

Abstract
Clonal hematopoiesis arises from somatic mutations that provide a fitness advantage to hematopoietic stem cells and the outgrowth of clones of blood cells. Clonal hematopoiesis commonly involves mutations in genes that are involved in epigenetic modifications, signaling and DNA damage repair. Clonal hematopoiesis has emerged as a major independent risk factor in atherosclerotic cardiovascular disease, thrombosis and heart failure. Studies in mouse models of clonal hematopoiesis have shown an increase in atherosclerosis, thrombosis and heart failure, involving increased myeloid cell inflammatory responses and inflammasome activation. Although increased inflammatory responses have emerged as a common underlying principle, some recent studies indicate mutation-specific effects. The discovery of the association of clonal hematopoiesis with cardiovascular disease and the recent demonstration of benefit of anti-inflammatory treatments in human cardiovascular disease converge to suggest that anti-inflammatory treatments should be directed to individuals with clonal hematopoiesis. Such treatments could target specific inflammasomes, common downstream mediators such as IL-1β and IL-6, or mutations linked to clonal hematopoiesis.
Clonal hematopoiesis (CH) occurs when somatic mutations in leukemogenic genes provide a selective advantage to haematopoietic stem and progenitor cells (HSPCs), leading to the expansion of mutant blood cells1. CH commonly involves mutations in genes mediating epigenetic modifications (TET2, DNMT3A and ASXL1), hematopoietic cytokine signaling (JAK2), DNA damage repair (PPM1D and TP53) or messenger RNA splicing (SF3B1 and SRSF2). Clonal hematopoiesis of indeterminate potential (CHIP) is a term used to describe the presence of a driver gene mutation with a variant allele frequency (VAF) of at least 2% in peripheral blood, in the absence of clinical criteria of hematological malignancy2. CHIP increases in frequency with aging and increases the risk of developing atherosclerotic cardiovascular disease (CVD) as well as hematological malignancies3. CHIP has also been associated with adverse outcomes in heart failure (HF), an increasing problem in cardiology practice4–6, and also following transcatheter aortic valve replacement of stenotic aortic valves, a common procedure in older people7.
Many excellent reviews on CHIP have been published, and CHIP has been associated with several other chronic diseases, including cirrhosis and chronic obstructive pulmonary disease1,8–11. This Review will focus on potential mechanisms that link CHIP to CVD and on how mechanistic and clinical studies may inform future potential therapies for CHIP-associated CVD. We will critically evaluate a recent proposal that the relationship between CHIP and atherosclerotic CVD may involve reverse causation12. Although there may be some shared general mechanisms for CHIP-associated CVD, such as macrophage inflammatory responses, a more granular view indicates that several mechanisms linking CHIP to CVD are mutation specific. Consequently, future therapies may target either common downstream pathways or mutation-specific mechanisms.
CHIP and atherosclerosis
Atherosclerosis develops as a result of the accumulation of cholesterol-rich low-density lipoprotein (LDL) particles in the artery wall, provoking an inflammatory response that eventually leads to the rupture or erosion of atherosclerotic plaques and blockage of arteries by thrombus13. Jaiswal et al.3 studied the relationship between CHIP mutations and the development of hematological neoplasms. Expectedly, CHIP increased the risk of developing hematological malignancy; unexpectedly, CHIP also predisposed to an increase in total mortality and to an increased risk of coronary artery disease (CAD) and thrombotic stroke, independently of traditional cardiovascular risk factors. In follow-up studies, Jaiswal et al. used whole-exome sequencing and a case–control design to assess the relationship between CHIP mutations and CVD3,14. People carrying genetic variants in TET2, DNMT3A, ASXL1 or JAK2 had a risk of CAD that was 1.9 times higher than that of non-carriers. In two retrospective case–control studies, participants with CHIP had a fourfold-higher risk of early-onset myocardial infarction. CHIP carriers also had increased coronary artery calcification, a marker of coronary atherosclerosis. These CHIP variants, as well as the DNA damage response (DDR) variants in TP53 and PPM1D, predict incident atherosclerotic disease in multiple vascular beds, including coronary, peripheral and mesenteric arteries15. CHIP has thus emerged as a major independent CVD risk factor, with comparable impact to traditional CVD risk factors, such as smoking or high LDL (Fig. 1). The causal relationship between CHIP and atherosclerosis has been established in hyperlipidemic mouse models transplanted with mixtures of wild-type (WT) and Tet2−/−, Jak2V617F or Tp53−/− bone marrow (BM) (Table 1).
Fig. 1 |. Somatic mutations and clonal hematopoiesis: at the crossroads of hematological malignancies and cardiovascular disease.
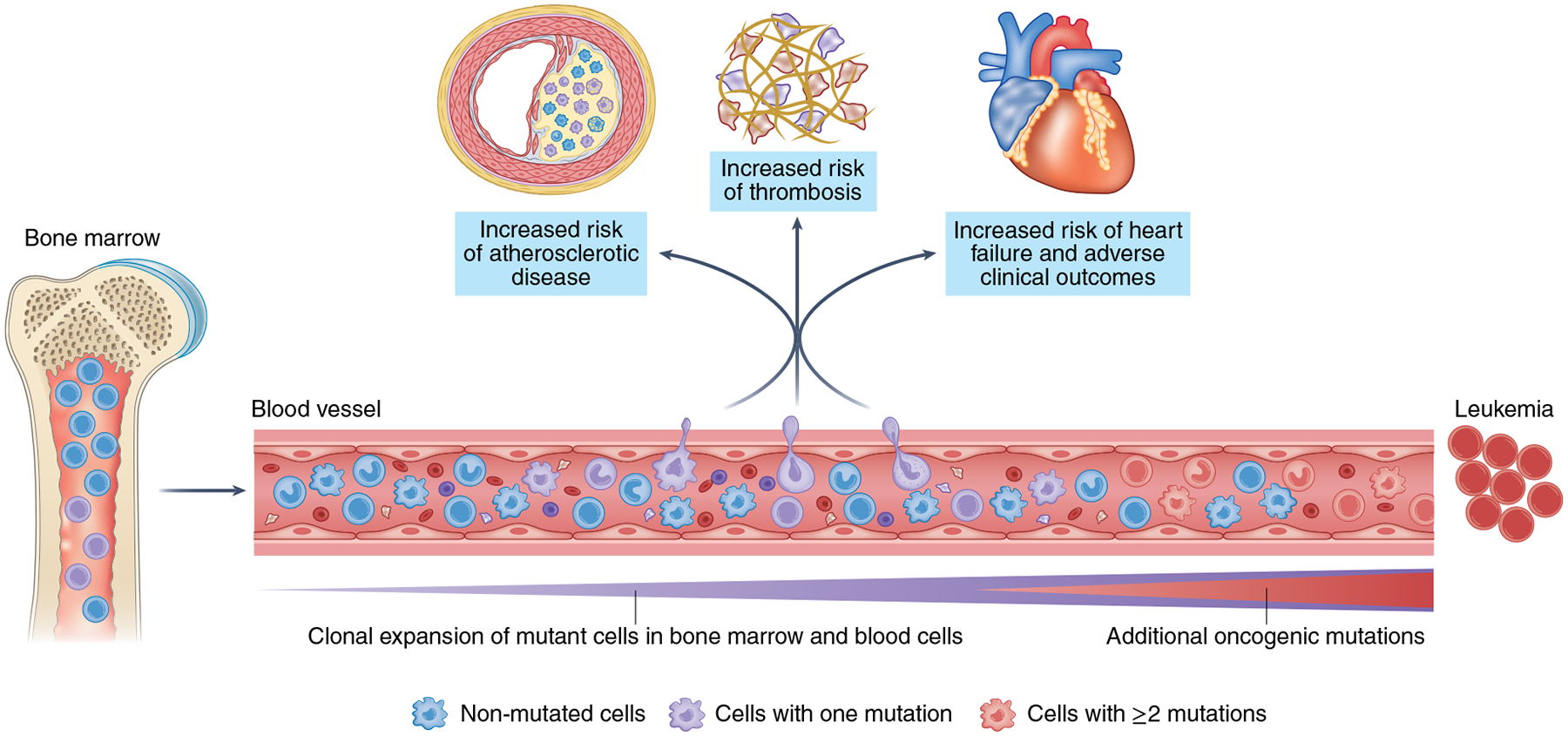
Hematopoietic stem cells accumulate somatic mutations continuously with aging. Some of these mutations confer a competitive advantage to the mutant cell, leading to its clonal expansion in bone marrow and blood. This clonal hematopoiesis is most frequently driven by one single mutation. While clonal hematopoiesis is associated with a large increase in the relative risk of developing a hematologic neoplasia, transition to malignancy typically requires the acquisition of multiple mutations, which is infrequent, even in individuals with clonal hematopoiesis. The main cause of death in individuals exhibiting clonal hematopoiesis is cardiovascular disease. Epidemiological and experimental evidence suggest that some clonal-hematopoiesis-related mutations can contribute to the development and clinical progression of atherosclerosis, heart failure and thrombosis. Heightened inflammatory responses mediated by tissue-infiltrating mutant immune cells are emerging as a central link between clonal hematopoiesis and cardiovascular disease.
Table 1 |.
Summary of experimental studies in mouse models that reported the effects of the most frequent CH candidate driver mutations on cardiovascular disease
| CH candidate gene | Experimental atherosclerosis phenotype | Experimental cardiac phenotype |
|---|---|---|
| DNMT3A | Accelerated atherosclerosis in Ldlr−/− mice after competitive BMT with 10% Dnmt3a−/− BM cells38 | Worse cardiac function and remodeling in angiotensin-II-infused mice bearing CRISPR-Cas9-edited Dnmt3a in hematopoietic cells39 |
| TET2 | Accelerated atherosclerosis in Ldlr−/− mice after competitive BMT with 10% Tet2−/− or Tet2+/− BM cells25 Accelerated atherosclerosis in Ldlr−/− mice with pan-hematopoietic ablation of Tet2 (ref.14) Accelerated atherosclerosis in Ldlr−/− with myeloid-restricted ablation of Tet2 (refs.14,25) |
Worse cardiac function and remodeling after LAD ligation or TAC in mice subjected to competitive BMT with 10% Tet2−/− or Tet2+/− BM cells54 Worse cardiac function and remodeling after LAD ligation or TAC in mice with myeloid-restricted Tet2 deficiency54 Worse cardiac function and remodeling in angiotensin-II-infused mice bearing CRISPR-Cas9-edited Tet2 in hematopoietic cells39 Worse cardiac function and remodeling in aged mice after non-conditioned BM transfer of Tet2−/− cells55 |
| ASXL1 | Unknown | Unknown |
| JAK2 | Accelerated atherosclerosis in Ldlr−/− mice with pan-hematopoietic expression of Jak2V617F (ref.31) Accelerated atherosclerosis in Ldlr−/− mice with competitive BMT or macrophage-specific expression of Jak2V617F (ref.32) |
Worse cardiac function and remodeling after LAD ligation or TAC in mice with lentivirus-mediated expression of Jak2V617F in myeloid cells56 |
| TP53 | Accelerated atherosclerosis in Ldlr−/− mice after competitive BMT with 20% Trp53−/− BM cells15 | Greater doxorubicin-induced cardiac toxicity in mice after non-conditioned BM transfer of Trp53+/− cells58 |
| PPM1D | Unknown | Worse cardiac function and remodeling in angiotensin-II-infused mice bearing hematopoietic cells expressing a CRISPR-Cas9-edited Ppm1d mutant59 |
LAD, left anterior descending artery; TAC, transverse aortic constriction
CHIP increases with aging and occurs in more than 10% of people over age 70. More sensitive DNA-sequencing methods can detect even higher frequencies of CH mutations16, but the clinical significance of very-low-burden CH mutations is poorly understood. Aging is also associated with altered methylation at a subset of CpGs17. Measurements of these modifications in epidemiological studies have been used to accurately predict chronological age in healthy individuals. Accelerated aging is inferred when measured methylation age is greater than chronological age. Epigenetic aging is associated with increased risk of CVD and all-cause mortality18. CHIP is strongly associated with epigenetic age acceleration19,20 and only the 40% of people with CHIP who had increased aging biomarkers also had increased CVD risk, suggesting that epigenetic aging could be used to identify people with CHIP who have greatest need for treatment20.
TET2.
Levine and colleagues showed that older people with clonal expansion of blood cells in the absence of concomitant hematological malignancy commonly had mutations in a potential driver gene, TET2 (ref.21). TET2 catalyzes the oxidation of 5-methylcytosine to 5-hydroxymethylcytosine, leading to widespread epigenetic modifications. TET2 deficiency increases HSPC self-renewal, leading to expansion of the HSPC compartment and increased production of blood cells, especially myeloid cells22,23. Loss of Tet2 resulted in the upregulation of several inflammatory mediators in macrophages, including IL-6, during the late-phase response to lipopolysaccharide challenge24. Tet2-deficient mice were more susceptible to endotoxin shock and dextran-sulfate-induced colitis, displaying a more severe inflammatory phenotype and increased IL-6 production compared with that of WT mice. The inflammatory repression mechanism involved TET2 recruitment of HDAC2 and repression of Il6 via histone deacetylation, independent of DNA methylation and hydroxymethylation24.
Fuster et al.25 showed that transplantation of Tet2−/− BM in a 10:90 ratio with WT BM resulted in an increased size of early atherosclerotic lesions in 9-week-old Ldlr−/− mice fed a Western-type diet compared with mice transplanted with 100% WT BM. Tet2-deficient cells expanded in the BM, spleen and blood but were not increased within atherosclerotic plaques relative to the bloodstream, and blood cell counts were unchanged. Although all Tet2-deficient blood cells were expanded, myeloid deficiency of Tet2 produced a similar increase in the size of atherosclerotic lesions, indicating a central role of macrophages or neutrophils in accelerated atherosclerosis. Tet2+/− BM cells modeling human TET2 CHIP also accelerated atherosclerosis. Early atherosclerotic lesions showed an increase in macrophage content, but there was no change in macrophage proliferation or apoptosis within plaques. Atherosclerotic plaques from Tet2−/− CH mice displayed prominently increased Il1b mRNA and increased IL-1β protein levels. The NLRP3 inhibitor MCC950 reduced atherosclerosis in Tet2−/− CH mice but not in controls, indicating a major role of the NLRP3 inflammasome in the accelerated progression of atherosclerosis in TET2-mutant CH. Inflammasome activation leads to the secretion of active IL-1β, which has the potential to increase recruitment of leukocytes into plaques. Accordingly, aortic endothelial P-selectin expression and monocyte recruitment to lesions were increased in Tet2−/− CH mice, with reversal of increased P-selectin by MCC950 treatment. On a mechanistic level, Tet2 deficiency resulted in increased Il1b and Il6 expression in response to inflammatory stimuli, which appeared to be independent of TET2 enzymatic activity on the basis of experiments with cultured macrophages expressing a catalytically inactive mutant TET2. This was associated with increased histone acetylation over the Il1b promoter and reversal of the difference by HDAC inhibitors, implicating increased histone acetylation. Tet2-deficient macrophages exhibited a widespread increase in the expression of inflammatory genes, including increased mRNA for Il1b and Nlrp3, indicating increased inflammasome priming, and secreted increased IL-1β in response to lipopolysacchardie and ATP, indicating increased inflammasome activation. These studies provide strong evidence for increased l1b transcription and NLRP3 inflammasome priming in Tet2-deficient macrophages and a central role of the NLRP3 inflammasome in Tet2−/− CH-accelerated atherosclerosis (Fig. 2a).
Fig. 2 |. Mutation-specific mechanisms linking clonal hematopoiesis to atherosclerotic cardiovascular disease.
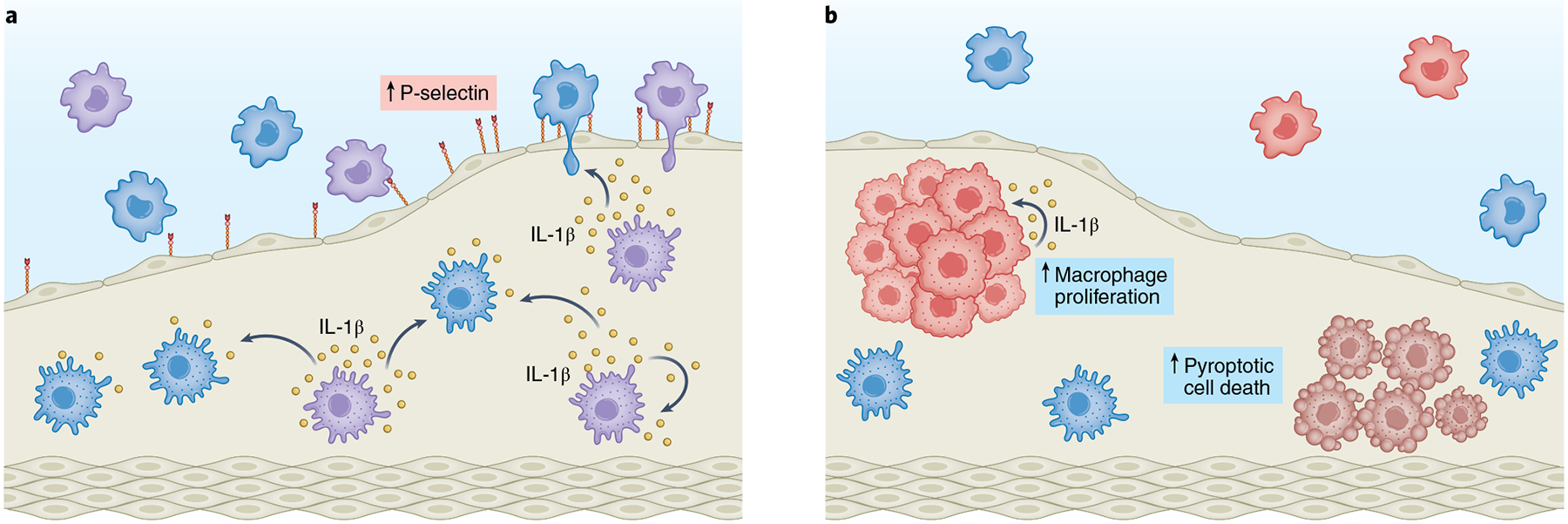
Emerging evidence supports that different mutant genes promote inflammation and atherosclerosis through diverse mechanisms. a, TET2-deficient macrophages (purple cells) exhibit increased IL-1β transcript levels and higher NLRP3 inflammasome-mediated IL-1β secretion. Heightened IL-1β levels in the plaque promote P-selectin expression and endothelial cell activation, leading to increased recruitment of circulating leukocytes. IL-1β may further stimulate its own expression in both TET2-mutant cells and WT macrophages (light blue cells) through an autoregulatory autocrine–paracrine feedback loop. b, JAK2VF expression in macrophages leads to an overactivation of the AIM2 inflammasome, which increases JAK2VF mutant macrophage proliferation (red cells) in a manner dependent on intrinsic JAK2VF function and IL-1β signaling. Increased AIM2 inflammasome activity also leads to greater pyroptotic cell death of mutant macrophages (darker red cells), which contributes to the expansion of necrotic cores within the atherosclerotic plaque.
JAK2VF.
JAK2V617F is a gain-of-function mutation that is associated with myeloproliferative neoplasms (MPNs) and an increased risk of atherothrombotic disease. In exome-wide sequencing studies, JAK2VF was associated with increased CAD despite lower levels of LDL26. Among the more frequent CHIP variants that associate with CVD, the JAK2 mutation (JAK2(V617F)) is less common but occurs at a younger age and has the greatest effect on CVD risk27. In a meta-analysis of four studies, JAK2VF endowed a 12-fold increase in risk of CAD compared with about a 2-fold increase for the other variants14. CHIP, especially that involving JAK2VF, has also been associated with increased risk of venous thrombo-embolic disease. In a general European population, JAK2VF was detected by droplet digital PCR in 3.1% of adults not selected for age28. JAK2VF increased thrombotic risk at allele frequency 1% or more28. In a study from Japan, JAK2VF allele frequencies of less than 2% were associated with increased CVD29. Reconstruction of clonal histories using whole-genome sequencing of single-cell hematopoietic stem cell (HSC)-derived colonies from people with MPNs has shown that Jak2VF mutations can be acquired several decades earlier than the appearance of MPN30, emphasizing the disease-potentiating effect of JAK2VF from the early years of life.
Mice with a conditional knock-in of Jak2VF and pan-hematopoietic expression of Jak2VF displayed accelerated atherosclerosis despite lower LDL-cholesterol levels31. Mice with macrophage-specific expression of Jak2VF and Jak2VF chimeric mice modeling CHIP displayed increased lesion area, increased proliferation of macrophages and larger necrotic cores32. In contrast to Tet2−/− CH mice25, there was little or no expansion of Jak2VF cells in blood. However, atherosclerotic lesions showed an enrichment of Jak2VF cells relative to the bloodstream, reflecting both increased recruitment of Jak2VF monocytes likely owing to increased surface expression of integrins33, as well as increased cell-autonomous proliferation of Jak2VF macrophages in lesions. Deletion of Casp1 and Casp11, which encode essential inflammasome components, in Jak2VF mice abrogated increased proliferation of macrophages in lesions; moreover, features of plaque stability, including macrophage accumulation, cap thickness, and necrosis, were markedly improved. Inflammasome activation in Jak2VF macrophages increased secretion of IL-1β, which increased AKT and ERK signaling and thereby led to both increased proliferation and pyroptotic cell death (Fig. 2b). Proliferating Jak2VF macrophages displayed increased glycolysis and mitochondrial respiration, as well as increased mitochondrial reactive oxygen species (ROS) and oxidized DNA. Increased mitochondrial ROS can lead to oxidation of DNA and activation of NLRP3 inflammasomes34 but can also promote damage of nuclear DNA, formation of double-stranded DNA breaks and activation of AIM2 inflammasomes35. Immunofluorescence staining of lesions from mice with Jak2VF CH showed accumulation of oxidized DNA and pγH2AX-positive macrophages, which suggests ROS-mediated oxidative DNA damage and replication stress. Jak2VF bone-marrow-derived macrophages (BMDMs) and human induced-pluripotent-stem-cell-derived macrophages showed increases in both polydAdT-mediated AIM2 activation and ATP-mediated NLRP3 inflammasome activation. However, AIM2 protein was increased, whereas NLRP3 was decreased. IFNγ-neutralizing antibodies reversed the increase in Aim2 expression in Jak2VF BMDMs, indicating a direct link between JAK2 gain-of-function, IFNγ signaling and Aim2 expression. Whereas Nlrp3 deficiency had no significant effect on lesion or necrotic core areas, Aim2 deficiency markedly reduced both. Consistent with cell culture studies, AIM2 levels were increased in Jak2VF lesions. Single-cell RNA sequencing (scRNA-seq) studies showed an increase in inflammatory, proliferative myeloid populations in Jak2VF lesions that were reduced by Gsdmd deficiency, but also revealed complex changes, with a reduced population of noninflammatory Trem2hi macrophages and a potentially adverse increase in monocytes and pyroptotic ‘zombie’ macrophages in Gsdmd−/− mice. Jak2VF chimeric mice treated with antibodies against IL-1β showed enhanced features of plaque stability, including decreased macrophage density, decreased necrotic cores and increased fibrous-cap thickness. These reciprocal changes did not result in an overall change in plaque size. Even though mice do not reliably rupture atherosclerotic plaques, such changes in macrophage density, necrotic core size and fibrous-cap thickness have been implicated in plaque stabilization in humans36.
DNMT3A.
The most common genetic variants in CH involve DNMT3A, which encodes a methyltransferase enzyme that catalyzes DNA methylation at CpG sites and is a critical epigenetic regulator of gene expression37. The majority of pathogenic mutations in DNMT3A are loss-of-function, including missense mutations in regulatory and catalytic domains as well as nonsense and dominant negative mutations. DNMT3A pathogenic variants enhance HSPC self-renewal and reduce differentiation potential. Paradoxically, DNMT3A and TET2 have opposing enzymatic activities with regard to DNA methylation while their reduced function in CHIP has parallel effects on atherosclerosis38 and heart failure4,14,39. Both Tet2−/− and Dnmt3a−/− CH increased atherosclerosis and promoted accumulation of a distinctive population of resident-like, inflammatory macrophages by scRNA-seq38. TET2 and DNMT3A CHIP both associate with epigenetic aging and have largely similar associations with reduced methylation of CpG islands20. However, there seems to be some differences in the mechanisms linking Tet2 and Dnmt3a CHIP to CVD. Monoallelic DNMT3A inactivation in macrophage-like cells (derived from embryonic stem cells) causes manifold complex alterations in immunomodulatory gene expression, with increased expression of many chemokine genes and IL6, but decreased expression of TLR4-responsive genes, including IL1B40. CHIP mutations have recently been associated with increased osteoporosis in the UK Biobank and in mouse models41. In Dnmt3a−/− CHIP mice, increased osteoporosis reflected increased osteoclastogenesis as a result of increased inflammatory gene expression and increased IL-20 secretion by Dnmt3a−/− macrophages. While ELISA assays from BMDM and RAW264.7 macrophage cultured medium showed increased IL-20, IL-1β levels were not increased in Dnmt3a−/− relative to the level in WT cultures. Together, these studies suggest distinctive effects of Tet2 and Dnmt3a mutations on macrophage inflammatory activities. Further studies are needed to resolve whether TET2-mutant CHIP and DNMT3A-mutant CHIP are impacting CVD by distinctive or convergent pathways.
PPM1D and TP53.
These DDR CHIP variants are increased in individuals who have been exposed to chemotherapy or radiation and are associated with increased risk of incident coronary and peripheral artery disease15. In mice, competitive bone marrow transplantation (BMT) of 20% Trp53−/− with 80% WT BM leads to an increase in early atherosclerotic lesions in the proximal aorta15. p53-deficient cells are moderately expanded in the BM and blood and incrementally increased in plaques relative to the bloodstream, likely as a result of increased p53-deficient macrophage proliferation within plaques. However, in contrast to findings with Tet2−/− and Jak2VF CHIP models, TP53-mutant CHIP plaques did not show evidence of increased Il1b or Il6 mRNA, providing further evidence for diverse mechanisms that increase atherosclerosis in murine CHIP models.
CHIP and thrombotic disease
It has long been known that JAK2VF individuals with myeloproliferative neoplasms have an increased risk of venous and arterial thrombosis, leading to thrombo-embolism and ischemic heart disease42. In mice, haematopoietic Jak2VF increased venous thrombosis in association with increased formation of neutrophil extracellular traps (NETs) within thrombi, whereas neutrophils from people with myeloproliferative disorders showed increased NETosis43. Genetic deficiency of PAD4, an essential enzyme in NET formation, appeared to decrease venous thrombosis. Another study identified pleckstrin-2 (PLEK2) as a downstream target of the JAK2–STAT5 pathway in erythroid and myeloid cells and showed that it is upregulated in a Jak2VF mouse model and in people with myeloproliferative disorders44. Loss of PLEF2 ameliorated JAK2VF-induced myeloproliferative phenotypes, thereby reverting the widespread vascular occlusions and lethality in Jak2VF knock-in mice. A reduction in red blood cell mass was the main contributing factor in the reversion of vascular occlusions. Together, these studies suggest that erythrocytosis, leukocytosis and intrinsic red blood cell and white blood cell abnormalities increase thrombotic risk in Jak2VF myeloproliferative disorders and could potentially also contribute to the increased thrombotic risk in JAK2VF CH.
LNK (also known as SH2B3) inhibits JAK–STAT signaling by hematopoietic cytokine receptors. Genome-wide association studies have shown the association of a common single nucleotide polymorphism in LNK (R262W, T allele) with neutrophilia, thrombocytosis and CAD45. LNK(TT) reduces LNK function and thus increases signaling downstream of JAK2(V617F) (ref.46). Lnk-deficient mice show accelerated atherosclerosis and arterial thrombosis. Increased arterial thrombosis reflected increased NET formation in thrombi47. NETosis was dependent on oxidized phospholipid and p-selectin released by activated platelets; consequently, accelerated thrombosis and atherosclerosis were reversed by an antibody targeting oxidized phospholipids. In the UK Biobank, individuals with the JAK2VF mutation only showed increased CAD when also carrying the LNK(R262W) allele47. Modeling CHIP in mice with all hematopoietic cells being Lnk+/– and with Jak2VF induced in a subset of hematopoietic cells showed accelerated thrombosis, but not atherosclerosis. This suggests that the increased CAD in JAK2VF individuals with LNK(R262W) might primarily reflect increased thrombotic risk.
CHIP and heart failure
HF arises when the heart cannot pump enough blood to meet the body’s needs. HF is a heterogeneous clinical syndrome that can be driven by a variety of etiologies, including hypertension and CAD, and it may involve infiltration of the myocardium with inflammatory cells and fibrotic remodeling. Recent epidemiological studies have demonstrated a robust link between CH and HF. In a meta-analysis of >56,000 participants in 5 cohorts who were free of HF at baseline, carriers of a CHIP mutation exhibited a 25% increased risk of incident HF, independent of traditional risk factors4. Single-gene-specific analyses suggested a differential effect of mutations in different genes in this context. Mutations in TET2, JAK2 and ASXL1 were separately associated with risk of incident HF that was increased by 1.5- to 2.5-fold, whereas the more prevalent mutations in DNMT3A were not associated with HF risk. CH has also been investigated in cohorts of people with established HF, in whom it is highly prevalent, being typically detected in approximately one-third of individuals with reduced left ventricular ejection fraction5,48,49. In this setting, carriers of CHIP-linked mutations exhibit a substantially higher risk of adverse clinical progression, defined as higher risk of death or composites of all-cause or HF-specific death and HF-related hospitalizations5,6,48,50,51. Both DNMT3A and TET2 mutations have been associated with worse outcomes in individuals with HF5,6,50, and emerging evidence suggests that less frequent CH variants in other genes may also have predictive value48,51. The association between CH and HF is similar in individuals with and without coronary heart disease4,5, which suggests a direct connection between CH and HF pathophysiology and argues against the possibility that these associations merely reflect the strong association between CH and atherosclerosis or effects of myocardial ischemia on the hematopoietic system. The observations in these epidemiological studies are also consistent with the idea that the clinical significance of CH may depend on the specific mutated gene and, in some cases, the specific context (that is, onset versus progression of HF).
Altered inflammatory responses contribute to the pathogenesis and progression of HF52, and CH has been shown to correlate with heightened systemic inflammation in people with HF. Circulating IL-6 levels are increased in individuals with HF with reduced ejection fraction and CH mutations in either DNMT3A or TET25. In addition, scRNA-seq studies have shown that blood monocytes from a small sample of individuals with HF carrying DNMT3A mutations exhibit increased expression of IL6, IL1B, IL8 and NLRP3, several chemokines and their receptors, and cell surface receptors involved in monocyte–T cell interactions53. An important question is whether the association between CH and HF differs between HF with reduced left ventricular ejection fraction and HF with preserved ejection fraction.
TET2.
TET2 is the CH driver gene most extensively studied in the context of HF. Sano et al. found that competitive transplantation of Tet2−/− BM cells in a 10:90 ratio with WT BM leads to greater left ventricular dysfunction after surgical ligation of the left anterior descending (LAD) artery54. This impaired cardiac function was paralleled by substantial increases in myocardial fibrosis, myocyte hypertrophy and macrophage content within the cardiac tissue. Tet2-deficient CH also worsened cardiac function and remodeling in an experimental model of non-ischemic pressure-overload-induced cardiac hypertrophy, achieved by transverse aortic constriction (TAC). In both experimental models, myeloid-restricted ablation of Tet2 was sufficient to aggravate cardiac dysfunction. Mechanistic studies indicated that overproduction of IL-1β by TET2-deficient macrophages and the ensuing exacerbation of cardiac inflammation has a central role in these aggravated HF phenotypes. Accordingly, the detrimental cardiac effects of hematopoietic TET2 deficiency, including increased fibrosis, were suppressed by the NLRP3 inhibitor MCC950. A study based on lentiviral-vector-mediated CRISPR–Cas9 editing of Tet2 corroborated the damaging effects of loss of function of TET2 in an experimental model of hypertension and cardiac dysfunction induced by angiotensin II infusion39. Furthermore, TET2-deficient CH has also been shown to accelerate age-related cardiac dysfunction and fibrotic remodeling in the absence of external insults in a model of CH achieved by non-conditioned BM transplantation55. Collectively, these studies provide strong evidence for a direct contribution of TET2-mutant CH to accelerated HF development and support a central role of NLRP3–IL-1β-driven inflammation, similar to the scenario in atherosclerosis.
DNMT3A.
The only report so far on the effects of DNMT3A-mutant CH on cardiac function showed that angiotensin II infusion induced greater cardiac dysfunction, fibrotic remodeling and immune cell infiltration into cardiac tissue in mice carrying Dnmt3a−/−-mutant cells than in controls39. However, these findings need to be interpreted with caution, as mutations were generated using a lentiviral-vector-mediated CRISPR–Cas9 approach, and the potential off-target effects of this intervention were not investigated. Furthermore, it remains unknown whether DNMT3A deficiency affects cardiac function in experimental conditions other than after chronic infusion of angiotensin II levels.
JAK2VF.
Similar to the strategy used in experimental atherosclerosis studies, myeloid-restricted JAK2VF expression has been used to model JAK2-mutant CH in the absence of major hematological abnormalities in experimental HF studies. Sano et al.56 used an ex vivo lentivirus-mediated approach to overexpress the JAK2VF mutation specifically in myeloid cells under control of a combined synthetic SP146 promoter–gp91 enhancer combination. In chronic LAD ligation and TAC conditions, myeloid JAK2VF-expressing mice showed accelerated cardiac dysfunction and fibrosis, as well as heightened expression of proinflammatory cytokines in cardiac tissue, supporting the notion that the JAK2VF mutation can contribute to accelerated HF development.
DDR gene mutations.
CHIP mutations affecting DDR genes are also being investigated in the context of cardiac disease, particularly as potential contributors to HF in cancer survivors, in whom these mutations are considerably more prevalent than in the general population owing to the selective pressure associated with genotoxic therapies57. Hematopoietic TP53 mutations have been reported to exacerbate doxorubicin-induced cardiac toxicity in mice, in parallel with increased neutrophil infiltration into the myocardium and elevated expression of IL6, IL1B and TNF58, which encode proinflammatory cytokines. CHIP mutations in the DDR-related phosphatase PPM1D have also been investigated in the context of experimental cardiac disease. These mutations typically result in a C-terminal-truncated protein that exhibits increased stability and activity, which has been investigated through an ex vivo lentivirus-mediated CRISPR–Cas9 approach coupled to BMT. Using this strategy, PPM1D-mutant-cell-carrying mice exhibit increased angiotensin-II-induced cardiac hypertrophy and fibrosis, as well as worsened cardiac function, compared with control mice59. Mechanistically, this cardiac phenotype has been linked to augmented expression of IL-1β and IL-18 expression in PPM1D-mutant myeloid cells.
CHIP and reverse causation
While mouse models mimicking CHIP variants show worsened atherosclerosis and HF, indicating causation, a recent study has suggested that atherosclerosis could accelerate expansion of CH mutant clones by promoting HSPC proliferation, indicating reverse causation12. Mathematical modeling showed that the combination of a CH variant that endows an intrinsic advantage to HSPCs with extrinsic factors that increase the rate of HSPC proliferation will inevitably amplify the CH allele burden over time. For example, inflammatory cytokines associated with accelerated atherosclerosis could promote proliferation of HSPCs, resulting in accelerated expansion of mutant cells. Accordingly, in Tet2−/− CHIP mice the expansion of Tet2−/− HSPCs was increased by hyperlipidemia and atherosclerosis and by sleep-fragmentation-accelerated atherosclerosis. Infectious stimuli can also promote CH. Chronic myco-bacterial infection increased expansion of mutant HSPCs in a Dnmt3a−/− CHIP model60. This effect was mediated through IFNγ and involved decreased differentiation and decreased apoptosis of Dnmt3a−/− HSPCs with widespread increases in gene methylation. Similarly, lipopolysaccharide treatment augmented the competitive advantage of TET2-deficient HSPCs in mice, owing to an overactivation of IL-6 signaling and improved TET2-deficient HSPC survival61. Consistent with bidirectional causation, a study in zebrafish suggests that enhanced fitness of ASXL1 mutant HSCs arises from increased expression of anti-inflammatory genes, which endows resistance to inflammatory signals arising from mutant mature cell progeny62.
Dyslipidemia involving elevated levels of atherogenic lipoproteins, decreased levels of HDL, or defects in cholesterol efflux genes Abca1 and Abcg1, could also lead to both increased atherosclerosis and expansion of CHIP mutant HSPCs63. Dyslipidemia acts both at the level of cells in atherosclerotic plaques and in BM HSPCs to stimulate their proliferation. In Apoe−/− mice, HSPC proliferation and myeloid bias increase production of monocytes that enter the artery wall in increased amounts, promoting atherosclerosis64. Hyperlipidemia promoted increased proliferation of HSPCs containing the Jak2VF mutation compared to chow-fed JAK2VF mice or WT controls31. In humans, a Mendelian randomization study showed that single nucleotide polymorphisms in genes associated with lower HDL levels are associated with higher WBC counts65, which in turn are associated with increased CVD in epidemiological studies66. CHIP variants are associated with type 2 diabetes14,67. Tet2−/− CHIP has been shown to foster insulin resistance in aging and obese mice with increased IL-1β in adipose tissue and reversal by the NLRP3 inhibitor MCC950 (ref.68). Together, these studies indicate the potential for a vicious cycle in which CHIP promotes insulin resistance and dyslipidemia that in turn induces expansion of Tet2−/− clones. Further prospective longitudinal studies in larger population samples are needed to ascertain the natural history of CH over time and to clarify the relationship between insulin resistance, dyslipidemia and emergence of CHIP.
Another potential example of reverse causation is the association of CHIP in later life with premature menopause and CVD69. In postmenopausal women, the presence of CHIP involving DNMT3A was associated with natural, but not surgical, premature menopause (age < 40). A genome-wide association study examining age of menopause revealed that the large majority of associations are for genes involved in DDR70. Causal inference analysis indicates that variants associated with early ovarian failure were associated with type 2 diabetes. This suggests a potential pathway in which genetic predisposition to early menopause promotes type 2 diabetes that in turn may facilitate the emergence of CH.
Although a vicious cycle connecting CHIP with atherosclerosis and other diseases is highly plausible71, the magnitude and consistency of this effect is uncertain72. In humans with atherosclerosis, the rate of HSPC proliferation over decades may not be as high as suggested by its measurement at a single time point in people of uncertain clinical status12. IL-6 is a key inflammatory factor in atherosclerosis that also increases HSPC proliferation73, but individuals with TET2 or DNMT3A CHIP and reduced-function IL-6R variants had reduced CVD without a change in CHIP allele burden74, even though TET2 and DNMT3A CHIP carriers have elevated IL-6 levels74. The finding that atherosclerosis promoted expansion of Tet2−/− BM progenitors12 conflicts with earlier studies in which Tet2−/− BM progenitor and blood cells expanded similarly in mice fed chow diets (without atherosclerosis) or fed high-cholesterol diets (with early atherosclerosis); moreover, inhibition of NLRP3 reduced atherosclerosis without diminishing the expansion of Tet2−/− cells25. Also, Jak2VF allele burden in blood cells did not increase over time in CHIP atherosclerotic mice32. Nonetheless, the possibility of bidirectional causation, especially over longer time periods or with more advanced atherosclerosis, remains likely. This highlights the importance of mechanistic studies in mouse models in which the CH variant is clearly initiating atherosclerosis and reverse causation can be assessed by measurements of HSPC and myeloid progenitor populations in bone marrow and blood under atherosclerotic or non-atherosclerotic conditions. Further research is needed to disentangle forward and reverse causation and the underlying mechanisms in the association between CHIP and CVD. From a therapeutic perspective, inflammatory cytokines such as IL-1β, IL-6 or IFNγ60 or dyslipidemia could be driving both CHIP emergence and atherosclerosis and are thus attractive targets for therapeutic control of atherosclerosis and potentially CHIP emergence.
Therapeutic approaches
Despite the success of LDL-lowering treatments, atherosclerotic CVD remains the major cause of morbidity and mortality in the United States and accounts for 28% of deaths75. Residual CVD remains substantial even in clinical trials with marked lowering of LDL cholesterol76–78, pointing to the need for development of new treatments. Recent clinical trials using anti-inflammatory therapies, notably IL-1β antibodies79 or colchicine80,81, have shown a reduction in CVD. IL-1β antibodies inactivate a major product of all inflammasomes, while colchicine inhibits the microtubule-dependent assembly of the NLRP3 inflammasome and IL-1β secretion82,83 and has other anti-inflammatory activities84. These studies have validated the importance of inflammation and inflammasomes in human atherothrombotic disease. However, IL-1β treatment caused an increase in fatal infections, whereas colchicine doubled the risk of pneumonia. IL-1β antibodies have not been marketed for CVD indications, and the therapeutic potential of colchicine may be limited85. This indicates that more precise targeting of anti-inflammatory treatments to individuals with higher levels of inflammatory CVD risk is essential to improve the benefit/risk ratio. The presence of CHIP mutations may help to identify such a population of people. CHIP carriers displayed increased aging biomarkers, suggesting that individuals with CHIP and increased aging biomarkers might represent a risk-enriched population that could be the target of anti-inflammatory therapies20. The identification of inherited variants that modulate CHIP-related CVD risk74 may also help identify high-risk individuals in this context. The treatments to reduce CVD risk in CHIP carriers could involve targeting upstream inflammatory factors, such as NLRP3 or AIM2 inflammasomes, downstream common mediators, such as IL-1β or IL-6, or specific driver gene mutations, such as those involving TET2 or JAK2.
Inhibition of inflammasome components.
In mouse CHIP models, Tet2 deficiency activated the NLRP3 inflammasome, while JAK2VF activated the AIM2 inflammasome. This suggests that specific inhibitors of NLRP3 or AIM2 inflammasomes may be required to treat CHIP-associated CVD according to CH status. A variety of NLRP3 inhibitors are in development including those that target the BRCC3/ABRO1 pathway that mediates specific deubiquitination and activation of NLRP386. Clinical trials using the NLRP3 inhibitor CRID3 (the same molecule as MCC950 in a different formulation) to treat rheumatoid arthritis were stopped owing to hepatotoxicity87. Inhibitors of the AIM2 inflammasome have been reported to increase features of plaque stability in Apoe−/− mice88; however, the specificity of such oligonucleotide inhibitors is uncertain. Whether direct targeting of inflammasomes reduces CVD risk without increasing infections remains to be determined.
Inhibition of pathways downstream of inflammasomes.
NLRP3 and AIM2 inflammasome activation that have been implicated in CHIP-promoted atherosclerosis leads to secretion of active IL-1β and IL-18. Plasma IL-1β is increased in TET2 CHIP, whereas IL-18 is increased in JAK2VF CHIP27. Given the positive outcome of CANTOS, and the results of preclinical studies cited above, IL-1β antagonism seems like a logical first choice for targeting TET2 and possibly JAK2VF CHIP-associated CVD. IL-18 has a major role in the production of IFNγ by T cells and natural killer cells89 and increases atherosclerosis in mice via release of IFNγ (ref.90) and other mechanisms91. IFNγ is potently atherogenic92 and has a specific role in JAK2VF-promoted atherosclerosis via upregulation of AIM2 (ref.32). The therapeutic potential of IL-18 inhibition in CHIP-associated atherosclerosis deserves further exploration.
IL-6 may be increased downstream of inflammasome activation; IL-1 and IL-6 levels were moderately increased in the three commonest forms of CHIP that associate with CVD (TET2, DNMT3A and ASXL1)27. However, this was not associated with increases in CRP levels. The increased CVD risk associated with TET2- and DNMT3A-mutant CHIP was abrogated by the D358A variant of IL-6R, pointing to a key role of IL-6 in the development of CVD in people with CHIP74. In another study, this variant did not impact total mortality or CHD in overall CHIP; however, protection was seen in the subgroup of individuals with TET2 CHIP and aging biomarkers20. Direct signaling of IL-6 occurs when it binds to its receptor (IL-6R) in the liver and WBCs. This leads to cleavage of the IL6R α-chain that forms a circulating complex with IL-6; the complex interacts with gp130 in a variety of cells to mediate trans-signaling93. The D358A variant is associated with reduced cell surface expression of IL-6R along with an increase in soluble IL6R receptor that has the potential to mediate trans-signaling94. Several studies have implicated IL-6 in the pathogenesis of atherosclerosis in mice95, especially the trans-signaling pathway96. However, knockout of the Il6 gene in Apoe−/− mice exacerbated hyperlipidemia and promoted advanced atherosclerosis93,97. Despite this complexity and apparent inconsistency, it remains plausible that targeting IL-6 might improve the net therapeutic benefit in selected individuals with atherosclerotic CVD93. One complication of IL-6 antagonism with tocilizumab in people with rheumatoid arthritis has been elevated levels of triglycerides, non-HDL-cholesterol and APOB98. The IL-6 antagonist ziltivekimab is being developed for chronic-kidney-disease associated atherosclerotic disease and was found to reduce inflammatory biomarkers in a phase 2 trial; however, this appeared to be accompanied by an increase in APOB levels99. Further assessment of IL-6 antagonism combined with rigorous control of plasma lipoprotein levels as a potential treatment option for CHIP-associated CVD is warranted.
CHIP-mutation-specific therapies.
Vitamin C metabolites activate TET2 and can mimic restoration of TET2 by enhancing 5-hydroxymethycytosine formation in TET2-deficient mice, leading to reversal of aberrant HSC self-renewal; this could represent a potential preventive treatment for TET2 CHIP carriers to prevent CVD100,101. JAK2VF, although less common than TET2 and DNMT3A variants, has important attributes that make it an attractive target for therapy to reduce CVD. JAK2VF increases venous and arterial thrombotic disease and atherosclerosis considerably more than the other CHIP variants, likely increases disease at lower allele burden than other variants, occurs at a younger age and is amenable to multiple approaches to inhibition that may prove beneficial for both MPN and CHIP. In JAK2VF CHIP, treatment with the JAK1/JAK2 inhibitor ruxolitinib reduced abnormal NET formation and deep vein thrombosis43. Ruxolitinib causes substantial increases in LDL cholesterol in JAK2VF mice and humans. In a JAK2VF mouse CHIP model, ruxolitinib treatment led to the development of slightly smaller atherosclerotic lesions with markedly increased necrotic cores and thinner caps32, which are features of unstable plaques in humans. By contrast, JAK2 inhibition by fedratinib in Apoe−/− mice suppressed myelopoiesis and the development of atherosclerosis102. Ruxolitinib inhibits both JAK1 and JAK2 and thus could have broad effects on signaling pathways that are required for cell survival. Ruxolitinib also reduces oxidative metabolism of glucose in Jak2VF cells, leading to depletion of NADPH and glutathione with the potential to promote oxidative cell death103. While one should exercise caution in extrapolating from mouse models to humans, it is notable that the Food and Drug Administration recently issued a regulatory warning concerning potential increased thrombosis and CAD in people taking the related JAK inhibitors baricitinib, tofacitinib and upadacitinib for treatment of arthritis or ulcerative colitis. This could indicate the need for more specific targeting of JAK2 or JAK2VF. To reduce athero-thrombotic risk, people carrying JAK2VF might benefit from more effective anti-thrombotic therapy, such as multiple dosing with aspirin, to compensate for increased platelet production104, or specific JAK2 inhibitors. Since platelet-associated oxidized phospholipids promote NETosis and arterial thrombosis in Lnk-deficient mice, anti-OxPL antibodies that reduced both atherosclerosis and arterial thrombosis in JAK2VF CVD could be a novel therapeutic approach47. LNK inhibits both JAK2 and JAK2VF signaling105. LNK increases the association of JAK2 with the E-3 ligase CBL, promoting its ubiquitination and degradation106. Thus, increasing the activity of the E-3 ligase CBL on JAK2VF, for example, via proteolysis targeting chimeras (PROTACs), could lead to increased JAK2VF degradation and thus a reduction in MPN and CVD.
Outlook
The convergence of recent evidence showing the efficacy of anti-inflammatory treatments in atherosclerotic CVD with the discovery that CHIP promotes atherosclerosis via increased plaque inflammation and inflammasome activation presents an important new therapeutic opportunity that could potentially lead to an acceptable risk/benefit ratio of anti-inflammatory therapy in individuals with CHIP. A variety of potential therapies may be considered, but those targeting common downstream factors, such as IL-1β or IL-6, and CHIP-mutation-specific therapies appear to be most promising. Future clinical trials enriched for individuals with CHIP and high atherosclerotic risk will be required to establish the efficacy and safety of such approaches.
Acknowledgements
A.T. and J.J.F. are supported by a grant from the Leducq Foundation (TNE-18CVD04). A.T. is supported by NIH grant 155431. We thank M. A. Zuriaga for assistance with figure design.
Footnotes
Competing interests
A.T. is a consultant for Amgen and CSL and is on the scientific advisory board of Staten Biotech and 1016 Biotech. J.J.F. declares no potential conflicts of interest.
References
- 1.Marnell CS, Bick A & Natarajan P Clonal hematopoiesis of indeterminate potential (CHIP): linking somatic mutations, hematopoiesis, chronic inflammation and cardiovascular disease. J. Mol. Cellular Cardiol 161, 98–105 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steensma DP et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 126, 9–16 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jaiswal S et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med 371, 2488–2498 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; These authors discovered that people with clonal hematopoiesis were at increased risk of developing both hamatological malignancies and atherosclerotic cardiovascular disease.
- 4.Yu B et al. Supplemental association of clonal hematopoiesis with incident heart failure. J. Am. Coll. Cardiol 78, 42–52 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; In a meta-analysis of different studies some, but not all, CHIP variants were found to be associated with an increased incidence of heart failure.
- 5.Pascual-Figal DA et al. Clonal hematopoiesis and risk of progression of heart failure with reduced left ventricular ejection fraction. J. Am. Coll. Cardiol 77, 1747–1759 (2021). [DOI] [PubMed] [Google Scholar]
- 6.Dorsheimer L et al. Association of mutations contributing to clonal hematopoiesis with prognosis in chronic ischemic heart failure. JAMA cardiology 4, 25–33 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mas-Peiro S et al. Clonal haematopoiesis in patients with degenerative aortic valve stenosis undergoing transcatheter aortic valve implantation. Eur. Heart J 41, 933–939 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jaiswal S & Ebert BL Clonal hematopoiesis in human aging and disease. Science 366, eaan4673 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jaiswal S & Libby P Clonal haematopoiesis: connecting ageing and inflammation in cardiovascular disease. Nat. Rev. Cardiol 17, 137–144 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Libby P et al. Clonal hematopoiesis: crossroads of aging, cardiovascular disease, and cancer: JACC review topic of the week. J. Am. Coll. Cardiol 74, 567–577 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yura Y, Sano S & Walsh K Clonal hematopoiesis: a new step linking inflammation to heart failure. JACC Basic Transl. Sci 5, 196–207 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heyde A et al. Increased stem cell proliferation in atherosclerosis accelerates clonal hematopoiesis. Cell 184, 1348–1361 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study provides evidence in mice and humans that atherosclerosis can promote the expansion of haematopoietic stem cells containing CH mutations suggesting reverse or bidirectional causation.
- 13.Libby P The changing landscape of atherosclerosis. Nature 592, 524–533 (2021). [DOI] [PubMed] [Google Scholar]
- 14.Jaiswal S et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. New Engl. J. Med 377, 111–121 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zekavat SM et al. TP53-mediated clonal hematopoiesis confers increased risk for incident peripheral artery disease. Preprint at medRxiv 10.1101/2021.08.22.21262430 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Young AL, Challen GA, Birmann BM & Druley TE Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat. Commun 7, 12484 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hannum G et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Molecular cell 49, 359–367 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Levine ME et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging 10, 573–591 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robertson NA et al. Age-related clonal haemopoiesis is associated with increased epigenetic age. Curr. Biol 29, R786–R787 (2019). [DOI] [PubMed] [Google Scholar]
- 20.Nachun D et al. Clonal hematopoiesis associated with epigenetic aging and clinical outcomes. Aging Cell 20, e13366 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Busque L et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat. Genet 44, 1179–1181 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ko M et al. Ten-eleven-translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. PNAS 108, 14566–14571 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moran-Crusio K et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 20, 11–24 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Q et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 525, 389–393 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fuster JJ et al. Clonal hematopoiesis associated with Tet2 deficiency accelerates atherosclerosis development in mice. Science 355, 842–847 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrated accelerated atherosclerosis in a mouse model of Tet2 deficiency CH and implicated NLRP3 inflammasome activation as a major underlying mechanism.
- 26.Liu DJ et al. Exome-wide association study of plasma lipids in >300,000 individuals. Nat. Genet 49, 1758–1766 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bick AG et al. Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature 586, 763–768 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cordua S et al. Prevalence and phenotypes of JAK2V617F and calreticulin mutations in a Danish general population. Blood 134, 469–479 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Yokokawa T et al. Crucial role of hematopoietic JAK2V617F in the development of aortic aneurysms. Haematologica 106, 1910–1922 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luque Paz D, Ashcroft P & Skoda RC Myeloproliferative neoplasms: the long wait for JAK2-mutant clone expansion. Cell Stem Cell 28, 359–361 (2021). [DOI] [PubMed] [Google Scholar]
- 31.Wang W et al. Macrophage inflammation, erythrophagocytosis, and accelerated atherosclerosis in Jak2 (V617F) mice. Circ. Res 123, e35–e47 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fidler TP et al. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature 592, 296–301 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; These authors implicate AIM2 inflammasome activation in the accelerated atherosclerosis of Jak2VF clonal hematopoiesis mice and show beneficial effects of treatment with IL-1β antibodies.
- 33.Edelmann B et al. JAK2-V617F promotes venous thrombosis through β1/β2 integrin activation. J. Clin. Invest 128, 4359–4371 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shimada K et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36, 401–414 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hu B et al. The DNA-sensing AIM2 inflammasome controls radiation-induced cell death and tissue injury. Science 354, 765–768 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nilsson J & Hansson GK The changing face of atherosclerotic plaque inflammation. J. Intern. Med 278, 430–432 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Challen GA & Goodell MA Clonal hematopoiesis: mechanisms driving dominance of stem cell clones. Blood 136, 1590–1598 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rauch PJ et al. Loss-of-function mutations in Dnmt3a and Tet2 lead to accelerated atherosclerosis and convergent macrophage phenotypes in mice. Blood 132, 745 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Sano S et al. CRISPR-mediated gene editing to assess the roles of Tet2 and Dnmt3a in clonal hematopoiesis and cardiovascular disease. Circ. Res 123, 335–341 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lim JY et al. DNMT3A haploinsufficiency causes dichotomous DNA methylation defects at enhancers in mature human immune cells. J. Exp. Med 218, e20202733 (2021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim PG et al. Dnmt3a-mutated clonal hematopoiesis promotes osteoporosis. J. Exp. Med 218, e20211872 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Falanga A & Marchetti M Thrombotic disease in the myeloproliferative neoplasms. Hematology/the Education Program of the American Society of Hematology. American Society of Hematology. Education Program 2012, 571–581 (2012). [DOI] [PubMed] [Google Scholar]
- 43.Wolach O et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci. Transl. Med 10, eaan8292 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao B et al. Loss of pleckstrin-2 reverts lethality and vascular occlusions in JAK2V617F-positive myeloproliferative neoplasms. J. Clin. Invest 128, 125–140 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gudbjartsson DF et al. Sequence variants affecting eosinophil numbers associate with asthma and myocardial infarction. Nat. Genet 41, 342–347 (2009). [DOI] [PubMed] [Google Scholar]
- 46.Wang W et al. LNK/SH2B3 loss of function promotes atherosclerosis and thrombosis. Circ. Res 119, e91–e103 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dou H et al. Oxidized phospholipids promote NETosis and arterial thrombosis in LNK(SH2B3) deficiency. Circulation 144, 1940–1954 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cremer S et al. Multiple somatic mutations for clonal hematopoiesis are associated with increased mortality in patients with chronic heart failure. Circ. Genom. Precis. Med 13, e003003 (2020). [DOI] [PubMed] [Google Scholar]
- 49.Palomo L et al. Prevalence and characteristics of clonal hematopoiesis in heart failure. Rev. Esp. Cardiol 74, 996–999 (2021). [DOI] [PubMed] [Google Scholar]
- 50.Assmus B et al. Clonal haematopoiesis in chronic ischaemic heart failure: prognostic role of clone size for DNMT3A- and TET2-driver gene mutations. Eur. Heart J 42, 257–265 (2021). [DOI] [PubMed] [Google Scholar]
- 51.Kiefer KC et al. Full spectrum of clonal haematopoiesis-driver mutations in chronic heart failure and their associations with mortality. ESC Heart Fail 8, 1873–1884 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Adamo L, Rocha-Resende C, Prabhu SD & Mann DL Reappraising the role of inflammation in heart failure. Nat. Rev. Cardiol 17, 269–285 (2020). [DOI] [PubMed] [Google Scholar]
- 53.Abplanalp WT et al. Clonal hematopoiesis-driver DNMT3A mutations alter immune cells in heart failure. Circ. Res 128, 216–228 (2021). [DOI] [PubMed] [Google Scholar]
- 54.Sano S et al. Tet2-mediated clonal hematopoiesis accelerates heart failure through a mechanism involving the IL-1β/NLRP3 inflammasome. J. Am. Coll. Cardiol 71, 875–886 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang Y et al. Tet2-mediated clonal hematopoiesis in nonconditioned mice accelerates age-associated cardiac dysfunction. JCI Insight 5, e135204 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sano S et al. JAK2V617F-mediated clonal hematopoiesis accelerates pathological remodeling in murine heart failure. JACC Basic Transl. Sci 4, 684–697 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bolton KL et al. Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat. Genet 52, 1219–1226 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sano S et al. TP53-mediated therapy-related clonal hematopoiesis contributes to doxorubicin-induced cardiomyopathy by augmenting a neutrophil-mediated cytotoxic response. JCI Insight 6, e146076 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yura Y et al. The cancer therapy-related clonal hematopoiesis driver gene Ppm1d promotes inflammation and non-ischemic heart failure in mice. Circ. Res 129, 684–698 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hormaechea-Agulla D et al. Chronic infection drives Dnmt3a loss-of-function clonal hematopoiesis via IFNγ signaling. Cell Stem Cell 28, 1428–1442(2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cai Z et al. Inhibition of inflammatory signaling in Tet2 mutant preleukemic cells mitigates stress-induced abnormalities and clonal hematopoiesis. Cell Stem Cell 23, 833–849 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Avagyan S et al. Resistance to inflammation underlies enhanced fitness in clonal hematopoiesis. Science 374, 768–772 (2021). [DOI] [PubMed] [Google Scholar]
- 63.Yvan-Charvet L et al. ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science 328, 1689–1693 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Murphy AJ et al. ApoE regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. J. Clin. Invest 121, 4138–4149 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Harslof M, Pedersen KM, Nordestgaard BG & Afzal S Low high-density lipoprotein cholesterol and high white blood cell counts: a mendelian randomization study. Arter. Thromb. Vasc. Biol 41, 976–987 (2021). [DOI] [PubMed] [Google Scholar]
- 66.Adamstein NH et al. The neutrophil–lymphocyte ratio and incident atherosclerotic events: analyses from five contemporary randomized trials. Eur. Heart J 42, 896–903 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bonnefond A et al. Association between large detectable clonal mosaicism and type 2 diabetes with vascular complications. Nat. Genet 45, 1040–1043 (2013). [DOI] [PubMed] [Google Scholar]
- 68.Fuster JJ et al. TET2-loss-of-function-driven clonal hematopoiesis exacerbates experimental insulin resistance in aging and obesity. Cell Rep. 33, 108326 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Honigberg MC et al. Premature menopause, clonal hematopoiesis, and coronary artery disease in postmenopausal women. Circulation 143, 410–423 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ruth KS et al. Genetic insights into biological mechanisms governing human ovarian ageing. Nature 596, 393–397 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lusis AJ A vicious cycle in atherosclerosis. Cell 184, 1139–1141 (2021). [DOI] [PubMed] [Google Scholar]
- 72.Sanchez-Cabo F & Fuster JJ Clonal haematopoiesis and atherosclerosis: a chicken or egg question? Nat. Rev. Cardiol 18, 463–464 (2021). [DOI] [PubMed] [Google Scholar]
- 73.Ogawa M Differentiation and proliferation of hematopoietic stem cells. Blood 81, 2844–2853 (1993). [PubMed] [Google Scholar]
- 74.Bick AG et al. Genetic IL-6 signaling deficiency attenuates cardiovascular risk in clonal hematopoiesis. Circulation 10.1161/CIRCULATIONAHA.119.044362 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; In people with TET2 or DNMT3A clonal hematopoiesis the increased risk of atherosclerotic CVD was reversed if they also carried a genetic variant in the IL-6 receptor that reduced its function.
- 75.Heron M Deaths: leading causes for 2015. Natl. Vital Stat. Rep 66, 1–76 (2017). [PubMed] [Google Scholar]
- 76.Vallejo-Vaz AJ et al. Triglyceride-rich lipoprotein cholesterol and risk of cardiovascular events among patients receiving statin therapy in the TNT trial. Circulation 138, 770–781 (2018). [DOI] [PubMed] [Google Scholar]
- 77.Cannon CP et al. Ezetimibe added to statin therapy after acute coronary syndromes. N. Engl. J. Med 372, 2387–2397 (2015). [DOI] [PubMed] [Google Scholar]
- 78.Sabatine MS et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N. Engl. J. Med 376, 1713–1722 (2017). [DOI] [PubMed] [Google Scholar]
- 79.Ridker PM et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med 377, 1119–1131 (2017). [DOI] [PubMed] [Google Scholar]
- 80.Nidorf SM et al. Colchicine in patients with chronic coronary disease. N. Engl. J. Med 383, 1838–1847 (2020). [DOI] [PubMed] [Google Scholar]
- 81.Tardif JC et al. Efficacy and safety of low-dose colchicine after myocardial infarction. N. Engl. J. Med 381, 2497–2505 (2019). [DOI] [PubMed] [Google Scholar]
- 82.Martinon F, Petrilli V, Mayor A, Tardivel A & Tschopp J Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–241 (2006). [DOI] [PubMed] [Google Scholar]
- 83.Misawa T et al. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat. Immunol 14, 454–460 (2013). [DOI] [PubMed] [Google Scholar]
- 84.Imazio M & Nidorf M Colchicine and the heart. Eur. Heart J 42, 2745–2760 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Newby LK Inflammation as a treatment target after acute myocardial infarction. N.Engl. J. Med 381, 2562–2563 (2019). [DOI] [PubMed] [Google Scholar]
- 86.Ren GM et al. Pharmacological targeting of NLRP3 deubiquitination for treatment of NLRP3-associated inflammatory diseases. Sci. Immunol 6, eabe2933 (2021). [DOI] [PubMed] [Google Scholar]
- 87.Mangan MSJ et al. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discovery 17, 588–606 (2018). [DOI] [PubMed] [Google Scholar]
- 88.Paulin N et al. Double-Strand DNA sensing Aim2 inflammasome regulates atherosclerotic plaque vulnerability. Circulation 138, 321–323 (2018). [DOI] [PubMed] [Google Scholar]
- 89.Dinarello CA, Novick D, Kim S & Kaplanski G Interleukin-18 and IL-18 binding protein. Front. Immunol 4, 289 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Whitman SC, Ravisankar P & Daugherty A Interleukin-18 enhances atherosclerosis in apolipoprotein E−/− mice through release of interferon-gamma. Circ. Res 90, E34–E38 (2002). [DOI] [PubMed] [Google Scholar]
- 91.Wang J et al. Interleukin 18 function in atherosclerosis is mediated by the interleukin 18 receptor and the Na-Cl co-transporter. Nat. Med 21, 820–826 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gupta S et al. IFN-γ potentiates atherosclerosis in ApoE knock-out mice. J. Clin. Invest 99, 2752–2761 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ridker PM & Rane M Interleukin-6 signaling and anti-interleukin-6 therapeutics in cardiovascular Disease. Circ. Res 128, 1728–1746 (2021). [DOI] [PubMed] [Google Scholar]
- 94.Galicia JC et al. Polymorphisms in the IL-6 receptor (IL-6R) gene: strong evidence that serum levels of soluble IL-6R are genetically influenced. Genes Immun. 5, 513–516 (2004). [DOI] [PubMed] [Google Scholar]
- 95.Huber SA, Sakkinen P, Conze D, Hardin N & Tracy R Interleukin-6 exacerbates early atherosclerosis in mice. Arter. Thromb. Vasc. Biol 19, 2364–2367 (1999). [DOI] [PubMed] [Google Scholar]
- 96.Schuett H et al. Transsignaling of interleukin-6 crucially contributes to atherosclerosis in mice. Arter. Thromb. Vasc. Biol 32, 281–290 (2012). [DOI] [PubMed] [Google Scholar]
- 97.Schieffer B et al. Impact of interleukin-6 on plaque development and morphology in experimental atherosclerosis. Circulation 110, 3493–3500 (2004). [DOI] [PubMed] [Google Scholar]
- 98.Pierini FS et al. Effect of tocilizumab on LDL and HDL characteristics in patients with rheumatoid arthritis. An observational study. Rheumatol. Ther 8, 803–815 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ridker PM et al. IL-6 inhibition with ziltivekimab in patients at high atherosclerotic risk (RESCUE): a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 397, 2060–2069 (2021). [DOI] [PubMed] [Google Scholar]
- 100.Blaschke K et al. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature 500, 222–226 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cimmino L et al. Restoration of TET2 function blocks aberrant self-renewal and leukemia progression. Cell 170, 1079–1095(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tang Y et al. Inhibition of JAK2 suppresses myelopoiesis and atherosclerosis in Apoe−/− mice. Cardiovasc. Drugs Ther 34, 145–152 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rao TN et al. JAK2-mutant hematopoietic cells display metabolic alterations that can be targeted to treat myeloproliferative neoplasms. Blood 134, 1832–1846 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rocca B et al. A randomized double-blind trial of 3 aspirin regimens to optimize antiplatelet therapy in essential thrombocythemia. Blood 136, 171–182 (2020). [DOI] [PubMed] [Google Scholar]
- 105.Bersenev A et al. Lnk constrains myeloproliferative diseases in mice. J. Clin. Invest 120, 2058–2069 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lv K et al. CBL family E3 ubiquitin ligases control JAK2 ubiquitination and stability in hematopoietic stem cells and myeloid malignancies. Genes Dev. 31, 1007–1023 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]