

Abstract
Three symmetrical bis-aryl-α,β-unsaturated ketone derivatives, 2,6-di((E)-benzylidene)-cyclohexan-1-one (DBC), 2,6-bis((E)-4-chlorobenzylidene)cyclohexan-1-one (BCC), and (1E,1′E,4E,4′E)-5,5′-(1,4-phenylene)bis(2-methyl-1-phenylpenta-1,4-dien-3-one) (PBMP), have been prepared using the aldol condensation approach toward ketones having two enolizable sites. The structures of DBC, BCC, and PBMP have been resolved via spectrometric methods. Moreover, the crystal structure of PBMP is determined by the single-crystal X-ray diffraction (SC-XRD) technique, which revealed that the PBMP molecular assembly is stabilized by the intermolecular C–H···O bonding and C–O···π and weak T-shaped offset π···π stacking interactions. The Hirshfeld surface analysis (HSA) of the PBMP crystal structure was performed as well, and the results were compared with the results of DBC and BCC. The density functional theory (DFT) study results revealed that the longer conjugated molecule of PBMP has smaller but still quite significant HOMO–LUMO gaps compared to the smaller molecules of BCC and DBC. The natural population analysis (NPA) and natural bonding orbital (NBO) analysis were performed. Accordingly, the hydrogen bonding and dipole–dipole interactions stabilize the crystal structures of these compounds. Additionally, the NBO analysis showed numerous high-energy stabilizing interactions for the PBMP compound due to the presence of numerous delocalized and relatively easily polarizable π-electrons, thus implying its significant thermodynamic stability. According to the global reactivity parameter (GRP) analysis, the compounds BCC and DBC are relatively stable in redox processes and have high thermodynamic stability and relatively lower reactivity in general. The molecular electrostatic potential (MEP) analysis results imply potential formation of the intermolecular hydrogen bonding and dispersion interactions, which stabilizes the crystal structures of these compounds.
1. Introduction
In synthetic organic chemistry, selecting facile reaction conditions together with a suitable precursor having the qualities of nonhazardous nature, tranquil handling, and easy accessibility has a vital role in the production of chemical entities of valuable interest, especially in the production of analytical reagents, pharmaceuticals, dyes, agriculture, and natural products. Aldol reaction is a simple carbon–carbon bond forming reaction for the production of valuable medicinal compounds in a facile manner under acid- or base-catalyzed conditions. Bis-aryl-α,β-unsaturated ketones are representatives of the broad class of pharmaceutically significant chemical architectures.1 These bis-aryl-α,β-unsaturated ketones are also known as monocarbonyl curcumin because of the only carbonyl functional group compared to the two carbonyl functional groups of the natural curcumin of the zingiberaceae family. These chemical building blocks, i.e., monocarbonyl curcumin, could be obtained via a simple aldol condensation approach that is a carbon–carbon bond forming a reaction between two carbonyl compounds with at least one of them having enolizable position.2 Bis-aryl-α,β-unsaturated ketones have been reported to demonstrate numerous biological actions, such as antimalarial,3 antitubercular,4 antiviral,5 antifertility,6 anticancer,7 antiparasitic,1a neuroprotective8 activities, etc. These compounds might exist in symmetrical or unsymmetrical form, but both are appreciated from medicinal perspectives.1a Besides the medicinal applications, these chemical building blocks have been explored for their photo-crosslinking properties, showing that these moieties have potential to be used as leading candidates for UV filters and sunscreens.9 The bis-aryl-α,β-unsaturated ketones could be obtained through utilization of ketones having two enolizable positions in a well-known acid- or base-catalyzed aldol condensation reaction (Figure 1).
Figure 1.

General acid- or base-catalyzed aldol condensation reaction for the preparation of bis-aryl-α,β-unsaturated ketones.
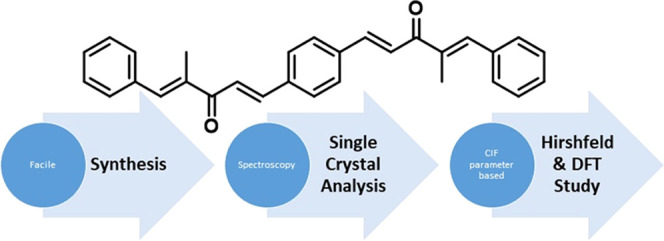
Recently, computational studies have been playing a fundamental role in understanding of structural stability, reaction mechanisms, potential reacting sites, electronic properties, etc. Theoretical studies can be used for the exploration of nonlinear optical (NLO) properties,10 noncovalent interactions (NCIs),11 natural bond orbital (NBO) properties,12 molecular electrostatic potential (MEP), etc., of different classes of organic compounds. In this study, we are presenting our findings regarding the synthesis of the symmetrical bis-aryl-α,β-unsaturated ketone derivatives DBC, BCC, and PBMP (Figure 2), their SC-XRD and spectroscopy-based structural details, and results of the DFT studies. The compounds DBC and BCC are synthesized in a single step, while the relative highly conjugated molecule, i.e., PBMP, has been synthesized in two steps, where the first step is catalyzed by an acid, while the second step is catalyzed by a base (Figure 3). Besides the medicinal importance of this class of compounds, computational exploration of the electronic properties will be a valuable addition in the field of experimental and theoretical chemistry.
Figure 2.

Symmetrical bis-aryl-α,β-unsaturated ketone derivatives considered in the study.
Figure 3.

Preparation of the compounds DBC and BCC (a) and PBMP (b).
2. Experimental Section
2.1. General Procedure
All of the chemicals employed were purchased from standard chemical companies and were used as received while solvents were purified by a simple distillation procedure. The reaction progress was monitored using precoated silica gel thin-layer chromatography (TCL) plates. The 1H and 13C NMR studies were accomplished using a Brüker AVANCE 400 spectrometer at 400 and 100 MHz, respectively. The single-crystal data of PBMP were collected on a Bruker KAPPA Apex-II diffractometer using APEX-II software.13 The absorption correction was performed using SADABS software.14 The structure was solved using SHELXT-2015 software15 and refined by SHELXL 2019/2 software.16 ORTEP-III17 and Mercury 4.018 softwares were employed for the graphical illustration of the SC-XRD results.
2.2. Preparation of the Compounds DBC and BCC
The compounds DBC and BCC were prepared according to the general procedure reported in the literature.19 According to the procedure, a mixture of cyclohexanone, 6 and 12 mmol of the appropriate aromatic aldehyde (benzaldehyde in the case of DBC and 4-chlorobenzaldehyde in the case of BCC) in ethanol (20 mL) was stirred at room temperature until both reagents were dissolved. To this reaction mixture, a solution of NaOH (14 mmol) in 20 mL of the water–ethanol mixture (1:1) was added dropwise. The reaction mixture turned yellow and solidified, next ice water (4 mL) was added, and the solid residue was collected by filtration, washed with cold water, and dried overnight (see Figure 3a).
2.3. Preparation of the Compound PBMP
Compound PBMP was prepared in the two-step procedure. In the first step, intermediate 3 was synthesized according to the general literature-reported procedure20 (Figure 3b). According to the procedure, an equimolar ratio mixture of benzaldehyde and 2-butanone was placed in a 50 mL two-neck round-bottom flask. Next, dry HCl gas was passed through the reaction mixture until it was saturated, and the solution turned red. The reaction mixture was stirred at room temperature for 6 h. The crude product was diluted with toluene and washed with the NaHSO3 solution. The organic layer was separated, dried with the anhydrous Na2SO4, and evaporated in vacuum. The residue was distilled under reduced pressure to give the pure compound. In the second step, the compound PBMP was prepared by reacting the intermediate compound 3 with terephthalaldehyde under basic conditions [2b].
2.4. Characterization of the Compound 3 [(E)-3-methyl-4-phenylbut-3-en-2-one]
The spectroscopic data were found to be in correspondence with the literature data.21 Percent yield: 56%, 1H NMR (400 MHz, CDCl3) δH 7.52 (d, 1H, J = 4 Hz), 7.41 (m, 3H), 7.39 (m, 1H), 7.32 (m, 1H), 2.47 (s, 3H), 2.06 (d, J = 1.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δC 129.7, 200.3, 139.7, 137.8, 135.9, 128.6, 128.5, 25.9, 12.9.
2.5. Characterization of the Compound DBC [2,6-di((E)-benzylidene)cyclohexan-1-one]
The spectroscopic data were found to be in correspondence with the literature data.22 M.P = 115–116 °C, 1H NMR (400 MHz, CDCl3) δ 7.84 (s, 2H), 7.52–7.48 (m, 4H), 7.46–7.41 (m, 4H), 7.39–7.35 (m, 2H), 3.00–2.93 (m, 4H), 1.86–1.78 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 190.41, 136.96, 136.21, 136.00, 130.39, 128.61, 128.41, 28.48, 23.03.
2.6. Characterization of the Compound BCC [2,6-bis((E)-4-chlorobenzylidene)cyclohexan-1-one]
The spectroscopic data were found to be in correspondence with the literature data.23 M.P = 108–110 °C, 1H NMR (400 MHz, CDCl3) δ 7.64 (d, J = 1.8 Hz, 2H), 7.35–7.23 (m, 8H), 2.86–2.77 (m, 4H), 1.76–1.68 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 189.85, 136.42, 135.78, 134.62, 134.31, 131.60, 128.69, 28.38, 22.81.
2.7. Characterization of the Compound PBMP [(1E,1′E,4E,4′E)-5,5′-(1,4-phenylene)bis(2-methyl-1-phenylpenta-1,4-dien-3-one)]
M.P = 297–298 °C, 1H NMR (400 MHz, CDCl3) δ 7.73–7.65 (m, 3H), 7.63–7.40 (m, 5H), 7.01–6.97 (m, 2H), 3.88 (s, 3H), 2.24 (d, J = 1.1 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 192.35, 160.02, 141.94, 139.05, 136.88, 136.62, 131.70, 128.69, 128.44, 122.87, 114.04, 55.36, 13.82. HRMS calc. for C30H27O2+ [M + H]+ 419.2006, found 419.2006. Also with sodium, C30H26NaO2+ [M + Na]+ 441.1830, found 441.1825 (Figure S10).
2.8. Computational Details
DFT studies were performed with Gaussian 09 software24a and Gaussian 16 software24b (note: we had to use two different computational facilities, in Brazil and in Chile, due to the scarcity of computational resources in the time of preparing the paper). Using as the starting geometries the structures from the SC-XRD analysis, we optimized the PBMP, BCC, and DBC molecules without any symmetry constraints and then performed frequency calculations to verify that the optimized structures are true energy minima. We checked both the singlet and triplet multiplicities for the molecules studied (vide infra). All calculations were performed with the hybrid density functional B3LYP25 and the triple-zeta split-valence polarized basis set 6-311+G(d,p)26,27 (one set of diffuse functions on C, N, O, Cl and two sets of polarization functions, both on hydrogens and heavier atoms). This approach is furthermore referred to as B3LYP/6-311+G(d,p). We did the computational studies and all analyses listed below with the B3LYP/6-311+G(d,p) approach and with the implicit effects from ethanol (dielectric constant ε = 24.852) and methanol (dielectric constant ε = 32.613) taken into account, employing the self-reliable IEF-PCM approach28 with the UFF default model as implemented in Gaussian 09/16 software, with the electrostatic scaling factor α = 1.0. Below, we compare the calculated structural parameters, natural population analysis (NPA) charges, natural bond orbital (NBO) interactions,29 and frontier molecular orbital (FMO) for the three compounds studied. It should be noticed that the results obtained in methanol were very close to the results obtained with ethanol (see below discussion for the relative energies of singlets and triplets); therefore, we decided to focus only on the results obtained using ethanol. Furthermore, we used the values of the energies of HOMO and LUMO to compute the global reactivity parameter (GRP)30−32 (see eqs 1–6). Eqs 1 and 2 were used to calculate the values of the ionization potential (IP) and electron affinity (EA)
| 1 |
| 2 |
For global hardness η and electronegativity X values, we used eqs 3 and 4
| 3 |
| 4 |
And the global electrophilicity ω value was calculated by eq 5
| 5 |
where  is the chemical potential of the system.
is the chemical potential of the system.
Finally, the global softness σ value was computed with eq 6
| 6 |
The open GL version of Molden 5.8.2 visualization program was used for the visualization of the structures and FMOs of the title compounds.33 Avogadro, version 1.1.1, was used to visualize the molecular electrostatic potential (MEP) maps.34,35
3. Results and Discussion
3.1. Single-Crystal Analysis
The Cambridge structural database (CSD 2021.2) confirms the novelty of the crystal structure of PBMP. The crystal structures of the compounds DBC and BCC were already reported in the literature.23,36 The experimental details of PBMP related to single-crystal analysis are provided in Table 1, whereas the selected bond lengths and bond angles determined experimentally are compared with their values determined in the DFT study (Table 2).
Table 1. Single-Crystal Analysis Data of PBMP.
| crystal data | PBMP |
|---|---|
| CCDC | 2156053 |
| chemical formula | C30H26O2 |
| Mr | 418.51 |
| crystal system, space group | monoclinic, P21/c |
| temperature (K) | 150 (2) |
| a, b, c (Å) | 5.6973 (6), 35.358 (4), 6.1245 (6) |
| α, β, γ (deg) | 90, 116.423 (2), 90 |
| V (Å3) | 1104.9 (2) |
| Z | 2 |
| density (calculated) g/cm–3 | 1.258 |
| F(000) | 444 |
| radiation type | MoKα |
| wavelength (λ) | 0.71073 |
| μ (mm–1) | 0.077 |
| crystal size (mm) | 0.57 × 0.32 × 0.09 |
| Data Collection | |
| diffractometer | Bruker APEX-II CCD diffractometer |
| absorption correction | multi-scan (SADABS; Bruker, 2007) |
| no. of measured, independent and observed [I > 2σ(I)] reflections | 21929, 2179, 2024 |
| Rint | 0.024 |
| theta range for data collection (deg) | 2.304 to 26.018 |
| index ranges | –6≤ h ≤7, –43≤ k ≤43, –7≤ l ≤6 |
| (sin θ/λ)max (Å–1) | 0.617 |
| Data Refinement | |
| R[F2 > 2σ(F2)], wR(F2), S | 0.039, 0.087, 1.11 |
| no. of reflections | 2179 |
| no. of parameters | 146 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin(e Å–3) | 0.18, −0.17 |
Table 2. Selected Bond Lengths (Å) and Bond Angles (deg) in PBMP.
| bond length | XRD | DFT | bond angles | XRD | DFT |
|---|---|---|---|---|---|
| C4-C5 | 1.3321 (18) | 1.347 | C4-C5-C6 | 121.87 (12) | 120.782 |
| C5-C6 | 1.4831 (18) | 1.485 | C5-C6-O1 | 121.00 (12) | 120.483 |
| C7-C8 | 1.4999 (18) | 1.506 | O1-C6-C7 | 120.69 (12) | 119.635 |
| C7-C9 | 1.3430 (18) | 1.355 | C6-C7-C8 | 116.16 (11) | 115.094 |
| C7-C6 | 1.4950 (18) | 1.498 | C8-C7-C9 | 125.69 (12) | 124.919 |
| C6-O1 | 1.2269 (15) | 1.233 | C9-C7-C6 | 117.77 (12) | 119.860 |
In PBMP (Figure 4, Table 1), the asymmetric unit contains half of the molecule, and the other half is generated by the symmetry transformations with the symmetry code (i) (−x, −y, 1–z). In the asymmetric unit (C1-C14/O1), the isoprene group A (C1–C5), propionaldehyde group B (C6–C8/O1), and benzyl group C (C9–C15) are planar with the root mean square (RMS) deviations of 0.0135, 0.0862, and 0.0094 Å, respectively. The central group B is oriented at a dihedral angle of 24.3(8) and 46.6(7)° with respect to the groups A and C, respectively. The selected bond lengths and bond angles are listed in Table 2, which conforms the nature of bonds and especially the double bond between the C6 atom and O1 atom. The molecular configuration is stabilized by the intramolecular C–H···O bonding. The molecules are interlinked by the nonclassical H-bonding of the type C–H···O in the form of dimers. As a result of dimerization, the R22(28) loop is formed. C5 infinite chains are formed through the C–H···O bonding that extends along a crystallographic axis (Figure 5). No strong classical H-bonding is present in the crystal packing. Very weak T-shaped π···π stacking interactions are present between phenyl rings of the symmetry-related molecules (x, 1/2 −y, 1/2 +z) with a distance of 5.0723(10) Å between the rings, whereas the dihedral angle between the plane of the interacting rings is 71.39(8)°. The further stabilization of the crystal packing is due to the C–O···π interactions with the O···π distances equal to 3.5116(13) Å as shown in Table 3. The C–O···π interactions along with weak T-shaped π···π stacking interactions interlink the molecules in the form of stairs (Figure S1).
Figure 4.

ORTEP diagram of PBMP drawn at a probability level of 50%. H-atoms are shown by small circles of arbitrary radii.
Figure 5.

Packing diagram of PBMP representing C5 chains formed by the C–H···O bonding. Only selected H-atoms are shown for clarity.
Table 3. Hydrogen-Bond Geometry (Å, deg) along with the C–O···π Interactions in PBMPa.
| D–H···A | D–H | H···A | D···A | ∠(D—H···A) (deg) |
|---|---|---|---|---|
| C8–H8C···O1 | 0.98 | 2.40 | 2.8467 (17) | 107 |
| C8–H8B···O1i | 0.98 | 2.71 | 3.6237 (18) | 156 |
| C–O···π | C–O | O···π | C···π | ∠(C–O···π) (deg) |
|---|---|---|---|---|
| C6–O1···Cg1ii | 1.2269(15) | 3.5116(13) | 3.6820(15) | 88.14(9) |
| C6–O1···Cg1iii | 1.2269(15) | 3.5116(13) | 3.6820(15) | 88.14(9) |
Symmetry codes: (i) x + 1, y, z; (ii) 1 + x, y, 1 + z; (iii) 1 – x, −y, 2–z. Cg1 is the centroid of phenyl ring (C10–C15).
3.2. Hirshfeld Surface Analysis
Hirshfeld surface (HS) analysis is a recently developed approach to explore the noncovalent interactions in the single crystal.37 Crystal Explorer version 21.538 was employed for the Hirshfeld surface analysis. The Hirshfeld surface concept arises from an attempt to divide the molecular electron density into small fragments to integrate it. Hirshfeld surfaces are employed not only to explore the H-bonding but also to explore the interactions weaker than the H-bonding. Cambridge structure database search provided two crystal structures: one with the reference code LEBPAK (CCDC number 268269)36 and another with the reference code QQQAQG02 (CCDC number 1433536) that are closely relevant to the compound PBMP. The HS was formed using the property dnorm to explore the short interatomic contacts.39 In other words, this HS provides useful knowledge about the H-bonding interactions. Red spots on the HS plotted over dnorm are the representatives on the interatomic contacts, where the distance between the interacting atoms is less than the sum of their van der Waals radii. White spots or regions represent the contacts for which the distance between the atoms is equal to the sum of their van der Waals radii (Figure 6a). Blue regions stand for the long-distance contacts that have negligible strength. The Hirshfeld surface formed using the shape index property was employed for the recognition of the π···π stacking interactions in the crystal packing (Figure 6b).40 The H-bond donors and acceptors can be separately recognized by plotting the HS over the electrostatic potential (Figure 6c). The H-bond donors are represented by blue spots, whereas the H-bond acceptors are represented by red spots on the HS.
Figure 6.

Hirshfeld surface plotted over (a) dnorm in the range −0.017 to 1.105 a.u., (b) shape index in the range −1 to 1 a.u., and (c) electrostatic potential in the range −0.057 to 0.032 a.u.
The 2D fingerprint plot analysis is a way to elaborate on the interatomic contacts in terms of their percentage contributions in the crystal packing.41,42 We are going to compare the important 2D fingerprint plots of PBMP with DBC (reference code LEBPAK) and BCC (reference code QQQAQG02). Figures 7a,e,i shows 2D plots for the overall interactions containing the contributions of all of the possible interatomic contacts for PBMP, DBC, and BCC, respectively. As the crystal structure contains a higher number of H-atoms as compared to other atoms, so the contribution of the H–H contact toward the crystal packing is higher than the other contacts in PBMP, DBC, and BCC. The percentage contribution of the H–H contact was found to be 53.1% in PBMP, 56.5% in DBC, and 33.7% in BCC. Like in PBMP, the intermolecular C–H···O bonding is present in DBC and BCC as well, so the contribution of the C–H and O–H contacts is also larger as compared to other contacts except for the H–H contacts. The percentage contributions of the C–H and O–H contacts are 30.6 and 9.2% in PBMP, 31.8 and 8.8% in DBC, and 27.2 and 6.9% in BCC. In BCC, the Cl–H contacts are important as this contact contribution is greater than 21% but the associated 2D fingerprint plot is not included in Figure 7 as the other crystal structures have no chlorine atoms. In PBMP, the contribution of the C–C contact (4.2%) is relatively smaller than the contribution of the C–H and O–H contacts as in the present case, the π···π stacking interaction is very weak and also the contribution of C–O contact is nonzero (2.9%) as there are the C–O···π interactions in the crystal packing of PBMP.
Figure 7.

Important 2D fingerprint plots for (a–d) PBMP, (e–h) DBC, and (i–l) BCC.
The ability of a pair of chemical species (X, Y) to form crystal packing interactions is determined by computing the enrichment ratio. All of the contacts are not equally favored in crystal packing. Short contacts are more favored than longer contacts. H-atoms are more numerous in the crystal structure as compared to other atoms; therefore, a large portion of the interaction area of the Hirshfeld surface is covered by H-atoms (73%, Table S1). The enrichment ratio is calculated by dividing the proportion of the actual contacts by the theoretical proportion of the random contacts.43 The values of the enrichment ratio for all of the possible pairs of chemical species are listed in Table S1. The C–O contact is the most favorable contact in the crystal packing of PBMP with the enrichment ratio 1.14. The other contacts that have a higher tendency to form crystal packing interactions are O–H, C–H, and H–H with the enrichment ratios of 1.04, 1.00, and 1.00, respectively.
The crystal packing environment of PBMP is further elaborated in terms of finding the interaction energy of molecular pairs. The calculations were performed using the B3LYP/6-31G(d,p) model coupled with appropriate scale factors for computing energies using the TONTO program embedded in Crystal Explorer 21.5. To perform the calculations of energy of molecular pairs, the cluster of molecules within 3.8 Å distance of the reference molecule is used. The molecule containing dark gray C-atoms is the reference molecule (Figure 8a). The total interaction energy between a pair of molecules is the sum of four energies named as electrostatic, polarization, dispersion, and repulsion.44 The scaled energies are listed in Figure S2. The energy values are reported to a minimum value of 0.1 kJ/mol but the authors of the Crystal Explorer program suggested that the reliability of the calculations is up to the minimum value of 1 kJ/mol. The major Etot energies (−60.2, −65, −51.2 kJ/mol) occur for the side-by-side interactions. The electrostatic energy is mostly attractive for the pair of molecules, but it may be repulsive for a pair of molecules. In the present case, the electrostatic energy is positive (1.3 kJ/mol) between a pair of molecules connected by (−x, 1/2 + y, −z + 1/2) symmetry transformations. The calculations infer that the dispersion energy is the major contributor to the total attractive energy contribution. The energy frameworks in terms of joining the centroids of the interacting molecules by the cylinders are displayed in Figure 8b–d for electrostatic, repulsive, and total energy, respectively, which are obtained using the interaction energy between the molecular pairs.45 The radius of the cylinder is proportional to the strength of the interactions. The cutoff energy is set to be 5 kJ/mol and the size of the cylinder is set to be 80 Å, and the cluster of molecules within 1 × 1 × 1 unit cell is used for generating the energy frameworks. The contribution of the repulsive energy is greater than the contribution of the electrostatic energy in defining the total energy as the radius of a cylinder for the repulsive energy framework (Figure 8c) is greater than the radius of the cylinder for the electrostatic energy framework.
Figure 8.

(a) Interaction energy between the molecular pairs falling in the range of 3.8 Å of the reference molecule for PBMP. Energy frameworks for (b) electrostatic energy, (c) repulsive energy, and (d) total energy.
Every single crystal has mechanical strength. Some single crystals have higher mechanical strength than the other single crystals. The factor that greatly influences the mechanical strength of the single crystals is the strength of the crystal packing. For a single crystal, the higher the strength of the crystal packing, the better the mechanical properties. To check the strength of the crystal packing, the void investigation was carried out for PBMP. The calculations were based on some assumptions: all of the atoms are assumed to spherically symmetric and electron densities of all of the atoms are added up to compute the void volume in the crystal packing.46 Two views of the voids in the crystal packing of PBMP are shown in Figure S3. The void volume in PBMP is 85.14 Å3, and the percentage space occupied by voids is just 7.7%, which indicates that the molecules are strongly packed.
3.3. Energetics and Structural Features
First, let us consider the relative energies of the singlet and triplet structures of the three compounds (see Table S2). It can be seen that the triplets are located very high in energy relative to the singlets, as is common for organic compounds, by ca. 42.3–42.9 kcal/mol in both implicit solvents. The singlet–triplet energy gap slightly increases from BCC to DBC and further to PBMP. For BCC and DBC, this gap is slightly larger in the implicit ethanol, whereas for PBMP, it is the same in both implicit solvents.
Figure 9 shows the optimized structures of the three compounds calculated at the B3LYP/6-311+G(d,p) level with the implicit effects from ethanol, and in Table 2, the selected bond lengths and bond angles obtained by the XRD and DFT approaches are compared for the PBMP compound. Furthermore, Table S3 in the Supporting Information provides the calculated values of the selected dihedral angles for the PBMP compound and Table S4 contains the calculated values of the selected bond distances, bond angles, and dihedral angles for BCC and DBC. Analysis of the data provided in Tables 2, S3, and S4 shows the following. (i) In PBMP, the calculated bond distances are generally slightly elongated compared to the experimentally determined bonds by ca. 0.003–0.015 Å. (ii) In PBMP, the calculated bond angles are either slightly smaller than the experimental bond angles by ca. 0.517–1.088° or slightly larger by 2.09° (∠C9-C7-C6). (iii) The absolute values of the corresponding dihedral angles show that the free PBMP compound molecule is not exactly symmetric (Table S3). This is not surprising due to the lack of restrictions existing in the crystal structure and quite high flexibility of the free PBMP molecule due to the numerous single C–C bonds. (iv) Both BCC and DBC molecules are symmetric, and their corresponding bond distances, valence angles, and dihedral angles are essentially the same or very close for both compounds. In both compounds, the cyclohexanone moiety is distorted, with the C1′ atom being out of the plane formed by the atoms O1–C1–C2–C2′, as can be seen from the values of the angles O1–C1–C1′ and C3–C2–C10–C1′, which are noticeably different from 180°, and the angle C2–C10–C1′–C10′, which is noticeably different from 0° (Table S3). Furthermore, the phenyl rings in both compounds are tilted relative to the central part of the molecule, as can be seen from the C9–C4–C3–C2 dihedral angle values, ca. 27° in both compounds. The Cl-atoms in BCC do not affect the molecular geometry compared to DBC. (v) Comparison of the C=O bond distances for the compounds PBMP, BCC, and DBC shows essentially no difference (cf. Tables 2 and S4).
Figure 9.

Global-minima structures of the PBMP (a), BCC (b), and DBC (c) compounds optimized at the B3LYP/6-311+G(d,p) level with the implicit effects from C2H5OH. PBMP atom numbering scheme corresponds to Figure 4. Color coding: brown for C, red for O, light gray for H, and bright green for Cl.
Thus, it can be seen that the DFT-optimized structure of the PBMP compound is very close to the experimentally obtained structure. Furthermore, both the BCC and DBC compounds show symmetric structures, with no influence of the Cl-atoms on the structure.
3.4. Frontier Molecular Orbital (FMO) Analysis
Figure 10 shows the plots of the HOMO and LUMO of the compounds PBMP, BCC, and DBC, and Table 4 provides the energies (eV) of their selected frontier MOs (HOMO – 2–LUMO + 2) along with the HOMO/LUMO gaps and TDDFT gaps (eV). Analysis of these results shows the following. (i) The PBMP HOMO is contributed by essentially the whole molecule, whereas the LUMO has stronger contributions from the central part of the molecule and smaller contributions from the outer phenyl rings (Figure 10a). (ii) On the contrary, the HOMOs and LUMOs of both BCC and DBC have contributions from the whole molecule. For both compounds, the FMOs are very similar. The BCC HOMO is stabilized compared to the PBMP HOMO by 0.10 eV, whereas the DBC HOMO is stabilized compared to the PBMP HOMO by 0.08 eV. The BCC LUMO is destabilized compared to the PBMP LUMO by 0.25 eV, whereas the DBC LUMO is destabilized compared to the PBMP LUMO by 0.34 eV. Thus, as can be seen, the chlorine atom incorporation in the BCC molecule significantly stabilizes its LUMO, whereas the BCC HOMO is stabilized slightly. The combined Cl-atom effect reduces the BCC HOMO/LUMO gap compared to the DBC HOMO/LUMO gap. (iii) The HOMO/LUMO gap of PBMP is quite large, 3.36 eV, which implies that this molecule is harder, less polarizable, and therefore less reactive and more stable. The TD-B3LYP calculated gap is noticeably smaller, 2.91 eV, although it can be still considered as quite significant. The TD-ωB97XD-calculated gap is by 0.54 eV larger than the TD-B3LYP-calculated gap and by 0.09 eV larger than the HOMO/LUMO gap, thus being considered as significant. (iv) The HOMO/LUMO gaps of the BCC and DBC are even larger than the PBMP HOMO/LUMO gap by 0.35 and 0.42 eV, respectively, which implies that their molecules are harder, less polarizable, less reactive, and more thermodynamically stable. Furthermore, the TD-B3LYP-calculated gaps for these compounds are larger as well by 0.20 and 0.23 eV. However, the TD-ωB97XD-calculated gaps are somewhat smaller by 0.04 and 0.03 eV. Thus, the presence of Cl-atoms in the BCC structure would make this compound somewhat more reactive than DBC. It should be noticed that the TDDFT-calculated gaps for both compounds are very close to each other. (v) For PBMP, the gaps in the pairs HOMO/HOMO – 1 and HOMO – 1/HOMO – 2 are relatively small, 0.27 and 0.08 eV, respectively, which implies that the MOs HOMO – 1 and HOMO – 2 would be able to participate in the redox reactions of the compound. On the contrary, the gaps in the pairs LUMO + 1/LUMO and LUMO + 2/LUMO + 1 are relatively large, 0.68 eV, which implies that the LUMO + 1 and LUMO + 2 are not likely to participate in the redox reactions of the compound. (vi) For BCC, the gap in the pair HOMO/HOMO – 1 is relatively small, 0.15 eV, whereas in the pair HOMO – 1/HOMO – 2, it is large, 0.54 eV, which implies that the HOMO – 1 of this compound would be able to participate in the redox reactions and the HOMO – 2 would be unlikely to participate in those. The gaps in the pairs LUMO + 1/LUMO and LUMO + 2/LUMO + 1 are significant, 1.30 and 0.52 eV, respectively, which implies that the LUMO + 1 and LUMO + 2 are not likely to participate in the redox reactions of the compound. For DBC, the gap in the pair HOMO/HOMO – 1 is also relatively small, 0.15 eV, whereas in the pair HOMO – 1/HOMO – 2, it is large, 0.49 eV, which implies that the HOMO – 1 of this compound would be able to participate in the redox reactions and the HOMO – 2 would be unlikely to participate in those. The gaps in the pairs LUMO + 1/LUMO and LUMO + 2/LUMO + 1 are significant, 1.38 and 0.62 eV, respectively, which implies that the LUMO + 1 and LUMO + 2 are not likely to participate in the redox reactions of the compound.
Figure 10.

Plots of the selected FMOs (HOMO and LUMO) of the PBMP (a), BCC (b), and DBC (c) compounds calculated at the B3LYP/6-311+G(d,p) level with the implicit effects from ethanol (isosurface value = 0.01).
Table 4. Energies of the Selected FMOs (HOMO – 2–LUMO + 2) of PBMP, BCC, and DBC (eV) along with the HOMO/LUMO Gaps and TDDFT Gaps (eV), Calculated at the B3LYP/6-311+G(d,p) Level with the Implicit Effects from Ethanol.
| MOs | energy (eV) | ΔE(HOMO/LUMO) (eV) | ΔE(HOMO/LUMO) (TDDFT) (eV) |
|---|---|---|---|
| PBMP | |||
| LUMO | –3.03 | 3.36 | 2.91/3.45a |
| HOMO | –6.39 | ||
| LUMO + 1 | –2.35 | 4.31 | |
| HOMO – 1 | –6.66 | ||
| LUMO + 2 | –1.67 | 5.07 | |
| HOMO – 2 | –6.74 | ||
| BCC | |||
| LUMO | –2.78 | 3.71 | 3.11/3.41a |
| HOMO | –6.49 | ||
| LUMO + 1 | –1.48 | 5.16 | |
| HOMO – 1 | –6.64 | ||
| LUMO + 2 | –0.96 | 6.22 | |
| HOMO – 2 | –7.18 | ||
| DBC | |||
| LUMO | –2.69 | 3.78 | 3.14/3.42a |
| HOMO | –6.47 | ||
| LUMO + 1 | –1.31 | 5.31 | |
| HOMO – 1 | –6.62 | ||
| LUMO + 2 | –0.69 | 6.42 | |
| HOMO – 2 | –7.11 | ||
Thus, as can be seen, the longer conjugated molecule of PBMP, as expected, has smaller (but still quite significant) HOMO/LUMO and TDDFT gaps compared to the smaller molecules of BCC and DBC. Furthermore, incorporation of the Cl-atoms in the molecule, in the case of BCC, decreases its HOMO/LUMO gap and TDDFT gaps due to stabilization of both HOMO and LUMO. So far, we are not aware about any experimental spectral studies of these three compounds; however, such studies would be planned by the experimental part of our research team, and the theoretical results obtained might provide good guidance for these studies. Moreover, in future research, more profound TDDFT studies could be performed, which was not the goal of the current study.
3.5. Natural Population Analysis (NPA)
Analysis of the NPA charges provided in Table 5 shows the following. (i) In the compound PBMP, the carbonyl oxygens of the molecule bear significant negative charges, ca. −0.63e. The carbons connected directly to these oxygens are significantly positively charged, ca. 0.5e. The carbons of the phenyls connecting them to the carbon–carbon chain, C3 and C10, carry relatively small negative charges, ca. −0.09e, and the carbons C1 and C2 of the central phenyl have quite noticeable negative charges, ca. −0.15–0.18e. The carbons of the methyls bear quite significant negative charges, close to the carbonyl oxygen charges, ca. −0.62e. Finally, the carbons of the carbon–carbon chain vary broadly within ca. −0.09 to −0.29e. (ii) In the compounds BCC and DBC, quite similar NPA charges on the same atoms can be seen. The more pronounced differences can be noticed for the carbonyl oxygen, which in BCC has slightly lower charge, by 0.005e, for the carbons connected to the Cl-atoms (C7/C7′), which in BCC have much lower negative charge, −0.031e compared to −0.196e in DBC, and for the C2/C2′ atoms, which in BCC have negative charge by 0.005e smaller than in DBC. The chlorines in BBC carry surprisingly small negative charges, merely −0.007e.
Table 5. NPA Charges, e, on the Selected Atoms of the Compounds PBMP, BCC, and DBC, Computed at the B3LYP/6-311+G(d,p) Level with the Implicit Effects from Ethanol (for numbering, see Figures S4 and S5).
| atom | charge (e) | atom | charge (e) | atom | charge (e) |
|---|---|---|---|---|---|
| PBMP | BCC | DBC | |||
| O1 | –0.629 | O1 | –0.638 | O1 | –0.643 |
| C6 | 0.503 | C1 | 0.525 | C1 | 0.525 |
| C5 | –0.286 | C2 | –0.100 | C2 | –0.105 |
| C4 | –0.094 | C3 | –0.108 | C3 | –0.106 |
| C3 | –0.091 | C4 | –0.093 | C4 | –0.096 |
| C2 | –0.178 | C7 | –0.031 | C7 | –0.196 |
| C1 | –0.152 | C10 | –0.421 | C10 | –0.420 |
| C7 | –0.078 | C1′ | –0.370 | C1′ | –0.370 |
| C8 | –0.616 | Cl1 | –0.007 | ||
| C9 | –0.134 | ||||
| C10 | –0.093 | ||||
Thus, from the results of the NPA analysis, it can be seen that molecules of the compounds under investigation carry significant varying charges on their atoms, which implies, on the one side, formation of the hydrogen bonding (cf. Table 3) and dipole–dipole interactions stabilizing the crystal structures of the title compounds, and, on the other side, would imply quite significant interactions of their molecules with polar solvents such as ethanol, methanol, water, etc. Moreover, these charges might imply potential interactions of these compounds with various electrophilic and nucleophilic species (see also discussion below).
3.6. Natural Bonding Orbital (NBO) Analysis
The NBO analysis allows us to obtain knowledge of orbital interactions of different types, both intra- and intermolecular.2b,2c,10,12,29,37,39,40,41c,42,49−61 Generally, the NBO analysis is done by considering all possible interactions among the filled, or donor, Lewis-type NBOs and empty, or acceptor, non-Lewis NBOs. Their energetic contributions in interactions are evaluated using second-order perturbation theory. These donor–acceptor interactions result in the decrease of the localized NBO occupancy in the idealized Lewis structure and increase of the occupancy of the empty non-Lewis orbitals. As a consequence, these interactions are referred to as “delocalization” corrections to the zeroth-order natural Lewis structure. The stronger donor–acceptor interactions have significant energies of stabilization. The second-order stabilization energy E(2) is computed using eq 7
| 7 |
In this equation, εi and εj are off-diagonal and Fi,j is the diagonal elements of the NBO Fock matrix, qi is the donor orbital possession, and E(2) is the energy of stabilization [29].
The representative values of the NBO analysis for the three compounds studied are provided in Table S5, and the numbering schemes are shown in Figures S4 and S5. Analysis of the data in Table S5 shows the following. (i) For the compound PBMP, the values of the stabilization energies vary very broadly from ca. 10 kcal/mol up to ca. 3071 kcal/mol. The donor–acceptor stabilizing interactions are represented by the types π(C-C) → π*(C-C), σ(C-C) → σ*(C-C), σ(C-C) → σ*(C-H), σ(C-C) → π*(C-C), σ(C-H) → σ*(C-H), σ(C-H) → π*(C-C), and LP(O) → σ*(C-C). Among them, the following several interactions with most significant stabilization energies, kcal/mol (given in red in Table S5), can be highlighted: σ(C40-C43) → σ*(C16-H21), 407.00, responsible for the interaction between the opposite sides of the molecule; σ(C42-H44) → π*(C49-C50), 113.37, responsible for the interaction within the same part of the molecule (the methyl and phenyl ring); σ(C42-H45) → π*(C49-C50), 115.07, also responsible for the interaction within the same part of the molecule (the methyl and phenyl ring); σ(C47-C53) → σ*(C3-H8), 182.88, responsible for the interaction between the opposite sides of the molecule; σ(C47-C53) → σ*(C22-H25), 121.19, also responsible for the interaction between the opposite sides of the molecule; σ(C47-C53) → σ*(C27-H32), 590.76, responsible for the interaction between the central phenyl and one side of the molecule; σ(C47-C53) → σ*(C28-C29), 177.22, also responsible for the interaction between the central phenyl and one side of the molecule; σ(C47-C53) → σ*(C49-C50), 210.55, responsible for the interaction within the same part of the molecule; σ(C47-C53) → π*(C49-C50), 416.36, responsible for the interaction within the same part of the molecule; σ(C52-C53) → σ*(C3-H8), 192.17, responsible for the interaction between the opposite sides of the molecule; σ(C52-C53) → σ*(C3-H8), 133.44, responsible for the interaction between the opposite sides of the molecule; σ(C52-C53) → σ*(C27-H32), 573.20, responsible for the interaction between the central phenyl and one side of the molecule; σ(C52-C53) → σ*(C28-C29), 184.38, responsible for the interaction between the central phenyl and one side of the molecule; σ(C52-C53) → σ*(C49-C50), 206.95, and σ(C52-C53) → π*(C49-C50), 403.77, both responsible for the interaction within the same part of the molecule; σ(C52-H57) → π*(C49-C50), 120.47, responsible for the interaction within the same part of the molecule; σ(C53-H58) → σ*(C3-H8), 424.18, σ(C53-H58) → σ*(C5-H10), 148.86, and σ(C53-H58) → σ*(C22-H25), 259.52, all three responsible for the interaction between the opposite sides of the molecule; σ(C53-H58) → σ*(C27-H32), 3071.17, and σ(C53-H58) → σ*(C28-C29), 355.51, both responsible for the interaction between the central phenyl and one side of the molecule; σ(C53-H58) → σ*(C49-C50), 464.29, and σ(C53-H58) → σ*(C52-H57), 137.71, both responsible for the interaction within the same part of the molecule. (ii) On the contrary, for BCC, the energies of the donor–acceptor interactions collected in Table S5 are relatively low, varying within ca. 10—23 kcal/mol. The donor–acceptor stabilizing interactions can be classified just into three types: π(C-C) → π*(C-C), σ(C-H) → σ*(C-H), and LP(Cl) → π*(C-C). (iii) For DBC, the situation becomes different: the energies of the donor–acceptor stabilization interactions vary within ca. 10—55 kcal/mol, and the interaction types are σ(C-C) → σ*(C-C), π(C-C) → π*(C-C), π(C-C) → π*(O-C), σ(C-H) → σ*(C-C), σ(C-H) → σ*(C-H), and LP(O) → σ*(C-C). Two stabilization interactions with the largest energies can be highlighted, σ(C2-C38) → σ*(C36-C38), 39.62 kcal/mol, σ(C38-H39) → σ*(C36-C38), 55.25 kcal/mol, both responsible for the stabilization of one of the phenyls.
Thus, the NBO analysis shows numerous high-energy stabilizing interactions for the PBMP compound due to the presence of many delocalized and relatively easily polarizable π-electrons, thus implying its significant thermodynamic stability. Furthermore, for two other compounds, the NBO analysis also shows the presence of stabilizing interactions, however, less numerous and with noticeably lower energies, especially for the compound BCC. This, in turn, might imply their somewhat lower stability or higher reactivity.
3.7. Global Reactivity Parameter (GRP) Analysis
The global reactivity parameters—ionization potential (IP), electron affinity (EA), global softness (σ), global electrophilicity (ω), global hardness (η), global electronegativity (X), and chemical potential (μ)—were computed using the FMO energies (Table 4) according to eqs 1–6 (see Computational Details) [30–32], and the computed values in eV are presented in Table 6.
Table 6. Calculated GRPs for the PBMP, BCC, and DBC Compounds (eV).
| IP | EA | gap | X | η | μ | σ | ω |
|---|---|---|---|---|---|---|---|
| PBMP | |||||||
| 6.39 | 3.03 | 3.36 | 4.71 | 1.68 | –4.71 | 0.298 | 6.602 |
| BCC | |||||||
| 6.49 | 2.78 | 3.71 | 4.635 | 1.855 | –4.635 | 0.270 | 5.791 |
| DBC | |||||||
| 6.47 | 2.69 | 3.78 | 4.58 | 1.89 | –4.58 | 0.265 | 5.549 |
Analysis of the GRP data given in Table 6 shows the following. (i) The IP and EA values of the PBMP compound are significant, 6.39 and 3.03 eV, respectively, which implies that this compound should be stable in the oxidation processes but might attach electrons in the reduction processes. This is also supported by the PBMP delocalized structure, which would mean that the charge attained could be easily delocalized over the molecule, thus stabilizing the anion formed. The global electronegativity X along with global electrophilicity ω has significantly high values, 4.71 and 6.602 eV, respectively, which would imply higher reactivity of the compound in reduction reactions. The significant negative value of the chemical potential, −4.71 eV, implies noticeable thermodynamic stability of the compound. A relatively low value of the global softness σ, 0.298 eV, along with a more significant value of the global hardness η, 1.68 eV, would also imply lower reactivity of this compound. (ii) The IP and EA values of BCC and DBC are also significant, the IP values being higher than the PBMP IP value and the EA values being lower than the PBMP EA value. This implies that these two compounds should be even less prone to both oxidation and reduction processes and thus more stable in the redox environment compared to the PBMP compound. Further, the global electronegativity X and global electrophilicity ω values for BCC and DBC are lower than those for PBMP, 4.635 and 4.58 eV, respectively, vs 4.71 eV, and 5.791 and 5.549 eV, respectively, vs 6.602 eV. This would also imply lower reactivity of these two compounds in redox processes. The chemical potentials of both BCC and DBC have quite significant negative values, −4.635 and −4.58 eV, respectively; however, these values are somewhat smaller than the PBMP value, which might imply somewhat lower thermodynamic stability of these two compounds. Relatively low values of the global softness σ, 0.270 and 0.265 eV, respectively, vs 0.298 eV for PBMP, along with even more significant values of the global hardness η, 1.855 and 1.89 eV, vs 1.68 eV for PBMP, would imply noticeably lower reactivity of these two compounds compared to PBMP.
Thus, the GRP analysis shows that all three compounds should be relatively stable in the redox processes, especially BCC and DBC, and also should have quite high thermodynamic stability and relatively low reactivity in general.
3.8. Molecular Electrostatic Potential (MEP) Mapping
The MEP plots of the PBMP, BCC, and DBC compounds provided in Figure 11 show the following. (i) In PBMP, accumulation of the negative potential, as indicated by the red color, can be seen at the carbonyl oxygen and, slightly, in the middle parts of the phenyls. The accumulation of the positive potential, as indicated by the blue color, can be seen on the hydrogens in the whole molecule. (ii) In BCC and DBC, the negative potential accumulates on the carbonyl oxygens (in a larger extent), inside the phenyl rings (in a smaller extent), and for BCC on the chlorines as well (in a smaller extent). Furthermore, some accumulation of the positive potential can be seen on the hydrogens in the whole molecule.
Figure 11.

Molecular electrostatic potential plots for the PBMP (a), BCC (b), and DBC (c) compounds calculated at the B3LYP/6-311+G(d,p) level with the implicit effects from ethanol. The bright red color corresponds to the negative MEP, −0.01 A.U., whereas the bright blue color corresponds to the positive MEP, +0.01 A.U.
These results imply potential formation of the intermolecular hydrogen bonding and dispersion interactions (cf. Tables 3 and 4), which would stabilize the crystal structures of these compounds (cf. with the Hirshfeld analysis above) and also would facilitate dissolution of these compounds in various polar solvents.
4. Conclusions
In this study, three compounds DBC, BCC, and PBMP were prepared and characterized by the spectroscopic method. Moreover, the chemical structure of PBMP is investigated by the single-crystal technique. The SC-XRD data revealed that the molecular configuration is stabilized by intramolecular C–H···O bonding, whereas the supramolecular assembly is stabilized by noncovalent interactions named as C–H···O, C–O···π, and weak T-shaped π···π stacking interactions. The strong as well as the comparatively weak noncovalent interactions of PBMP are explored by the Hirshfeld surface analysis and compared with DBC and BCC. The crystal packing environment is further explored by the interaction energy calculations between the molecular pairs and energy framework plots. DFT results corroborate the experimental data. The PBMP DFT-optimized structure is very close to the experimentally obtained structure, and both the BCC and DBC compounds show symmetric structures, with no influence of the Cl-atoms on the structure. The FMO analysis showed that the longer conjugated molecule of PBMP, as expected, has smaller but still quite significant HOMO/LUMO and TDDFT gaps compared to the smaller molecules of BCC and DBC. Furthermore, incorporation of the Cl-atoms in the molecule, in the case of BCC, decreases its HOMO/LUMO gap and TDDFT gaps due to stabilization of both HOMO and LUMO. The NPA analysis showed that molecules of the compounds under investigation carry significant varying charges on their atoms, which implies, on the one side, formation of the hydrogen bonding and dipole–dipole interactions stabilizing the crystal structures of the title compounds, and, on the other side, would imply quite significant interactions of their molecules with polar solvents, such as ethanol, methanol, water, etc. The NBO analysis shows numerous high-energy stabilizing interactions for the PBMP compound due to the presence of many delocalized and relatively easily polarizable π-electrons, thus implying its significant thermodynamic stability. Furthermore, for two other compounds, the NBO analysis also shows the presence of stabilizing interactions, however, less numerous and with noticeably lower energies, especially for the compound BCC. The GRP analysis shows that all three compounds should be relatively stable in redox processes, especially BCC and DBC, and also should have quite high thermodynamic stability and relatively lowered reactivity in general. The MEP analysis results imply potential formation of intermolecular hydrogen bonding and dispersion interactions, which would stabilize the crystal structures of these compounds and also would facilitate dissolution of these compounds in various polar solvents.
Acknowledgments
A.A. gratefully acknowledges the support of TWAS-CNPq and HEC Pakistan. A.K. appreciates the financial support of USM and computational facilities of the Department of Chemistry, ITA, Brazil. Powered@NLHPC: This research was partially supported by the supercomputing infrastructure of the NLHPC (ECM-02). M.A.A. appreciates the support of the Research Center for Advanced Materials Science (RCAMS) at King Khalid University Abha, Saudi Arabia, through grant KKU/RCAMS/22.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c05441.
Graphical representation of the stair-like pattern formed by C–O···π and weak T-shaped π···π stacking interactions; enrichment ratio of pair of chemical species of PBMP; summary of the interaction energies of a reference molecule with its nearest neighbors; voids in the crystal packing of PBMP, a view along (a) c-axis and (b) a-axis; relative energies for singlets and triplets; selected bond angles and dihedral angles, degrees, for the PBMP, BCC, and DBC compounds, B3LYP/6-311+G(d,p), implicit C2H5OH; representative values of the NBO analysis; numbering scheme for the NBO analysis of PBMP, BCC, and DBC compounds; 1H and 13C NMR spectra of compounds 3, PBMP, BCC, and DBC; HRMS spectrum of the compound PBMP; coordinates of the atoms of molecules around the reference molecule; checkCIF/PLATON report (PDF)
Crystallographic data for PBMP (CIF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Braga S. F. P.; Alves É. V. P.; Ferreira R. S.; Fradico J. R. B.; Lage P. S.; Duarte M. C.; Ribeiro T. G.; Júnior P. A. S.; Romanha A. J.; Tonini M. L.; Steindel M.; Coelho E. F.; Oliveira R. B. Synthesis and evaluation of the antiparasitic activity of bis-(arylmethylidene) cycloalkanones. Eur. J. Med. Chem. 2014, 71, 282–289. 10.1016/j.ejmech.2013.11.011. [DOI] [PubMed] [Google Scholar]; b Lazarin-Bidóia D.; Desoti V. C.; Martins S. C.; Ribeiro F. M.; Din Z. U.; R -Filho E.; U-Nakamura T.; Nakamura C. V.; Silva S. O. Dibenzylideneacetones are potent trypanocidal compounds that affect the Trypanosoma cruzi redox system. Antimicrob. Agents Chemo. 2016, 60, 890–903. 10.1128/AAC.01360-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Nielsen A. T.; Houlihan W. J. The Aldol Condensation. Org. Reactions 1968, 16, 1–438. 10.1002/0471264180.or016.01. [DOI] [Google Scholar]; b Khalid M.; Ali A.; Adeel M.; Din Z. U.; Tahir M. N.; Filho E. R.; Iqbal J.; Khan M. U. Facile preparation, characterization, SC-XRD and DFT/TDDFT study of diversely functionalized unsymmetrical bis-aryl-α, β-unsaturated ketone derivatives. J. Mol. Struct. 2020, 1206, 127755. [Google Scholar]; c Ali A.; Din Z. U.; Khalid M.; Tahir M. N.; Filho E. R.; Ali B.; Asim S.; Muhammad S. Crystal and Quantum Chemical Exploration of the Potent Monocarbonyl Curcuminoids to Unveil Their Structural and Intriguing Electronic Properties. ChemistrySelect 2020, 5, 3735–3745. 10.1002/slct.201904757. [DOI] [Google Scholar]
- Franco L. L.; de Almeida M. V.; E Silva L. F. R.; Vieira P. P. R.; Pohlit A. M.; Valle M. S. Synthesis and antimalarial activity of dihydroperoxides and tetraoxanes conjugated with bis(benzyl)acetone derivatives. Chem. Biol. Drug Des. 2012, 79, 790–797. 10.1111/j.1747-0285.2012.01345.x. [DOI] [PubMed] [Google Scholar]
- Aher R. B.; Wanare G.; Kawathekar N.; Kumar R. R.; Kaushik N. K.; Sahal D.; Chauhan V. S. Dibenzylideneacetone analogues as novel Plasmodium falciparum inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 3034–3036. 10.1016/j.bmcl.2011.03.037. [DOI] [PubMed] [Google Scholar]
- Lv Y.; Lan A.; Li C.; Lei N.; Wu C.; Liu L. Activity of curcumin against human cytomegalovirus in vitro. African. J. Phar. Pharmacol. 2012, 6, 30–35. 10.5897/ajpp11.439. [DOI] [Google Scholar]
- Hall I. H.; Carlson G. L.; Abernethy G. S.; Piantadosi C. Cycloalkanones. Antifertility Activity. J. Med. Chem. 1974, 17, 1253–1257. 10.1021/jm00258a004. [DOI] [PubMed] [Google Scholar]
- Prasad S.; Yadav V. R.; Ravindran J.; Aggarwal B. B. ROS and CHOP are critical for dibenzylideneacetone to sensitize tumor cells to TRAIL through induction of death receptors and downregulation of cell survival proteins. Cancer Res. 2011, 71, 538–549. 10.1158/0008-5472.CAN-10-3121. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Leong S. W.; Faudzi S. M. M.; Abas F.; Aluwi M. F. F. M.; Rullah K.; Wai L. K.; Bahari M. N. A.; Ahmad S.; Tham C. L.; Shaari K.; Lajis N. H. Synthesis and Sar Study of Diarylpentanoid Analogues as New Anti-Inflammatory Agents. Molecules 2014, 19, 16058–16081. 10.3390/molecules191016058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidharthan J.; Peter Amaladhas T. Synthesis and characterization of photo-crosslinkable liquid crystalline copolyesters containing arylidene-keto and chalcone moieties. J. Polym. Res. 2017, 24, 53. 10.1007/s10965-017-1206-7. [DOI] [Google Scholar]
- a Ali A.; Khalid M.; Tahir M. N.; Imran M.; Ashfaq M.; Hussain R.; Assiri M. A.; Khan I. Synthesis of Diaminopyrimidine Sulfonate Derivatives and Exploration of Their Structural and Quantum Chemical Insights via SC-XRD and the DFT Approach. ACS Omega 2021, 6, 7047–7057. 10.1021/acsomega.0c06323. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Khalid M.; Khan M. U.; Shafiq I.; Hussain R.; Mahmood K.; Hussain A.; Jawaria R.; Hussain A.; Imran M.; Assiri M. A.; Ali A.; ur-Rehman M. F.; Sun K.; Li Y. NLO potential exploration for D–p–A heterocyclic organic compounds by incorporation of various p-linkers and acceptor units. Arab. J. Chem. 2021, 14, 103295. [Google Scholar]; c Ali A.; Khalid M.; Din Z. U.; Asif H. M.; Imran M.; Tahir M. N.; Ashfaq M.; Filho E. R. Exploration of structural, electronic and third order nonlinear optical properties of crystalline chalcone systems: Monoarylidene and unsymmetrical diarylidene cycloalkanones. J. Mol. Struct. 2021, 1241, 130685. 10.1016/j.molstruc.2021.130685. [DOI] [Google Scholar]; d Concepcion O.; Ali A.; Khalid M.; de la Torre A. F.; Khan M. U.; Raza A. R.; Kamal G. M.; ur Rehman M. F.; Alam M. M.; Imran M.; Braga A. A. C.; Pertino M. W. Facile Synthesis of Diversely Functionalized Peptoids, Spectroscopic Characterization, and DFT-Based Nonlinear Optical Exploration. ACS Omega 2021, 6, 26016–26025. 10.1021/acsomega.1c02962. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Ashfaq M.; Tahir M. N.; Muhammad S.; Munawar K. S.; Ali A.; Bogdanov G.; Alarfaji S. S. Single-Crystal Investigation, Hirshfeld Surface Analysis, and DFT Study of Third-Order NLO Properties of Unsymmetrical Acyl Thiourea Derivatives. ACS Omega 2021, 6, 31211–31225. 10.1021/acsomega.1c04884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Khalid M.; Ali A.; Khan M. U.; Tahir M. N.; Ahmad A.; Ashfaq M.; Hussain R.; Morais S. F. A.; Braga A. A. C. Non-covalent interactions abetted supramolecular arrangements of N-Substituted benzylidene acetohydrazide to direct its solid-state network. J. Mol. Struct. 2021, 1230, 129827. 10.1016/j.molstruc.2020.129827. [DOI] [Google Scholar]; b Khalid M.; Ali A.; Din Z. U.; Tahir M. N.; Morais S. F. A.; Braga A. A. C.; Akhtar M. N.; Imran M.; Filho E. R. β-Hydroxy Carbonyl compounds via aldol reaction: Single crystal investigation and quantum chemical exploration for the unveiling of supramolecular behavior. J. Mol. Struct. 2021, 1241, 130650. 10.1016/j.molstruc.2021.130650. [DOI] [Google Scholar]; c Ashfaq M.; Munawar K. S.; Bogdanov G.; Ali A.; Tahir M. N.; Ahmed G.; Ramalingam A.; Alam M. M.; Imran M.; Sambandam S.; Munir B. Single crystal inspection, Hirshfeld surface investigation and DFT study of a novel derivative of 4-fluoroaniline: 4-((4-fluorophenyl) amino)-4-oxobutanoic acid (BFAOB). J. Iran. Chem. Soc. 2022, 19, 1953–1961. 10.1007/s13738-021-02432-4. [DOI] [Google Scholar]
- a Ashfaq M.; Ali A.; Kuznetsov A.; Tahir M. N.; Khalid M. DFT and single-crystal investigation of the pyrimethamine-based novel co-crystal salt: 2,4-diamino-5-(4-chlorophenyl)-6-ethylpyrimidin-1- ium-4-methylbenzoate hydrate (1:1:1) (DEMH). J. Mol. Struct. 2021, 1228, 129445. 10.1016/j.molstruc.2020.129445. [DOI] [Google Scholar]; b Khalid M.; Ali A.; Haq S.; Tahir M. N.; Iqbal J.; Braga A. A. C.; Ashfaq M.; Akhtar S. U. H. O-4-Acetylamino-benzenesulfonylated pyrimidine derivatives: synthesis, SC-XRD, DFT analysis and electronic behaviour investigation. J. Mol. Struct. 2021, 1224, 129308. 10.1016/j.molstruc.2020.129308. [DOI] [Google Scholar]; c Ali A.; Kuznetsov A.; Ashfaq M.; Tahir M. N.; Khalid M.; Imran M.; Irfan A. Synthesis, single-crystal exploration, and theoretical insights of arylsulfonylated 2-amino-6-methylpyrimidin derivatives. J. Mol. Struct. 2021, 1243, 130789. 10.1016/j.molstruc.2021.130789. [DOI] [Google Scholar]
- APEX2, Bruker AXS Inc.: Madison, Wisconsin, USA, 2013. [Google Scholar]
- Sheldrick G. M.. SADABS, Version 2.03; University of Göttingen: Germany, 2002, 1600–5368. Acta Crystallogr. Sec. E: Struct. Reps.
- Sheldrick G. M. SHELXT–Integrated space-group and crystal-structure determination. Acta Crystallogr. Sec. A: Found. Adv. 2015, 71, 3–8. 10.1107/S2053273314026370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick G. M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrugia L. J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. 10.1107/S0021889812029111. [DOI] [Google Scholar]
- Macrae C. F.; Sovago I.; Cottrell S. J.; Galek P. T. A.; McCabe P.; Pidcock E.; Platings M.; Shields G. P.; Stevens J. S.; Towler M.; Wood P. A. Mercury 4.0: from visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. 10.1107/S1600576719014092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotfy G.; Said M. M.; Ashry E. S. H. E.; Tamany E. S. H. E.; Aziz Y. M. A.; Soliman S. M.; Barakat A. Synthesis, structure combined with conformational analysis, biological activities and docking studies of bis benzylidene cyclohexanone derivatives. J. Saudi Chem. Soc. 2017, 21, 619–632. 10.1016/j.jscs.2017.04.002. [DOI] [Google Scholar]
- Ud Din Z.; Fill T. P.; de Assis F. F.; Bidóia D. L.; Kaplum V.; Garcia F. P.; Nakamura C. V.; de Oliveira K. T.; Filho E. R. Unsymmetrical 1,5-diaryl-3-oxo-1,4-pentadienyls and their evaluation as antiparasitic agents. Bioorg. Med. Chem. 2014, 22, 1121–1127. 10.1016/j.bmc.2013.12.020. [DOI] [PubMed] [Google Scholar]
- a Ashfaq M.; Munawar K. S.; Tahir M. N.; Dege N.; Yaman M.; Muhammad S.; Alarfaji S. S.; Kargar H.; Arshad M. U. Synthesis, crystal structure, Hirshfeld surface analysis, and computational study of a novel organic salt obtained from benzylamine and an acidic component. ACS Omega 2021, 6, 22357–22366. 10.1021/acsomega.1c03078. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kargar H.; Ashfaq M.; Fallah-Mehrjardi M.; Behjatmanesh-Ardakani R.; Munawar K. S.; Tahir M. N. Synthesis, characterization, SC-XRD, HSA and DFT study of a novel copper(I) iodide complex with 2-(thiophen-2-yl)-4, 5-dihydro-1H-imidazole ligand: An experimental and theoretical approach. J. Mol. Struct. 2022, 1253, 132264. 10.1016/j.molstruc.2021.132264. [DOI] [Google Scholar]; c Fallah-Mehrjardi M.; Kargar H.; Behjatmanesh-Ardakani R.; Ashfaq M.; Munawar K. S.; Tahir M. N. Symmetrical Pd(II) and Ni(II) Schiff base complexes: Synthesis, crystal structure determination, spectral characterization, and theoretical studies. J. Mol. Struct. 2022, 1251, 132037. 10.1016/j.molstruc.2021.132037. [DOI] [Google Scholar]
- Badal M. M. R.; Islam H. M. A.; Maniruzzaman M.; Yousuf M. A. Acidochromic Behavior of Dibenzylidene Cyclohexanone-Based Bischalcone: Experimental and Theoretical Study. ACS Omega 2020, 5, 22978–22983. 10.1021/acsomega.0c02556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotfy G.; Said M. M.; Ashry E. S. H. E.; Tamany E. S. H. E.; Aziz Y. M. A.; Soliman S. M.; Al-Majid A. M.; Ghabbour H. A.; Barakat A. Syntheses and X-ray crystal structures combined with conformational and Hirshfeld analyses of chalcones based on a cyclohexanone scaffold. J. Mol. Struct. 2019, 1198, 126873 10.1016/j.molstruc.2019.126873. [DOI] [Google Scholar]
- a Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas Ö.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09, Revision B.01Gaussian Inc.: Wallingford, 2010.; b Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas Ö.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 16, Revision B.01Gaussian, Inc.: Wallingford CT, 2016.
- Becke A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- McLean A. D.; Chandler G. S. Contracted Gaussian-basis sets for molecular calculations. 1. 2nd row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. 10.1063/1.438980. [DOI] [Google Scholar]
- Krishnan R.; Binkley J. S.; Raymond J. S.; People J. A. Self-Consistent Molecular Orbital Methods. 20. Basis set for correlated wave-functions. J. Chem. Phys. 1980, 72, 650–654. 10.1063/1.438955. [DOI] [Google Scholar]
- Tomasi J.; Mennucci B.; Cammi R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. 10.1021/cr9904009. [DOI] [PubMed] [Google Scholar]
- Reed A. E.; Curtiss L. A.; Weinhold F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. 10.1021/cr00088a005. [DOI] [Google Scholar]
- Geerlings P.; De Proft F.; Langenaeker W. Conceptual density functional theory. Chem. Rev. 2003, 103, 1793–1874. 10.1021/cr990029p. [DOI] [PubMed] [Google Scholar]
- Chakraborty A.; Pan S.; Chattaraj P. K.. Biological Activity and Toxicity: A Conceptual DFT approach. In Structure and Bonding, 2013; Vol. 150, pp 143–180. [Google Scholar]
- Jorio S.; Salah M.; Abou El Makarim H.; Tabyaoui M. Reactivity indices related to DFT theory, the electron localization function (ELF) and non-covalent interactions (NCI) calculations in the formation of the non-halogenated pyruvic esters in solution. Mediterr. J. Chem. 2019, 8, 476–485. 10.13171/mjc8619072612sj. [DOI] [Google Scholar]
- Schaftenaar G.; Noordik J. H. Molden: a pre- and post-processing program for molecular and electronic structures. J. Comput.-Aided Mol. Design. 2000, 14, 123–134. 10.1023/A:1008193805436. [DOI] [PubMed] [Google Scholar]
- Hanwell M. D.; Curtis D. E.; Lonie D. C.; Vandermeersch T.; Zurek E.; Hutchison G. R. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J. Cheminformatics 2012, 4, 1–17. 10.1186/1758-2946-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avogadro: an open-source molecular builder and visualization tool. Version 1.XX. http://avogadro.cc/.
- Zhang L.; Wang S.; Sheng E.; Zhou S. A solvent-free synthesis of α,α′-9-bis(substituted benzylidene) cycloalkanones catalyzed by lanthanide amides [(Me3Si)2N]3Ln(μ-Cl)Li(THF)3 under microwave irradiation. Green Chem. 2005, 7, 683–686. 10.1039/b504890g. [DOI] [Google Scholar]
- Ashfaq M.; Bogdanov G.; Ali A.; Tahir M. N.; Abdullah S. Pyrimethamine-Based Novel Co-Crystal Salt: Synthesis, Single-Crystal Investigation, Hirshfeld surface analysis and DFT inspection of the 2,4-diamino-5-(4-chlorophenyl)-6-ethylpyrimidin-1-ium 2,4-dichlorobenzoate (1:1) (DECB). J. Mol. Struct. 2021, 1235, 130215 10.1016/j.molstruc.2021.130215. [DOI] [Google Scholar]
- Spackman P. R.; Turner M. J.; McKinnon J. J.; Wolff S. K.; Grimwood D. J.; Jayatilaka D.; Spackman M. A. CrystalExplorer: a program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Cryst. 2021, 54, 1006–1011. 10.1107/S1600576721002910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Spackman M. A.; Jayatilaka D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. 10.1039/B818330A. [DOI] [Google Scholar]; b Mehmood H.; Khalid M.; Haroon M.; Akhter T.; Ashfaq M.; Tahir M. N.; Khan M. U.; Imran M.; Braga S A. A. C.; Woodward Synthesis, Characterization and DFT Calculated Properties of Electron-rich Hydrazinylthiazoles: Experimental and Computational Synergy. J. Mol. Struct. 2021, 1245, 131043. 10.1016/j.molstruc.2021.131043. [DOI] [Google Scholar]; c Madni M.; Ahmed M. N.; Hafeez M.; Ashfaq M.; Tahir M. N.; Gil D. M.; Galmés B.; Hameed S.; Frontera A. Recurrent π–π stacking motifs in three new 4,5-dihydropyrazolyl–thiazole–coumarin hybrids: X-ray characterization, Hirshfeld surface analysis and DFT calculations. New J. Chem. 2020, 44, 14592–14603. 10.1039/D0NJ02931A. [DOI] [Google Scholar]
- Kargar H.; Ashfaq M.; Fallah-Mehrjardi M.; Behjatmanesh-Ardakani R.; Munawar K. S.; Tahir M. N. Synthesis, crystal structure, spectral characterization, theoretical and computational studies of Ni(II), Cu(II) and Zn(II) complexes incorporating Schiff base ligand derived from 4-(diethylamino) salicylaldehyde. Inorg. Chim. Acta 2022, 536, 120878. 10.1016/j.ica.2022.120878. [DOI] [Google Scholar]
- a McKinnon J. J.; Jayatilaka D.; Spackman M. A. Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces. Chem Commun. 2007, 3814–3816. 10.1039/b704980c. [DOI] [PubMed] [Google Scholar]; b Ashfaq M.; Tahir M. N.; Muhammad S.; Munawar K. S.; Ali S.; Ahmed G.; Al-Sehemi A. G.; Alarfaji S. S.; Khan M. E. I. Shedding Light on the Synthesis, Crystal Structure, Characterization, and Computational Study of Optoelectronic Properties and Bioactivity of Imine derivatives. ACS Omega 2022, 7, 5217–5230. 10.1021/acsomega.1c06325. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ahmed M. N.; Ghias M.; Shah S. W. A.; Shoaib M.; Tahir M. N.; Ashfaq M.; Ibrahim M. A. A.; Andleeb H.; Gil D. M.; Frontera A. X-ray characterization, Hirshfeld surface analysis, DFT calculations, in vitro and in silico lipoxygenase inhibition (LOX) studies of dichlorophenyl substituted 3-hydroxy-chromenones. New J. Chem. 2021, 45, 19928–19940. 10.1039/D1NJ04340D. [DOI] [Google Scholar]
- a Ahmed M. N.; Madni M.; Anjum S.; Andleeb S.; Hameed S.; Khan A. M.; Ashfaq M.; Tahir M. N.; Gil D. M.; Frontera A. Crystal engineering with pyrazolyl-thiazole derivatives: Structure-directing role of π-stacking and σ-hole interactions. CrystEngComm 2021, 23, 3276–3287. 10.1039/D1CE00256B. [DOI] [Google Scholar]; b Khalid M.; Ali A.; Abid S.; Tahir M. N.; Khan M. U.; Ashfaq M.; Imran M.; Ahmad A. Facile Ultrasound-Based Synthesis, SC-XRD, DFT Exploration of the Substituted Acyl-Hydrazones: An Experimental and Theoretical Slant towards Supramolecular Chemistry. ChemistrySelect 2020, 5, 14844–14856. 10.1002/slct.202003589. [DOI] [Google Scholar]; c Ashfaq M.; Tahir M. N.; Kuznetsov A.; Mirza S. H.; Khalid M.; Ali A. DFT and single crystal analysis of the pyrimethamine-based novel co-crystal salt: 2,4-diamino-5-(4-chloro-phenyl)-6-ethylpyrimidin-1-ium:4-hydroxybenzoate:methanol:hydrate (1:1:1:1) (DEHMH). J. Mol. Struct. 2020, 1199, 127041. 10.1016/j.molstruc.2019.127041. [DOI] [Google Scholar]; d Khalid M.; Ali A.; Tariq J.; Tahir M. N.; Aliabad H. A. R.; Hussain I.; Ashfaq M.; Khan M. U. Stabilization of Supramolecular Assembly of N-Substituted Benzylidene Acetohydrazide Analogs by Non-Covalent Interactions: A Concise Experimental and Theoretical Approach. ChemistrySelect 2020, 5, 10618–10631. 10.1002/slct.202002653. [DOI] [Google Scholar]; e Ali A.; Khalid M.; ur Rehman M. F.; Haq S.; Ali A.; Tahir M. N.; Ashfaq M.; Rasool F.; Braga A. A. C. Efficient Synthesis, SC-XRD, and Theoretical Studies of O -Benzenesulfonylated Pyrimidines: Role of Noncovalent Interaction Influence in Their Supramolecular Network. ACS Omega 2020, 5, 15115–15128. 10.1021/acsomega.0c00975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelsch C.; Ejsmont K.; Huder L. The enrichment ratio of atomic contacts in crystals, an indicator derived from the Hirshfeld surface analysis. IUCrJ 2014, 1, 119–128. 10.1107/S2052252514003327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner M. J.; Grabowsky S.; Jayatilaka D.; Spackman M. A. Accurate and efficient model energies for exploring intermolecular interactions in molecular crystals. J. Phys. Chem. Lett. 2014, 5, 4249–4255. 10.1021/jz502271c. [DOI] [PubMed] [Google Scholar]
- Turner M. J.; Thomas S. P.; Shi M. W.; Jayatilaka D.; Spackman M. A. Energy frameworks: insights into interaction anisotropy and the mechanical properties of molecular crystals. Chem. Commun. 2015, 51, 3735–3738. 10.1039/C4CC09074H. [DOI] [PubMed] [Google Scholar]
- a Turner M. J.; McKinnon J. J.; Jayatilaka D.; Spackman M. A. Visualisation and characterisation of voids in crystalline materials. CrystEngComm 2011, 13, 1804–1813. 10.1039/C0CE00683A. [DOI] [Google Scholar]; b Ashfaq M.; Munawar K. S.; Bogdanov G.; Ali A.; Tahir M. N.; Ahmed G.; Ramalingam A.; Alam M. M.; Imran M.; Sambandam S.; Munir B. Single crystal inspection, Hirshfeld surface investigation and DFT study of a novel derivative of 4-fluoroaniline: 4-((4-fluorophenyl) amino)-4-oxobutanoic acid (BFAOB). J. Iran. Chem. Soc. 2022, 19, 1953–1961. 10.1007/s13738-021-02432-4. [DOI] [Google Scholar]
- Bauernschmitt R.; Ahlrichs R. Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory. Chem. Phys. Lett. 1996, 256, 454–464. 10.1016/0009-2614(96)00440-X. [DOI] [Google Scholar]
- Chai J.-D.; Head-Gordon M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. 10.1039/B810189B. [DOI] [PubMed] [Google Scholar]
- Khalid M.; Ullah M. A.; Adeel M.; Khan M. U.; Tahir M. N.; Braga A. A. C. Synthesis, crystal structure analysis, spectral IR, UV–Vis, NMR assessments, electronic and nonlinear optical properties of potent quinoline based derivatives: interplay of experimental and DFT study. J. Saudi Chem. Soc. 2019, 23, 546–560. 10.1016/j.jscs.2018.09.006. [DOI] [Google Scholar]
- Tahir M. N.; Mirza S. H.; Khalid M.; Ali A.; Khan M. U.; Braga A. A. C. Synthesis, single-crystal X-ray diffraction analysis and DFT based computational studies of 2,4-diamino-5-(4-chlorophenyl)-6-ethylpyrim idin-1-ium 3,4,5-trihydroxybenzoate-methanol (DETM). J. Mol. Struct. 2019, 1180, 119–126. 10.1016/j.molstruc.2018.11.089. [DOI] [Google Scholar]
- Haroon M.; Khalid M.; Akhtar T.; Tahir M. N.; Khan M. U.; Saleem M M.; Jawaria R. Synthesis, spectroscopic, SC-XRD characterizations and DFT based studies of ethyl-2-(substituted-(2-benzylidenehydrazinyl))thiazole-4-carboxylate derivatives. J. Mol. Struct. 2019, 1187, 164–171. 10.1016/j.molstruc.2019.03.075. [DOI] [Google Scholar]
- Rafiq M.; Khalid M.; Tahir M. N.; Ahmad M. U.; Khan M. U.; Naseer M. M.; Braga A. A. C.; Muhammad S.; Shafiq Z. Synthesis, XRD, spectral (IR, UV–Vis, NMR) characterization and quantum chemical exploration of benzoimidazole-based hydrazones: a synergistic experimental-computational analysis. Appl. Organomet. Chem. 2019, 33, e5182 10.1002/aoc.5182. [DOI] [Google Scholar]
- Ali A.; Khalid M.; Abid S.; Iqbal J.; Tahir M. N.; Raza A. R.; Zukerman-Schpector J.; Paixão M. W. Facile synthesis, crystal growth, characterization and computational study of new pyridine-based halogenated hydrazones: unveiling the stabilization behavior in terms of noncovalent interactions. Appl. Organomet. Chem. 2020, 34, e5399 10.1002/aoc.5399. [DOI] [Google Scholar]
- Khan E.; Khalid M.; Gul Z.; Shahzad A.; Tahir M. N.; Asif H. M.; Asim S.; Braga A. A. C. Molecular structure of 1,4-bis(substituted-carbonyl)benzene: a combined experimental and theoretical approach. J. Mol. Struct. 2020, 1205, 127633 10.1016/j.molstruc.2019.127633. [DOI] [Google Scholar]
- Ashfaq M.; Ali A.; Kuznetsov A.; Tahir M. N.; Khalid M. DFT and single-crystal investigation of the pyrimethamine-based novel co-crystal salt: 2,4-diamino-5-(4-chlorophenyl)-6-ethylpyrimidin-1-ium-4-methylbenzoate hydrate (1:1:1) (DEMH). J. Mol. Struct. 2021, 1228, 129445 10.1016/j.molstruc.2020.129445. [DOI] [Google Scholar]
- Kargar H.; Behjatmanesh-Ardakani R.; Fallah-Mehrjardi M.; Torabi V.; Munawar K. S.; Ashfaq M.; Tahir M. N. Ultrasound-based synthesis, SC-XRD, NMR, DFT, HSA of new Schiff bases derived from 2-aminopyridine: Experimental and theoretical studies. J. Mol. Struct. 2021, 1233, 130105 10.1016/j.molstruc.2021.130105. [DOI] [Google Scholar]
- Kargar H.; Ardakani A. A.; Tahir M. N.; Ashfaq M.; Munawar K. S. Synthesis, spectral characterization, crystal structure and antibacterial activity of nickel (II), copper (II) and zinc (II) complexes containing ONNO donor Schiff base ligands. J. Mol. Struct. 2021, 1233, 130112 10.1016/j.molstruc.2021.130112. [DOI] [Google Scholar]
- Kargar H.; Fallah-Mehrjardi M.; Behjatmanesh-Ardakani R.; Munawar K. S.; Ashfaq M.; Tahir M. N. Synthesis, spectral characterization, SC-XRD, HSA, DFT and catalytic activity of novel dioxovanadium (V) complex with aminobenzohydrazone Schiff base ligand: An experimental and theoretical approach. Inorg. Chim. Acta 2021, 526, 120535 10.1016/j.ica.2021.120535. [DOI] [Google Scholar]
- Kargar H.; Fallah-Mehrjardi M.; Behjatmanesh-Ardakani R.; Munawar K. S.; Ashfaq M.; Tahir M. N. Experimental and theoretical studies of new dioxomolybdenum complex: Synthesis, characterization and application as an efficient homogeneous catalyst for the selective sulfoxidation. Inorg. Chim. Acta 2021, 527, 120568 10.1016/j.ica.2021.120568. [DOI] [Google Scholar]
- Ali A.; Kuznetsov A.; Khan M. U.; Tahir M. N.; Ashfaq M.; Raza A. R.; Muhammad S. 2-Amino-6-Methylpyridine Based Co-Crystal Salt Formation Using Succinic Acid: Single-Crystal Analysis and Computational Exploration. J. Mol. Struct. 2021, 1230, 129893 10.1016/j.molstruc.2021.129893. [DOI] [Google Scholar]
- Ashfaq M.; Ali A.; Tahir M. N.; Kuznetsov A.; Munawar K. S.; Muhammad S. Synthesis, single-crystal exploration, hirshfeld surface analysis, and DFT investigation of the thiosemicarbazones. J. Mol. Struct. 2022, 1262, 133088 10.1016/j.molstruc.2022.133088. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.