Tools and Database (http://chemoinfo.ipmc.cnrs.fr/MOLDB/index.html) was performed utilizing AutoDock Vina in PyRx, as done by Dalal et al.26−28 The best drug molecules with higher binding energies were re-docked by utilizing HADDOCK (https://alcazar.science.uu.nl/services/HADDOCK2.2/) and AutoDockTools.26,29−31 The molecular grid in virtual screening and molecular docking was set around the active site residues (Ser127, Lys130, Tyr211, and Asp213) of FmtA. The generated docking conformations were evaluated, and figures were made in Maestro and PyMOL.32−34
2.2. Molecular Dynamics
Molecular dynamics for FmtA–Native and FmtA–inhibitor(s) complexes were carried out using GROMACS software package, as performed by Dalal et al.26 The protein coordinates and topology files were created utilizing the GROMOS96 43a1 force field, and the protonation states were updated at pH 7.2 by the H++ server.26,35−38 The ligands (gemifloxacin, paromomycin, streptomycin, and tobramycin) were prepared using PRODRG (http://davapc1.bioch.dundee.ac.uk/cgi-bin/prodrg), and the charges were corrected by the 6-311G (d,p) basis set of DFT/B3LYP in Gaussian.39−42 The systems were neutralized by substituting the counterions (Cl–) and solvated in triclinic boxes (Table S1). The systems were equilibrated for 1 ns and minimized for 50,000 steps, as done by Dalal et al.26 The molecular dynamics simulation was run for 100 ns for all the systems, and the files were updated every 10 ps to evaluate the root-mean-square fluctuations (RMSF), root-mean-square deviation (RMSD), hydrogen-bond numbers, radius of gyration (Rg), and solvent-accessible surface area (SASA).43,44
2.3. MMPBSA Binding Free Energy
The molecular mechanics Poisson–Boltzmann surface area (MMPBSA) method was utilized to determine the binding affinity of FmtA–drug(s) complexes.45−47 Here, a total of 2000 snapshots from the last 20 ns of molecular dynamics were utilized to calculate the binding energy of FmtA–gemifloxacin, FmtA–paromomycin, FmtA–streptomycin, and FmtA–tobramycin complexes.48 The per residue decomposition analysis was performed to calculate the binding energy contribution of active site residues with the ligand in FmtA–drug(s) complexes.49,50
2.4. Quantum Mechanics and Quantum Mechanics/Molecular Mechanics
DFT calculation was done to evaluate the polarity of drugs (gemifloxacin, paromomycin, streptomycin, and tobramycin).39 The molecular electrostatic potential and molecular orbital energies were assessed by using B3LYP with the basis set of 6-311G (d,p), as done by Dalal et al.26,39,40,50,51 Furthermore, quantum mechanics/molecular mechanics (QM/MM) estimations for FmtA–drug(s) complexes were conducted using our own N-layered integrated molecular orbital and molecular mechanics (ONIOM) method, as done by Dalal et al.24,26,52,53 The inhibitor and side chain atoms of Ser127, Lys130, Tyr211, and Asp213 were considered in the QM layer, whereas the remaining part of the protein was put in the MM layer. The enthalpy (ΔH), relative Gibbs free energy (ΔG), and entropy (ΔS) were assessed by ONIOM (B3LYP/6-311G(d,p): AMBER).
2.5. Cloning and Purification of FmtAΔ80
fmtAΔ80 was amplified utilizing the oligonucleotide primers having a restriction site for enzymes NdeI and HindIII. The gene was cloned in pET24a (+) vector, and the recombinant plasmid was transformed into E. coli BL21 (DE3).54 Further, the transformed cell was allowed to grow in Luria–Bertani (LB) broth having 30 μg/mL kanamycin overnight at 37 °C. The overnight-grown culture was diluted in fresh LB media, complemented with 0.4 M d-sorbitol, 2.5 mM β-betaine, and 30 μg/mL of kanamycin, and incubated at 37 °C until OD600 reached 0.6. The expressed recombinant protein was induced with 1 mM IPTG (isopropyl β-d-1-thiogalactopyranoside) and incubated for 16 h at 25 °C, followed by harvesting the cells. The harvested cells were allowed to resuspend in 50 mM sodium phosphate buffer (pH 7.2), and they underwent lysis at 20 kpsi pressure using a cell disruptor (Constant Systems Ltd., Daventry, England). Subsequently, the cell lysate was centrifuged at 12000 g at 4 °C for 60 min, and the supernatant was recovered. The protein lysate was subjected to cation exchange. The recombinant protein was purified with a linear gradient of 0–1.0 M sodium chloride in 50 mM sodium phosphate buffer (pH 7.2), and the purity of the eluted fractions was evaluated by 12.5% SDS-PAGE. The eluents having FmtAΔ80 were concentrated and subjected to a HiLoad 16/600 Superdex 200 pg size-exclusion column (GE Life Sciences) to remove the remaining impurities. The eluents were evaluated by 12.5% SDS-PAGE to check the purity of the protein. The pure fractions of FmtAΔ80 were pooled and concentrated utilizing a 10 kDa Amicon filter.
2.6. Fluorescence Spectroscopy
Fluorescence spectroscopy was performed by utilizing a HORIBA FluoroMax spectrofluorometer using a 1.0 cm quartz cuvette at 25 °C to assess the structural alterations in FmtAΔ80 (2 μM in 50 mM sodium phosphate pH 7.2) upon inhibitor interactions (gemifloxacin, paromomycin, streptomycin, and tobramycin). The excitation wavelength of FmtAΔ80 was 295 nm, while the emission spectra were recorded between 300 and 450 nm. Furthermore, fluorescence quenching was performed using the same protein concentration, followed by its titration with the consecutive addition of drugs. Three different scans of every titration were collected, and the control (with no protein) was subtracted for précising the fluorescence data, and the results were examined using the Stern Volmer equation:
where Fo and F represent the fluorescence intensities of FmtAΔ80 in the absence and presence of a quencher (Q); Ksv represents the Stern–Volmer quenching constant; kq is the bimolecular quenching rate constant; and τo denotes the fluorescence lifetime without a quencher.
Moreover, using the modified form of the Stern–Volmer equation, the number of binding sites (N) and the equilibrium binding constant (Kb) were computed. The inverse of Kb (i.e., 1/Kb), which is the dissociation constant (Kd), is also calculated.
2.7. Isothermal Titration Calorimetry
Isothermal titration calorimetry (ITC) studies were performed, employing a MicroCal-iTC 200 unit (GE Healthcare) operating at 25 °C temperature. The association of FmtAΔ80 with four identified drugs (gemifloxacin, paromomycin, streptomycin, and tobramycin) was studied. Buffer I (50 mM sodium phosphate pH 7.2) was kept in the reference cell. The titration reactions were performed by placing the protein FmtAΔ80 (50 μM) in the sample cell and the ligands (gemifloxacin, paromomycin, streptomycin, and Ttbramycin) in the syringe, keeping the stirring speed at 700 rpm and the initial delay of 60 s. A total of 20 injections were performed, keeping the first injection of 0.5 μL and other injections of 2.0 μL. Each consecutive injection had a time interval of 150 s, and the reference cell power was adjusted to 8 μW. The resulting data were evaluated by the difference in the control and arranged to a one-site binding model using Origin 7.0 software for calculating the values of the entropy change (ΔS), enthalpy change ΔH, stoichiometry (n) and the association constant (Ka).
2.8. Circular Dichroism Spectroscopy
The far-UV circular dichroism (CD) spectral measurements were performed with a J-Model-1500 spectrophotometer (JASCO) to analyze the conformational alterations that occurred in the protein in the presence of drugs, as done by Dalal et al.26 The spectral measurements were recorded for FmtAΔ80 in the absence and presence of drugs ranging from 190 to 250 nm using a quartz cuvette having an optical path length of 0.1 cm at 25 °C.
3. Results
3.1. Virtual Screening and Molecular Docking
To determine the novel potent inhibitors against FmtA, virtual screening was conducted by AutoDock Vina in PyRx0.8. The molecules possessing a higher binding affinity than −7 kcal/mol were selected and analyzed in PyMOL. Further, to deduce the binding affinity and molecular interactions of FmtAΔ80 with the inhibitors (gemifloxacin, paromomycin, streptomycin, and tobramycin), molecular docking studies were performed utilizing HADDOCK and AutoDockTools. The binding affinities depicted by HADDOCK were also in accordance with the AutoDock results (Tables S2 and S3). Gemifloxacin makes a lower energy complex (FmtA–gemifloxacin) in comparison to other inhibitor molecules revealed by HADDOCK and AutoDockTools. Gemifloxacin forms hydrogen bonds with Ser127, Lys130, Tyr211, Asp213, and Asn343 with a binding affinity (−7.1 kcal/mol), as presented in Figure 2A. The FmtA–gemifloxacin complex is also supported by polar and hydrophobic interactions via Ser127, Tyr211, Asp213, Tyr282, Arg 341, Asn343, Gly345, and Phe346, as depicted in Figure S1A. Moreover, HADDOCK revealed a score (−60.6 ± 7.4) with an electrostatic energy of −153.6 ± 17.9 kcal/mol for the FmtA–gemifloxacin complex, as presented in Table S3. AutoDockTools showed that paromomycin, tobramycin, and streptomycin have binding affinities of −6.9, −6.8, and −6.5 kcal/mol, respectively. Paromomycin exhibited H bonds with the residues Ser127, Lys130, Tyr211, Asp213, Tyr180, and Ser182 and was also stabilized by polar and hydrophobic interactions through the residues Ser127, Lys130, Ser182, Tyr 180, Tyr211, Asp213, Leu274, Tyr282, Asn343, Phe346, and Gly345 (Figures 2B and S1B). Streptomycin formed H-bond, polar, and hydrophobic interactions with the residues Ser127, Lys130, Tyr211, Asp213, Tyr282, Leu274, Tyr282, Gly345, Phe346, Phe347, Gly348, and Lys368 (Figures 2C and S1C). Tobramycin sustained H bonds with the residues Ser127, Lys130, Ser182, Tyr211, and Asp213 and showed polar and hydrophobic interactions with the residues Ser127, Lys130, Tyr211, Asn212, Asp213, Lys179, Tyr180, Ser182, Tyr282, Phe346, and Gly345 (Figures 2D and S1D).
Figure 2.

Molecular docking of FmtA with gemifloxacin, paromomycin, streptomycin, and tobramycin. (A) Gemifloxacin (pink color), (B) paromomycin (yellow color), (C) streptomycin (orange color), and (D) tobramycin (cyan color). The interacting residues of FmtA are displayed in the stick format, and atoms like carbon, nitrogen, oxygen, and fluorine are in green, blue, red, and cyan colors, respectively. The interactions are represented by broken gray lines.
3.2. Molecular Dynamics
The FmtA protein, along with drugs, was analyzed by molecular dynamics to study the stability of the protein–drug(s) complexes. The RMSD values along the backbone atoms were calculated for FmtA in the absence or presence of drug(s) (Figure 3A). After 35 ns, a lesser RMSD is shown by FmtA–drug(s) complexes (0.27 to 0.33 nm) than FmtA–Native (0.35 to 0.37 nm). The average RMSD values for FmtA–Native, FmtA–gemifloxacin, FmtA–paromomycin, FmtA–streptomycin, and FmtA–tobramycin complexes were 0.34 ± 0.01, 0.30 ± 0.02, 0.31 ± 0.01, 0.31 ± 0.02, and 0.31 ± 0.02 nm, respectively, as presented in Table S4. The RMSD values for gemifloxacin, paromomycin, streptomycin, and tobramycin depicted that the interaction of these inhibitors with FmtA made stable FmtA–drug(s) complexes (Figure 3B). The residue-wise fluctuation of FmtA upon binding with the drug was estimated to deduce the stability of FmtA–drug(s) complexes (Figure S2). FmtA–Native, FmtA–gemifloxacin, FmtA–paromomycin, FmtA–streptomycin, and FmtA–tobramycin complexes indicated average RMSF values of 0.13 ± 0.09, 0.11 ± 0.07, 0.11 ± 0.07, 0.11 ± 0.05, and 0.11 ± 0.07 nm, respectively, as presented in Table S4. The RMSD and RMSF plots showed that drugs were significantly fitted in the active sites of FmtA and generated stable FmtA–inhibitor(s) complexes.
Figure 3.
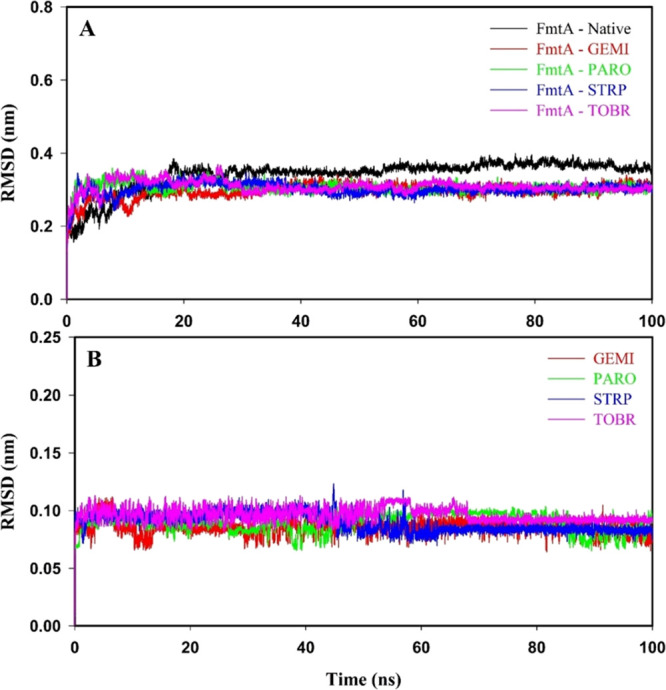
RMSD values of protein, protein–inhibitor(s) complexes, and drugs alone: gemifloxacin (GEMI), paromomycin (PARO), streptomycin (STRP), and tobramycin (TOBR) for the time span of 100 ns of molecular dynamics. (A) FmtA–Native (black color), FmtA–gemifloxacin (red color), FmtA–paromomycin (green color), FmtA–streptomycin (blue color), and FmtA–tobramycin (pink color) complexes. (B) Gemifloxacin (red color), paromomycin (green color), streptomycin (blue color), and tobramycin (pink color).
The solvent-accessible surface area (SASA) and radius of gyration (Rg) were analyzed to explore the stability of FmtA–inhibitor(s) complexes. Rg plots revealed a lesser Rg of FmtA–inhibitor(s) complexes as compared to FmtA–Native (Figure 4). The average Rg values of FmtA–Native, FmtA–gemifloxacin, FmtA–paromomycin, FmtA–streptomycin, and FmtA–tobramycin complexes were 2.03 ± 0.01, 2.00 ± 0.01, 2.00 ± 0.01, 2.01 ± 0.01, and 2.01 ± 0.01 nm, respectively (Table S4). From Figure S3, it can be illustrated that drug-bound FmtA had lesser SASA values than unbound FmtA. FmtA–Native, FmtA–gemifloxacin, FmtA–paromomycin, FmtA–streptomycin, and FmtA–tobramycin complexes had average SASA values of 164.43 ± 2.54, 160.58 ± 2.96, 160.03 ± 3.25, 160.10 ± 3.24, and 160.56 ± 3.31 nm2, respectively (Table S4). The Rg and SASA results depicted that the interaction of drug(s) to FmtA leads to the formation of stable FmtA–drug(s) complexes.
Figure 4.

Radius of gyration (Rg) graph of FmtA–Native and FmtA–inhibitor(s) complexes for the molecular simulation of 100 ns. The Rg analysis of FmtA in the absence or presence of gemifloxacin (GEMI), paromomycin (PARO), streptomycin (STRP), and tobramycin (TOBR) showed the compactness and stability of protein–drug(s) complexes.
Further, hydrogen bonds were calculated to explore the stability of drug(s) with FmtA during the molecular dynamics calculation. FmtA–drug(s) complexes displayed more intraprotein hydrogen-bond numbers than FmtA–Native (Figure S4A). FmtA–Native, FmtA–gemifloxacin, FmtA–paromomycin, FmtA–streptomycin, and FmtA–tobramycin complexes revealed the average intraprotein hydrogen-bond numbers of 288.9 ± 9.3, 294.2 ± 8.7, 293.8 ± 9.7, 294.1 ± 9.1, and 294.1 ± 8.8, respectively (Table S4). The intraprotein hydrogen-bond distribution patterns for FmtA–Native and FmtA– inhibitor(s) complexes are presented in Figure S4B. FmtA–inhibitor(s) complexes were stabilized by the presence of a minimum of five intermolecular hydrogen bonds (Figure S5A). The hydrogen-bond distances between the acceptor and donor atoms ranged from 0.23 to 0.35 nm (Figure S5B). Hydrogen-bond analysis supported the results of other MD studies that the FmtA interaction with drug(s) tends to form stable and compact FmtA–inhibitor(s) complexes.
3.3. MMPBSA Binding Free Energy
The binding free energy of inhibitor(s) with FmtA was determined by the molecular mechanics Poisson–Boltzmann surface area (MMPBSA) method by g_mmpbsa tool. The binding free energies of FmtA–gemifloxacin, FmtA–paromomycin, FmtA–streptomycin, and FmtA–tobramycin complexes were −158.82 ± 7.73, −154.15 ± 8.42, −152.53 ± 9.03, and −156.13 ± 8.91 kJ/mol, as presented in Figure S6. Further, the per residue decomposition analysis revealed that Ser127, Lys130, Tyr211, Asp213, Tyr282, Gly345, and Phe346 of FmtA are crucial in the stabilization of FmtA–drug(s) complexes (Figure S7). Conclusively, MMPBSA showed that drug(s)-bound complexes were stable.
3.4. Quantum Mechanics and Quantum Mechanics/Molecular Mechanics
The DFT calculation by B3LYP/6-311G (d,p) was utilized to study the molecular and electrostatic properties such as MEP, HOMO, LUMO, energy, hardness, and electronegativity of drugs. The high and low electron density regions of gemifloxacin, paromomycin, streptomycin, and tobramycin are presented in Figure 5. The HOMO and LUMO energy gaps for gemifloxacin, paromomycin, streptomycin, and tobramycin were 3.22, 2.47, 2.43, and 2.67 eV, as presented in Figure S8 and Table S5. ONIOM (B3LYP/6-311G(d,p): AMBER) method was conducted to determine the Gibbs free energy (ΔG) and thermodynamics parameters (entropy and enthalpy) of FmtA–inhibitor(s) complexes. FmtA–gemifloxacin, FmtA–paromomycin, FmtA–streptomycin, and FmtA–tobramycin showed Gibbs free energies of −49.52, −50.45, −47.59, and −45.23 kcal/mol, respectively (Table 1). The negative entropy values for the drug(s) with FmtA depicted strong noncovalent bonds in the FmtA–drug(s) complexes.55 QM and QM/MM analysis confirmed that all the identified drug(s) were reactive in nature and bound at the active site residues of FmtA to generate the stable protein–inhibitor(s) complexes.
Figure 5.

Molecular electrostatic potential (MEP) results for gemifloxacin (A), paromomycin (B), streptomycin (C), and tobramycin (D). The colors for electron-rich to poor regions are red < orange < yellow < green < blue.
Table 1. Binding Free Energy (ΔG), Entropy (ΔS), and Enthalpy (ΔH) of Gemifloxacin, Paromomycin, Streptomycin, and Tobramycin with FmtA Calculated from Our Own N-Layered Integrated Molecular Orbital and Molecular Mechanics (ONIOM) Method Using B3LYP/6-311G (d,p): AMBER.
| complex | ΔG (kcal/mol) | ΔH (kcal/mol) | ΔS (cal/mol/K) |
|---|---|---|---|
| FmtA–gemifloxacin | –49.52 | –68.99 | –65.28 |
| FmtA–paromomycin | –50.45 | –69.22 | –62.94 |
| FmtA–streptomycin | –47.59 | –65.32 | –59.45 |
| FmtA–tobramycin | –45.23 | –64.08 | –63.23 |
3.5. Cloning and Purification of FmtAΔ80
The cloning of fmtAΔ80 was performed using the bacterial expression vector pET24a (+). The amplification was performed using primers with restriction sites for NdeI and HindIII, and the clone was checked by restriction digestion with NdeI and HindIII, as shown in Figures S9A and S9B, respectively. Furthermore, the recombinant plasmid having fmtAΔ80 gene was overexpressed in E. coli BL21. The purification of the recombinant protein was achieved by cation exchange chromatography (SP sepharose column) using 50 mM sodium phosphate buffer (pH 7.2), and the purity of the protein was visualized through SDS-PAGE gel, as shown in Figure S9C. The fractions having the protein were subjected to gel filtration using a pre-equilibrated 200 pg HiLoad 16/600 Superdex column. Figure S9D reveals the purity and molecular weight of the protein, which is observed to be 36.1 kDa, which is in accordance with the calculated value.
3.6. Fluorescence-Based Binding Studies
Fluorescence spectroscopy is often utilized to provide insights into the binding between proteins and their ligands.56 The fluorescence spectra of purified FmtAΔ80 with different concentrations of gemifloxacin, paromomycin, streptomycin, and tobramycin are shown in Figure 6. The results illustrated that the binding of ligands to the protein leads to the subsequent decrease in fluorescence intensity, which indicates an alteration in the microenvironment of the active site residue. Further, the fluorescence spectra were evaluated using the equations provided in Material and Methods, and the determined binding parameters, equilibrium binding constant (Kb), Stern–Volmer constant (Ksv), bimolecular quenching rate constant (kq), dissociation constant (Kd), including the number of the binding sites (n), are listed in Table 2. The Stern–Volmer plots of FmtAΔ80 with all four drug molecules are shown in Figure S10. All protein–ligand interactions have kq values greater than 2.0 × 1010 M–1 s–1, indicating a “static quenching” mechanism for the binding of the ligand to the protein.57 The dissociation constant was observed in the range of 11–85 μM with a single binding site (Table 2), which suggests that all ligands bind significantly to the protein and form stable protein–ligand(s) complexes.
Figure 6.

Fluorescence emission spectra of FmtAΔ80 with different concentrations (2–20 μM) of ligands. (A) Gemifloxacin, (B) paromomycin, (C) streptomycin, and (D) tobramycin.
Table 2. Stern–Volmer Constants and Binding Constants Calculated for the Interactions of FmtAΔ80 with Different Ligands.
| compound | Ksv (× 103 M–1) | kq (× 1011 M–1 s–1) | Kb (× 104 M–1) | Kd (μM) | N |
|---|---|---|---|---|---|
| gemifloxacin | 11.830 | 11.83 | 8.904304 | 11.23 | 1.18 |
| parmomycin | 18.474 | 18.5 | 2.552701 | 39.17 | 1.034 |
| streptomycin | 14.548 | 14.55 | 1.898016 | 52.91 | 1.09 |
| tobramycin | 15.269 | 15.27 | 1.174898 | 85.11 | 0.9729 |
3.7. Isothermal Titration Calorimetry Measurements
All ligands (gemifloxacin, paromomycin, streptomycin, and tobramycin) bind substantially to FmtAΔ80, as determined by in silico and fluorescence experiments. Further, ITC studies were performed to determine the thermodynamic parameters associated with the protein–ligand(s) interaction. Figure 7 shows the typical binding isotherms and fitting curves for the binding of gemifloxacin, paromomycin, streptomycin, and tobramycin to FmtAΔ80. The upward trend in the binding isotherms illustrates the endothermic binding interaction of FmtAΔ80 with gemifloxacin and streptomycin (Figure 7A,C), while the downward trend in the binding isotherms (Figure 7B,D) shows the exothermic binding interactions of FmtAΔ80 with paromomycin and tobramycin. This demonstrates that the binding of gemifloxacin and streptomycin to FmtAΔ80 was governed by entropy change (ΔS > 0), whereas the binding of paromomycin and tobramycin was governed by enthalpy change (ΔH < 0). The thermodynamic parameters obtained by fitting all the isotherms with a one-site model are summarized in Table 3. All four ligands have a KD value in the micromolar range, indicating a high affinity for FmtAΔ80. Hence, the ITC data also indicate that all ligands have a high affinity for FmtAΔ80.
Figure 7.

Isothermal titration calorimetry profile obtained upon the titration of 500 μM ligands into 50 μM FmtAΔ80. (A) Gemifloxacin, (B) paromomycin, (C) streptomycin, and (D) tobramycin. The bottom panel depicts the binding isotherm obtained by integrating the peak area and normalizing it to obtain the molar enthalpy change.
Table 3. Thermodynamic Parameters Determined by Colorimetric Titration of Different Ligands with FmtAΔ80.
| compound | enthalpy change (cal/mol) | entropy change (cal/mol/K) | free-energy change (cal/mol) | N | KD (μM) |
|---|---|---|---|---|---|
| gemifloxacin | 3.9 × 104 | 158 | –7.88 × 103 | 1.87 | 1.7 |
| parmomycin | –1.2 × 104 | –15.6 | –7.17 × 103 | 0.868 | 8.56 |
| streptomycin | 6.9 × 103 | 45.9 | –6.75 × 103 | 1.62 | 11.13 |
| tobramycin | –1.0 × 106 | –3360 | –7.72 × 103 | 0.750 | 25.77 |
3.8. CD Spectroscopy
CD studies were conducted to examine the conformational changes in the FmtAΔ80 protein caused by the interaction of inhibitors. The far-UV CD spectra of FmtAΔ80 display a negative peak at 207 nm and a positive peak at 196 nm, which indicate the α-helices in the native protein (Figure 8). The broad minima in the region of 210–220 nm indicate the existence of β-sheets. Figure 8 illustrates the far-UV CD spectra of FmtAΔ80 in the presence of different ligands. The binding of ligands have resulted in alterations to the far-UV spectra of FmtAΔ80, the most notable of which is the loss of the helical content at 209 nm, as seen by the ellipticity decrease at these wavelengths. The changes were also observed in the β-sheet region (210–220 nm).
Figure 8.

Far-UV CD spectra of native FmtAΔ80 and with ligands at the wavelength of 190–250 nm.
4. Discussion
FmtA is associated with the biosynthesis of peptidoglycan and is described as a component of methicillin resistance in S. aureus. It contains two out of three conserved motifs of β-lactamases and penicillin-binding proteins (PBPs). However, FmtA shows similarities with penicillin-recognizing proteins (PRPs), namely d-aminopeptidase (DAP), d-esterase, and d, d-endopeptidase, and so forth, which do not possess PBP and β-lactamase activities. Rahman et al. found that FmtA exhibits an esterase activity that eliminates d-alanine from the teichoic acids.11 FmtA esterase activity is linked to teichoic acid charge regulation and hence can be associated with S. aureus autolysis, colonization, and cell division.11,54 Moreover, the reported crystal structure of FmtA and mutational studies revealed that Ser-Lys-Asp may function as a catalytic triad for catalyzing WTA in FmtA.23 The conserved residues of the catalytic triad have different roles: Ser functions as a nucleophile, Lys works in acylation/deacylation, while Tyr holds the substrate in the course of hydrolysis of WTA.25 The QM/MM study suggested that the release of d-Alanine from WTA involves the formation of an acyl–enzyme complex and two tetrahedral intermediate complexes, which are stabilized via oxyanion hole residues (backbone amide of Ser127 and Gly344).24 Therefore, in FmtA, the first motif residues (Ser127 and Lys130) were involved in catalysis and charge neutralization; second motif (Tyr211 and Asp213) in holding the substrate; and third motif (Asn343 and Gly344) is crucial for the stability and charge neutralization of WTA.24
The present study aims to find the potential inhibitors against FmtAΔ80 of S. aureus. Virtual screening and molecular docking studies indicated that four drug molecules (gemifloxacin, paromomycin, streptomycin, and tobramycin) exhibited good binding affinities (−7.6 to −7.2 kcal/mol for AutoDock Vina and −7.1 to −6.5 kcal/mol for AutoDockTools) due to the hydrogen bonds and hydrophobic interactions with FmtA. The structures of the selected compounds are displayed in Figure S11. The drug molecules occupied the active sites of FmtA, as shown in Figure S12. Additionally, BLASTP (https://blast.ncbi.nlm.nih.gov/Blast.cgi) against the human proteome showed no significant similarity for humans, suggesting that FmtA can be chosen as a drug target for S. aureus. Virulence factor database (VFDB) (http://www.mgc.ac.cn/VFs/) revealed no similarity of FmtA with the S. aureus virulence factors.49,58
Gemifloxacin, paromomycin, tobramycin and streptomycin are aminoglycoside antibacterial agents which are known to treat a wide variety of bacterial infections.59−63 Gemifloxacin functions by preventing DNA synthesis by hampering both DNA gyrase and topoisomerase IV, which are crucial for microbial life cycle.64 Paromomycin binds to 16S ribosomal RNA and produces defective polypeptide chains or inhibits protein synthesis.65 Tobramycin binds to the 30S ribosomal subunit and subsequently inhibits the protein synthesis in bacteria.66 Streptomycin functions by disrupting the membrane permeability and inhibiting the protein synthesis.67
Furthermore, to determine the stability of gemifloxacin, paromomycin, streptomycin, and tobramycin along with FmtA, molecular dynamics simulation of FmtA–inhibitor(s) complexes was conducted. RMSD and RMSF analyses indicated that the FmtA–inhibitor(s) complexes were more stable than FmtA–Native. SASA and Rg displayed that FmtA–inhibitor(s) complexes were stable and compact. Hydrogen-bond analysis displayed that hydrogen bonds in the FmtA–inhibitor(s) complexes stabilized the complexes. MMPBSA results supported that the selected inhibitors interacted with Ser127, Lys130, Tyr211, Asp213, Tyr282, Gly345, and Phe346 of FmtA and formed lower energy stable complexes. QM/MM analysis showed that the inhibitor(s) interacts with the active site residues of FmtA for generating stable FmtA–inhibitor(s) complexes. MD, MMPBSA, and QM/MM results deduced that the selected inhibitor(s) interacts with the active site residues of the protein, leading to the generation of low-energy and stable FmtA–inhibitor(s) complexes.
To study the interactions of ligands with the protein, we performed fluorescence quenching measurements. According to the FmtAΔ80 structure, two internal fluorophores are present: Trp158 is present at the distal end and thereby does not contribute much to the fluorescence intensity, whereas tyrosine (Tyr211) is present in the active site of protein.11,25,26 Tyrosine is generally considered as a simple fluorophore, but under certain conditions, it can also show dominated fluorescence behavior such as in neurophysin, ribonuclease A, peptide hormones, spinach photosystem II, and mitochondrial malate dehydrogenase.68−73 It has been reported that protein–drug interaction induces a change in the microenvironments around the Tyr residues in a protein; as a result, the fluorescence emission behavior of the protein will be affected.70,74−76 Here, we have observed that in the presence of drug molecules, the fluorescence intensity declined, which revealed that the modification occurred in the microenvironment of Tyr211 and stabilized the protein–ligand(s) complex. All the ligands have a Kd value in the micromolar range, although gemifloxacin has a high binding affinity in comparison to other ligands (Table 2). We supplemented these binding affinities by using ITC experiments to estimate the thermodynamic parameters of each protein–ligand complex (Figure 7 and Table 3). We have measured the heat exchange during complex formation at a constant temperature and determined the binding process or intermolecular interactions. The free-energy change with a negative value indicates that the binding reactions are spontaneous in nature, and the obtained dissociation constants for all the reactions are very consistent with the findings from fluorescence experiments. Thus, ITC measurements also supported the fluorescence data that gemifloxacin has a strong binding affinity for FmtAΔ80, followed by paromomycin, streptomycin, and tobramycin. Further, far-UV CD data revealed that all binding ligands have resulted in the conformational changes in the secondary structure of FmtAΔ80, which are consistent with our fluorescence and ITC results.
5. Conclusions
In the present study, we have evaluated the inhibitory potential of four antibacterial molecules, namely gemifloxacin, paromomycin, tobramycin, and streptomycin, against FmtA of Staphylococcus aureus. These compounds were found using virtual screening against the FmtA crystal structure. The molecular docking analysis depicted that the drugs bind with the protein active site residues through hydrophobic interactions and hydrogen bonding. The MD simulation, MM-PBSA, and QM/MM analyses results revealed that FmtA forms compact and stable FmtA–drug(s) complexes. Fluorescence spectroscopy results demonstrated the good binding affinity of drugs with FmtAΔ80, with the Kd values ranging from 11 to 85 μM. ITC experiments showed spontaneous thermodynamic parameters, with gemifloxacin having the highest binding affinity for FmtA, accompanied by paromomycin, streptomycin, and tobramycin. The binding of gemifloxacin, paromomycin, tobramycin, and streptomycin to FmtAΔ80 resulted in the conformational changes in the secondary structure of FmtAΔ80. Conclusively, the screened drug molecules are a good starting point and can provide a path for further innovation and exploration of antibacterial molecules against S. aureus in the near future.
Acknowledgments
PK acknowledges the Council of Scientific and Industrial Research (CSR-1459-BIO). PK is thankful to Translational Bioinformatics Center at IIT Roorkee, Department of Biotechnology (BT/PR40141/BTIS/137/16/2021), and Macromolecular Crystallographic Unit (MCU), a Central Facility at Institute Instrumentation Centre (IIC), IIT Roorkee, for providing the computational facility.
Glossary
Abbreviations
- MRSA
Methicillin-resistant Staphylococcus aureus
- PBPs
penicillin-binding proteins
- PRE
penicillin-recognizing protein
- MD
molecular dynamics
- RMSF
root-mean-square fluctuation
- RMSD
root-mean-square deviation
- Rg
radius of gyration
- SASA
solvent-accessible surface area
- MMPBSA
molecular mechanics Poisson–Boltzmann surface area
- DFT
density functional theory
- HOMO
highest occupied molecular orbital
- LUMO
lowest unoccupied molecular orbital
- ITC
isothermal titration calorimetry
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c03671.
MD details of FmtA–drug complexes; summary of molecular docking and hydrogen-bond interactions;, RMSD, RMSF, Rg, and SASA values; DFT calculations; 2D representation of interactions between FmtA and drug(s); RMSF plot of FmtA–drug(s); SASA profile of FmtA–drug(s); intraprotein hydrogen-bond plots for FmtA–drug(s) complexes; interhydrogen-bond numbers; MMPBSA binding energies; amino acid residue decomposition analysis of FmtA; highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) analysis of drugs; cloning and purification of FmtAΔ80; Stern–Volmer plots of FmtAΔ80 quenching studies; structures of drugs; and superimposition of drugs along with FmtAΔ80 (PDF)
Author Contributions
§ V.S., P.D. and V.D. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Saïd-Salim B.; Mathema B.; Kreiswirth B. N. Community-acquired methicillin-resistant Staphylococcus aureus: an emerging pathogen. Infect. Control Hosp. Epidemiol. 2003, 24, 451–455. 10.1086/502231. [DOI] [PubMed] [Google Scholar]
- Lowy F. D. Antimicrobial resistance: the example of Staphylococcus aureus. J. Clin. Invest. 2003, 111, 1265–1273. 10.1172/JCI18535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S. M.; Webb S. Nosocomial bacterial infections in Intensive Care Units. I: Organisms and mechanisms of antibiotic resistance. Anaesthesia 2005, 60, 887–902. 10.1111/j.1365-2044.2005.04220.x. [DOI] [PubMed] [Google Scholar]
- Cox R.; Mallaghan C.; Conquest C.; King J. Epidemic methicillin-resistant Staphylococcus aureus: controlling the spread outside hospital. Infect. Control Hosp. Epidemiol. 1995, 29, 107–119. 10.1016/0195-6701(95)90192-2. [DOI] [PubMed] [Google Scholar]
- Holden M. T.; Feil E. J.; Lindsay J. A.; Peacock S. J.; Day N. P.; Enright M. C.; Foster T. J.; Moore C. E.; Hurst L.; Atkin R.; Barron A.; Bason N.; Bentley S. D.; Chillingworth C.; Chillingworth T.; Churcher C.; Clark L.; Corton C.; Cronin A.; Doggett J.; Dowd L.; Feltwell T.; Hance Z.; Harris B.; Hauser H.; Holroyd S.; Jagels K.; James K. D.; Lennard N.; Line A.; Mayes R.; Moule S.; Mungall K.; Ormond D.; Quail M. A.; Rabbinowitsch E.; Rutherford K.; Sanders M.; Sharp S.; Simmonds M.; Stevens K.; Whitehead S.; Barrell B. G.; Spratt B. G.; Parkhill J. Complete genomes of two clinical Staphylococcus aureus strains: evidence for the rapid evolution of virulence and drug resistance. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 9786–9791. 10.1073/pnas.0402521101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald J. R.; Sturdevant D. E.; Mackie S. M.; Gill S. R.; Musser J. M. Evolutionary genomics of Staphylococcus aureus: insights into the origin of methicillin-resistant strains and the toxic shock syndrome epidemic. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 8821–8826. 10.1073/pnas.161098098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugliese G.; Favero M. S.; Bartley J. First case of VRSA identified in Michigan. Infect. Control Hosp. Epidemiol. 2002, 23, 480–480. 10.1017/s0195941700082333. [DOI] [PubMed] [Google Scholar]
- Brown D. F.; Reynolds P. E. Intrinsic resistance to β-lactam antibiotics in Staphylococcus aureus. FEBS Lett. 1980, 122, 275–278. 10.1016/0014-5793(80)80455-8. [DOI] [PubMed] [Google Scholar]
- Sobral R.; Ludovice A.; De Lencastre H.; Tomasz A. Role of murF in cell wall biosynthesis: isolation and characterization of a murF conditional mutant of Staphylococcus aureus. J. Bacteriol. 2006, 188, 2543–2553. 10.1128/JB.188.7.2543-2553.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balibar C. J.; Shen X.; Tao J. The mevalonate pathway of Staphylococcus aureus. J. Bacteriol. 2009, 191, 851–861. 10.1128/JB.01357-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman M. M.; Hunter H. N.; Prova S.; Verma V.; Qamar A.; Golemi-Kotra D. The Staphylococcus aureus methicillin resistance factor FmtA is a d-amino esterase that acts on teichoic acids. J. Mol. Biol. 2016, 7, e02070–e02015. 10.1128/mBio.02070-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsuzawa H.; Sugai M.; Ohta K.; Fujiwara T.; Nakashima S.; Suzuki J.; Lee C. Y.; Suginaka H. Cloning and characterization of the fmt gene which affects the methicillin resistance level and autolysis in the presence of triton X-100 in methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 1997, 41, 2355–2361. 10.1128/AAC.41.11.2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utaida S.; Dunman P.; Macapagal D.; Murphy E.; Projan S.; Singh V.; Jayaswal R.; Wilkinson B. Genome-wide transcriptional profiling of the response of Staphylococcus aureus to cell-wall-active antibiotics reveals a cell-wall-stress stimulon. Microbiology 2003, 149, 2719–2732. 10.1099/mic.0.26426-0. [DOI] [PubMed] [Google Scholar]
- McCallum N.; Spehar G.; Bischoff M.; Berger-Bächi B. Strain dependence of the cell wall-damage induced stimulon in Staphylococcus aureus. Biochim. Biophys. Acta, Gen. Subj. 2006, 1760, 1475–1481. 10.1016/j.bbagen.2006.06.008. [DOI] [PubMed] [Google Scholar]
- Okazaki S.; Suzuki A.; Komeda H.; Yamaguchi S.; Asano Y.; Yamane T. Crystal structure and functional characterization of a D-stereospecific amino acid amidase from Ochrobactrum anthropi SV3, a new member of the penicillin-recognizing proteins. J. Mol. Biol. 2007, 368, 79–91. 10.1016/j.jmb.2006.10.070. [DOI] [PubMed] [Google Scholar]
- Neuhaus F. C.; Baddiley J. A continuum of anionic charge: structures and functions of D-alanyl-teichoic acids in gram-positive bacteria. Microbiol. Mol. Biol. Rev. 2003, 67, 686–723. 10.1128/MMBR.67.4.686-723.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qamar A.; Golemi-Kotra D. Dual roles of FmtA in Staphylococcus aureus cell wall biosynthesis and autolysis. Antimicrob. Agents Chemother. 2012, 56, 3797–3805. 10.1128/AAC.00187-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boles B. R.; Thoendel M.; Roth A. J.; Horswill A. R. Identification of genes involved in polysaccharide-independent Staphylococcus aureus biofilm formation. PLoS One 2010, 5, e10146 10.1371/journal.pone.0010146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown S.; Xia G.; Luhachack L. G.; Campbell J.; Meredith T. C.; Chen C.; Winstel V.; Gekeler C.; Irazoqui J. E.; Peschel A.; Walker S. Methicillin resistance in Staphylococcus aureus requires glycosylated wall teichoic acids. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 18909–18914. 10.1073/pnas.1209126109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner U. G.; Petersen E. I.; Schwab H.; Kratky C. EstB from Burkholderia gladioli: a novel esterase with a β-lactamase fold reveals steric factors to discriminate between esterolytic and β-lactam cleaving activity. Protein Sci. 2002, 11, 467–478. 10.1110/ps.33002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne D. G.; Riddles P.; Jones G. J.; Smith W.; Blakeley R. L. Characterisation of a gene cluster involved in bacterial degradation of the cyanobacterial toxin microcystin LR. Environ. Toxicol. 2001, 16, 523–534. 10.1002/tox.10013. [DOI] [PubMed] [Google Scholar]
- Delmarcelle M.; Boursoit M. C.; Filée P.; Baurin S. L.; Frère J. M.; Joris B. Specificity inversion of Ochrobactrum anthropi D-aminopeptidase to a D,D-carboxypeptidase with new penicillin binding activity by directed mutagenesis. Protein Sci. 2005, 14, 2296–2303. 10.1110/ps.051475305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komeda H.; Asano Y. Gene cloning, nucleotide sequencing, and purification and characterization of the D-stereospecific amino-acid amidase from Ochrobactrum anthropi SV3. Eur. J. Biochem. 2000, 267, 2028–2035. 10.1046/j.1432-1327.2000.01208.x. [DOI] [PubMed] [Google Scholar]
- Dalal V.; Golemi-Kotra D.; Kumar P. Quantum Mechanics/Molecular Mechanics Studies on the Catalytic Mechanism of a Novel Esterase (FmtA) of Staphylococcus aureus. J. Chem. Inf. Model. 2022, 62, 2409. 10.1021/acs.jcim.2c00057. [DOI] [PubMed] [Google Scholar]
- Dalal V.; Kumar P.; Rakhaminov G.; Qamar A.; Fan X.; Hunter H.; Tomar S.; Golemi-Kotra D.; Kumar P. Repurposing an ancient protein core structure: Structural studies on FmtA, a novel esterase of Staphylococcus aureus. J. Mol. Biol. 2019, 431, 3107–3123. 10.1016/j.jmb.2019.06.019. [DOI] [PubMed] [Google Scholar]
- Dalal V.; Dhankhar P.; Singh V.; Rakhaminov G.; Golemi-Kotra D.; Kumar P. Structure-based identification of potential drugs against FmtA of Staphylococcus aureus: Virtual screening, molecular dynamics, MM-GBSA, and QM/MM. Protein J. 2021, 40, 148–165. 10.1007/s10930-020-09953-6. [DOI] [PubMed] [Google Scholar]
- Dallakyan S.; Olson A. J.. Small-molecule library screening by docking with PyRx. In Chem. Biol.; Springer, 2015; pp 243–250. [DOI] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurkcuoglu Z.; Koukos P. I.; Citro N.; Trellet M. E.; Rodrigues J.; Moreira I. S.; Roel-Touris J.; Melquiond A. S.; Geng C.; Schaarschmidt J.; Xue L. C.; Vangone A.; Bonvin A. M. J. J. Performance of HADDOCK and a simple contact-based protein–ligand binding affinity predictor in the D3R Grand Challenge 2. J. Comput.-Aided Mol. Des. 2018, 32, 175–185. 10.1007/s10822-017-0049-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini G.; Dalal V.; Savita B. K.; Sharma N.; Kumar P.; Sharma A. K. Molecular docking and dynamic approach to virtual screen inhibitors against Esbp of Candidatus Liberibacter asiaticus. J. Mol. Graphics Modell. 2019, 92, 329–340. 10.1016/j.jmgm.2019.08.012. [DOI] [PubMed] [Google Scholar]
- Release S.2: Maestro, Schrödinger; LLC, New York, NY, 2017. Received: February 2016, 21, 2018. [Google Scholar]
- DeLano W. L.The pymol molecular graphics system (2002) http://www.pymol.org, 2002, 10.1016/s1052-5149(03)00068-6. [DOI] [Google Scholar]
- Singh N.; Dalal V.; Mahto J. K.; Kumar P. Biodegradation of phthalic acid esters (PAEs) and in silico structural characterization of mono-2-ethylhexyl phthalate (MEHP) hydrolase on the basis of close structural homolog. J. Hazard. Mater. 2017, 338, 11–22. 10.1016/j.jhazmat.2017.04.055. [DOI] [PubMed] [Google Scholar]
- van Gunsteren W. F.; Billeter S.; Eising A.; Hünenberger P.; Krüger P.; Mark A.; Scott W.; Tironi I.. Biomolecular simulation: the GROMOS96 manual and user guide; Vdf Hochschulverlag AG an der ETH Zürich: Zürich, 1996; Vol. 86, pp 1–1044. [Google Scholar]
- Anandakrishnan R.; Aguilar B.; Onufriev A. V. H++ 3.0: automating p K prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40, W537–W541. 10.1093/nar/gks375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari N.; Dalal V.; Kumar P.; Rath S. N. Antagonistic interaction between TTA-A2 and paclitaxel for anti-cancer effects by complex formation with T-type calcium channel. J. Mol. Graphics Modell. 2022, 40, 2395–2406. 10.1080/07391102.2020.1839558. [DOI] [PubMed] [Google Scholar]
- Dhankhar P.; Dalal V.; Singh V.; Tomar S.; Kumar P. Computational guided identification of novel potent inhibitors of N-terminal domain of nucleocapsid protein of severe acute respiratory syndrome coronavirus 2. J. Mol. Graphics Modell. 2022, 40, 4084–4099. 10.1080/07391102.2020.1852968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Schüttelkopf A. W.; Van Aalten D. M. PRODRG: a tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 1355–1363. 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- Kumar P.; Dalal V.; Kokane A.; Singh S.; Lonare S.; Kaur H.; Ghosh D. K.; Kumar P.; Sharma A. K. Mutation studies and structure-based identification of potential inhibitor molecules against periplasmic amino acid binding protein of Candidatus Liberibacter asiaticus (CLasTcyA). Int. J. Biol. Macromol. 2020, 147, 1228–1238. 10.1016/j.ijbiomac.2019.09.250. [DOI] [PubMed] [Google Scholar]
- Dhankhar P.; Dalal V.; Singh V.; Sharma A. K.; Kumar P. Structure of dye-decolorizing peroxidase from Bacillus subtilis in complex with veratryl alcohol. Int. J. Biol. Macromol. 2021, 193, 601–608. 10.1016/j.ijbiomac.2021.10.100. [DOI] [PubMed] [Google Scholar]
- Singh N.; Dalal V.; Kumar P. Structure based mimicking of Phthalic acid esters (PAEs) and inhibition of hACMSD, an important enzyme of the tryptophan kynurenine metabolism pathway. Int. J. Biol. Macromol. 2018, 108, 214–224. 10.1016/j.ijbiomac.2017.12.005. [DOI] [PubMed] [Google Scholar]
- Kumari R.; Kumar R.; Consortium O. S. D. D.; Lynn A. g_mmpbsa— A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- Dhankhar P.; Dalal V.; Mahto J. K.; Gurjar B. R.; Tomar S.; Sharma A. K.; Kumar P. Characterization of dye-decolorizing peroxidase from Bacillus subtilis. Arch. Biochem. Biophys. 2020, 693, 108590 10.1016/j.abb.2020.108590. [DOI] [PubMed] [Google Scholar]
- Malik A.; Dalal V.; Ankri S.; Tomar S. Structural insights into Entamoeba histolytica arginase and structure-based identification of novel non-amino acid based inhibitors as potential antiamoebic molecules. FEBS J. 2019, 286, 4135–4155. 10.1111/febs.14960. [DOI] [PubMed] [Google Scholar]
- Dhankhar P.; Dalal V.; Golemi-Kotra D.; Kumar P. In-silico approach to identify novel potent inhibitors against GraR of S. aureus. Front. Biosci.-Landmark 2020, 25, 1337–1360. 10.2741/4859. [DOI] [PubMed] [Google Scholar]
- Singh V.; Dhankhar P.; Dalal V.; Tomar S.; Kumar P. In-silico functional and structural annotation of hypothetical protein from Klebsiella pneumonia: A potential drug target. J. Mol. Graphics Modell. 2022, 116, 108262 10.1016/j.jmgm.2022.108262. [DOI] [PubMed] [Google Scholar]
- Gupta D. N.; Dalal V.; Savita B. K.; Dhankhar P.; Ghosh D. K.; Kumar P.; Sharma A. K. In-silico screening and identification of potential inhibitors against 2Cys peroxiredoxin of Candidatus Liberibacter asiaticus. J. Biomol. Struct. Dyn. 2021, 1–15. 10.1080/07391102.2021.1916597. [DOI] [PubMed] [Google Scholar]
- Saini G.; Dalal V.; Gupta D. N.; Sharma N.; Kumar P.; Sharma A. K. A molecular docking and dynamic approach to screen inhibitors against ZnuA1 of Candidatus Liberibacter asiaticus. Mol. Simul. 2021, 47, 510–525. 10.1080/08927022.2021.1888948. [DOI] [PubMed] [Google Scholar]
- Vreven T.; Morokuma K.; Farkas Ö.; Schlegel H. B.; Frisch M. J. Geometry optimization with QM/MM, ONIOM, and other combined methods. I. Microiterations and constraints. J. Comput. Chem. 2003, 24, 760–769. 10.1002/jcc.10156. [DOI] [PubMed] [Google Scholar]
- Dapprich S.; Komáromi I.; Byun K. S.; Morokuma K.; Frisch M. J. A new ONIOM implementation in Gaussian98. Part I. The calculation of energies, gradients, vibrational frequencies and electric field derivatives. J. Mol. Struct. 1999, 461, 1–21. 10.1016/S0166-1280(98)00475-8. [DOI] [Google Scholar]
- Fan X.; Liu Y.; Smith D.; Konermann L.; Siu K. M.; Golemi-Kotra D. Diversity of Penicillin-binding Proteins. J. Biol. Chem. 2007, 282, 35143–35152. 10.1074/jbc.M706296200. [DOI] [PubMed] [Google Scholar]
- Ryde U. A fundamental view of enthalpy–entropy compensation. Med. Chem. Commun. 2014, 5, 1324–1336. 10.1039/C4MD00057A. [DOI] [Google Scholar]
- Mocz G.; Ross J. A. Fluorescence techniques in analysis of protein-ligand interactions. Methods Mol. Biol. 2013, 1008, 169–210. 10.1007/978-1-62703-398-5_7. [DOI] [PubMed] [Google Scholar]
- Ishtikhar M.; Khan S.; Badr G.; Mohamed A. O.; Khan R. H. Interaction of the 5-fluorouracil analog 5-fluoro-2′-deoxyuridine with ‘N’and ‘B’isoforms of human serum albumin: a spectroscopic and calorimetric study. Mol. Syst. 2014, 10, 2954–2964. 10.1039/c4mb00306c. [DOI] [PubMed] [Google Scholar]
- Chen L.; Yang J.; Yu J.; Yao Z.; Sun L.; Shen Y.; Jin Q. VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 2005, 33, D325–D328. 10.1093/nar/gki008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatz A.; Bugle E.; Waksman S. A. Streptomycin, a Substance Exhibiting Antibiotic Activity Against Gram-Positive and Gram-Negative Bacteria*. Exp. Biol. Med. 1944, 55, 66–69. 10.3181/00379727-55-14461. [DOI] [PubMed] [Google Scholar]
- Waksman S. A. Streptomycin: background, isolation, properties, and utilization. Science 1953, 118, 259–266. 10.1126/science.118.3062.259. [DOI] [PubMed] [Google Scholar]
- Neu H. C. Tobramycin: an overview. J. Infect. Dis. 1976, S3–S19. 10.1093/infdis/134.Supplement_1.S3. [DOI] [PubMed] [Google Scholar]
- Lowe M. N.; Lamb H. M. Gemifloxacin. Drugs 2000, 59, 1137–1147. 10.2165/00003495-200059050-00009. [DOI] [PubMed] [Google Scholar]
- Davidson R. N.; den Boer M.; Ritmeijer K. Paromomycin. Trans. R. Soc. Trop. Med. Hyg. 2009, 103, 653–660. 10.1016/j.trstmh.2008.09.008. [DOI] [PubMed] [Google Scholar]
- Blondeau J. M.; Missaghi B. Gemifloxacin: a new fluoroquinolone. Expert Opin. Pharmacother. 2004, 5, 1117–1152. 10.1517/14656566.5.5.1117. [DOI] [PubMed] [Google Scholar]
- Fourmy D.; Yoshizawa S.; Puglisi J. D. Paromomycin binding induces a local conformational change in the A-site of 16 S rRNA. J. Mol. Biol. 1998, 277, 333–345. 10.1006/jmbi.1997.1551. [DOI] [PubMed] [Google Scholar]
- Brogden R. N.; Pinder R. M.; Sawyer P. R.; Speight T. M.; Avery G. S. Tobramycin: a review of its antibacterial and pharmacokinetic properties and therapeutic use. Drugs 1976, 12, 166–200. 10.2165/00003495-197612030-00002. [DOI] [PubMed] [Google Scholar]
- Cox E. C.; White J. R.; Flaks J. G. Streptomycin action and the ribosome. Proc. Natl. Acad. Sci. U. S. A. 1964, 51, 703. 10.1073/pnas.51.4.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir M.; Qureshi M. A.; Javed S. Biomolecular interactions and binding dynamics of tyrosine kinase inhibitor erdafitinib, with human serum albumin. J. Biomol. Struct. Dyn. 2021, 39, 3934–3947. 10.1080/07391102.2020.1772880. [DOI] [PubMed] [Google Scholar]
- Sethuraman S.; Rajendran K. Multicharacteristic Behavior of Tyrosine Present in the Microdomains of the Macromolecule Gum Arabic at Various pH Conditions. ACS Omega 2018, 3, 17602–17609. 10.1021/acsomega.8b02928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander Ross J. B.; Laws W. R.; Rousslang K. W.; Wyssbrod H. R.. Tyrosine Fluorescence and Phosphorescence from Proteins and Polypeptides. In Top. Fluoresc. Spectrosc., Lakowicz J. R., Ed.; Springer US: Boston, MA, 2002; pp 1–64. [Google Scholar]
- Lux B.; Gerard D.; Laustriat G. Tyrosine fluorescence of S8 and S15 Escherichia coli ribosomal proteins. FEBS Lett. 1977, 80, 66–70. 10.1016/0014-5793(77)80408-0. [DOI] [PubMed] [Google Scholar]
- Liu X. Y.; Cottrell K. O.; Nordlund T. M. Spectroscopy and fluorescence quenching of tyrosine in lima bean trypsin/chymotrypsin inhibitor and model peptides. Photochem. Photobiol. 1989, 50, 721–731. 10.1111/j.1751-1097.1989.tb02902.x. [DOI] [PubMed] [Google Scholar]
- Lux B.; Baudier J.; Gerard D. Tyrosyl fluorescence spectra of proteins lacking tryptophan: effects of intramolecular interactions. Photochem. Photobiol. 1985, 42, 245–251. 10.1111/j.1751-1097.1985.tb08938.x. [DOI] [PubMed] [Google Scholar]
- Feitelson J. On the mechanism of fluorescence quenching. Tyrosine and similar compounds. J. Phys. Chem. 1964, 68, 391–397. 10.1021/j100784a033. [DOI] [Google Scholar]
- Brun F.; Toulme J. J.; Helene C. Interactions of aromatic residues of proteins with nucleic acids. Fluorescence studies of the binding of oligopeptides containing tryptophan and tyrosine residues to polynucleotides. Biochemistry 1975, 14, 558–563. 10.1021/bi00674a015. [DOI] [PubMed] [Google Scholar]
- Lim B. T.; Kimura T. Conformation prediction and spectral studies on adrenodoxin. The accessibility of the tyrosine residue at position 82 in the polypeptide. J. Biophys. Chem. 1981, 256, 4400–4406. 10.1016/S0021-9258(19)69448-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.