

Abstract

Alzheimer’s disease is characterized by the presence of distinct amyloid-β peptide (Aβ) assemblies with diverse sizes, shapes, and toxicity. However, the primary determinants of Aβ aggregation and neurotoxicity remain unknown. Here, the N-terminal amino acid residues of Aβ42 that distinguished between humans and rats were substituted. The effects of these modifications on the ability of Aβ to aggregate and its neurotoxicity were investigated using biochemical, biophysical, and cellular techniques. The Aβ-derived diffusible ligand, protofibrils, and fibrils formed by the N-terminal mutational peptides, including Aβ42(R5G), Aβ42(Y10F), and rat Aβ42, were indistinguishable by conventional techniques such as size-exclusion chromatography, negative-staining transmission electron microscopy and silver staining, whereas the amyloid fibrillation detected by thioflavin T assay was greatly inhibited in vitro. Using circular dichroism spectroscopy, we discovered that both Aβ42 and Aβ42(Y10F) generated protofibrils and fibrils with a high proportion of parallel β-sheet structures. Furthermore, protofibrils formed by other mutant Aβ peptides and N-terminally shortened peptides were incapable of inducing neuronal death, with the exception of Aβ42 and Aβ42(Y10F). Our findings indicate that the N-terminus of Aβ is important for its fibrillation and neurotoxicity.
Introduction
One of the most notable clinical hallmarks of Alzheimer’s disease (AD) is the abnormal accumulation of Aβ.1 Aβ is generated by the successive cleavage of amyloid precursor protein through β-secretase and γ-secretase, resulting in a plethora of Aβ peptides ranging in length from 37 to 43 amino acids and subsequently forming oligomers, protofibrils, fibrils and plaques.2 Aβ monomers and their assemblies may deteriorate the homeostasis of neurons and glia through receptor-mediated actions. Microglia, for example, may recognize and phagocytose Aβ plaques to promote the production of amyloid dense-core plaques.3 Several receptors on the surface of nerve cells, including PrPc, the insulin receptor, the NGF receptor and the NMDA receptor, have been involved in the binding of Aβ oligomers.4 In clinical trials, numerous antibodies that neutralize Aβ monomeric and/or fibrillar forms in the brains of AD patients were unsuccessful. Instead, accumulating data suggests that Aβ oligomers, but not mature amyloid fibrils, are closely associated with AD severity.5 Consequently, some researchers believe that Aβ oligomers have already begun to accumulate prior to the development of AD, leading to amyloid aggregation,6 inflammatory response,7 oxidative damage,8 mitochondrial dysfunction9 and finally gliosis and neuronal death.10 Furthermore, a number of studies demonstrate that Aβ fibrils extracted from the brains of AD patients are significantly distinct from synthetic Aβ fibrils.11,12 Therefore, it is crucial to investigate the structure and biochemical properties of Aβ aggregates.
Aβ aggregates are a heterogeneous combination of species with various sizes, stabilities, biological and toxic properties.13 Previous study has revealed that Aβ oligomers contain both parallel and antiparallel β-sheet structures, but mature fibrils only have parallel β-sheet structures.14 Rapid Aβ α-oligomers may accelerate the shift to β-oligomers to initiate the amyloid assembly process.15 The conformational transition from intermolecular β-sheet to in-register parallel β-sheet may also contribute to the transformation of hazardous Aβ oligomer to Aβ fibrils.16 In the lag phase of amyloid aggregation, there is evidence that some forms of Aβ oligomer with a nonstandard α-sheet secondary structure could form harmless β-sheet fibrils.17 Thus, the interconversion between the Aβ oligomer and its higher-order assemblies complicates the research of Aβ. In addition, current research suggests that distinct types of soluble Aβ42 aggregates may produce toxicity in multiple ways.18 For example, a smaller Aβ oligomer may impair membrane permeability, whereas a larger oligomer induces inflammation in microglial cells. In conclusion, the precise mechanism of Aβ aggregation and toxicity remains unclear.
Previous studies have shown that Aβ42 aggregates more readily and is more toxic than Aβ40, implying that the C-terminal region of Aβ42 is crucial for its aggregation and toxicity.19 There are three amino acid modifications between the N-terminal portions of human and rat Aβ (R5G, Y10F, and H13R, numbered according to the human Aβ sequence). Given that wild-type rodents exhibit little or no Aβ deposition,20 it is reasonable to hypothesize that the Aβ’s N-terminal region regulates its own aggregation.
Here, the effects of N-terminal mutation on Aβ’s aggregation ability and neurotoxicity were investigated using biophysical and cellular analyses. Briefly, the size and shape of monomers, Aβ-derived diffusible ligand (ADDL), protofibrils, and fibrils formed by human Aβ40 and Aβ42 were compared to those of rat Aβ42. In addition, the amyloid fibrillation capability and secondary structure content of human Aβ42 variants containing the substituted amino acids of the rat Aβ42 version were also analyzed. Finally, the neurotoxicity of these Aβ42 variants was evaluated. Collectively, we found that the N-terminal region of Aβ may influence amyloid fibrillation, secondary structure formation and neurotoxicity. Our findings shed light on the formation and toxicity of Aβ fibrils, as well as why wild-type rats do not develop amyloid plaques.
Results
N-terminal Domain of Aβ May Impose a Weaker Effect on Aggregation States
The factors that impact Aβ aggregation and Aβ-induced neurotoxicity remain unclear. In comparison to humans, rodents are naturally less susceptible to an amyloid burden, even as they age, which may be attributed to a discrepancy of three-amino-acid residues in the N-terminal region of rodents Aβ. Therefore, we hypothesized that variations in amino acid residues in the N-terminal domain of Aβ might affect their ability to aggregate. To verify this idea, we customized rat Aβ42, human Aβ42/Aβ40 and their mutants with multiple N-terminal amino acid substitutions (R5G, Y10F, and H13R) (Figure 1A). Monomers and protofibrils derived from rat Aβ42 and two human Aβ40 and Aβ42 peptides were examined through size-exclusion chromatography (SEC) in nondenaturing and nondisaggregating buffers using a Superdex 75 column. In addition to a dominating peak of Aβ monomers, a minor peak was observed in the void volume of the chromatographed column for these Aβ monomers, highlighting the potential of Aβ peptides to aggregate (Figure 1B). Moreover, the molecular weights of Aβ monomers and aggregates from rats and humans were comparable. We also found that these three Aβ peptides formed protofibrils all exhibited a major and minor peak, corresponding to monomers and aggregates (Figure 1B). Transmission electron microscopy (TEM) further revealed that rat Aβ42 formed monomers, ADDL, protofibrils and fibrils that were morphologically identical to those produced by human Aβ40 and Aβ42 at low resolution (Figure 1C), confirming previous findings.21 Due to the limited resolution of SEC and TEM, it may be preferable to discover the structural features with X-ray diffraction or reconstruct the structure with cryo-EM. Collectively, the size and morphology of varied aggregated forms of rat Aβ42 may be comparable to that of human Aβ, implying that the Aβ N-terminal region exerts less influence on the final aggregated states.
Figure 1.

Comparison of human and rat Aβ amino acid sequences and aggregation states. (A) The Aβ peptide variants were used in this study. Note the differences between the human and rat Aβ sequences at positions 5, 10, and 13. (B) SEC analysis of the aggregation states of Aβ monomers and protofibrils in solution using a Superdex 75 column. (C) TEM examination of the morphology of Aβ monomers, ADDL, protofibrils and fibrils using negative staining techniques.
N-terminal Region of Aβ Affects Amyloid Fibrillation In Vitro
Even though the morphology of aggregated human and rat Aβ peptides are likely similar, we are yet unclear regarding whether N-terminal variations affect fibrillation dynamics during amyloid aggregation. To do this, we applied silver staining to analyze the formation of aggregates at various time points, which were produced by rat Aβ, human Aβ and their variants with the appropriate rat amino acid at the fifth and tenth residues, designated Aβ42(R5G) and Aβ42(Y10F), respectively (Figure 1A). This study did not include Aβ42(H13R) because, for unexplained reasons, commercial companies were unable to purify it. Our results showed that except for scrambled Aβ42, all of the investigated Aβ peptides formed oligomers, including trimers and tetramers, to varied degrees at the observed time points (Figure 2A,B). Notably, rat Aβ42 was more prone to oligomerization compared to human Aβ42 and its variants (Figure 2A,B). Because mutations of arginine or histidine may alter the electric charge of Aβ peptides, we assume that the presence of SDS during electrophoresis led to the formation of these oligomers, based on their similar quantities at various time points. Furthermore, after 7 days incubation, two types of SDS-stable aggregates were clearly observed: one with a molecular weight more than 26 kDa and the other at the interface of the stacking gel and the separating gel (Figure 2A,B). As determined by thioflavin T (ThT) assay, human Aβ42 fibrillated faster than Aβ40 and rat Aβ42 (Figure 2C). Furthermore, the fibrillation process was significantly reduced by the N-terminal variants Aβ42(R5G) and Aβ42(Y10F) (Figure 2C). Additionally, Aβ42(Y10F) was found fibrillated preceding Aβ42(R5G) (Figure 2C). These Aβ fibrils formed during the ThT experiment were examined further by TEM, which showed that they seemed identical (Figure 2D). Taken together, these results suggest that the Aβ N-terminal region plays a vital role in shaping the amyloid fibrillation dynamics.
Figure 2.
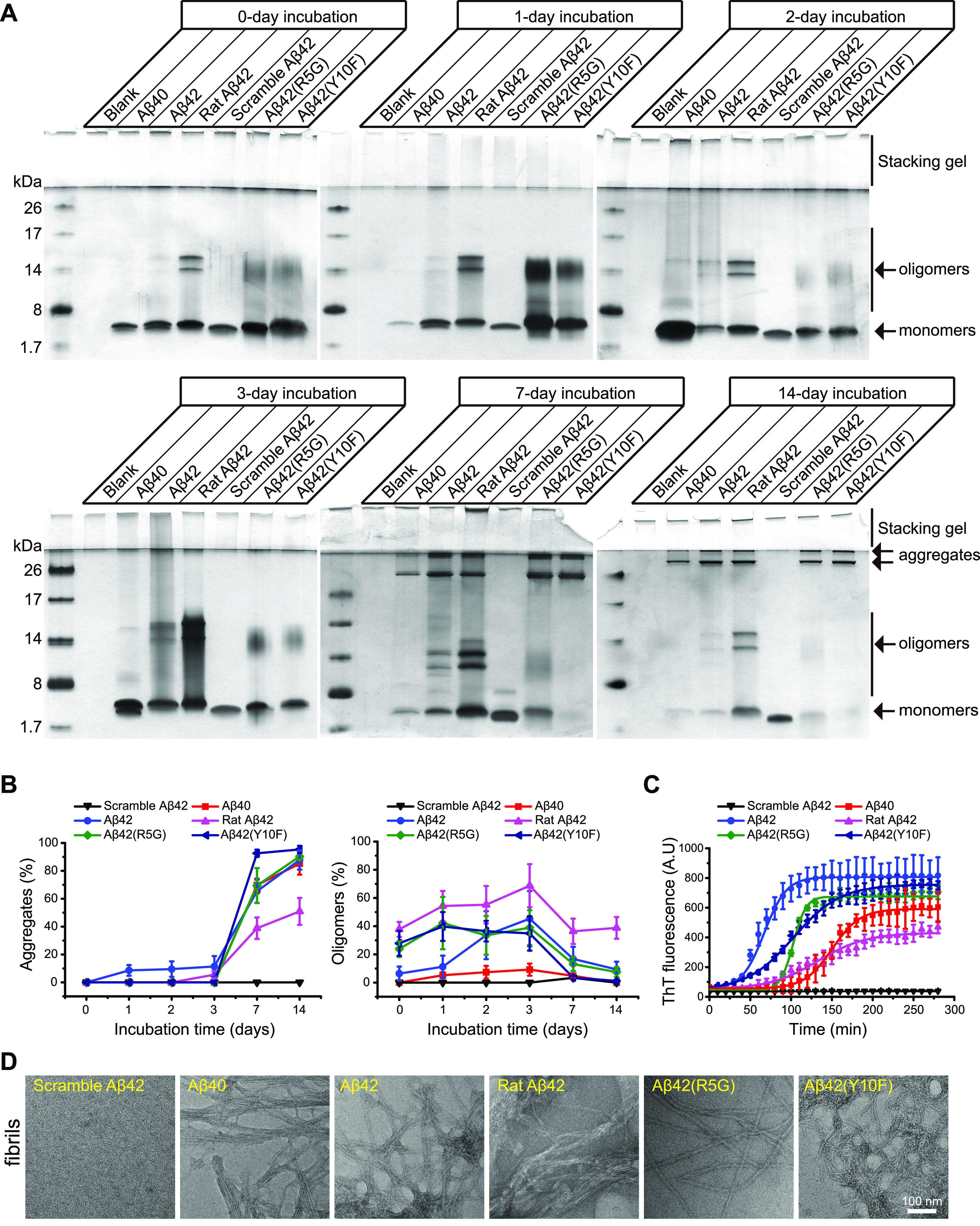
Human Aβ42 N-terminal mutations alter amyloid fibrillation. (A) Silver staining of Aβ fibril preparations at the time intervals indicated. (B) Semi-quantitative analysis of aggregates and oligomers from Aβ fibril preparations in (A). Each measurement was performed in three independent biological replicates. (C) ThT assays for measuring Aβ fibrillation. The raw data was fitted with the mathematical model of Boltzmann’s sigmoidal equation. Each measurement was performed in three independent biological replicates. (D) Representative TEM images of Aβ fibrils in (C).
N-terminal Domain Regulates Secondary Structure Formation in Aβ Species
Numerous studies have shown that mutations in Aβ peptides can result in the rearrangement of secondary structures in their assemblies. Here, we examined the effects of Aβ N-terminal mutations on secondary structure formation during amyloid fibrillation using circular dichroism spectroscopy (CD), a method for assessing the composition of Aβ secondary structure elements (Figure 3A,B).22 Although Aβ40 and Aβ42 differed by two amino acids at the C-terminus, their monomers showed comparable amounts of the helix and antiparallel structure. Aβ40 protofibrils contained an 8% parallel β-sheet structure and a 30% antiparallel structure, whereas Aβ42 protofibrils had 21 and 16% of the same structure, respectively. In fibrils, Aβ40 and Aβ42 had an identical level of parallel structure. Moreover, Aβ42 fibrils showed a 28% extra turn structure. Furthermore, rat Aβ42 monomers had almost half the turn structure, rat Aβ42 protofibrils had a high antiparallel structure content (81%), and rat Aβ42 fibrils had 27% turn structure. We also found that Aβ42(R5G) had nearly identical secondary structure content in monomers, protofibrils and fibrils to its rat Aβ42 counterparts. In contrast, Aβ42(Y10F) displayed a high fraction of parallel structure in all three investigated forms, resembling Aβ40 and Aβ42 rather than rat Aβ42 and Aβ42(R5G). Secondary structures of Aβ42(11–42) and Aβ42(17–42) were comparable to those of rat Aβ42 and Aβ42(R5G). Collectively, the Aβ N-terminus, in particular the fifth arginine (R5), is crucial for regulating secondary structure formation.
Figure 3.

The N-terminal region of human Aβ42 modulates the secondary structural composition of Aβ monomers, protofibrils and fibrils. (A,B) CD spectroscopy was used to determine the secondary structure composition of the various Aβ aggregates (200 μM) (A), which was afterwards interpreted using the BeStSel online tools (http://bestsel.elte.hu/index.php) (B).
Neurotoxicity of Aβ42 Protofibrils is Partly Determined by the Fifth Arginine (R5) in the N-terminal Domain
Prior research has shown that Aβ species may exert their toxicity through distinct mechanisms. For example, Aβ42 oligomers can diminish the synaptic density in mouse hippocampal neurons, inhibit long-term potentiation (LTP), and impair animal learning and memory.23 Since the N-terminal segment can influence the contents of secondary structure of Aβ aggregates, we wanted to see if changing Aβ’s N-terminus would affect the neurotoxicity of its assembly. We treated primary hippocampal neurons with multiple Aβ protofibrils at various concentrations and counted MAP2 positive neurons to determine neurotoxicity (Figure 4A,B). Aβ40 protofibrils did not alter neuronal survival compared to Aβ42 protofibrils, supporting the hypothesis that the C-terminal part of Aβ is required for neuronal toxicity. Only protofibrils made by Aβ42 and Aβ(Y10F) caused significant neuron loss at a concentration of 30 μM, which is consistent with the fact that Aβ42 and Aβ(Y10F) had similar secondary structure, as opposed to protofibrils formed by Aβ(R5G) and rat Aβ42. Likewise, Aβ(11–42) and Aβ(17–42) protofibrils were less lethal for survival. Therefore, mutations or shortenings in the N-terminal region of human Aβ42, such as Aβ(R5G), Aβ(11–42) and Aβ(17–42), would substantially reduce neurotoxicity. The fifth arginine (R5) of Aβ42 is particularly important for its neurotoxicity.
Figure 4.
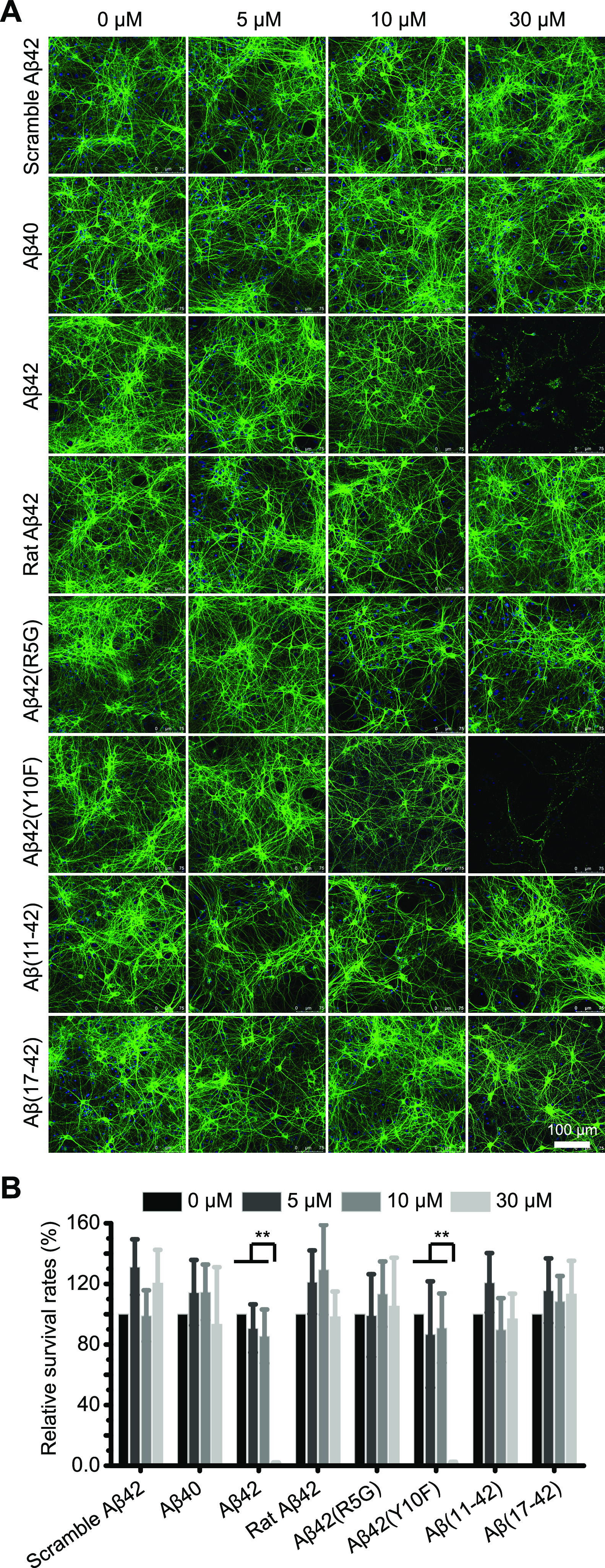
The R5G mutation of human Aβ42 reduces neuronal death caused by protofibrils. (A) Representative images of primary hippocampal neurons treated with the corresponding Aβ protofibrils. MAP2 expression was used to identify the surviving neurons. (B) Survival rates of neurons treated with the indicated Aβ protofibrils were calculated. Each measurement was performed in three independent biological replicates. Statistical significance was assessed by the unpaired Student’s t-test; *: P < 0.01; **: P < 0.001.
Discussion
The types of Aβ species and their aggregates are intimately linked to toxicity, while the underlying mechanism is still the subject of intense debate. Here, we analyzed the biochemistry and morphology of monomers, ADDL, protofibrils and fibrils derived from human Aβ40, Aβ42 and rat Aβ42. The amyloid fibrillation, secondary structure content and neurotoxicity of these peptides and their N-terminal variants were further evaluated. We found that mutations in the N-terminal region of human Aβ42 substantially lowered the amyloid fibrillation process. Furthermore, human Aβ42 N-terminal mutations that reduce the number of parallel β-sheet structures in protofibrils would improve neuronal survival significantly. In conclusion, the N-terminus of Aβ42 is required for the formation of secondary structures, amyloid fibrillation and neurotoxicity.
The N-terminal domain influences the structure and toxicity of Aβ oligomers. For instance, A2T, H6R, and D7N mutations can alter the secondary structure composition, oligomerization and neurotoxicity.24 Metal ions, such as Zn2+ and Cu2+, have also been shown to modulate oligomerization by binding to the N-terminal histidine.25 Previous research indicates that rodent Aβ improved the solubility of human Aβ aggregates in vivo.20 Nonetheless, we discovered that rat Aβ42 formed more oligomers in sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) than human Aβ42 (Figure 2A,B). Therefore, we hypothesized that SDS could increase the solubility of human Aβ42 by interacting with arginine or histidine at the N-terminus. Thus, the R5G mutation may prevent SDS binding in part and promote oligomerization.
The role of the N-terminus of Aβ in fibril formation remains uncertain. Numerous studies have shown that the N-terminus of Aβ forms a disordered structure, suggesting that it may have little impact on Aβ fibril formation.26 However, investigations utilizing high resolution cryo-electron microscopy and solid-state nuclear magnetic resonance demonstrate that Ala2 and Phe4 are essential for the stabilization of hydrophobic clusters, as well as Asp1 coupled with Lys28, and Asp7 coupled with Arg5 form salt-bridges in fibril assembly.27 Furthermore, replacing His13 with arginine could prevent the formation of a kink around Tyr10.27 Besides, the N-terminal portion of Aβ(1–10) forms a β-sheet structure by binding with Aβ(12–22) in fibrils.11
We discovered that the morphology of fibrils formed by Aβ42(R5G) and rat Aβ42 was comparable to that of human Aβ42 (Figure 1), but that their fibrillation (Figure 2), secondary structure components (Figure 3) and toxicity (Figure 4) were dramatically different. The R5G mutation delayed amyloid fibrillation kinetics significantly more than the Y10F mutation (Figure 2C). Furthermore, the R5G mutations, which include rat Aβ42 and Aβ42(R5G) protofibrils and fibrils, result in a shift from parallel to antiparallel β-sheet conformations (Figure 3). This change may be the result of mutations in the fifth arginine, which may prevent the formation of salt bridges.28 Accordingly, this R5G mutation prevented the neuronal death caused by Aβ42 protofibrils (Figure 4). However, the substitution of Tyr10 with phenylalanine, as in Aβ42(Y10F), resulted in only a minor modification to the molecular structure because these two amino acids share a similar benzene ring. Consequently, Aβ42(Y10F) did not differ significantly from Aβ42 in terms of amyloid fibrillation, secondary structure content and neurotoxicity. Moreover, the N-terminal truncation identified in the AD brain region affects Aβ solubility and aggregation as well.29 We found that the N-terminally truncated peptides Aβ(11–42) and Aβ(17–42) had comparable secondary structure contents and neurotoxicity in protofibrils (Figures 3 and 4), which is consistent with the findings that fibril formation rate and morphology differed from Aβ42.30
Conclusions
In this study, we discovered that the Aβ N-terminus influences the dynamics of amyloid fibrillation, the formation of secondary structures and neuronal death. Our findings contribute to a better understanding of the regulatory role of Aβ’s N-terminus in amyloid fibril formation and toxicity.
Materials and Methods
Reagents
The synthetic human Aβ40, human Aβ42, Aβ42(R5G), Aβ11–42, Aβ17–42, scramble Aβ42, and rat Aβ42 peptides were purchased from the rPeptide Company (Athens, GA, USA). All of these peptides are purer above 97%. The peptide Aβ42(Y10F) was obtained from ChinaPeptides Co., Ltd Company (Jiangsu, Suzhou, China). ThT, 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP), and dimethylsulfoxide were purchased from Sigma-Aldrich (St. Louis, MO, USA). Cell culture media (DMEM, DMEM/F12) were purchased from Thermo Fisher Scientific (Waltham, MA, USA). BrainPhys Neuronal Medium and SM1 Kit (#5795) were purchased from STEMCELL Technologies China Company (Shanghai, China). Poly-d-lysine hydrobromide (#P0899) was purchased from Sigma (St Louis, MO, USA). Mouse monoclonal [HM-2] to MAP2 (#ab11267) was purchased from Abcam Company (Cambridge, UK). Penicillin–streptomycin (#03-031-1B) was purchased from BI Company (Israel).
Preparation of Aβ Aggregates
The various Aβ42 peptides were treated as previously described, with some modifications.31 Briefly, 1 mg Aβ peptides were combined with HFIP, vortexed for 2 min and incubated overnight at 4 °C with gentle shaking. The Aβ solution was aliquoted and stored at −80 °C after being evaporated in an N2 flow.
For ADDL preparation, Aβ was dissolved in F12 medium at a final concentration of 100 μM and incubated at 4 °C for 24 h.
For protofibril and fibril preparation, Tris buffer (50 mM Tris, 100 mM NaCl, pH 7.5) was added to solubilize Aβ peptides at a final concentration of 200 μM, and the solution was incubated at room temperature for 24 h with moderate shaking to produce Aβ protofibrils. To achieve mature fibrils, a two-week incubation period was required.
Tricine-SDS-PAGE and Silver Staining
Electrophoresis and silver staining were performed as described previously.32 Briefly, a 4% Tris-tricine concentrating gel and a 16.5% Tris-tricine separating gel were used in this experiment. After the protein marker began to separate, a constant voltage of 40 V was applied for 15–20 min, followed by an increase in voltage to 60 V. For silver staining, the gel was fixed with 10% acetic acid and 30% ethanol.
SEC Analysis
The SEC operation method was modified slightly according to a previous study.33 The preassembled Superdex 75 10/300 GL column was rinsed at a flow rate of 0.75 mL/min. The Aβ sample was centrifuged at 4 °C for 15,000g × 10 min to remove the insoluble materials. A total of 50 μL of sample (200 μM) was loaded. Each sample was detected three times at 210 nm with a shunting speed of 0.75 mL/min.
TEM Analysis
Copper grids (3.05 mm in diameter and 18 μm in thickness; 300 mesh) were purchased from Electron Microscopy Sciences (Hatfield, PA, USA) and coated with continuous carbon films. About 5 μL of Aβ42 solution was applied to discharged carbon films and incubated for 1 min at room temperature. After being washed with ddH2O, the carbon films were negatively stained with 1% sodium phosphotungstate for 1 min. The carbon films were air-dried following the absorption of any excess liquids. A Tecnai G20 microscope (FEI, Hillsboro, OR, USA) with a 200 kV accelerating voltage was used to examine the sample. Denka LaB6 Cathodes were utilized (Ted Pella, Inc., Redding, CA, USA).
ThT Assay
The amount of Aβ fibril production was quantified using ThT fluorescence detection. The fluorescence of ThT was measured with a PerkinElmer LS-55 Spectrometer following the manufacturer’s instructions. The ThT powder was dissolved to a concentration of 50 mM in ultrapure water. The solution was stored in aliquots in tightly sealed vials at −20 °C. The aliquots may be used for up to one month. They were brought to room temperature for at least 1 h before use. For detection, the ThT stock solution was added to a Tris buffer (50 mM Tris, 100 mM NaCl, pH 7.5) containing the tested Aβ. At room temperature, 10 μM of Aβ peptide was incubated with 50 μM of ThT with constant stirring. The fluorescence detection conditions employed were Ex440 nm, slit 10 nm and Em490 nm, slit 10 nm. Each Aβ peptide assay was conducted for 280 min at 10 min intervals, and the dynamics of Aβ fibril formation were studied.
CD Analysis
The experiment was conducted using a CD spectrometer (UK) at room temperature. The experimental conditions are: 1 nm step, 1 nm bandwidth, 10 s collecting time per step, and a 190–250 nm wavelength range. The concentration of Aβ used was 0.45 μg/μL. The signals were detected after adding Aβ samples, and the online BeStSel software (http://bestsel.elte.hu/index.php) was used to analyze the secondary structure contents.22
Primary Culture of Mouse Hippocampal Neurons
All experimental protocols were conducted under the supervision and approval of the Animal Welfare and Ethics Committees of Xizang Minzu University, and University of Electronic Science and Technology of China. Primary neuronal cultures were performed on newborn C57BL/6 mice, which were purchased from Dossy experimental animals (Chengdu, China). To isolate the hippocampus tissue, the brain was decapitated and the meninges were removed. The dissected hippocampus was placed in a 1.5 mL Eppendorf tube, 500 μL of 0.25% trypsin was added, and the mixture was digested for 15 min in a 37 °C water bath. After washing with 2% FBS to stop the digestion, triturate the hippocampus tissue into single cells in BrainPhys Neuronal Medium and SM1 Kit medium. Inoculate cells into a poly-d-lysine coated 24-well culture plate and culture at 37 °C in a 5% CO2 cell incubator. The neurons were used for tests after 14 days of cultivation.
Immunofluorescence Analysis
The primary neurons in culture were fixed for 20 min in 4% paraformaldehyde and then permeabilized for 20 min at room temperature with 0.25% Triton X-100. The cells were blocked with 1% bovine serum albumin in PBST for 30 min before being incubated overnight at 4 °C with the MAP2 antibody. After washing, the cells were incubated for 60 min at 37 °C with secondary antibodies conjugated to Alexa Fluor 488. Fluorescent signals were detected using a laser confocal microscope (Leica, Germany).
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (grant no. 32160215); Natural Science Foundation of Tibet Autonomous Region (grant no. XZ202001ZR0017G); Fund for Tibetan and Qin scholars (Distinguished Young Scholars) of Xizang Minzu University. This work was also supported by Natural Science Foundation of Sichuan Province (grant no. 2022NSFSC0607).
Author Contributions
J.-M.S. and Y.Z. planned and designed the experiments. J.-M.S., H.-Y.L., H.L., L.Z., Y.B.G., J.P., H.A., Y.-S.L., S.D.L. and Z.-Y.Z performed the experiments. J.-M.S. and Y.Z. analyzed experimental results and wrote the manuscript with input from all authors.
The authors declare no competing financial interest.
References
- Pannuzzo M. Beta-amyloid pore linked to controlled calcium influx into the cell: A new paradigm for Alzheimer’s Disease. Alzheimer’s Dementia 2022, 18, 191–196. 10.1002/alz.12373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Sevigny J.; Chiao P.; Bussière T.; Weinreb P. H.; Williams L.; Maier M.; Dunstan R.; Salloway S.; Chen T.; Ling Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. 10.1038/nature19323. [DOI] [PubMed] [Google Scholar]; b Sasaguri H.; Nilsson P.; Hashimoto S.; Nagata K.; Saito T.; De Strooper B.; Hardy J.; Vassar R.; Winblad B.; Saido T. C. APP mouse models for Alzheimer’s disease preclinical studies. EMBO J 2017, 36, 2473. 10.15252/embj.201797397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.; Happonen K. E.; Burrola P. G.; O’Connor C.; Hah N.; Huang L.; Nimmerjahn A.; Lemke G. Microglia use TAM receptors to detect and engulf amyloid β plaques. Nat. Immunol. 2021, 22, 586–594. 10.1038/s41590-021-00913-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin L.; Harris D. A. Aβ receptors specifically recognize molecular features displayed by fibril ends and neurotoxic oligomers. Nat. Commun. 2021, 12, 3451. 10.1038/s41467-021-23507-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline E. N.; Bicca M. A.; Viola K. L.; Klein W. L. The Amyloid-β Oligomer Hypothesis: Beginning of the Third Decade. J. Alzheimer’s Dis. 2018, 64, S567–S610. 10.3233/JAD-179941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlmann R. E.; Rother C.; Rasmussen J.; Schelle J.; Bergmann C.; Ullrich Gavilanes E. M.; Fritschi S. K.; Buehler A.; Baumann F.; Skodras A.; et al. Acute targeting of pre-amyloid seeds in transgenic mice reduces Alzheimer-like pathology later in life. Nat. Neurosci. 2020, 23, 1580–1588. 10.1038/s41593-020-00737-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ransohoff R. M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. 10.1126/science.aag2590. [DOI] [PubMed] [Google Scholar]; b LaRocca T. J.; Cavalier A. N.; Roberts C. M.; Lemieux M. R.; Ramesh P.; Garcia M. A.; Link C. D. Amyloid beta acts synergistically as a pro-inflammatory cytokine. Neurobiol. Dis. 2021, 159, 105493. 10.1016/j.nbd.2021.105493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown M. R.; Radford S. E.; Hewitt E. W. Modulation of β-Amyloid Fibril Formation in Alzheimer’s Disease by Microglia and Infection. Front. Mol. Neurosci. 2020, 13, 609073. 10.3389/fnmol.2020.609073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins H. M.; Troutwine B. R.; Menta B. W.; Manley S. J.; Strope T. A.; Lysaker C. R.; Swerdlow R. H. Mitochondrial Membrane Potential Influences Amyloid-β Protein Precursor Localization and Amyloid-β Secretion. J. Alzheimer’s Dis. 2022, 85, 381–394. 10.3233/JAD-215280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a George A. A.; Vieira J. M.; Xavier-Jackson C.; Gee M. T.; Cirrito J. R.; Bimonte-Nelson H. A.; Picciotto M. R.; Lukas R. J.; Whiteaker P. Implications of Oligomeric Amyloid-Beta (oAβ42) Signaling through α7β2-Nicotinic Acetylcholine Receptors (nAChRs) on Basal Forebrain Cholinergic Neuronal Intrinsic Excitability and Cognitive Decline. J. Neurosci. 2021, 41, 555–575. 10.1523/JNEUROSCI.0876-20.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Huffels C. F. M.; Osborn L. M.; Hulshof L. A.; Kooijman L.; Henning L.; Steinhäuser C.; Hol E. M. Amyloid-β plaques affect astrocyte Kir4.1 protein expression but not function in the dentate gyrus of APP / PS1 mice. Glia 2022, 70, 748–767. 10.1002/glia.24137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollmer M.; Close W.; Funk L.; Rasmussen J.; Bsoul A.; Schierhorn A.; Schmidt M.; Sigurdson C. J.; Jucker M.; Fändrich M. Cryo-EM structure and polymorphism of Aβ amyloid fibrils purified from Alzheimer’s brain tissue. Nat. Commun. 2019, 10, 4760. 10.1038/s41467-019-12683-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.; Arseni D.; Zhang W.; Huang M.; Lövestam S.; Schweighauser M.; Kotecha A.; Murzin A. G.; Peak-Chew S. Y.; Macdonald J.; et al. Cryo-EM structures of amyloid-β 42 filaments from human brains. Science 2022, 375, 167–172. 10.1126/science.abm7285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willem M.; Fändrich M. A molecular view of human amyloid-β folds. Science 2022, 375, 147–148. 10.1126/science.abn5428. [DOI] [PubMed] [Google Scholar]
- Yu L.; Edalji R.; Harlan J. E.; Holzman T. F.; Lopez A. P.; Labkovsky B.; Hillen H.; Barghorn S.; Ebert U.; Richardson P. L.; et al. Structural Characterization of a Soluble Amyloid β-Peptide Oligomer. Biochemistry 2009, 48, 1870–1877. 10.1021/bi802046n. [DOI] [PubMed] [Google Scholar]
- Misra P.; Kodali R.; Chemuru S.; Kar K.; Wetzel R. Rapid α-oligomer formation mediated by the Aβ C terminus initiates an amyloid assembly pathway. Nat. Commun. 2016, 7, 12419. 10.1038/ncomms12419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y.; Matsuda I.; Inoue M.; Sasahara T.; Hoshi M.; Ishii Y. NMR-based site-resolved profiling of β-amyloid misfolding reveals structural transitions from pathologically relevant spherical oligomer to fibril. J. Biol. Chem. 2020, 295, 458–467. 10.1074/jbc.RA119.008522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shea D.; Hsu C. C.; Bi T. M.; Paranjapye N.; Childers M. C.; Cochran J.; Tomberlin C. P.; Wang L.; Paris D.; Zonderman J.; et al. α-Sheet secondary structure in amyloid β-peptide drives aggregation and toxicity in Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 2019, 116, 8895–8900. 10.1073/pnas.1820585116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De S.; Wirthensohn D. C.; Flagmeier P.; Hughes C.; Aprile F. A.; Ruggeri F. S.; Whiten D. R.; Emin D.; Xia Z.; Varela J. A.; et al. Different soluble aggregates of Aβ42 can give rise to cellular toxicity through different mechanisms. Nat. Commun. 2019, 10, 1541. 10.1038/s41467-019-09477-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed M.; Davis J.; Aucoin D.; Sato T.; Ahuja S.; Aimoto S.; Elliott J. I.; Van Nostrand W. E.; Smith S. O. Structural conversion of neurotoxic amyloid-β1-42 oligomers to fibrils. Nat. Struct. Mol. Biol. 2010, 17, 561–567. 10.1038/nsmb.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky J. L.; Younkin L. H.; Gonzales V.; Fadale D. J.; Slunt H. H.; Lester H. A.; Younkin S. G.; Borchelt D. R. Rodent Aβ Modulates the Solubility and Distribution of Amyloid Deposits in Transgenic Mice. J. Biol. Chem. 2007, 282, 22707–22720. 10.1074/jbc.M611050200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M.; Sachse C.; Richter W.; Xu C.; Fändrich M.; Grigorieff N. Comparison of Alzheimer Aβ(1-40) and Aβ(1-42) amyloid fibrils reveals similar protofilament structures. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 19813–19818. 10.1073/pnas.0905007106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micsonai A.; Bulyáki E.; Kardos J. BeStSel: From Secondary Structure Analysis to Protein Fold Prediction by Circular Dichroism Spectroscopy. Methods Mol. Biol. 2021, 2199, 175–189. 10.1007/978-1-0716-0892-0_11. [DOI] [PubMed] [Google Scholar]
- Lambert M. P.; Barlow A. K.; Chromy B. A.; Edwards C.; Freed R.; Liosatos M.; Morgan T. E.; Rozovsky I.; Trommer B.; Viola K. L.; et al. Diffusible, nonfibrillar ligands derived from Aβ 1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 6448–6453. 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]; , Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S
- a Benilova I.; Gallardo R.; Ungureanu A. A.; Castillo Cano V.; Snellinx A.; Ramakers M.; Bartic C.; Rousseau F.; Schymkowitz J.; De Strooper B. The Alzheimer Disease Protective Mutation A2T Modulates Kinetic and Thermodynamic Properties of Amyloid-β (Aβ) Aggregation. J. Biol. Chem. 2014, 289, 30977–30989. 10.1074/jbc.M114.599027. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Viet M. H.; Nguyen P. H.; Ngo S. T.; Li M. S.; Derreumaux P. Effect of the Tottori Familial Disease Mutation (D7N) on the Monomers and Dimers of Aβ40 and Aβ42. ACS Chem. Neurosci. 2013, 4, 1446–1457. 10.1021/cn400110d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Minicozzi V.; Stellato F.; Comai M.; Serra M.; Potrich C.; Meyer-Klaucke W.; Morante S. Identifying the Minimal Copper- and Zinc-binding Site Sequence in Amyloid-β Peptides. J. Biol. Chem. 2008, 283, 10784–10792. 10.1074/jbc.M707109200. [DOI] [PubMed] [Google Scholar]; b Shin B. K.; Saxena S. Substantial Contribution of the Two Imidazole Rings of the His13–His14 Dyad to Cu(II) Binding in Amyloid-β(1–16) at Physiological pH and Its Significance. J. Phys. Chem. A 2011, 115, 9590–9602. 10.1021/jp200379m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Thacker D.; Sanagavarapu K.; Frohm B.; Meisl G.; Knowles T. P. J.; Linse S. The role of fibril structure and surface hydrophobicity in secondary nucleation of amyloid fibrils. Proc. Natl. Acad. Sci. U.S.A. 2020, 117, 25272–25283. 10.1073/pnas.2002956117. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ghosh U.; Thurber K. R.; Yau W. M.; Tycko R. Molecular structure of a prevalent amyloid-β fibril polymorph from Alzheimer’s disease brain tissue. Proc. Natl. Acad. Sci. U.S.A. 2021, 118, e2023089118 10.1073/pnas.2023089118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gremer L.; Schölzel D.; Schenk C.; Reinartz E.; Labahn J.; Ravelli R. B. G.; Tusche M.; Lopez-Iglesias C.; Hoyer W.; Heise H.; et al. Fibril structure of amyloid-β(1-42) by cryo-electron microscopy. Science 2017, 358, 116–119. 10.1126/science.aao2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vemulapalli S. P. B.; Becker S.; Griesinger C.; Rezaei-Ghaleh N. Combined High-Pressure and Multiquantum NMR and Molecular Simulation Propose a Role for N-Terminal Salt Bridges in Amyloid-Beta. J. Phys. Chem. Lett. 2021, 12, 9933–9939. 10.1021/acs.jpclett.1c02595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Shinohara M.; Koga S.; Konno T.; Nix J.; Shinohara M.; Aoki N.; Das P.; Parisi J. E.; Petersen R. C.; Rosenberry T. L.; et al. Distinct spatiotemporal accumulation of N-truncated and full-length amyloid-β42 in Alzheimer’s disease. Brain 2017, 140, 3301–3316. 10.1093/brain/awx284. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bouter Y.; Dietrich K.; Wittnam J. L.; Rezaei-Ghaleh N.; Pillot T.; Papot-Couturier S.; Lefebvre T.; Sprenger F.; Wirths O.; Zweckstetter M.; Bayer T. A.; et al. N-truncated amyloid β (Aβ) 4-42 forms stable aggregates and induces acute and long-lasting behavioral deficits. Acta Neuropathol. 2013, 126, 189–205. 10.1007/s00401-013-1129-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J. M.; Zhang L.; Liu E. Q. Dissecting the behaviour of β-amyloid peptide variants during oligomerization and fibrillation. J. Pept. Sci. 2017, 23, 810–817. 10.1002/psc.3028. [DOI] [PubMed] [Google Scholar]
- Shi J. M.; Zhu L.; Lan X.; Zhao D. W.; He Y. J.; Sun Z. Q.; Wu D.; Li H. Y. Endocytosis Is a Key Mode of Interaction between Extracellular β-Amyloid and the Cell Membrane. Biophys. J. 2020, 119, 1078–1090. 10.1016/j.bpj.2020.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J.-M.; Zhang L.; Liu E. Q. Dissecting the behaviour of β-amyloid peptide variants during oligomerization and fibrillation. J. Pept. Sci. 2017, 23, 810. 10.1002/psc.3028. [DOI] [PubMed] [Google Scholar]
- Jan A.; Hartley D. M.; Lashuel H. A. Preparation and characterization of toxic Aβ aggregates for structural and functional studies in Alzheimer’s disease research. Nat. Protoc. 2010, 5, 1186–1209. 10.1038/nprot.2010.72. [DOI] [PubMed] [Google Scholar]