

Abstract
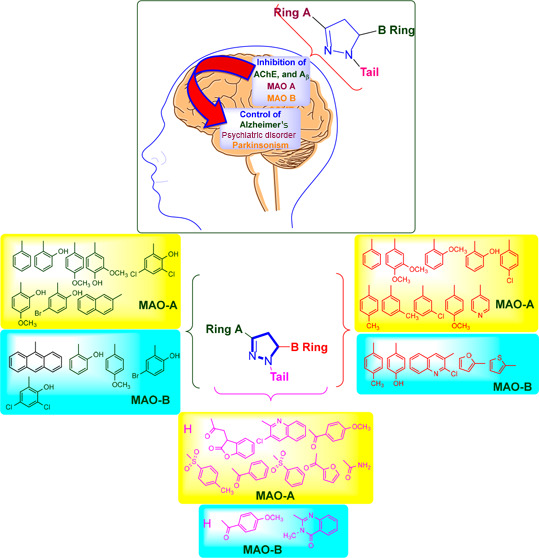
Pyrazolines are a significant class of heterocyclic compounds with essential biological activities. They are quite stable, which has inspired medicinal chemists to experiment with the ring’s structure in many different ways to create a variety of pharmacological activities. The structures of numerous commercially available therapeutic agents contain a pyrazoline ring. Pyrazolines are well-known for their ability to treat neurodegenerative diseases. The neurodegenerative diseases that affect huge populations globally include Alzheimer’s disease (AD), Parkinson’s disease (PD), and psychiatric disorders. The neuroprotective properties of pyrazolines published since 2003 are covered in the current review. Structure–activity relationships (SARs), molecular docking simulation, anticholinesterase (anti-AChE), and monoamine oxidase (MAO A/B) inhibitory actions are all covered in this article. Pyrazolines were discovered to have beneficial effects in the management of AD and were revealed to be inhibitors of acetylcholine esterase (AChE) and beta-amyloid (Aβ) plaques. They were discovered to be efficient against PD and also targeted MAO B and COMT. It was discovered that the pyrazolines block MAO A to treat psychiatric diseases. Pyrazolines are significant heteroaromatic scaffolds with a variety of biological functions. They were discovered to be remarkably stable and serve as an indispensable anchor for the development of new drugs. By blocking AChE and MAOs, they may be used to treat neurodegenerative diseases. The discussion outlined here is an essential and helpful resource for medicinal chemists who are investigating and applying pyrazolines in neurodegenerative research initiatives as well as to expedite future research programs on neurodegenerative disorders.
1. Introduction
Pyrazolines are five-membered heterocyclic rings with two nitrogen atoms in their structure. As illustrated in Figure 1, they exist in three isomeric forms: 1-pyrazoline, 2-pyrazoline, and 3-pyrazoline.1 In 2-pyrazolines, two or three substitutions were carried out by medicinal chemists to explore their biological potentials. Additionally, they serve as isosteres for a variety of heterocyclic rings, including imidazole, thiazole, and oxazole.2 The pyrazolines are highly stable, which has inspired chemists to make several structural changes to the ring to prepare a variety of compounds with different pharmacological activities.3−5 Pyrazoline scaffolds have been thoroughly investigated by numerous experts in the field of medicinal chemistry to treat a variety of ailments. Pyrazolines possess diverse biological activities including anticancer,6,7 anti-Alzheimer’s,8 antitubercular,9 anticonvulsant,10,11 anti-inflammatory,12 antidepressant,13 antimicrobial,14 anti-HIV,14 antimalarial,15 monoamine oxidase (MAO) inhibitory,16 anticholinesterase,8 cannabinoid antagonist,17 anti-EGFR,18 antiparkinsonian,19 and many more. Pyrazolines are a significant class of heterocyclic compounds that are found in the chemical structure of numerous therapeutic agents. For pyrazoline based drugs, Axitinib is a selective vascular endothelial growth factor receptor (VEGFR) inhibitor used for the treatment of metastatic renal cell carcinoma,20 Ibipinabant is used clinically as a selective cannabinoid CB1 receptor inverse agonist,21,22 and ibrutinib as a first line treatment for chronic lymphocytic leukemia.23 Antipyrin, aminophenazone, famoprofazone, edaravone, metamizole, morazone, phenylbutazone, oxyphenbutazone, ramifenazone, and sulfinpyrazone are just a few of the commercially available pyrazoline medications that are now used in clinical practice.5,24,25 The biological and synthetic importance of pyrazolines has been the subject of several review studies.5,26−30 Some of the pyrazoline incorporated molecules are in clinical developmental stages. Pyrazoline containing anticancer molecules pictilisib dimethanesulfonate (GDC-0941), pyrazoloacridine (NSC 366140), and 3-(5′-hydroxymethyl-2′-furyl)-1-benzyl indazole (YC-1) are in phase II clinical trials.31−33 Enficoxib (E-6087), an anti-inflammatory, and SLV-330, a cannabinoid antagonist, are pyrazoline containing molecules in phase I clinical trials.34,17 Pyrazoline based clinically approved and investigational new drugs are given in Table 1. Pyrazolines can be prepared in two steps (Scheme 1). In the first step, chalcone (α,β-unsaturated carbonyl compound) is prepared from aromatic or aliphatic aldehydes and ketone in alkaline medium (Claisen–Schmidt condensation), and the second step involves allowing hydrazine and chalcone to react in a suitable solvent to produce pyrazoline. 5-Substituted pyrazolines possess at least one chiral carbon, and hence a minimum of two isomers can be possible.
Figure 1.

Different isomers of pyrazoline and substitution at positions 1, 3, and 5 on 2-pyrazoline.
Table 1. Pyrazoline Based Clinically Approved and Investigational New Drugs.


Scheme 1. General Method for the Synthesis of 2-Pyrazolines.

Pyrazolines have well-established therapeutic benefits for neurodegenerative diseases.35−48 The present investigation considered the beneficial effects of pyrazolines reported since 2003 on certain neurodegenerative disorders, including Alzheimer’s disease (AD), Parkinson’s disease (PD), and psychiatric disorders. AD is one of the most common neurodegenerative disorders in elderly people and accounts for more than 80% of the world’s cases of dementia.49 By 2050, one new case of AD is predicted to occur every 33 s, or nearly a million new cases every year, with an approximate total prevalence of 13.8 million to be expected.50 β-Amyloid (Aβ) plaques and inadequate levels of the essential neurotransmitter acetylcholine (ACh) are associated with AD.51,52 β-Amyloid (Aβ) containing extracellular plaques and tau-containing intracellular neurofibrillary tangles are the hallmarks of AD.53−55 AD is a complex and multifactorial neurodegenerative disorder, as the disease’s pathogenesis is still poorly understood, and the current AD medications only mildly alleviate symptoms.56,57 Aducanumab, an Aβ aggregate-targeting antibody, has recently generated a lot of interest in the field due to its potential to reduce amyloid plaques and improve cognitive decline in AD patients.54,58 It was also reported that transcription factor EB (TFEB)-mediated autophagy reduces Tau pathology and enhances memory in animal models of AD.59,60 The first cholinergic hypothesis cannot be disproved because one of the most successful treatments for AD, donepezil, is a cholinesterase inhibitor.56,61 Parkinsonism disease (PD) is another neurodegenerative disorder mainly associated with movement impairments such as rigidity, slowness, and tremors affect 2–3% of the total population.62,63 The loss of dopaminergic nigrostriatal neurons is the hallmark of PD.64 Monoamine oxidase B (MAO B) and catechol-O-methyl transferase (COMT) are two most important neurotransmitters that play an important role in PD.65−67 Monoamine oxidase enzyme exists as two isoforms MAO A and MAO B, and they play an important role in psychiatric and neurological disorders, respectively.65,67
2. Pyrazolines as ACh, Aβ, and Tau Inhibitors
AD is one of the most common neurodegenerative disorders in elderly people and accounts for more than 80% of the world’s cases of dementia.49 AD is a complex and multifactorial neurodegenerative disorder as the disease’s pathogenesis is still poorly understood, and the current AD medications only mildly alleviate symptoms.56,57 β-Amyloid (Aβ) containing extracellular plaques and tau-containing intracellular neurofibrillary tangles are the hallmarks of AD.53−55 Inhibition of ACh, Aβ plaques, and tau-containing intracellular neurofibrillary tangles could be a target for the treatment of AD. Pyrazolines were reported as an effective scaffold to inhibit the degradation of ACh and Aβ plaques and hence could have a salutary effect in the treatment of AD.
Yamali et al. prepared eight new 4-(3-(difluorophenyl)-5-(dimethoxyphenyl)-4,5-dihydropyrazol-1-yl)benzenesulfonamides (Scheme 2) and evaluated their acetyl cholinesterase (AChE) inhibitory activity by Ellman’s method. The novel compounds significantly inhibit AChE at the nanomolecular level. 4-(3-(2,5-Difluorophenyl)-5-(3,4-dimethoxyphenyl)-4,5-dihydropyrazol-1-yl)benzenesulfonamide (A01) exhibited the most promising anti-AChE with an IC50 value of 6.36 nM and Ki value of 3.31 ± 1.44 nM, even better than the standard drug tacrine (IC50 = 43.21 nM; Ki = 22.99 ± 4.07 nM). The structure–activity relationship (SAR) suggested that 2,5-difluoro substituents (in the phenyl ring) were found to be more active against AChE inhibitory activity than 3,5-difluoro. It was found that shifting the fluorine atom from position 3 to position 2 resulted in a greater inhibition of AChE.68 A molecular docking study was conducted against the most potent compound (A01) in the series against VX-phosphonylated hAChE (PDB ID: 6U37) to assess the binding affinity. With a docking score of −6.845 kcal/mol and interactions with the residues Glu292 (H-bond) and Tyr341 (π–π-stacking), compound A01 was found to display efficient binding within the active site. The AChE inhibitory activity and 2D molecular docking pose compound A01 are given in Figure 2.
Scheme 2. Synthesis of 4-(3-(Difluorophenyl)-5-(dimethoxyphenyl)-4,5-dihydropyrazol-1-yl)benzenesulfonamides.

Figure 2.

AChE inhibitory activity and 2D molecular docking pose compound A01.
Singh and Bansal prepared a new series of 16,17-N′-(alkyl/arylsulfonyl)pyrazolinyl steroids (Scheme 3) and evaluated their neuroprotective activity. 1′-(4-Fluorobenzenesulfonyl)-5′-(pyridin-4-yl)-androst-5-en-[17,16-c]-1′H-pyrazoline-3β-ol (A02) showed promising suppression of AChE and lipid peroxidation [escape latency 47.1 ± 2.1(s) (time spent in target quadrant (TSTQ) 47.3 ± 2.9(s) and % initial transfer latency (ITL) 21.4 ± 2.8 (s)] and inhibited TNF-α levels in brain serum of LPS-treated rats with 59 ± 1.4 pg per mg of protein.69 A molecular docking study was conducted against the most potent compound (A02) in the series against VX-phosphonylated hAChE (PDB ID: 6U37) to assess the binding affinity. With a docking score of −6.885 kcal/mol and interactions with the residues Arg296 (H-bond), Phe295 (H-bond), and Tyr72 (π–π-stacking), compound A02 was found to display an efficient binding within the active site of hAChE. The AChE inhibitory activity and 2D molecular docking pose compound A02 are given in Figure 3.
Scheme 3. Synthesis of Pyrazoline from Steroids.

Figure 3.

AChE inhibitory activity and 2D molecular docking pose of compound A02.
Ozgun et al. prepared a novel series of 4-(3-substitutedphenyl-5-polymethoxyphenyl-4,5-dihydro-1H-pyrazol-1-yl)benzenesulfonamides (Scheme 4) and evaluated their anti-AChE activity. 4-(3-(4-Bromophenyl)-5-(2,4-dimethoxyphenyl)4,5-dihydro-1H-pyrazol-1-yl)benzenesulfonamide (A03) emerged as the best AChE inhibitor among the series with a Ki value of 37.7 ± 14.4 nM. The anti-AChE activity of 4-(3-substitutedphenyl-5-polymethoxyphenyl-4,5-dihydro-1H-pyrazol-1-yl)benzenesulfonamides and 4-(3-(difluorophenyl))-5-(dimethoxyphenyl)-4,5-dihydropyrazol-1-yl)benzenesulfonamides was found to be respectively 5.9–2.2 and 6.8–2.9 times more potent than Tacrine.70 A molecular docking study was conducted against the most potent compound (A03) in the series against VX-phosphonylated hAChE (PDB ID: 6U37) to assess the binding affinity. With a docking score of −6.708 kcal/mol and interactions with the residues Glu292 (H-bond), and Tyr124 (π–π-stacking), compound A03 was found to display an efficient binding within the active site of hAChE. The AChE inhibitory activity and 2D molecular docking pose compound A03 are given in Figure 4.
Scheme 4. Synthesis of Pyrazoline Benzene Sulfonamides.

Figure 4.

AChE inhibitory activity and 2D molecular docking pose of compound A03.
Singh et al. prepared 16,17-pyrazolinyl dehydroepiandrosterone analogues (Scheme 5) and evaluated their neuroprotective activity. 1′-(Acetyl)-5′-(pyridin-4-yl)-androst-5-en-[17,16-c]-1′H-pyrazoline-3β-yl acetate (A04) showed promising activity with escape latency 52.9 ± 1.5 (s), TSTQ 43 ± 3 (s), and % ITL 26.1 ± 3.8 (s) in terms of restoration of learning and memory functions. The compound A04 inhibited AChE with 0.0028 ± 0.0006 μM of AChE/min/mg protein and TNF-α levels in brain serum of LPS-treated rats with 69.6 ± 2.5 pg per mg of protein. A 4-pyridyl moiety at 5-position of the heterocyclic ring was found to be most promising for activity.19 A molecular docking study was conducted against the most potent compound (A04) in the series against VX-phosphonylated hAChE (PDB ID: 6U37) to assess the binding affinity. With a docking score of −6.714 kcal/mol and interactions with the residues Phe295 (H-bond), Thr95 (H-bond), and Tyr341 (π–π-stacking), compound A04 was found to display an efficient binding within the active site of hAChE. The AChE inhibitory activity and 2D molecular docking pose compound A04 are given in Figure 5.
Scheme 5. Synthesis of of 16,17-Pyrazolinyl Steroidal Analogues.

Figure 5.

AChE inhibitory activity and 2D molecular docking pose of compound A04.
Mishra and Sasmal studied the virtual screening of 18000 molecules and synthesized the top 10 HITS (anthracene incorporated pyrazolines) (Scheme 6) and reported their AChE inhibitory activity.71 4-(3-(Anthracen-9-yl)-4,5-dihydro-1H-pyrazol-5-yl)benzene-1,3-diol (A05) displayed promising AChE inhibitory activity with Ki of 7.2 ± 0.2 nM comparable to the standard drug donepezil (Ki = 7.2 ± 0.4 nM).72 A molecular docking studies was conducted against the most potent compound (A05) in the series against VX-phosphonylated hAChE (PDB ID: 6U37) to assess the binding affinity. With a docking score of −6.468 kcal/mol and interactions with the residues Phe295 (H-bond), Arg296 (H-bond), and Tyr341 (π–π-stacking), compound A05 was found to display an efficient binding within the active site of hAChE. The AChE inhibitory activity and 2D molecular docking pose compound A05 are given in Figure 6.
Scheme 6. Synthesis of 3-(Anthracene-9-yl)-5-aryl Pyrazolines.

Figure 6.

AChE inhibitory activity and 2D molecular docking pose of compound A05.
Gokhan et al. prepared a series of 12 1-N-substituted thiocarbamoyl-3-phenyl-5-thienyl-2-pyrazolines (Scheme 7) through the intermediate formation of hydrazone.73 Ucar et al. reported their anti-AChE activity, and 1-N-methylthiocarbamoyl-3-phenyl-5-thienyl-2-pyrazoline (A06) was found to be promising among the series, displaying promising AChE inhibitory activity with an IC50 value of 0.09 ± 0.004 μM (erythrocyte), which was found to be more potent than rivastigmine (IC50 = 12.23 ± 1.34 μM).74 A molecular docking studies was conducted against the most potent compound (A06) in the series against VX-phosphonylated hAChE (PDB ID: 6U37) to assess the binding affinity. With a docking score of −7.461 kcal/mol and interactions with the residues Phe295 (H-bond), and Tyr337 (π–π-stacking), compound A06 was found to display an efficient binding within the active site of hAChE. The AChE inhibitory activity and 2D molecular docking pose compound A06 are given in Figure 7.
Scheme 7. Synthesis of 1-N-Substituted Thiocarbomoyl-3-phenyl-5-thienyl-2-pyrazolines.

Figure 7.

AChE inhibitory activity and 2D molecular docking pose of compound A06.
Tan et al. prepared a series of 2-pyrazolines (Scheme 8) and reported their AChE inhibitory activity. 1-(5-(4-(4-Methylpiperazin-1-yl)phenyl)-3-(3,4-dimethoxyphenyl)-4,5-dihydro-1H-pyrazol-1-yl)ethanone (A07) inhibited AChE with an IC50 value of 6.5 ± 0.1 μM and also reduced the Aβ1–42 aggregation levels significantly 72.6%.8 A molecular docking study was conducted against the most potent compound (A07) in the series against VX-phosphonylated hAChE (PDB ID: 6U37) to assess the binding affinity. With a docking score of −9.011 kcal/mol and interactions with the residues Arg296 (H-bond), and Tyr124 and Tyr337 (π–π-stacking), compound A07 was found to be an efficient binding within the active site of hAChE. The AChE inhibitory activity and 2D molecular docking pose compound A07 are given in Figure 8.
Scheme 8. Synthesis of 2-Pyrazoline Analogues.

Figure 8.

AChE inhibitory activity and 2D molecular docking pose of compound A07.
Mishra et al. reported the β-amyloid (Aβ) inhibitory activity of the synthesized pyrazolines (as shown in Scheme 6). Aβ aggregation was inhibited by the compound A08, observed through a thioflavin-T assay (IC50 = 60 μM) and scanning electron microscopy (no fibril formation). Marked defense against scopolamine and Aβ1–42-induced memory impairments has been demonstrated by the compound A08. In the brains of the Aβ1–42-induced mice model, it also minimized neuronal loss and amyloid plaque deposition.75 A molecular docking study was conducted against the most potent compound (A08) in the series against VX-phosphonylated hAChE (PDB ID: 6U37) to assess the binding affinity. With a docking score of −6.874 kcal/mol and interactions with the residues Glu292 (H-bond), and Tyr72 and Trp286 (π–π-stacking), compound A08 was found to display an efficient binding within the active site of hAChE. The AChE inhibitory activity and 2D molecular docking pose compound A08 are given in Figure 9.
Figure 9.

AChE inhibitory activity and 2D molecular docking pose of compound A08.
Zitouni et al. prepared 5 pyrazolines (Scheme 9) and reported their anti-AChE activity. 1-[5-(Benzo[d][1,3]dioxol-5-yl)-3-(thien-2-yl)-4,5-dihydro-1H-pyrazol-1-yl]-2-[(((4-(diethylamino)butyl)amino)methyl)thio]ethan-1-one (A09) showed AChE inhibitory activity with an IC50 value of 18.53 μM.76 A molecular docking studies was conducted against the most potent compound (A08) in the series against VX-phosphonylated hAChE (PDB ID: 6U37) to assess the binding affinity. With a docking score of −8.896 kcal/mol and interactions with the residues Phe295 (H-bond), Phe297 (π–π-stacking), and His281 (π-cation), compound A09 was found to display an efficient binding within the active site of hAChE. The AChE inhibitory activity and 2D molecular docking pose compound A09 is given in Figure 10.
Scheme 9. Synthesis of 2-[(((4-(Diethylamino)butyl)amino)methyl)thio]-1-[5-phenyl-3-(thien-2-yl)-4,5-dihydro-1H-pyrazol-1-yl]ethan-1-ones.

Figure 10.

AChE inhibitory activity and 2D molecular docking pose of compound A09.
Server et al. prepared a series of pyrazolines by molecular hybridization of thiazole and pyrazoline scaffolds (Scheme 10) and reported their anti-AChE activity. Three pyrazoline (A10) analogues showed promising AChE inhibitory activity with IC50 values ranging from 14.37 ± 0.21 to 17.96 ± 0.31 nM. The structure–activity relationship of thiazolyl-pyrazoline showed the following order: 4-chlorophenyl > phenyl >4-trifluoromethylphenyl > 4-bromophenyl > 4-fluorophenyl > 4-cyanophenyl > 4-methylsulfonylphenyl > 4-(2-naphthyl) > 4-methoxyphenyl > 4-nitrophenyl > 4-methylphenyl.77 A molecular docking studies was conducted against the most potent compound (A10c) in the series against VX-phosphonylated hAChE (PDB ID: 6U37) to assess the binding affinity. With a docking score of −7.136 kcal/mol and interactions with the residues Tyr341 (π–π-stacking), compound A10c was found to display efficient binding within the active site of hAChE. The AChE inhibitory activity compound A10 and 2D molecular docking pose compound A10c are given in Figure 11.
Scheme 10. Synthesis of Thiazolyl-pyrazoline Analogues.

Figure 11.

AChE inhibitory activity and 2D molecular docking pose of compound A10c.
Altinlop et al. synthesized a new series of pyrazolines via the reaction of 1-(chloroacetyl)-3-(2-furyl)-5-aryl-2-pyrazolines with sodium salts of N,N-disubstituted dithiocarbamic acids (Scheme 11). 1-[[(4-(2-Dimethylaminoethyl)piperazin-1-yl)-thiocarbamoylthio]acetyl]-3-(2-furyl)-5-(3,4-methylenedioxyphenyl)-2-pyrazoline (A11) was found to be the most potent compound among the series showed AChE inhibitory activity with 96.22 ± 1.65% inhibition at 80 μg/mL and an IC50 value of 0.72 ± 0.06 μg/mL.78 A molecular docking studies was conducted against the most potent compound (A11) in the series against VX-phosphonylated hAChE (PDB ID: 6U37) to assess the binding affinity. With a docking score of −8.610 kcal/mol and interactions with the residues Tyr124 (H-bond) and Phe297 (π–π-stacking), compound A11 was found to display efficient binding within the active site of hAChE. The AChE inhibitory activity and 2D molecular docking pose compound A11 are given in Figure 12.
Scheme 11. Synthesis of Pyrazoline Analogues.

Figure 12.

AChE inhibitory activity and 2D molecular docking pose of compound A11.
Tugruk et al. synthesized [2-(3-(4-methoxyphenyl)-5-aryl-4,5-dihydro-1H-pyrazol-1-yl)benzo[d]thiazoles (Scheme 12) and reported their anti-AChE activity. 2-(3-(4-Methoxyphenyl)-5-phenyl-4,5-dihydro-1H-pyrazol-1-yl)benzo[d]thiazole (A12) exhibited promising anti-AChE activity with Ki value of 0.13 ± 0.004 μM and IC50 value of 1.3 μM.79 The unsubstituted phenyl at position 5 showed maximum anti-AChE activity. Among the halogenated substitution in the phenyl ring at 5 position, 4-fluoro substitution showed more anti-AChE activity than 4-chloro and 4-bromo, and additionally 3-bromo was found to be more effective than 2/4-bromo substitution.79 A molecular docking study was conducted against the most potent compound (A12) in the series against VX-phosphonylated hAChE (PDB ID: 6U37) to assess the binding affinity. With a docking score of −5.934 kcal/mol and interactions with the residues Trp286 (π–π-stacking), compound A12 was found to display an efficient binding within the active site of hAChE. The AChE inhibitory activity and 2D molecular docking pose compound A12 are given in Figure 13.
Scheme 12. Synthesis of [2-(3-(4-Methoxyphenyl)-5-aryl-4,5-dihydro-1H-pyrazol-1-yl)benzo[d]thiazoles.

Figure 13.

AChE inhibitory activity and 2D molecular docking pose of compound A12.
Jayaprakash et al. reported the anti-AChE activity of 3,5-diaryl-2-pyrazoline 1-carbothioamides. 5-(4-Chlorophenyl)-3-(2-hydroxyphenyl)-N-phenyl-4,5-dihydro-1H-pyrazole-1-carbothioamide (A13) exhibited promising anti-AChE activity with an IC50 value of 23.47 ± 1.17 nM.80 A molecular docking studies was conducted against the most potent compound (A13) in the series against VX-phosphonylated hAChE (PDB ID: 6U37) to assess the binding affinity. With a docking score of −6.547 kcal/mol and interactions with the residues Tyr341 (π–π-stacking), compound A13 was found to display an efficient binding within the active site of hAChE. The AChE inhibitory activity and 2D molecular docking pose compound A13 are given in Figure 14.
Figure 14.

AChE inhibitory activity and 2D molecular docking pose of compound A13.
Zondagh et al. reported a series of edaravone-N-benzyl pyridinium hybrid pyrazolinones (Scheme 13) and reported their anti-AChE. 1-[(2-Fluorophenyl)methyl]-4-({[4–(3-methyl-5-oxo-4Hpyrazol-1-yl)phenyl]foramido}methyl)pyridin-1-ium bromide (A14) displayed the most promising activity inhibiting AChE and an IC50 value of 1.2 μM. The selectivity index (selectivity against AChE over BuChE) for the compound A14 was found to be >833.81 A molecular docking study was conducted against the most potent compound (A14) in the series against VX-phosphonylated hAChE (PDB ID: 6U37) to assess the binding affinity. With a docking score of −7.626 kcal/mol and interactions with the residues Asp74 (H-bond), and Tyr124, Tyr341, and Trp286 (π–π-stacking), compound A14 was found to display an efficient binding within the active site of hAChE. The AChE inhibitory activity and 2D molecular docking pose compound A14 are given in Figure 15.
Scheme 13. Synthesis of Edaravone-N-benzyl Pyridinium Hybrid Pyrazolinone.

Figure 15.

AChE inhibitory activity and 2D molecular docking pose of compound A14.
Narayan et al. synthesized and reported the anti-AChE activity of coumarin-pyrazolinone (Scheme 14). (E)-2-(2-((3-Bromo-4-methyl-2-oxo-2H-chromen-7-yl)oxy)acetyl)-4-(2-(2-bromophenyl)hydrazono)-5-methyl-2,4-dihydro-3H-pyrazol-3-one (A15) displayed significant anti-AChE activity.82 A molecular docking study was conducted against the most potent compound (A15) in the series against VX-phosphonylated hAChE (PDB ID: 6U37) to assess the binding affinity. With a docking score of −6.813 kcal/mol and interactions with the residues Hip447 and Phe295 (H-bond), and Tyr124 and Tyr337 (π–π-stacking), compound A15 was found to display an efficient binding within the active site of hAChE. The AChE inhibitory activity and 2D molecular docking pose compound A15 are given in Figure 16.
Scheme 14. Synthesis of Coumarin-pyrazolinone.

Figure 16.

AChE inhibitory activity and 2D molecular docking pose of compound A15.
Temel et al. synthesized thiazolyl-pyrazoline derivatives via a ring closure reaction of 3-(2-furyl)-5-(1,3-benzodioxol-5-yl)-1-thiocarbamoyl-4,5-dihydro-1H-pyrazole with 2-bromo-1-arylethanones (Scheme 15). 2-[5-(1,3-Benzodioxol-5-yl)-3-(2-furyl)-4,5-dihydro-1H-pyrazol-1-yl]-4-(naphthalen-2-yl)thiazole (A16) was found to be the most effective AChE inhibitor (38.5 ± 2.85%), at 80 μg/mL.83 A molecular docking study was conducted against the most potent compound (A16) in the series against VX-phosphonylated hAChE (PDB ID: 6U37) to assess the binding affinity. With a docking score of −5.675 kcal/mol and interactions with the residues Tyr341 (π–π-stacking), compound A16 was found to display an efficient binding within the active site of hAChE. The AChE inhibitory activity and 2D molecular docking pose compound A16 are given in Figure 17.
Scheme 15. Synthesis of Thiazolyl-pyrazoline Derivatives.

Figure 17.

AChE inhibitory activity and 2D molecular docking pose of compound A16.
The most potent compounds A01–A16, in each series showed encouraging anti-AChE action at nanomolar (nM) or micromolar (μM) concentrations. A few compound (A08) also displayed anti-Aβ aggregation in a thioflavin-T assay as well as displayed a marked defense against scopolamine and Aβ1–42 induced memory impairment in mice. Some of the pyrazolines A02 and A04 displayed promising neuroprotective activity in terms of restoration of learning and memory functions. Furthermore, all of these compounds in molecular docking studies were found to display an efficient binding with the docking score of −5.675 to −9.011 kcal/mol, against the active site of VX-phosphonylated hAChE (PDB ID: 6U37) and exhibited three types of interactions: H-bond, π–π-stacking, and π-cation interactions. All of these compounds exhibited promising anti-AChE, and a few active against Aβ plaques, and some of the pyrazolines displayed neuroprotective activities, and they displayed an efficient binding against VX-phosphonylated hAChE. Aβ-amyloid (Aβ) plaques and low levels of the vital neurotransmitter acetylcholine (ACh) are thought to be associated with AD, suggesting that these substances may have a beneficial therapeutic efficacy in the treatment of AD.
3. Pyrazolines Act as MAO-B and COMT Inhibitors
The therapeutic benefit of pyrazoline as MAO inhibition was identified and explored by Parmar et al. in 1974.2 Monoamine oxidase B (MAO B) and catechol-O-methyl transferase (COMT) are two most important neurotransmitters that play an important role in Parkinson’s disease (PD).65−67 Singh and Bansal prepared 16,17-N′-(alkyl/arylsulfonyl)pyrazolinyl steroids (Scheme 3) and evaluated their neuroprotective activity. 1′-(4-Fluorobenzenesulfonyl)-5′-(pyridin-4-yl)-androst-5-en-[17,16-c]-1′H-pyrazoline-3β-ol (A02) (Figure 18) also displayed promising anti-Parkinsonian activity with ambulation 221.3 ± 2.3 (s), rearing 77 ± 3.4 (s), degree of catatonia 0.19 ± 0.04 (score).69 Singh et al. prepared 16,17-pyrazolinyl dehydroepiandrosterone analogues (Scheme 4) and evaluated their neuroprotective activity. 1′-(Acetyl)-5′-(pyridin-4-yl)-androst-5-en-[17,16-c]-1′H-pyrazoline-3β-yl acetate (A04) (Figure 18) displayed promising anti-Parkinsonian activity with locomotory activity of 211 ± 3.0 (ambulation (min)) and 70 ± 1.5 (Rearing (min)) as well as a catatonic response of 0.26 ± 0.11 (degree of Catatonia (score)) together with 3.5 ± 0.7 μM of H2O2 decomposed/min/mg protein in a catalase assay. MAO B inhibitors were found to be productive in the treatment of PD and AD because selegiline, an MAO B inhibitor, showed neuronal protection from oxidative stress in AD and PD patients.19
Figure 18.

Compounds A02 and A04 and their anti-Parkinsonism activity.
Gokhan et al. prepared a series of 12 1-N-substituted thiocarbamoyl-3-phenyl-5-thienyl-2-pyrazoline derivatives (Scheme 7),73 and Ucar et al. evaluated their anti-Alzheimer and MAO B inhibitory activities. The compound P01 showed MAO- B (IC50 = 180.0 ± 20.00 μM (0 min) and 91.5 ± 1.10 μM (60 min)) inhibitory activity.74 A molecular docking study was conducted against the most potent compound (P01) in the series against hMAO B (PDB ID: 2BYB) to access the binding affinity. With a docking score of −7.526 kcal/mol and interactions with the residues Tyr326 (π–π-stacking), compound P01 was found to display an efficient binding within the active site of hMAO B. The hMAO B inhibitory activity and 2D molecular docking pose compound P01 are given in Figure 19.
Figure 19.

hMAO B inhibitory activity and 2D molecular docking pose of compound P01.
On the basis of structure based virtual screening on an in-house library, top 10 HTS were synthesized (Scheme 5) and evaluated for their MAO inhibitory activity by Mishra and Sasmal. All of the compounds were found to have selective, reversible, and 100 times more potent inhibition than selegeline toward MAO B with Ki values in nM. The compounds A08 (Figure 4) and P02 showed MAO B inhibitory activity with Ki values of 0.31 and 1.70 nM against human enzyme while 0.45 and 0.60 nM against rat enzyme respectively.71 A molecular docking study was conducted against the most potent compounds (P01 and A08) in the series against hMAO B (PDB ID: 2BYB) to access the binding affinity. With a docking score of −9.665 (P02) and −9.507 (A08) kcal/mol and interactions with the residues Ile198 (H-bond) and Phe343, Tyr398, and Tyr435 (π–π-stacking), for the compound P02, while interactions with the residues Trp388 (π–π-stacking) and Lys296 (π-cation) for the compound P08, both compounds were found to display an efficient binding within the active site of hMAO B. The hMAO B inhibitory activity and 2D molecular docking pose compound P02 and A08 are given in Figure 20.
Figure 20.

hMAO-B inhibitory activity and 2D molecular docking pose of compounds A08 and P02.
Gokhan et al. prepared a series of 12 pyrazolines (Scheme 6) and evaluated their MAO inhibitory activity. Four compounds, P03, P04, P05, and P06 selectively and irreversibly inhibited MAO B in a classical noncompetitive manner with IC50 values in the range of 22.00–91.50 μM. The compound P04 showed promising MAO B inhibitory activity with an IC50 value of 22.00 ± 0.9 μM.73 A molecular docking studies was conducted against the most potent compounds (P03, P04, P05, and P06) in the series against hMAO B (PDB ID: 2BYB) to access the binding affinity. With a docking score of −8.359 (P03), −6.948 (P04), −8.284 (P05), and −7.620 (P06) kcal/mol, the compounds efficiently bind within the active site of hMAO B. The compound P04 was found to interact with the residues Gly57 (H-bond) via water molecules. The hMAO B inhibitory activity of the compounds P03, P04, P05, and P06 and 2D molecular docking pose compound P04 are given in Figure 21.
Figure 21.

hMAO-B inhibitory activity of the compounds P03, P04, P05, and P06 and 2D molecular docking pose of compounds P04.
Hitge et al. prepared a series of pyrazoline (Scheme 16) and reported the MAO and COMT inhibitory activities of nitro catechol derivatives of chalcone and pyrazoline. The compound P07 exhibited the most potent COMT inhibitory activity with an IC50 value of 0.048 μM and moderately inhibited MAO B with an IC50 value of 83.2 ± 14.5 μM. The compound P07 was found to be a more potent COMT inhibitor than entacapone (IC50 = 0.23 μM).84 A molecular docking study was conducted against the most potent compound (P07) in the series against hMAO-B (PDB ID: 2BYB) to access the binding affinity. With a docking score of −8.879 kcal/mol and interactions with the residues Gln206 (H-bond), Trp388 (π-cation), and Tyr398 (π–π-stacking), compound P07 was found to display an efficient binding within the active site of hMAO-B. The hMAO B inhibitory activity and 2D molecular docking pose compound P07 are given in Figure 22.
Scheme 16. Synthesis of Nitro Catechol Derivatives of Pyrazolines.

Figure 22.

hMAO-B inhibitory activity and 2D molecular docking pose of compounds P07.
Zaib et al. prepared 28 pyrazolines (Scheme 17) and reported their hMAO inhibitory activity. The compound P08 showed excellent inhibitory activity against MAO B having an IC50 value of 0.217 ± 0.02 μM.85 A molecular docking study was conducted against the most potent compound (P08) in the series against hMAO-B (PDB ID: 2BYB) to access the binding affinity. With a docking score of −9.113 kcal/mol and interactions with the Tyr398 (π–π-stacking), compound P08 was found to display an efficient binding within the active site of hMAO B. The hMAO B inhibitory activity and 2D molecular docking pose compound P08 are given in Figure 23.
Scheme 17. Synthesis of Pyrazolines Containing Quinoline Heterocycle.

Figure 23.

hMAO-B inhibitory activity and 2D molecular docking pose of compounds P08.
Aksoz et al. prepared 13 pyrazolines (Scheme 18) and reported their MAO inhibitory activity. The compound P09 appeared to be the most selective MAO-B inhibitor with a Ki value of 29.66 ± 1.90 μM.86 A molecular docking studies was conducted against the most potent compound (P09) in the series against hMAO-B (PDB ID: 2BYB) to access the binding affinity. With a docking score of −8.883 kcal/mol and interactions with the residue Tyr435 (H-bond), compound P09 was found to display an efficient binding within the active site of hMAO B. The hMAO B inhibitory activity and 2D molecular docking pose compound P09 are given in Figure 24.
Scheme 18. Synthesis of Pyrazoline Derivatives.

Figure 24.

hMAO B inhibitory activity and 2D molecular docking pose of compounds P09.
Sahoo et al. prepared a series of 10 pyrazoline analogues (Scheme 19) and reported the MAO inhibitory activity of 10 novel pyrazolines. Most of the compounds were found to be a selective and reversible inhibitor of MAO B. The compound P10 showed competitive and reversible inhibition of MAO B with a Ki value of 0.60 μM.87 A molecular docking studies was conducted against the most potent compound (P10) in the series against hMAO B (PDB ID: 2BYB) to access the binding affinity. With a docking score of −8.604 kcal/mol and interactions with the residue Ser59 and Tyr60 (H-bond), and Tyr326, 398, and 435 (π–π-stacking) compound P10 was found to display an efficient binding within the active site of hMAO B. The hMAO B inhibitory activity and 2D molecular docking pose compound P10 are given in Figure 25.
Scheme 19. Synthesis of Pyrazoline Analogues.

Figure 25.

hMAO-B inhibitory activity and 2D molecular docking pose of compounds P10.
Kelekci et al. prepared 12 pyrazolines (Scheme 20) and evaluated their MAO inhibitory activity. 2-[(3-(4-Methoxyphenyl)-50-furyl-20-pyrazoline-1-yl]-3-methyl-4(3H)-quinazolinone (P12) showed potent selective inhibition of MAO B with Ki and IC50 values of 1.48 ± 0.66 and 2.03 ± 0.27 μM respectively, and was found to be more active than that of the standard drug Pargyline (Ki = 3.36 ± 0.27; IC50 = 3.85 ± 0.23). The compound P11 displayed selective competitive inhibition of MAO B of rat liver MAO isoforms with a Ki value of 0.98 μM.88 A molecular docking studies was conducted against the most potent compound (P11) in the series against hMAO B (PDB ID: 2BYB) to access the binding affinity. With a docking score of −7.757 kcal/mol and interactions with the residue Gly57 (H-bond) via water molecule, and Trp388 and Tyr398 (π–π-stacking), compound P11 was found to display an efficient binding within the active site of hMAO B. The hMAO B inhibitory activity and 2D molecular docking pose compound P11 are given in Figure 26.
Scheme 20. Synthesis of Pyrazoline Bearing 4-(3H)-Quinazolinone Analogues.

Figure 26.

hMAO-B inhibitory activity and 2D molecular docking pose of compounds P11.
Jayaprakash et al. prepared a library of 32 pyrazolines (Scheme 21) and evaluated their rat liver MAO inhibitory activity. The compound P12 (Scheme 21) showed MAO B inhibitory activity with IC50 values of 45.13 ± 3.20 μM (preincubation at 0 min) and 19.45 ± 1.02 μM (preincubation at 60 min). The compound P12 was also found to be a selective inhibitor of rat liver MAO-B. A molecular docking investigation was carried out against hMAO-B (PDB ID: 2BYB) for all of the compounds. The most potent compound (P12) displayed an efficient binding with a docking score of −8.69 kcal/mol and interactions with the residues Asn181 and Ser209 (H-bond).89
Scheme 21. Synthesis of Pyrazoline Analogues.

The most potent compounds P01–P12, in each series showed encouraging MAO B inhibitory action at nanomolar (nM) or micromolar (μM) concentrations. Some of the compounds (P07) showed COMT inhibitory action in addition to MAO B inhibitory activity, while a couple compounds (A02 and A04) showed neuroprotective potential in the catalase experiment. Furthermore, all of these compounds in molecular docking studies were found to display an efficient binding with a docking score of −6.948 to −9.665 kcal/mol, against the active site of hMAO B (PDB ID: 2BYB) and exhibited three types of interactions: H-bond, π–π-stacking, and π-cation interactions. All of these compounds exhibited promising MAO B inhibitory activity, a few active against COMT, while some of the pyrazolines displayed neuroprotective activities, and they also displayed efficient binding against hMAO B. MAO B and COMT are important neurotransmitters that play an important role in PD, and their inhibition could be a potential target to treat PD, suggesting that these substances may have beneficial therapeutic efficacy in the treatment of PD.
4. Pyrazolines as MAO A Inhibitor
Inhibition of MAO A inhibitors is useful in psychiatric disorders.65−67 Pyrazolines are important heterocyclics having a substantial benefit in the treatment of psychiatric disorders by inhibiting MAO A. Aksoz et al. prepared 13 pyrazolines (Scheme 18) and reported their MAO inhibitory activity. The compound D01 appeared to be the most selective MAO A inhibitor with a Ki value of 0.13 ± 0.007 μM.86 A molecular docking study was conducted against the most potent compound (D01) in the series against hMAO A (PDB ID: 2BXR) to assess the binding affinity. With a docking score of −7.649 kcal/mol and interactions with the residues Tyr344 and Ser209 (H-bond) and Tyr69 (π–π-stacking), compound D01 was found to display an efficient binding within the active site of hMAOA. The hMAOA inhibitory activity and 2D molecular docking pose compound D01 are given in Figure 27.
Figure 27.

HMAOA inhibitory activity and 2D molecular docking pose compound D01.
Hitge et al. prepared (Scheme 16) and reported the MAO A inhibitory activity of nitro catechol derivatives of chalcone and pyrazoline. The compound D02 moderately inhibited MAO A with an IC50 value of 70.6 ± 7.47 μM.84 A molecular docking study was conducted against the most potent compound (D02) in the series against hMAO A (PDB ID: 2BXR) to assess the binding affinity. With a docking score of −9.405 kcal/mol and interactions with the residues Tyr444, Tyr69, and Met445 (H-bond) and Tyr444 (π–π-stacking), compound D02 was found to display an efficient binding within the active site of hMAOA. The hMAOA inhibitory activity and 2D molecular docking pose compound D02 are given in Figure 28.
Figure 28.

HMAOA inhibitory activity and 2D molecular docking pose compound D02.
Zitouni et al. prepared five pyrazolines (Scheme 9) and reported their MAO A inhibitory activity. 1-[5-(Benzo[d][1,3]dioxol-5-yl)-3-(thien-2-yl)-4,5-dihydro-1H-pyrazol-1-yl]-2-[(((4-(diethylamino)butyl)amino)methyl)thio]ethan-1-one (D03) showed MAO-B inhibitory activity with an IC50 value of 20.34 μM.76 A molecular docking studies was conducted against the most potent compound (D03) in the series against hMAO A (PDB ID: 2BXR) to assess the binding affinity. With a docking score of −10.536 kcal/mol and interactions with the residues Ser209 (H-bond), Glu216 (salt-bridge), Tyr407 (π–π-stacking), and Phe209 (π-cation), compound D03 was found to display an efficient binding within the active site of hMAOA. The compound D03 accommodated well within the active site of MAO A and demonstrated four types of interactions. The hMAOA inhibitory activity and 2D molecular docking pose compound D03 are given in Figure 29.
Figure 29.

HMAOA inhibitory activity and 2D molecular docking pose compound D03.
Sahoo et al. prepared (Scheme 19) and reported the MAO inhibitory activity of 10 novel pyrazolines. Most of the compounds are a selective and reversible inhibitor of MAO A. The compounds D04 showed MAO A inhibition with a Ki value of 0.36 μM.87 A molecular docking study was conducted against the most potent compound (D04) in the series against hMAO A (PDB ID: 2BXR) to assess the binding affinity. With a docking score of −9.768 kcal/mol and interactions with the residues Tyr444 and Tyr69 (H-bond) and Phe208 and Tyr407 (π–π-stacking), compound D04 was found to display an efficient binding within the active site of hMAOA. The hMAOA inhibitory activity and 2D molecular docking pose compound D04 are given in Figure 30.
Figure 30.
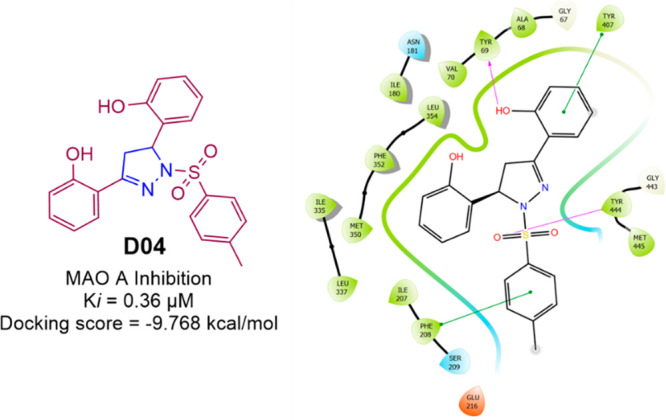
HMAOA inhibitory activity and 2D molecular docking pose compound D04.
Kelekci et al. prepared 12 pyrazolines (Scheme 20) and evaluated their MAO inhibitory activity. The compound D05 showed potent selective inhibition of MAO A with Ki and IC50 values of 0.90 ± 0.07 and 1.02 ± 0.09 μM, more active than that of the standard drug Clorgyline (Ki = 1.20 ± 0.09; IC50 = 2.05 ± 0.19) and Moclobemide (Ki = 5.53 ± 0.27; IC50 = 3.90 ± 0.19).88 A molecular docking studies was conducted against the most potent compound (D05) in the series against hMAO A (PDB ID: 2BXR) to assess the binding affinity. With a docking score of −7.130 kcal/mol and interactions with the residues Tyr444 and Tyr407 (π–π-stacking), compound D05 was found to display an efficient binding within the active site of hMAOA. The hMAOA inhibitory activity and 2D molecular docking pose compound D05 are given in Figure 31.
Figure 31.
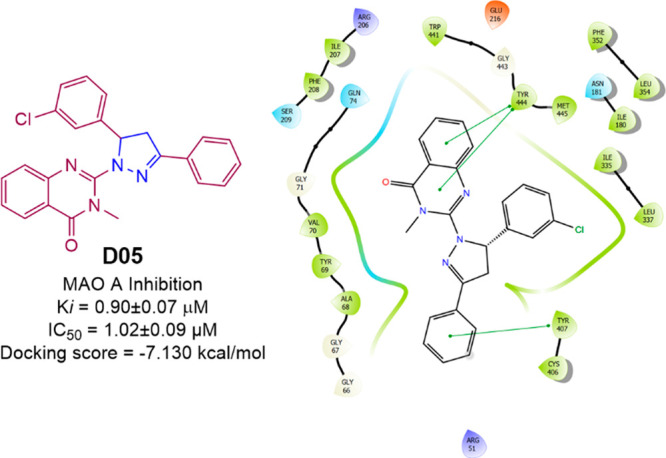
hMAO A inhibitory activity and 2D molecular docking pose compound D05.
Jayaprakash et al. prepared a library of 32 pyrazolines (Scheme 21) and evaluated their rat liver MAO inhibitory activity. The compound D06 showed MAO A inhibitory activity with IC50 values of 39.20 ± 2.30 μM (preincubation at 0 min) and 2.84 ± 0.19 μM preincubation at 60 min).89 A molecular docking study was conducted against the most potent compound (D06) in the series against hMAO A (PDB ID: 2BXR) to assess the binding affinity. With a docking score of −5.890 kcal/mol and interactions with the residues Ser209 (H-bond), compound D06 was found to display an efficient binding within the active site of hMAOA. The hMAOA inhibitory activity and 2D molecular docking pose compound D06 are given in Figure 32.
Figure 32.

hMAO A inhibitory activity and 2D molecular docking pose compound D06.
Zaib et al. prepared 28 pyrazolines (Scheme 17) and reported their hMAO inhibitory activity. The compound D07 exhibited most potent MAO A inhibition with an IC50 value of 0.079 ± 0.003 μM.85 A molecular docking studies was conducted against the most potent compound (D07) in the series against hMAO A (PDB ID: 2BXR) to assess the binding affinity. With a docking score of −8.949 kcal/mol and interactions with the residues Ile207 (H-bond), and Tyr444 (π–π-stacking), compound D07 was found to display an efficient binding within the active site of hMAOA. The hMAOA inhibitory activity and 2D molecular docking pose compound D07 are given in Figure 33.
Figure 33.

HMAOA inhibitory activity and 2D molecular docking pose compound D07.
Goksen et al. prepared and resolved pyrazoline to R and S forms of pyrazolines (Scheme 22) and evaluated their MAO A inhibitory activity. 1-[2-(2-Benzoxazolinone-3-yl)acetyl]-3-phenyl-5-(3,4-dimethoxyphenyl)-4,5-dihydro-(1H)-pyrazole (D08) showed a promising MAO A inhibitory effect. The racemic form exhibited hMAO A inhibitory activity with a Ki value of 0.00091 ± 0.0001 μM. The R (Ki 0.00085 ± 0.00005 μM) form was found to be more potent than the S (Ki = 0.184 ± 0.007 μM) form.90 A molecular docking study was conducted against the most potent compound (D08) in the series against hMAO A (PDB ID: 2BXR) to assess the binding affinity. With a docking score of −7.825 kcal/mol and interactions with the residue Tyr444 (π–π-stacking), compound D08 was found to display an efficient binding within the active site of hMAOA. The hMAOA inhibitory activity and 2D molecular docking pose compound D08 are given in Figure 34.
Scheme 22. Synthesis of 1-[2-(2-Benzoxazolinone-3-yl)acetyl]-3-phenyl-5-(3,4-dimethoxyphenyl)-4,5-dihydro-1H-pyrazole.
Figure 34.

HMAOA inhibitory activity and 2D molecular docking pose compound D08.
Nath et al. prepared a series of pyrazolines based on the structure of curcumin (Scheme 23) and evaluated their MAO A inhibitory activity. The compound D09 exhibited competitive reversible hMAO A inhibitory activity with an inhibition constant (Ki) value of 0.08 μM and a strong hMAO A selectivity (Ki(hMAO-B)/Ki(hMAOA) > 1751). The compound D09 was also found to be nontoxic and displayed ∼90% viability for HepG2 cell lines at 25 μM concentration.91 A molecular docking study was conducted against the most potent compound (D09) in the series against hMAO A (PDB ID: 2BXR) to assess the binding affinity. With a docking score of −7.750 kcal/mol and interactions with the residues Tyr69 and Asn181 (H-bond), compound D09 was found to display an efficient binding within the active site of hMAO A. The hMAO A inhibitory activity and 2D molecular docking pose compound D09 are given in Figure 35.
Scheme 23. Synthesis of Curcumin-Based Pyrazoline Analogues.

Figure 35.

hMAO A inhibitory activity and 2D molecular docking pose compound D09.
Aksoz et al. prepared another series of 12 pyrazolines (Scheme 24) and reported their hMAO inhibitory activity. The compounds D10 and D11 showed potent competitive and reversible hMAO A inhibition more than the standard drug moclobemide with Ki values of 0.004 ± 0.001 and 0.005 ± 0.001 μM respectively. The SI values of the compounds D10 and D11 were found to be 5.55 × 10–5 and 0.003 respectively. These two compounds were also found to be nontoxic in cytotoxicity screening.92 A molecular docking study was conducted against the most potent compounds (D10 and D11) in the series against hMAO A (PDB ID: 2BXR) to assess the binding affinities. With a docking scores of −7.787 (D10) and −7.459 (D11) kcal/mol and interactions with the residues Ser209 and Tyr 444 (H-bond) and Tyr69 and Tyr407 (π–π-stacking), for the compound D10, while with the residues Tyr444 and Phe352 (π–π-stacking), for the compound D11 and efficiently bind within the active site of hMAOA was observed. The docking score and hMAO A inhibition of compound D10 was found to be higher than that of the compound D11. The hMAOA inhibitory activity and 2D molecular docking pose compound D10 and D11 are given in Figure 36.
Scheme 24. Synthesis of Curcumin-Based Pyrazoline Analogues.

Figure 36.

hMAO A inhibitory activity and 2D molecular docking pose compounds D10 and D11.
Tong et al. prepared 2-pyrazoline-1-ethanones (Scheme 25) and reported their hMAO inhibitory activity. 1-(5-(5-Chloro-2-hydroxy-phenyl)-3-(4-fluoro-phenyl)-4,5-dihydro-pyrazol-1-yl)ethanone-2-piperazine (D12) (IC50 = 2.40 ± 0.14 μM) and 1-(5-(3,5-dibromo-2-hydroxyphenyl)-3-methyl-4,5-dihydropyrazol-1-yl)ethanone-2-piperazidine (D13) (IC50 = 2.00 ± 0.11 μM) exhibited the best inhibitory activity and selectivity against hMAO A, surpassing that of the positive control Clorgyline (IC50 = 2.76 ± 0.10 μM). The structure–activity relationship (SAR) analysis showed that the order of N1 substituent contribution was bromo > piperidinyl > morpholinyl > imidazolyl, and compounds with electron-withdrawing substituents (−F, −Cl) at C3 or C5 phenyl ring of 2-pyrazoline nucleus dedicated stronger MAO A inhibitory activity as shown in Figure 7.93 A molecular docking study was conducted against the most potent compounds (D12 and D13) in the series against hMAO A (PDB ID: 2BXR) to assess the binding affinity. With a docking scores of −8.915 (D12) and −7.241 (D13) kcal/mol and interactions with the residues Asn181 (H-bond), Tyr407 (π–π-stacking), and Glu216 (salt-bridge) for the compound D12, while with the residues Tyr444 (H-bond), Tyr407 (π–π-stacking), Glu216 (salt-bridge), and Gly443, Tyr69, and Ala68 (Halogen bond) for the compound D13. An efficient binding within the active site of hMAOA was observed for both the compounds. The docking score of compound D12 was found to be higher than that of the compound D13; however, the hMAO A inhibitory activity of compound D13 was found slightly higher than D12. The hMAO A inhibitory activity and 2D molecular docking pose compound D12 and D13 are given in Figure 37.
Scheme 25. Synthesis of 2-Pyrazoline-1-ethanone Derivatives.

Figure 37.

hMAO A inhibitory activity and 2D molecular docking pose compounds D12 and D13.
Badavath et al. prepared 12 pyrazolines and reported their hMAO inhibitory activity. The compound D14 showed MAO A inhibitory activity with a Ki value of 0.151 μM.94 A molecular docking study was conducted against the most potent compound (D14) in the series against hMAO A (PDB ID: 2BXR) to assess the binding affinity. With a docking score of −5.914 kcal/mol and interactions with the residues Tyr407 (H-bond) and Tyr444 (π–π-stacking), compound D14 was found to display an efficient binding within the active site of hMAOA. The hMAOA inhibitory activity and 2D molecular docking pose compound D14 are given in Figure 38.
Figure 38.

HMAOA inhibitory activity and 2D molecular docking pose compound D14.
Aksoz et al. prepared another series of 10 diaryl-2-pyrazolines (Scheme 26) and reported their MAO inhibitory activity with Ki values ranging between 0.005 ± 0.001 and 14.445 ± 1.087 μM. (E)-1-(3,5-Dichloro-2-hydroxyphenyl)-3-p-tolylprop-2-en-1-one (D15) showed hMAO A selective competitive reversible inhibitory activity with a Ki value of 0.005 ± 0.001 μM. (3-(3,5-Dichloro-2-hydroxyphenyl)-5-p-tolyl-4,5-dihydropyrazole-1-yl)(4-methoxyphenyl)methanone (D16) showed inhibited hMAO A inhibitory activity with a Ki value of 0.123 ± 0.012 μM and was found to be highly selectivity than the standard drug moclobemide against hMAO A.95 A molecular docking study was conducted against the most potent compounds (D15 and D16) in the series against hMAO A (PDB ID: 2BXR) to assess the binding affinity. With a docking score of −8.074 (D15) and −7.913 (D16) kcal/mol and interactions with the residues Tyr444 (H-bond), Tyr444 (π–π-stacking), and Met445 (halogen bond), compound D15 was found to display an efficient binding within the active site of hMAOA. The docking score as well as hMAO A inhibitory activity of compound D15 was found to be higher than that of the compound D16. hMAO A inhibitory activity and 2D molecular docking pose compound D14 are given in Figure 39.
Scheme 26. Synthesis of Pyrazoline Derivatives.

Figure 39.

hMAO A inhibitory activity and 2D molecular docking pose compounds D15 and D16.
Goksen et al. prepared a series of pyrazolines (Scheme 27) and reported their MAO inhibitory activity. The compounds D17, D18, and D19 were found to be more selective MAO A inhibitors with Ki values of 0.003 ± 0.00001, 0.010 ± 0.001, and 0.050 ± 0.002 μM.96 A molecular docking study was conducted against the most potent compounds (D17–D18) in the series against hMAO A (PDB ID: 2BXR) to assess the binding affinity. The docking scores of compounds D17, D18, and D19 were found to be −8.344, −7.519, and −7.242 kcal/mol respectively. The docking score and hMAO A inhibitory activity of compound D17 were found to be higher than that of the compounds D18 and D19. The D17 interacted with residues Ser209 (H-bond), and Tyr407 and Phe352 (π–π-stacking). hMAO A inhibitory activity and 2D molecular docking pose compounds D17–D19 are given in Figure 40.
Scheme 27. Synthesis of Benoxazolin-3-one Pyrazoline Derivatives.

Figure 40.

hMAO A inhibitory activity and 2D molecular docking pose compounds D17–D19.
Nayak et al. prepared a series of pyrazolines (Scheme 28) and reported their selective MAO inhibitory activity. Phenyl-3-(2-hydroxyphenyl)-5-(4-methylphenyl)-4,5-dihydro-1H-pyrazole-1-carboxylate (D20) (MAO A; Ki = 4.96 ± 0.21 nM) was found to be as active as Moclobemide (KiMAO-A; 5.01 ± 0.13 nM) but with the highest selectivity index (8.86 × 10–5). The molecular docking studies with R & S conformer of N1 revealed that S-enantiomer is better than R-enantiomer. It was further proposed that VdW’s radii of the substitution (bulkiness) in ring B determine the potency of phenyl carbamates.97 A molecular docking study was conducted against the most potent compound (D20) in the series against hMAO A (PDB ID: 2BXR) to assess the binding affinity. The docking scores of compounds D20 were found to be −7.278 kcal/mol and interacted with the residues Tyr69 (H-bond), and Phe352, Tyr407, and Try444 (π–π-stacking). hMAO A inhibitory activity and 2D molecular docking pose compound D20 are given in Figure 41.
Scheme 28. Synthesis of Benoxazolin-3-one Pyrazoline Derivatives.

Figure 41.

hMAO A inhibitory activity and 2D molecular docking pose compound D20.
Jagrat et al. prepared 22 pyrazolines (Scheme 29) and reported their MAO inhibitory activity. All of the pyrazolines were reversible and competitive inhibitors of MAO A. The compounds, D21 and D22 showed potent MAO A inhibitory activity with a Ki value of 90.45 ± 4.73 nM and 0.15 ± 0.01 μM. The compounds, D21 and D22 were found to be selective toward MAO A with a selectivity index (SI) of 1.02 × 10–4 and 2.26 × 10–4 respectively. SAR within this series reveals that (i) 2-hydroxy substitution in ring B is better than 4-hydroxy, (ii) methoxy substitution in ring C is better than methyl substitution, and (iii) meta substitution was found to be favorable when the above two factors exist. Ortho hydroxy substitution in ring B increases potency and selectivity index toward MAO A when there is methoxy substitution in the meta position of ring C. Whereas para hydroxy substitution in ring B increases potency and selectivity toward MAO A, with both the methoxy (favorable at the para > meta position) and methyl (favorable at the meta > para position) group at either para or meta positions as shown in Figure 42.98 A molecular docking study was conducted against the most potent compounds (D21 and D22) in the series against hMAO A (PDB ID: 2BXR) to assess the binding affinity. The docking scores of compounds D21 and D22 were found to be −8.472 and −7.979 kcal/mol and interacted with the residues Ile207 (H-bond), and Tyr407 and Try444 (π–π-stacking) for compound D21 interacted with the residues Tyr69 and Met445 (H-bond), and Tyr407 (π–π-stacking) for compound D22. hMAO A inhibitory activity and 2D molecular docking pose compound D20 are given in Figure 43.
Scheme 29. Synthesis of Benoxazolin-3-one Pyrazoline Derivatives.

Figure 42.

SAR of pyrazoline for MAO A inhibitory activity.
Figure 43.

hMAO A inhibitory activity and 2D molecular docking pose compounds D21 and D22.
Karuppasamy et al. prepared (Scheme 30) and reported the MAO inhibitory activity of 3,5-diaryl pyrazolines analogs. All of the compounds exhibited reversible and selective inhibition of MAO A with a selectivity index with a magnitude of 103–105. The compound D23 showed potent competitive and reversible MAO A inhibitory activity with a Ki value of 150.10 ± 10.05 nM.99 A molecular docking study was conducted against the most potent compounds (D23) in the series against hMAO A (PDB ID: 2BXR) to assess the binding affinity. The molecular docking score of the compound D23 was found to be −7.278 kcal/mol and interacted with the residues Tyr69 (H-bond), and Phe352, Tyr407, and Try444 (π–π-stacking). hMAO A inhibitory activity and 2D molecular docking pose compound D23 are shown in Figure 44.
Scheme 30. Synthesis of 3,5-Diaryl Pyrazolines Analogues.

Figure 44.

hMAO A inhibitory activity and 2D molecular docking pose compound D23.
Chimenti et al. prepared a library of 42 pyrazolines (Scheme 31) and evaluated their MAO inhibitory activity. The most active compounds show inhibitory activity toward MAO A in the 1.0 × 10–8 to 8.6 × 10–9 M range. The compound D24 selectively inhibits MAO A with an IC50 value of 8.6 ± 0.01 nM. A molecular docking study was conducted against the most potent compounds (D24) in the series against hMAO A (PDB ID: 2BXR) to assess the binding affinity. The molecular modeling study showed the compound D24 strongly and selectively inhibited the MAO A isoform.100 The molecular docking score of the compound D24 was found to be −9.388 kcal/mol and interacted with the residues Tyr69 and Tyr444 (H-bond), and Tyr407 and Try444 (π–π-stacking). hMAO A inhibitory activity and 2D molecular docking pose compound D24 are shown in Figure 45.
Scheme 31. Synthesis of 4,5-Dihydro-(1H)-pyrazolines.

Figure 45.

hMAO A inhibitory activity and 2D molecular docking pose compound D24.
Badavath et al. designed and prepared a series of 13 new pyrazoline (Scheme 32) based on the structure of curcumin. The yield of chalcone prepared by 4-hydroxy-3-methoxy acetophenone by Claisen–Schmidt was found to be 23%, and hence the scheme was modified by protecting the hydroxyl group of acetophenone with 3,4-dihydro-α-pyran, and the yield was increased to 90% as shown in Scheme 31. 2-Methoxy-4-(5-phenyl-1-tosyl-4,5-dihydro-1H-pyrazol-3-yl)-phenol (D25) was found to be a potent inhibitor (competitive and reversible) of hMAO A with a Ki = 0.06 ± 0.003 μM and had a selectivity index of (SI = 1.02 × 10–5). It was found to be better than the standard drug Moclobemide (hMAO A with a Ki = 0.11 ± 0.01 μM) with a selectivity index SI = 0.049.101 A molecular docking study was conducted against the most potent compounds (D25) in the series against hMAO A (PDB ID: 2BXR) to assess the binding affinity. The molecular docking score of the compound D25 was found to be −8.086 kcal/mol and interacted with the residues Tyr444 (H-bond), and Phe208, Tyr407, and Try444 (π–π-stacking). hMAO A inhibitory activity and 2D molecular docking pose compound D25 are shown in Figure 46.
Scheme 32. Synthesis of Curcumin-Based Pyrazoline Analogues.

Figure 46.

hMAO A inhibitory activity and 2D molecular docking pose compound D25.
Most of the potent compounds in each series, D01–D25, showed encouraging MAO A inhibitory action at nanomolar (nM) or micromolar (μM) concentrations. All of these compounds in molecular docking studies were found to display an efficient binding against the active site of hMAO A (PDB ID: 2BXR) with a docking score of −5.890 to −10.536 kcal/mol, and exhibited three types of interactions: H-bond, π–π-stacking, and π-cation interactions. All of these compounds exhibited promising MAO A inhibition, and they also displayed an efficient binding against hMAO-A. MAO A is an important neurotransmitter that play an important role in psychiatric disorders, and its inhibition could be a potential target to treat depression, suggesting that these substances may have beneficial therapeutic efficacy in the treatment of psychiatric disorders (depression).
After carefully analyzing the MAO A and MAO B activity, we have concluded the following important points about the substituents’ effect on bioactivity as shown in Figure 47.
Figure 47.

Effect of substituents for selectivity of MAO A and MAO B activities.
Ring A
-
1.
For MAO A activity, ring A should contain phenyl, 2-hydroxyphenyl, 4-methoxyphenyl, 4-hydroxy-3-methoxyphenyl, 3,5-dichloro-2-hydroxyphenyl, 2-hydroxy-3-methoxyphenyl, 5-bromo-2-hydroxyphenyl, and 2-naphthyl for promising activity.
-
2.
For MAO B activity, ring A should contain 2-hydroxyphenyl, 4-methoxyphenyl, 3,5-dichloro-2-hydroxyphenyl, 5-bromo-2-hydroxyphenyl, and anthracen-9-yl for promising activity.
Ring B
-
3.
For MAO A, activity ring B should contain phenyl, 2-hydroxyphenyl, 3,4-dimethoxyphenyl, 4-methoxyphenyl, 2-methoxyphenyl, 2-hydroxyphenyl, 4-chlorophenyl, 4-methylphenyl, 3-chlorophenyl, 3-methylphenyl, and 4-pyridyl for promising activity.
-
4.
For MAO B activity, ring B should contain 4-hydroxyphenyl, 4-methoxyphenyl, 2-furyl, 2-thiophenyl, and 2-chloro-quinolin-3-yl for promising activity.
Tail
-
5.
For MAO A activity, ring B should contain hydrogen (H), 2-(2-benzoxazolinone-3-yl)acetyl, 3-chloro-quinolin-2-yl, 4-methoxybenzoyl, 2-furanacetyl, benzoyl, benzene sulphonyl, 4-methylbenzene sulphonyl, and aminoacetyl, for promising activity.
-
6.
For MAO B activity, ring B should contain hydrogen (H), 3-methyl-4(3H)-quinazolinon-2-yl, and 4-methoxybenzoyl, for promising activity.
5. Conclusion
Pyrazolines are important heterocyclic nucleus with broad spectrum biological activities. They are very stable and serve as an important anchor for drug design and exploration by substitution at two or more positions in the pyrazoline nucleus. We described herein the bioactivity of pyrazolines as an inhibitor of AChE, Aβ plaque aggregation MAO A/B, and COMT. Alzheimer’s disease (AD) is a complex and multifactorial neurodegenerative disorder as the disease’s pathogenesis is still poorly understood, and current AD medications only mildly alleviate symptoms. Aβ-amyloid (Aβ) plaques and low levels of the vital neurotransmitter acetylcholine (ACh) are thought to be associated with AD, suggesting that pyrazoline activity against AChE and Aβ may have beneficial therapeutic efficacy in the treatment of AD. Some of pyrazolines were reported as MAO B inhibitors, and a few were reported as COMT inhibitors. MAO B and COMT are important neurotransmitters that play an important role in Parkinsonism’s disease (PD), and their inhibition could be a potential target to treat PD. Similarly, pyrazolines were also reported as MAO A inhibitors. MAO A is an important neurotransmitter that plays an important role in psychiatric disorders, and its inhibition could be a potential target to treat psychiatric disorders like depression. The molecular docking studies also supported that pyrazolines showed an efficient binding against VX-phosphonylated hAChE (PDB ID: 6U37), human monoamine oxidase B (MAO B; PDB ID: 2BYB), and human monoamine oxidase A (MAO A; PDB ID: 2BXR). Therefore, it can be concluded that pyrazolines may act as a potential lead structure for the fabrication of new molecules targeting neurodegenerative disorders and may escalate neurological drug discovery programs in the near future.
6. Materials and Methods
6.1. Review of Literature
The literature related to pyrazoline as anti-AChE and MAO A/B inhibitors were searched from a database, and a total of 80 articles were found.102 Out of 80 only 61 articles were found to be related to pyrazolines and their anti-AChE and MAO A/B inhibitory activities. The value of the inhibition constant, or Ki, describes the binding affinity between an enzyme and its inhibitor. The binding affinity increases with decreasing Ki values, and less drug is required to block the activity of that enzyme. The IC50 is the molar concentration of drug required to reduce the enzyme’s activity to 50%.
6.2. Molecular Docking Study against AChE
The X-ray crystal structure of AChE with PDB ID: 6U37 (structure of VX-phosphonylated hAChE in complex with oxime reactivator RS194B) was retrieved from a protein database.103 An X-ray crystal structure of AChE (PDB: 6U37), with a resolution of 2.25° A; R-value: 0.208 (obs.), was obtained from the Protein Data Bank (Research Collaboratory for Structural Bioinformatics (RCSB) (https://www.rcsb.org/structure/6u37).103 The molecular docking was carried out as per the docking protocol given.77,104
6.3. Molecular Docking Study against MAO B
The X-ray crystal structure of MAO B with PDB ID: 2BYB (human monoamine oxidase B in complex with Deprenyl) was retrieved from a protein database.105 An X-ray crystal structure of MAO B (PDB: 2BYB), with a resolution of 2.20° A; R-value: 0.239 (obs.), was obtained from the Protein Data Bank (RCSB (https://www.rcsb.org/structure/2BYB)).105 The molecular docking was carried out as per the docking protocol given.101,106
6.4. Molecular Docking Study against MAO A
An X-ray crystal structure of MAO A with PDB ID: 2BXR (human monoamine oxidase A in complex with Clorgyline) was retrieved from a protein database.107 An X-ray crystal structure MAO A (PDB: 2BXR), with a resolution of 3.00 Å; R-value: 0.195 (obs.), was obtained from the Protein Data Bank (RCSB (https://www.rcsb.org/structure/2bxr)).107 The molecular docking was carried out as per the docking protocol given.101,106
Acknowledgments
The author would like to express his gratitude to Maharishi Arvind College of Pharmacy, Jaipur, India.
Glossary
List of Abbreviations
- AChE
acetylcholine esterase
- AD
Alzheimer’s disease
- Aβ
β-amyloid
- CB
cannabinoid
- COMT
catechol-O-methyltransferase
- IC50
inhibition concentration required to reduce the rate of an enzymatic reaction by 50%
- ITL
initial transfer latency
- Ki
inhibition constant
- MAO
monoamine oxidase
- μM
micromolar
- nM
nanomolar
- PD
Parkinson’s disease
- SAR
structure–activity relationship
- TSTQ
time spent in target quadrant
- VEGFR
vascular endothelial growth factor receptor
The authors declare no competing financial interest.
References
- Matiadis D.; Sagnou M. Pyrazoline Hybrids as Promising Anticancer Agents: An Up-to-Date Overview. Int. J. Mol. Sci. 2020, 21, 5507. 10.3390/ijms21155507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmar S. S.; Pandey B. R.; Dwivedi C.; Harbison R. D. Anticonvulsant activity and monoamine oxidase inhibitory properties of 1,3,5-trisubstituted pyrazolines. J. Pharm. Sci. 1974, 63, 1152–1155. 10.1002/jps.2600630730. [DOI] [PubMed] [Google Scholar]
- Manna K.; Banik U.; Ghosh P.; Das M. A review on synthesis and pharmacological diversity of isoxazoles and pyrazolines. Pharmaceut. Sci. 2014, 1, 37–49. [Google Scholar]
- Marella A.; Ali M. R.; Alam M. T.; Saha R.; Tanwar O.; Akhter M.; Shaquiquzzaman M.; Alam M. M. Pyrazolines: a biological review. Mini. Rev. Med. Chem. 2013, 13, 921–931. 10.2174/1389557511313060012. [DOI] [PubMed] [Google Scholar]
- Nehra B.; Rulhania S.; Jaswal S.; Kumar B.; Singh G.; Monga V. Recent advancements in the development of bioactive pyrazoline derivatives. Eur. J. Med. Chem. 2020, 205, 112666. 10.1016/j.ejmech.2020.112666. [DOI] [PubMed] [Google Scholar]
- Tok F.; Abas B. I.; Cevik O.; Kaymakcioglu B. K. Design, synthesis and biological evaluation of some new 2-Pyrazoline derivatives as potential anticancer agents. Bioorg. Chem. 2020, 102, 104063. 10.1016/j.bioorg.2020.104063. [DOI] [PubMed] [Google Scholar]
- Ahsan M. J. Synthesis and anticancer activity of 3a,4-dihydro-3H-indeno[1,2-c]pyrazole-2-carboxamide/ carbothioamide analogues. Lett. Drug Des. Dis. 2012, 9, 823–827. 10.2174/157018012803307996. [DOI] [Google Scholar]
- Unsal-Tan O.; Tuylu Kucukkılınc T.; Ayazgok B.; Balkan A.; Ozadali-Sari K. Synthesis, molecular docking, and biological evaluation of novel 2-pyrazoline derivatives as multifunctional agents for the treatment of Alzheimer’s disease. Med. Chem. Commun. 2019, 10, 1018–1026. 10.1039/C9MD00030E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahsan M. J.; Samy J. G.; Khalilullah H.; Bakht M. A.; Hassan M. Z. Synthesis and antimycobacterial evaluation of 3a,4-dihydro-3H-indeno[1,2-c]pyrazole-2-carboxamide analogues. Eur. J. Med. Chem. 2011, 46, 5694–5697. 10.1016/j.ejmech.2011.09.035. [DOI] [PubMed] [Google Scholar]
- Ahsan M. J.; Khalilullah H.; Stables J. P.; Govindasamy J. Synthesis and anticonvulsant activity of 3a,4-dihydro-3H-indeno[1,2-c]pyrazole-2-carboxamide/carbothioamide analogues. J. Enzy. Inh. Med. Chem. 2013, 28, 644–650. 10.3109/14756366.2012.663364. [DOI] [PubMed] [Google Scholar]
- Ahsan M. J. Anticonvulsant activity and neuroprotection assay of 3-substituted-N-aryl-6,7-dimethoxy-3a,4-dihydro-3H-indeno[1,2-c]pyrazole-2-carboxamide analogues. Arab. J. Chem. 2017, 10, S2762–S2766. 10.1016/j.arabjc.2013.10.023. [DOI] [Google Scholar]
- Akhtar M.; Marella A.; Alam M. M.; Khan M. F.; Akhtar M.; Anwer T.; Khan F.; Naematullah M.; Azam F.; Rizvi M. A.; Shaquiquzzaman M. Design and synthesis of pyrazole–pyrazoline hybrids as cancer-associated selective COX-2 inhibitors. Arch. Pharm. 2020, e2000116. 10.1002/ardp.202000116. [DOI] [PubMed] [Google Scholar]
- Tripathi A. C.; Upadhyay S.; Paliwal S.; Saraf S. K. N1-benzenesulfonyl-2-pyrazoline hybrids in neurological disorders: Syntheses, biological screening and computational studies. EXCLI J. 2018, 17, 126–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahsan M. J.; Govindasamy J.; Khalilullah H.; Mohan G.; Stables J. P.; Pannecouque C.; Clercq E. D. POMA analyses as new efficient bioinformatics’ platform to predict and optimize bioactivity of synthesized 3a,4-dihydro-3H-indeno[1,2-c]pyrazole-2-carboxamide/carbothioamide analogues. Bioorg. Med. Chem. Lett. 2012, 22, 7029–7035. 10.1016/j.bmcl.2012.09.108. [DOI] [PubMed] [Google Scholar]
- Kumar G.; Tanwar O.; Kumar J.; Akhter M.; Sharma S.; Pillai C. R.; Alam M. M.; Zama M. S. Pyrazole-pyrazoline as promising novel antimalarial agents: A mechanistic study. Eur. J. Med. Chem. 2018, 149, 139–147. 10.1016/j.ejmech.2018.01.082. [DOI] [PubMed] [Google Scholar]
- Badavath V. N.; Baysal İ.; Ucar G.; Sinha B. N.; Jayaprakash V. Monoamine Oxidase Inhibitory Activity of Novel Pyrazoline Analogues: Curcumin Based Design and Synthesis. ACS Med. Chem. Lett. 2016, 7, 56–61. 10.1021/acsmedchemlett.5b00326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruin N. M. W. J.; Prickaerts J.; Lange J. H. M.; Akkerman S.; Andriambeloson E.; de Haan M.; Wijnen J.; van Drimmelen M.; Hissink E.; Heijink L.; Kruse C. G. SLV330, a cannabinoid CB1 receptor antagonist, ameliorates deficits in the T-maze, object recognition and Social Recognition Tasks in rodents. Neurobiol. Learn. Mem. 2010, 93, 522–531. 10.1016/j.nlm.2010.01.010. [DOI] [PubMed] [Google Scholar]
- Nawaz F.; Alam O.; Perwez A.; Rizvi M. A.; Naim M. J.; Siddiqui N.; Firdaus J. U.; Rahman S.; Jha M.; Sheikh A. A. Design, Synthesis, Molecular Docking and Anticancer Evaluation of Pyrazole Linked Pyrazoline Derivatives with Carbothioamide tail as EGFR Kinase Inhibitors. Anti-Cancer Agents Med. Chem. 2020, 21, 42–60. 10.2174/1871520620666200727093613. [DOI] [PubMed] [Google Scholar]
- Singh R.; Thota S.; Bansal R. Studies on 16,17-pyrazoline substituted hetero steroids as anti-Alzheimer and anti-Parkinsonian agents using LPS induced neuroinflammation models of mice and rats. ACS Chem. Neurosci. 2018, 9, 272–283. 10.1021/acschemneuro.7b00303. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Tortorici M. A.; Garrett M.; Hee B.; Klamerus K. J.; Pithavala Y. K. Clinical pharmacology of axitinib. Clin. Pharmacokinet. 2013, 52, 713–725. 10.1007/s40262-013-0068-3. [DOI] [PubMed] [Google Scholar]
- Schirris T. J. J.; Ritschel T.; Renkema G. H.; Willems P. H. G. M.; Smeitink J. A. M.; Russel F. G. M. Mitochondrial ADP/ATP exchange inhibition: a novel off-target mechanism underlying ibipinabant-induced myotoxicity. Sci. Rep. 2015, 5, 14533. 10.1038/srep14533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chorvat R. J.; Berbaum J.; Seriacki K.; McElroy J. F. JD-5006 and JD-5037: peripherally restricted (PR) cannabinoid-1 receptor blockers related to SLV-319 (Ibipinabant) as metabolic disorder therapeutics devoid of CNS liabilities. Bioorg. Med. Chem. Lett. 2012, 22, 6173–6180. 10.1016/j.bmcl.2012.08.004. [DOI] [PubMed] [Google Scholar]
- Byrd J. C.; Furman R. R.; Coutre S. E.; Flinn I. W.; Burger J. A.; Blum K. A.; Grant B.; Sharman J. P.; Coleman M.; Wierda W. G.; et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 2013, 369, 32–42. 10.1056/NEJMoa1215637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R. S.; Arif I. A.; Ahamed A.; Idhayadhulla A. Anti-inflammatory and antimicrobial activities of novel pyrazole analogues. Saudi J. Biol. Sci. 2016, 23, 614–620. 10.1016/j.sjbs.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamogawa E.; Sueishi Y. A multiple free-radical scavenging (MULTIS) study on the antioxidant capacity of a neuroprotective drug, edaravone as compared with uric acid, glutathione, and trolox. Bioorg. Med. Chem. Lett. 2014, 24, 1376–1379. 10.1016/j.bmcl.2014.01.045. [DOI] [PubMed] [Google Scholar]
- Yusuf M.; Jain P. Synthetic and biological studies of pyrazolines and related heterocyclic compounds. Arab. J. Chem. 2014, 7, 553–596. 10.1016/j.arabjc.2011.09.013. [DOI] [Google Scholar]
- Alex J. M.; Kumar R. 4,5-Dihydro-1H-pyrazole: An indispensable scaffold. J. Enzyme Inhib. Med. Chem. 2014, 29, 427–442. 10.3109/14756366.2013.795956. [DOI] [PubMed] [Google Scholar]
- Shaaban M. R.; Mayhoub A. S.; Farag A. M. Recent advances in the therapeutic applications of pyrazolines. Expert Opin. Ther. Patents 2012, 22, 253–291. 10.1517/13543776.2012.667403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S.; Bawa S.; Drabu S.; Kumar R.; Gupta H. Biological activities of pyrazoline derivatives–A recent development. Recent Pat. Antiinfect. Drug Discovery 2009, 4, 154–163. 10.2174/157489109789318569. [DOI] [PubMed] [Google Scholar]
- Marella A.; Ali R.; Alam T.; Saha R.; Tanwar O.; Akhter M.; Shaquiquzzaman; Alam M. Pyrazolines: A biological review. Mini-Rev. Med. Chem. 2013, 13, 921–931. 10.2174/1389557511313060012. [DOI] [PubMed] [Google Scholar]
- Schoffski P.; Cresta S.; Mayer I. A.; Wildiers H.; Damian S.; Gendreau S.; Rooney I.; Morrissey K. M.; Spoerke J. M.; Ng V. W.; Singel S. M.; Winer E. A phase Ib study of pictilisib (GDC-0941) in combination with paclitaxel, with and without bevacizumab or trastuzumab, and with letrozole in advanced breast cancer. Breast Cancer Res. 2018, 20, 109. 10.1186/s13058-018-1015-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy B.; Mrozek E.; Kuebler J. P.; Bekaii-Saab T.; Kraut E. H. Phase II trial of pyrazoloacridine (NSC#366140) in patients with metastatic breast cancer. Invest. N. Drugs 2011, 29, 347–351. 10.1007/s10637-009-9338-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L. C.; Lin H. Y.; Tsai M. T.; Chou R. H.; Lee F. Y.; Teng C. M.; Hsieh M. T.; Hung H. Y.; Huang L. J.; Yu Y. L.; Kuo S. C. YC-1 inhibits proliferation of breast cancer cells by down-regulating EZH2 expression via activation of c-Cbl and ERK. Br. J. Pharmacol. 2014, 171, 4010–4025. 10.1111/bph.12708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iniguez M. A.; Punzon C.; Cacheiro-Llaguno C.; Díaz-Munoz M. D.; Duque J.; Cuberes R.; Alvarez I.; Andres E. M.; Buxens J.; Buschmann H.; Vela J. M.; Fresno M. Cyclooxygenase-independent inhibitory effects on T cell activation of novel 4,5-dihydro-3 trifluoromethyl pyrazole cyclooxygenase-2 inhibitors. Int. Immunopharm. 2010, 10, 1295–1304. 10.1016/j.intimp.2010.07.013. [DOI] [PubMed] [Google Scholar]
- Mathew B.; Suresh J.; Anbazhagan S.; Mathew G. Pyrazoline: a promising scaffold for the inhibition of monoamine oxidase. Cent. Nerv. Syst. Agents Med. Chem. 2014, 13, 195–206. 10.2174/1871524914666140129122632. [DOI] [PubMed] [Google Scholar]
- Secci D.; Carradori S.; Bolasco A.; Bizzarri B.; D’Ascenzio M.; Maccioni E. Discovery and Optimization of Pyrazoline Derivatives As Promising Monoamine Oxidase Inhibitors. Current Topics Med. Chem. 2012, 12, 2240–2257. 10.2174/156802612805220057. [DOI] [PubMed] [Google Scholar]
- Ardiansah B. Some bioactivities of heterocyclic compounds containing pyrazoline moiety - a short review. Int. J. ChemTechnol. Res. 2018, 11, 22–26. 10.20902/IJCTR.2018.111004. [DOI] [Google Scholar]
- Neudorfer C.; Shanab K.; Jurik A.; Schreiber V.; Neudorfer C.; Vraka C.; Schirmer E.; Holzer W.; Ecker G.; Mitterhauser M.; Wadsak W.; Spreitzer H. Development of potential selective and reversible pyrazoline based MAO-B inhibitors as MAO-B PET tracer precursors and reference substances for the early detection of Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2014, 24, 4490–4495. 10.1016/j.bmcl.2014.07.085. [DOI] [PubMed] [Google Scholar]
- Secci D.; Bolasco A.; Chimenti P.; Carradori S. The state of the art of pyrazole derivatives as monoamine oxidase inhibitors and antidepressant/anticonvulsant agents. Curr. Med. Chem. 2011, 18, 5114–5144. 10.2174/092986711797636090. [DOI] [PubMed] [Google Scholar]
- Chimenti F.; Bolasco A.; Manna F.; Secci D.; Chimenti P.; Granese A.; Befani O.; Turini P.; Cirilli R.; La Torre F.; Alcaro S.; Ortuso F.; Langer T. Synthesis, biological evaluation and 3D-QSAR of 1,3,5-trisubstituted-4,5-dihydro-(1H)-pyrazole derivatives as potent and highly selective monoamine oxidase A inhibitors. Curr. Med. Chem. 2006, 13, 1411–1428. 10.2174/092986706776872907. [DOI] [PubMed] [Google Scholar]
- Keisuke M.; Toshiyuki A.; Yoko A.; Masaichi L.. Antioxidant comprising pyrazole or pyrazoline compound, and use thereof. PCT Int. Appl., WO 2020158225 A1 20200806, 2020.
- Timothy A. M.; Dennis L. C.; Stephen T. F.; Yao J.. Pyrazoline dihydroquinolones, pharmaceutical compositions, and uses. PCT Int. Appl., WO 2014210456 A1 20141231, 2014.
- Antonio T. J.; Susana Y. M.. Indoline-substituted pyrazoline derivatives, their preparation and use as medicaments. U.S. patent, US 8106085 B2 20120131, 2012.
- Antonio T. J.; Susana Y. M.. Cycloalkane-substituted pyrazoline derivatives, their preparation and use as medicaments. U.S. patent, US 7994200 B2 20110809, 2011.
- Arnold V. L.; Wouter I. B. I.; Axel S.; Agatha R. A. M.; Jennifer V.; Martina V.NA.W.; Martin D. H.; Cornelis K. G.. Arylsulfonyl pyrazoline carboxamidine derivatives as 5-HT6 antagonists and their preparation, pharmaceutical compositions and use in the treatment of diseases. PCT Int. Appl., WO 2009115515 A1 20090924, 2009.
- Antonio T. J.; Jose M. P.; Heinrich B. H.; Ramon Q. R. J.; Alberto D. Z.. Carbonyl-substituted pyrazoline compounds, their preparation and use as medicaments. Eur. Pat. Appl., EP 1743888 A1 20070117, 2007.
- Antonio T. J.; Jose M. P.. Quaternary ammonium salts of substituted pyrazoline compounds, their preparation and use as medicaments. PCT Int. Appl., WO 2007009697 A1 20070125, 2007.
- Michela G.; Kay L. P.; Joel S. C.; Thorsten M.. Prevention and treatment of cognitive impairment using (R)-(−)-5-methyl-1-nicotinoyl-2-pyrazoline (MNP) and analogs thereof. U.S. Pat. Appl. Publ., US 20060014801 A1 20060119, 2006.
- Anand R.; Gill K. D.; Mahdi A. A. Therapeutics of Alzheimer’s disease: past, present and future. Neuropharmacology 2014, 76, 27–50. 10.1016/j.neuropharm.2013.07.004. [DOI] [PubMed] [Google Scholar]
- Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2014, 10, 47–92. [DOI] [PubMed] [Google Scholar]
- Farlow M. R.; Miller M. L.; Pejovic V. Treatment options in Alzheimer’s disease: maximizing benefit, managing expectations. Dement. Geriatr. Cogn. Disord. 2008, 25, 408–22. 10.1159/000122962. [DOI] [PubMed] [Google Scholar]
- Kumar A.; Singh A.; Ekavali A review on Alzheimer’s disease pathophysiology and its management: an update. Pharmacol. Rep. 2015, 67, 195–203. 10.1016/j.pharep.2014.09.004. [DOI] [PubMed] [Google Scholar]
- Knopman D. S.; Amieva H.; Petersen R. C.; Chetelat G.; Holtzman D. M.; Hyman B. T.; Nixon R. A.; Jones D. T. Alzheimer disease. Nat. Rev. Dis. Primers 2021, 7, 33. 10.1038/s41572-021-00269-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyaswamy A.; Wang X.; Krishnamoorthi S.; Kaliamoorthy V.; Sreenivasmurthy S. G.; Durairajan S. S. K.; Song J. X.; Tong B. C.; Zhu Z.; Su C. F.; Liu J.; Cheung K. H.; Lu J. H.; Tan J. Q.; Li H.; Wong W. M. S.; Li M. Theranostic F-SLOH mitigates Alzheimer’s disease pathology involving TFEB and ameliorates cognitive functions in Alzheimer’s disease models. Redox Biol. 2022, 51, 102280. 10.1016/j.redox.2022.102280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Iyaswamy A.; Xu D.; Krishnamoorthi S.; Sreenivasmurthy S. G.; Yang Y.; Li Y.; Chen C.; Li M.; Li H. W.; Wong M. S.. Real-Time Detection and Visualization of Amyloid-β Aggregates Induced by Hydrogen Peroxide in Cell and Mouse Models of Alzheimer’s Disease. ACS Appl. Mater. Interfaces 2022, 10.1021/acsami.2c07859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGirr S.; Venegas C.; Swaminathan A. Alzheimer’s Disease: A Brief Review. J. Exp. Nerol. 2020, 1, 89–98. [Google Scholar]
- Barage S. H.; Sonawane K. D. Amyloid cascade hypothesis: Pathogenesis and therapeutic strategies in Alzheimer’s disease. Neuropeptides 2015, 52, 1–8. 10.1016/j.npep.2015.06.008. [DOI] [PubMed] [Google Scholar]
- Sevigny J.; Chiao P.; Bussiere T.; Weinreb P. H.; Williams L.; Maier M.; Dunstan R.; Salloway S.; Chen T.; Ling Y.; O’Gorman J.; Qian F.; Arastu M.; Li M.; Chollate S.; Brennan M. S.; Quintero-Monzon O.; Scannevin R. H.; Arnold H. M.; Engber T.; Rhodes K.; Ferrero J.; Hang Y.; Mikulskis A.; Grimm J.; Hock C.; Nitsch R. M.; Sandrock A. The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. 10.1038/nature19323. [DOI] [PubMed] [Google Scholar]
- Yang C.; Liu J.; Tong B. C.; Wang Z.; Zhu Z.; Su C.; Sreenivasmurthy S. G.; Wu J.; Iyaswamy A.; Krishnamoorthi S.; Huang S.; Cheung K.; Song J.; Tan J.; Lu J.; Li M. TFEB, a master regulator of autophagy and biogenesis, unexpectedly promotes apoptosis in response to the cyclopentenone prostaglandin 15d-PGJ2. Acta Pharmacol. Sin. 2022, 43, 1251–1263. 10.1038/s41401-021-00711-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C.; Su C.; Iyaswamy A.; Krishnamoorthi S. K.; Zhu Z.; Yang S.; Tong B. C.; Liu J.; Sreenivasmurthy S. G.; Guan X.; Kan Y.; Wu A. J.; Huang A. S.; Tan J.; Cheung K.; Song J.; Li M. Celastrol enhances transcription factor EB (TFEB)-mediated autophagy and mitigates Tau pathology: Implications for Alzheimer’s disease therapy. Acta Pharm. Sin. B 2022, 12, 1707–1722. 10.1016/j.apsb.2022.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard R.; McShane R.; Lindesay J.; Ritchie C.; Baldwin A.; Barber R.; Burns A.; Dening T.; Findlay D.; Holmes C.; Hughes A.; Jacoby R.; Jones R.; Jones R.; McKeith I.; Macharouthu A.; O’Brien J.; Passmore P.; Sheehan B.; Juszczak E.; Katona C.; Hills R.; Knapp M.; Ballard C.; Brown R.; Banerjee S.; Onions C.; Griffin M.; Adams J.; Gray R.; Johnson T.; Bentham P.; Phillips P. Donepezil and Memantine for Moderate-to-Severe Alzheimer’s Disease. N. Engl. J. Med. 2012, 366, 893–903. 10.1056/NEJMoa1106668. [DOI] [PubMed] [Google Scholar]
- Armstrong M. J.; Okun M. S. Diagnosis and Treatment of Parkinson Disease A Review. JAMA 2020, 323, 548–560. 10.1001/jama.2019.22360. [DOI] [PubMed] [Google Scholar]
- Koller W. C.; Busenbark K. L.. Essential tremor. In Movement Disorders; Watts R. L., Koller W. C., Eds.; McGraw-Hill: New York, 1997; pp 365–385. [Google Scholar]
- Zhu Z.; Liu L.; Su C.; Liu J.; Tong B. C.; Iyaswamy A.; Krishnamoorthi S.; Sreenivasmurthy S. G.; Guan X.; Kan Y.; Xie W.; Zhao C.; Cheung K.; Lu J.; Tan J.; Zhang H.; Song J.; Li M. Corynoxine B derivative CB6 prevents Parkinsonian toxicity in mice by inducing PIK3C3 complex-dependent autophagy. Acta Pharmacol. Sin. 2022, 43, 2511–2526. 10.1038/s41401-022-00871-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finberg J. P. M.; Rabey J. M. Inhibitors of MAO-A and MAO-B in Psychiatry and Neurology. Front. Pharmacol. 2016, 7, 340. 10.3389/fphar.2016.00340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacio M. J.; Palma P. N.; Almeida L.; Soares-da-Silva P. Catechol-O-methyltransferase and its inhibitors in Parkinson’s disease. CNS Drug Rev. Fall 2007, 13, 352–379. 10.1111/j.1527-3458.2007.00020.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston J. P. Some observations upon a new inhibitor of monoamine oxidase in brain tissue. Biochem. Pharmacol. 1968, 17, 1285–1297. 10.1016/0006-2952(68)90066-X. [DOI] [PubMed] [Google Scholar]
- Yamali C.; Gul H. I.; Kazaz C.; Levent S.; Gulcin I. Synthesis, structure elucidation, and in vitro pharmacological evaluation of novel polyfluoro substituted pyrazoline type sulfonamides as multi-target agents for inhibition of acetylcholinesterase and carbonic anhydrase I and II enzymes. Bioorg. Chem. 2020, 96, 103627. 10.1016/j.bioorg.2020.103627. [DOI] [PubMed] [Google Scholar]
- Singh R.; Bansal R. 16,17-N′-(alky/arylsulfonyl)pyrazoline substituted neuroprotective heterosteroids: Synthesis, molecular docking and preclinical efficacy/toxicity studies in rodents. Steroids 2019, 148, 114–124. 10.1016/j.steroids.2019.05.002. [DOI] [PubMed] [Google Scholar]
- Ozgun D. O.; Gul H. I.; Yamali C.; Sakagami H.; Gulcin I.; Sukuroglu M.; Supuran C. T. Synthesis and bioactivities of pyrazoline benzensulfonamides as carbonic anhydrase and acetylcholinesterase inhibitors with low cytotoxicity. Bioorg. Chem. 2019, 84, 511–517. 10.1016/j.bioorg.2018.12.028. [DOI] [PubMed] [Google Scholar]
- Mishra N.; Sasmal D. Development of selective and reversible pyrazoline based MAO-B inhibitors: Virtual screening, synthesis and biological evaluation. Bioorg. Med. Chem. Lett. 2011, 21, 1969–1973. 10.1016/j.bmcl.2011.02.030. [DOI] [PubMed] [Google Scholar]
- Mishra N.; Sasmal D. Additional acetyl cholinesterase inhibitory property of diaryl pyrazoline derivatives. Bioorg. Med. Chem. Lett. 2013, 23, 702–705. 10.1016/j.bmcl.2012.11.100. [DOI] [PubMed] [Google Scholar]
- Gökhan N.; Yesilada A.; Uçar G.; Erol K.; Bilgin A. A. 1-N-Substituted Thiocarbamoyl-3-phenyl-5-thienyl-2-pyrazolines: Synthesis and Evaluation as MAO Inhibitors. Arch. Pharm. Pharm. Med. Chem. 2003, 336, 362–371. 10.1002/ardp.200300732. [DOI] [PubMed] [Google Scholar]
- Ucar G.; Gokhan N.; Yesilada A.; Bilgin A. A. 1-N-Substituted thiocarbamoyl-3-phenyl-5-thienyl-2-pyrazolines: A novel cholinesterase and selective monoamine oxidase B inhibitors for the treatment of Parkinson’s and Alzheimer’s diseases. Neurosci. Lett. 2005, 382, 327–331. 10.1016/j.neulet.2005.03.028. [DOI] [PubMed] [Google Scholar]
- Mishra N.; Sasmal D.; Singh K.K. Attenuating Aβ1–42-Induced Toxicity By A Novel Acetylcholinesterase Inhibitor. Neuroscience 2013, 250, 309–319. 10.1016/j.neuroscience.2013.07.014. [DOI] [PubMed] [Google Scholar]
- Zitouni G. T.; Hussein W.; Sağlık B. N.; Baysal M.; Kaplancıklı Z. A. Fighting Against Alzheimer’s Disease: Synthesis of New Pyrazoline and Benzothiazole Derivatives as New Acetylcholinesterase and MAO Inhibitors. Lett. Drug Des. Dis. 2017, 14, 1–14. 10.2174/1570180814666170704144917. [DOI] [Google Scholar]
- Sever B.; Türkeş C.; Altıntop M. D.; Demir Y.; Beydemir Ş. Thiazolyl-pyrazoline derivatives: In vitro and in silico evaluation as potential acetylcholinesterase and carbonic anhydrase inhibitors. Int. J. Bio. Macrmol. 2020, 163, 1970–1988. 10.1016/j.ijbiomac.2020.09.043. [DOI] [PubMed] [Google Scholar]
- Altintop M. D.; Ozdemir A.; Kaplancikli Z. A.; Turan-Zitouni G.; Temel H. E.; Ciftci G. A. Synthesis and Biological Evaluation of Some Pyrazoline Derivatives Bearing a Dithiocarbamate Moiety as New Cholinesterase Inhibitors. Arch. Pharm. Chem. Life Sci. 2013, 346, 189–199. 10.1002/ardp.201200384. [DOI] [PubMed] [Google Scholar]
- Tuğrak M.; Gül H. İ.; Gülçin İ. Acetylcholinesterase Inhibitory Potency of New Pyrazoline Derivatives. J. Res. Pharm. 2020, 24, 464–471. [Google Scholar]
- Jayaprakash V.; Yabanoglu S.; Sinha B. N.; Ucar G. Pyrazoline-based Mycobactin Analogues as Dual Inhibitors of MAO/Cholinesterase. Turk. J. Biochem. 2010, 35, 91–98. [Google Scholar]
- Zondagh L. S.; Malan S. F.; Joubert J. Design, synthesis and biological evaluation of edaravone derivatives bearing the N-benzyl pyridinium moiety as multifunctional anti-Alzheimer’s agents. J. Enzy. Inhib. Med. Chem. 2020, 35, 1596–1605. 10.1080/14756366.2020.1801673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan S. E.; Narayanan H.; Mukundan M.; Balan S.; Vishnupriya C. P.; Gopinathan A.; Rajamma R. G.; Marathakam A. Design, synthesis and biological evaluation of substituted pyrazoles endowed with brominated 4-methyl 7-hydroxy coumarin as new scaffolds against Alzheimer’s disease. Fut. J. Pharm. Sci. 2021, 7, 161. 10.1186/s43094-021-00278-4. [DOI] [Google Scholar]
- Temel H. E.; Altintop M. D.; Özdemir A. Synthesis and Evaluation of a New Series of Thiazolyl-pyrazoline Derivatives as Cholinesterase Inhibitors. Turk. J. Pharm. Sci. 2018, 15, 333–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitge R.; Smit S.; Petzer A.; Petzer J. P. Evaluation of nitrocatechol chalcone and pyrazoline derivatives as inhibitors of catechol-O-methyltransferase and monoamine oxidase. Bioorg. Med. Chem. Lett. 2020, 30, 127188. 10.1016/j.bmcl.2020.127188. [DOI] [PubMed] [Google Scholar]
- Zaib S.; Rizvi S. U. F.; Aslam S.; Ahmad M.; al-Rashida M.; Iqbal J. Monoamine oxidase inhibition and molecular modeling studies of piperidyl-thienyl and 2-pyrazoline derivatives of chalcones. Med. Chem. 2015, 11, 497–505. 10.2174/1573406410666141229101130. [DOI] [PubMed] [Google Scholar]
- Aksoz B. E.; Ciftci S. Y.; Ucar G.; Yelekci K.; Ertan R. Synthesis of some novel hydrazone and 2-pyrazoline derivatives: Monoamine oxidase inhibitory activities and docking studies. Bioorg. Med. Chem. Lett. 2014, 24, 3278–3284. 10.1016/j.bmcl.2014.06.015. [DOI] [PubMed] [Google Scholar]
- Sahoo A.; Yabanoglu S.; Sinha B. N.; Ucar G.; Basu A.; Jayaprakash V. Towards development of selective and reversible pyrazoline based MAO-inhibitors: Synthesis, biological evaluation and docking studies. Bioorg. Med. Chem. Lett. 2010, 20, 132–136. 10.1016/j.bmcl.2009.11.015. [DOI] [PubMed] [Google Scholar]
- Kelekci N. G.; Koyunoglu S.; Yabanoglu S.; Yelekci K.; Ozgen O.; Ucar G.; Erol K.; Kendi E.; Yesilada A. New pyrazoline bearing 4(3H)-quinazolinone inhibitors of monoamine oxidase: Synthesis, biological evaluation, and structural determinants of MAO-A and MAO-B selectivity. Bioorg. Med. Chem. 2009, 17, 675–689. 10.1016/j.bmc.2008.11.068. [DOI] [PubMed] [Google Scholar]
- Jayaprakash V.; Sinha B. N.; Ucar G.; Ercan A. Pyrazoline-based mycobactin analogues as MAO-inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 6362–6368. 10.1016/j.bmcl.2008.10.084. [DOI] [PubMed] [Google Scholar]
- Goksen U. S.; Sarigul S.; Bultinck P.; Herrebout W.; Dogan I.; Yelekci K.; Ucar G.; Kelekci N. G. Absolute configuration and biological profile of pyrazoline enantiomers as MAO inhibitory activity. Chirality 2019, 31, 21–33. 10.1002/chir.23027. [DOI] [PubMed] [Google Scholar]
- Nath C.; Badavath V. N.; Thakur A.; Ucar G.; Acevedo O.; Siddique M. U. M.; Jayaprakash V. Curcumin-Based Pyrazoline Analogues as Selective Inhibitors of Human Monoamine Oxidase A. Med. Chem. Commun. 2018, 9, 1164–1171. 10.1039/C8MD00196K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aksoz B. E.; Ucar G.; Yelekci K. Design synthesis and hMAO inhibitory activity of novel 2-pyrazoline analogues. Comb. Chem. High. Screen. 2017, 20, 510–521. 10.2174/1386207320666170504114208. [DOI] [PubMed] [Google Scholar]
- Tong X.; Chen R.; Zhang T. T.; Han Y.; Tang W. J.; Liu X. H. Design and synthesis of novel 2-pyrazoline-1-ethanone derivatives as selective MAO inhibitors. Bioorg. Med. Chem. 2015, 23, 515–525. 10.1016/j.bmc.2014.12.010. [DOI] [PubMed] [Google Scholar]
- Badavath V. N.; Sinha B. N.; Jayaprakash V. Design, in-silico docking and predictive ADME properties of novel pyrazoline derivatives with selective human MAO inhibitory activity. Int. J. Pharm. Pharm. Sci. 2015, 7, 277–282. [Google Scholar]
- Aksoz E. B.; Baysal I.; Ciftci S. Y.; Djikic T.; Yelekci K.; Ucar G.; Ertan R. Synthesis and Screening of Human Monoamine Oxidase-A Inhibitor Effect of New 2-Pyrazoline and Hydrazone Derivatives. Arch. Pharm. Chem. Life Sci. 2015, 348, 743–756. 10.1002/ardp.201500212. [DOI] [PubMed] [Google Scholar]
- Goksen U. S.; Ciftci S. Y.; Ercan A.; Yelekci K.; Ucar G.; Kelekci N. G. Evaluation of selective human MAO inhibitory activities of some novel pyrazoline derivatives. J. Neural. Transm. 2013, 120, 863–873. 10.1007/s00702-013-0980-6. [DOI] [PubMed] [Google Scholar]
- Nayak B. V.; Yabanoglu S. C.; Jadav S. S.; Jagrat M.; Sinha B. N.; Ucar G.; Jayaprakash V. Monoamine oxidase inhibitory activity of 3,5-biaryl-4,5-dihydro-1Hpyrazole-1-carboxylate derivatives. Eur. J. Med. Chem. 2013, 69, 762–767. 10.1016/j.ejmech.2013.09.010. [DOI] [PubMed] [Google Scholar]
- Jagrat M.; Behera J.; Yabanoglu S.; Ercan A.; Ucar G.; Sinha B. N.; Sankaran V.; Basu A.; Jayaprakash V. Pyrazoline based MAO inhibitors: Synthesis, biological evaluation and SAR studies. Bioorg. Med. Chem. Lett. 2011, 21, 4296–4300. 10.1016/j.bmcl.2011.05.057. [DOI] [PubMed] [Google Scholar]
- Karuppasamy M.; Mahapatra M.; Yabanoglu S.; Ucar G.; Sinha B. N.; Basu A.; Mishra N.; Sharon A.; Kulandaivelu U.; Jayaprakash V. Development of selective and reversible pyrazoline based MAO-A inhibitors: Synthesis, biological evaluation and docking studies. Bioorg. Med. Chem. 2010, 18, 1875–1881. 10.1016/j.bmc.2010.01.043. [DOI] [PubMed] [Google Scholar]
- Chimenti F.; Bolasco A.; Manna F.; Secci D.; Chimenti P.; Granese A.; Befani O.; Turini P.; Alcaro S.; Ortuso F. Synthesis and Molecular Modelling of Novel Substituted-4,5-dihydro-(1H)-pyrazole Derivatives as Potent and Highly Selective Monoamine Oxidase-A Inhibitors. Chem. Biol. Drug Des. 2006, 67, 206–214. 10.1111/j.1747-0285.2006.00367.x. [DOI] [PubMed] [Google Scholar]
- Badavath V. N.; Baysal İ.; Ucar G.; Sinha B. N.; Jayaprakash V. Monoamine Oxidase Inhibitory Activity of Novel Pyrazoline Analogues: Curcumin Based Design and Synthesis. ACS Med. Chem. Lett. 2016, 7 (1), 56–61. 10.1021/acsmedchemlett.5b00326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SciFinder: https://scifinder-n.cas.org/ (Retrieved on 12 December 2021).
- X-ray crystal structure of VX-phosphonylated hAChE in complex with oxime reactivator RS194B: https://www.rcsb.org/structure/6u37 (Retrieved on 09 June 2022).
- Gorecki L.; Gerlits O.; Kong X.; Cheng X.; Blumenthal D. K.; Taylor P.; Ballatore C.; Kovalevsky A.; Radić Z. Rational design, synthesis and evaluation of uncharged, “smart” bis-oxime antidotes of organophosphate-inhibited human acetylcholinesterase. J. Bio. Chem. 2020, 295, 4079–4092. 10.1074/jbc.RA119.012400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- X-ray crystal structure of human monoamine oxidase B in complex with Deprenyl https://www.rcsb.org/structure/2BYB (Retrieved on 16 August 2022).
- De Colibus L.; Li M.; Binda C.; Lustig A.; Edmondson D. E.; Mattevi A. Three-dimensional structure of human monoamine oxidase A (MAO A): relation to the structures of rat MAO A and human MAO B. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 12684–9. 10.1073/pnas.0505975102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- X-ray crystal structure of human monoamine oxidase A in complex with Clorgyline: https://www.rcsb.org/structure/2bxr (Retrieved on 14 August 2022).