

Abstract
Human urine is traditionally considered to be sterile, and whether the urine harbours distinct microbial communities has been a matter of debate. Potential links between female urine and reproductive tract microbial communities is currently not clear. Here, we collected urine samples from 147 Chinese women of reproductive age and explored the nature of colonization by 16S rRNA gene amplicon sequencing, quantitative real-time PCR, and live bacteria culture. To demonstrate the utility of this approach, the intra-individual Spearman’s correlation was used to explore the relationship between urine and multiple sites of the female reproductive tract. PERMANOVA was also performed to explore potential correlations between the lifestyle and various clinical factors and urinary bacterial communities. Our data demonstrated distinct bacterial communities in urine, indicative of a non-sterile environment. Streptococcus-dominated, Lactobacillus-dominated, and diverse type were the three most common urinary bacterial community types in the cohort. Detailed comparison of the urinary microbiota with multiple sites of the female reproductive tract microbiota demonstrated that the urinary microbiota were more similar to the microbiota in the cervix and uterine cavity than to those of the vagina in the same women. Our data demonstrate the potential connectivity among microbiota in the female urogenital system and provide insight and resources for exploring diseases of the urethra and genital tract.
Data Description
Purpose of data acquisition
The role of microbiota in the vaginal environment has received a lot of attention over the past decade, while the female upper reproductive tract was traditionally believed to be sterile and mostly studied in the context of infections or incontinence [1]. Despite continued controversy, the presence of microorganisms beyond the cervix (i.e. the female upper reproductive tract) is increasingly recognized even in non-infectious conditions [2]. Like the female upper reproductive tract, the sterile hypothesis of urine has also been overturned by emerging evidence that indicates the existence of microorganisms in the urinary tract by culturing or sequencing approaches [3, 4]. A recent study using an expanded quantitative urine culture in combination with whole-genome sequencing has isolated and sequenced the genomes of 149 bacterial strains from catheterized urine of both symptomatic and asymptomatic peri-menopausal women [5]. It also showed highly similar strains of commensal bacteria in both the bladder and vagina of the same individual [5]. Another study analysed the urinary microbiota of 189 individuals using 16S rRNA gene amplicon sequencing and suggested that the urethra and bladder can harbour microbial communities distinct from the vagina [6]. However, the relationship between female urine microbiota and the upper reproductive tract microbiota has so far not been studied.
Here, we present a dataset of the urinary microbiota for a relatively large cohort of 147 women of reproductive age. Together with our recently published study of peritoneal fluid, uterine, and vaginal samples from the same individuals [2], this data shows that although urinary microbiota contain larger populations of Lactobacillus and Streptococcus, they are more similar to the microbiota of the cervix and uterine cavity, in accordance with the anatomical opening of the bladder. Together with a wealth of metadata, we demonstrate that these data are useful for exploring the potential of the urinary microbiota for clinical diagnosis.
Methods
A protocol collection including methods for DNA extraction, bioinformatics analysis and quantitative real-time PCR is available via protocols.io (Figure 1) [7].
Figure 1.

Protocol collection for sequencing and analysing female urinary microbiota. https://www.protocols.io/widgets/doi?uri=dx.doi.org/10.17504/protocols.io.bp3wmqpe
Sample collection
In this study, a total of 147 reproductive age women (age 22–48) were recruited by Peking University Shenzhen Hospital [8]. All participants were reproductive age women who underwent hysteroscopy and/or laparoscopy for conditions without infections, such as hysteromyoma, adenomyosis, endometriosis, or salpingemphraxis. Subjects with other related diseases, such as vaginal inflammation, severe pelvic adhesion, endocrine or autoimmune disorders were removed. Pregnant women, breastfeeding women, and menstruating women at the time of sampling were also excluded. None of the subjects received any antibiotic treatments or vaginal medications within two weeks of sampling. In addition, no cervical treatment was performed within the previous 7 days, no vaginal douching was performed within 5 days, and no sexual activity was performed within at least 2 days.
137 self-sampling morning mid-stream urine samples were collected between December 2013 and July 2014 prior to the surgery (sample_metadata.csv [8]), and then stored at −80 °C until they were transported on dry ice to BGI-Shenzhen for sequencing. The samples from an additional 10 women were collected for validation purposes by a doctor during the surgery in July 2017. For each operation, a urine catheter was inserted into the disinfected urethra to collect mid-stream urine. For each sample of urine collected through a catheter, an identical volume of saline solution was set as the control sample. The samples were then placed at 4 °C, transported to BGI-Shenzhen, and processed within 6 hours. A portion of each sample was used for culturing live bacteria and the rest was used for sequencing.
DNA extraction and 16S rRNA amplicon sequencing
Genomic DNA extraction was carried out following the protocol [9]. The primers 515F and 907R were utilized for PCR amplification of the hypervariable regions V4-V5 of the bacterial 16S rRNA gene. The 907R primer includes a unique barcoded fusion. The primer sequences were: 515F: 5′-GTGCCAGCMGCCGCGGTAA-3′ and 907R: 5′-CCGTCAATTCMTTTRAGT-3′, where M denotes A or C and R denotes purine. The conditions for PCR amplification were: 3 min of denaturation at 94 °C, followed by 25 cycles of 45 s at 94 °C (denaturing), 60 s at 50 °C (annealing), and 90 s at 72 °C (elongation), followed by a final elongation for 10 min at 72 °C. The amplification products were purified by the AxyPrep™ Mag PCR Clean-Up Kit (Axygen, USA). The amplicon libraries were constructed with an Ion Plus Fragment Library Kit (Thermo Fisher Scientific Inc.) [10], then sequenced by the Ion PGM™ Sequencer with the Ion 318™ Chip v2 with a read length of 400 bp (Thermo Fisher Scientific Inc., Ion PGM™ Hi-Q™ OT2 Kit, Cat.No: A27739; Ion PGM™ Hi-Q™ Sequencing Kit, Cat.No: A25592) [11]. All experiments were performed in the laboratory of BGI-Shenzhen.
Processing of sequencing reads
The raw sequencing reads were first subjected to Mothur (Mothur, RRID:SCR_011947; V1.33.3) [12] for filtering out the low-quality reads meeting the following criteria: (1) reads shorter than 200 bp; (2) reads not matching the degenerated PCR primers for up to two errors; (3) reads with an average quality score less than 25. A total of 8,812,607 reads, with an average of 57,225 reads per sample (a minimum of 1113 reads and a maximum of 194,564 reads) were obtained. Subsequently, the sequences with identity greater than 97% were clustered into Operational Taxonomic Units (OTUs) using the QIIME (QIIME, RRID:SCR_008249; V1.8.0) uclust programme [13], where each cluster was thought of as representing a species. The seed sequences of each OTU were aligned against the Greengene reference sequences (gg_13_8_otus) for annotation using Mothur. The detailed analysis workflow was deposited in protocols.io [14].
We also calculated the Unifrac distance using QIIME based on taxonomic abundance profiles at the OTU level [11].
PERMANOVA on the influence of phenotypes
Permutational multivariate analysis of variance (PERMANOVA) was used to assess the effect of different covariates based on the relative abundances of OTUs of the samples [15, 16] using Bray-Curtis and UniFrac distance and 9999 permutations from the vegan package (vegan, RRID:SCR_011950) in R [16, 17].
Quantitative real-time PCR
We quantified the four Lactobacillus species, including L.iners, L jensenii, L. crispatus and L. gasseri using the modified qPCR protocol [18]. SYBR Premix Ex Taq GC (TAKARA) was used and the reactions were run on a StepOnePlus Real-time PCR System (Life Technologies). Each PCR reaction mixture contained 10 μl of 2×SYBR Premix Ex Taq GC, 0.2 μM forward primer, 0.2 μM reverse primer, 1.6 μl of DNA sample, and 8.2 μl of ultrapure water to make up the final reaction volume of 20 μl. Each run included a standard curve and all samples were amplified in triplicate. Ultrapure water was used as the blank control template.
To construct the standard curves, the sequencing-confirmed plasmids of four species were used after quantification with a Qubit Fluorometer and serial 10-fold dilutions. The amplification efficiency was (100 ±10)% and linearity values were all ≥0.99. The detailed procedure was deposited in protocols.io [19].
Bacterial culturing
The urine samples and controls from 10 additional subjects were cultivated in the laboratory by spreading 100 μl of sample on different agars containing 5% horse blood, such as PYG agar (DSMZ 104 medium), BHI agar, and EG agar. The plates were incubated in both aerobic and anaerobic conditions at 37 °C for 72 hours. To keep the medium anaerobically during culture, resazurin and cysteine-HCl were added as reducing agents. The genomic DNA of the isolates was extracted by the Bacterial DNA Kit (OMEGA) and then underwent 16S rRNA gene amplification using the universal primers 27F/1492R [20]. The amplicons were purified and sent for Sanger sequencing. The generated sequences were then submitted to BLAST on the EzBioCloud [21] for identification.
Preliminary analysis and validation
Microbiota composition of the urine
To explore the urinary microbiota in this dataset, morning midstream urine (UR) was self-collected prior to surgery from an exploratory cohort of 137 Chinese women recruited for the study (median age 31.6, range 22–48). As with our previous vagino-uterine microbiota study [2], all volunteers had conditions that were not known to involve infections [8]. From 95 women in the cohort, six locations within the female reproductive tract, including the lower third of the vagina (CL), the posterior fornix (CU), cervical mucus (CV), endometrium (ET), left and right fallopian tubes (FLL and FRL), and peritoneal fluid (PF) were also sampled. Their vagino-uterine microbiota information have been published previously [2]. After 16S rRNA gene amplicon sequencing, the sequencing reads were pre-processed for quality control and filtering, then clustered into OTUs (Methods, Table 1 and OTU_table_urine.biom.hdf5 [8]).
Table 1.
Sequencing and annotation of the 137 samples from the exploratory cohort.
| Sample name | Sequencing amount | % of reads annotated to taxa | Archive accession number | |||
|---|---|---|---|---|---|---|
| #raw reads | #clean reads | #filtered reads | Genus | Species | ||
| C001UR | 52506 | 11326 | 9347 | 100.00% | 75.51% | SAMEA5042945 |
| C002UR | 55955 | 14529 | 5930 | 100.00% | 65.36% | SAMEA5042987 |
| C003UR | 61367 | 21181 | 16831 | 100.00% | 91.06% | SAMEA5043040 |
| C004UR | 54585 | 18506 | 3325 | 100.00% | 41.59% | SAMEA5042979 |
| C005UR | 52177 | 22856 | 20683 | 100.00% | 92.62% | SAMEA5043003 |
| C007UR | 50766 | 15468 | 8737 | 100.00% | 71.10% | SAMEA5043001 |
| C008UR | 53748 | 14062 | 6169 | 100.00% | 64.05% | SAMEA5043004 |
| C009UR | 53383 | 12247 | 11327 | 100.00% | 96.97% | SAMEA5043046 |
| C011UR | 47814 | 13058 | 11292 | 100.00% | 66.76% | SAMEA5042941 |
| C012UR | 55279 | 16923 | 7484 | 100.00% | 55.46% | SAMEA5042938 |
| C014UR | 55713 | 15175 | 8818 | 100.00% | 64.31% | SAMEA5043060 |
| C016UR | 73372 | 22054 | 17669 | 100.00% | 57.04% | SAMEA5043009 |
| C018UR | 69142 | 26505 | 23581 | 100.00% | 12.12% | SAMEA5043006 |
| C019UR | 72249 | 17868 | 14440 | 100.00% | 44.63% | SAMEA5043054 |
| C020UR | 54574 | 19391 | 5452 | 100.00% | 63.83% | SAMEA5042942 |
| C021UR | 58118 | 17294 | 12123 | 100.00% | 55.33% | SAMEA5042998 |
| C023UR | 47476 | 18452 | 16795 | 100.00% | 16.09% | SAMEA5042947 |
| C026UR | 46583 | 16741 | 3267 | 100.00% | 83.96% | SAMEA5042969 |
| C028UR | 88245 | 26955 | 19268 | 100.00% | 19.93% | SAMEA5042984 |
| C033UR | 90431 | 31998 | 26496 | 100.00% | 66.99% | SAMEA5043062 |
| C035UR | 63773 | 27044 | 24115 | 100.00% | 97.31% | SAMEA5043037 |
| C038UR | 55562 | 10165 | 9208 | 100.00% | 84.56% | SAMEA5042972 |
| C039UR | 77957 | 18891 | 15748 | 100.00% | 33.38% | SAMEA5042963 |
| C040UR | 58940 | 12555 | 5438 | 100.00% | 69.49% | SAMEA5043021 |
| C041UR | 60028 | 15361 | 9366 | 100.00% | 78.38% | SAMEA5043000 |
| C042UR | 74086 | 14402 | 11088 | 100.00% | 67.53% | SAMEA5042955 |
| C043UR | 74146 | 23691 | 18730 | 100.00% | 60.19% | SAMEA5043032 |
| C045UR | 61249 | 17801 | 10367 | 100.00% | 2.96% | SAMEA5043048 |
| C047UR | 47742 | 11940 | 3506 | 100.00% | 54.25% | SAMEA5043024 |
| C048UR | 35550 | 1100 | 816 | 100.00% | 62.75% | SAMEA5042931 |
| C050UR | 51565 | 18902 | 290 | 100.00% | 72.76% | SAMEA5042936 |
| C051UR | 58783 | 10403 | 8234 | 100.00% | 50.77% | SAMEA5042983 |
| C053UR | 32311 | 1653 | 26 | 100.00% | 73.08% | SAMEA5043035 |
| C055UR | 45054 | 13326 | 6184 | 100.00% | 56.40% | SAMEA5043016 |
| C056UR | 69173 | 24652 | 8282 | 100.00% | 86.78% | SAMEA5043023 |
| C057UR | 64417 | 27033 | 24444 | 100.00% | 98.11% | SAMEA5043059 |
| C058UR | 42089 | 1415 | 912 | 100.00% | 4.28% | SAMEA5042935 |
| C059UR | 53642 | 12618 | 577 | 100.00% | 74.70% | SAMEA5042930 |
| C060UR | 73930 | 22110 | 19192 | 100.00% | 20.17% | SAMEA5043008 |
| C062UR | 63220 | 19932 | 17112 | 100.00% | 79.58% | SAMEA5043012 |
| C063UR | 44974 | 1201 | 790 | 100.00% | 53.42% | SAMEA5043039 |
| C064UR | 63505 | 15051 | 7134 | 100.00% | 82.38% | SAMEA5042981 |
| C065UR | 53884 | 15094 | 13794 | 100.00% | 52.97% | SAMEA5043027 |
| C066UR | 63269 | 16157 | 12090 | 100.00% | 45.86% | SAMEA5042985 |
| C067UR | 55812 | 19047 | 2481 | 100.00% | 86.86% | SAMEA5042986 |
| C068UR | 54396 | 17456 | 15352 | 100.00% | 86.72% | SAMEA5042937 |
| T000UR | 57607 | 11995 | 9166 | 100.00% | 37.51% | SAMEA5043045 |
| T001UR | 47924 | 13474 | 2849 | 100.00% | 48.58% | SAMEA5043014 |
| T002UR | 63839 | 18381 | 3623 | 100.00% | 49.71% | SAMEA5042975 |
| T003UR | 70242 | 19166 | 5255 | 100.00% | 51.67% | SAMEA5042988 |
| T004UR | 67280 | 20578 | 2947 | 100.00% | 57.24% | SAMEA5042943 |
| T005UR | 52820 | 12868 | 4931 | 100.00% | 57.92% | SAMEA5043019 |
| T006UR | 79409 | 19472 | 13710 | 100.00% | 19.58% | SAMEA5043017 |
| T007UR | 34173 | 1403 | 797 | 100.00% | 50.06% | SAMEA5042999 |
| T008UR | 30074 | 1346 | 1044 | 100.00% | 84.77% | SAMEA5043031 |
| T009UR | 58440 | 10386 | 7936 | 100.00% | 81.92% | SAMEA5042950 |
| T010UR | 65382 | 17191 | 9801 | 100.00% | 47.54% | SAMEA5042967 |
| T011UR | 38550 | 1163 | 464 | 100.00% | 65.52% | SAMEA5043061 |
| T012UR | 75848 | 21956 | 4605 | 100.00% | 52.62% | SAMEA5043049 |
| T013UR | 26872 | 1383 | 203 | 100.00% | 74.38% | SAMEA5042996 |
| T014UR | 23298 | 1741 | 518 | 100.00% | 93.44% | SAMEA5043042 |
| T015UR | 40653 | 2052 | 1470 | 100.00% | 29.25% | SAMEA5042953 |
| T016UR | 58448 | 16261 | 3993 | 100.00% | 58.75% | SAMEA5043053 |
| T017UR | 58703 | 19270 | 1929 | 100.00% | 54.12% | SAMEA5042990 |
| T018UR | 54726 | 12668 | 7152 | 100.00% | 17.18% | SAMEA5042970 |
| T019UR | 67711 | 14153 | 1606 | 100.00% | 58.90% | SAMEA5042954 |
| T020UR | 89936 | 22579 | 17919 | 100.00% | 68.74% | SAMEA5043052 |
| T021UR | 66094 | 14761 | 5276 | 100.00% | 29.66% | SAMEA5042951 |
| T022UR | 28712 | 803 | 422 | 100.00% | 74.88% | SAMEA5042940 |
| T023UR | 27738 | 1338 | 385 | 100.00% | 30.65% | SAMEA5042961 |
| T024UR | 19345 | 824 | 55 | 100.00% | 90.91% | SAMEA5042948 |
| T025UR | 29739 | 1578 | 1214 | 100.00% | 76.85% | SAMEA5042995 |
| T026UR | 79923 | 17606 | 6269 | 100.00% | 37.09% | SAMEA5042959 |
| T027UR | 61145 | 10093 | 6169 | 100.00% | 59.51% | SAMEA5042949 |
| T028UR | 71755 | 19529 | 16118 | 100.00% | 7.61% | SAMEA5042965 |
| T029UR | 58776 | 11505 | 8300 | 100.00% | 6.04% | SAMEA5043018 |
| T030UR | 57098 | 7000 | 5119 | 100.00% | 70.23% | SAMEA5042966 |
| T031UR | 49283 | 16113 | 1636 | 100.00% | 38.63% | SAMEA5043063 |
| T032UR | 46822 | 15041 | 1187 | 100.00% | 53.24% | SAMEA5042927 |
| T033UR | 63044 | 14272 | 10097 | 100.00% | 80.89% | SAMEA5043043 |
| T035UR | 50618 | 12403 | 1122 | 100.00% | 76.20% | SAMEA5043002 |
| T036UR | 78781 | 22492 | 17075 | 100.00% | 84.36% | SAMEA5042982 |
| T038UR | 73752 | 15237 | 11784 | 100.00% | 1.14% | SAMEA5043022 |
| T039UR | 58904 | 22286 | 19836 | 100.00% | 95.82% | SAMEA5043015 |
| T040UR | 77039 | 15332 | 8059 | 100.00% | 40.76% | SAMEA5043028 |
| T041UR | 58382 | 13735 | 11893 | 100.00% | 97.26% | SAMEA5043056 |
| T042UR | 53948 | 17112 | 1392 | 100.00% | 29.74% | SAMEA5043026 |
| T043UR | 72662 | 15446 | 10584 | 100.00% | 16.18% | SAMEA5042956 |
| T044UR | 59818 | 19779 | 724 | 100.00% | 73.48% | SAMEA5042993 |
| T045UR | 63627 | 21438 | 19282 | 100.00% | 39.34% | SAMEA5043033 |
| T046UR | 58142 | 20606 | 912 | 100.00% | 56.69% | SAMEA5042991 |
| T047UR | 24190 | 736 | 26 | 100.00% | 73.08% | SAMEA5042978 |
| T048UR | 10255 | 441 | 53 | 100.00% | 79.25% | SAMEA5043036 |
| T049UR | 63640 | 22520 | 20236 | 100.00% | 98.73% | SAMEA5043010 |
| T051UR | 22322 | 1066 | 101 | 100.00% | 77.23% | SAMEA5042997 |
| T052UR | 57909 | 11757 | 6419 | 100.00% | 67.85% | SAMEA5043058 |
| T053UR | 63637 | 24574 | 21178 | 100.00% | 99.17% | SAMEA5043034 |
| T054UR | 59194 | 18986 | 17148 | 100.00% | 15.87% | SAMEA5042929 |
| T055UR | 70744 | 14983 | 2724 | 100.00% | 84.07% | SAMEA5042962 |
| T056UR | 58876 | 19486 | 846 | 100.00% | 65.84% | SAMEA5043051 |
| T057UR | 53598 | 13889 | 12456 | 100.00% | 96.76% | SAMEA5042939 |
| T058UR | 18302 | 739 | 45 | 100.00% | 64.44% | SAMEA5043044 |
| T059UR | 59974 | 15542 | 12378 | 100.00% | 23.53% | SAMEA5042964 |
| T060UR | 21736 | 500 | 203 | 100.00% | 89.16% | SAMEA5042946 |
| T061UR | 33002 | 1153 | 503 | 100.00% | 56.46% | SAMEA5043011 |
| T062UR | 64983 | 10519 | 6302 | 100.00% | 52.19% | SAMEA5042933 |
| T063UR | 53347 | 12793 | 4023 | 100.00% | 40.00% | SAMEA5043041 |
| T064UR | 68122 | 23665 | 21094 | 100.00% | 79.99% | SAMEA5042934 |
| T065UR | 51210 | 17070 | 1242 | 100.00% | 65.30% | SAMEA5042977 |
| T066UR | 64589 | 26532 | 1911 | 100.00% | 56.04% | SAMEA5042968 |
| T067UR | 67938 | 16248 | 4974 | 100.00% | 50.12% | SAMEA5043005 |
| T068UR | 70192 | 28890 | 3698 | 100.00% | 40.37% | SAMEA5043013 |
| T069UR | 60564 | 21236 | 17683 | 100.00% | 43.97% | SAMEA5042957 |
| T070UR | 83453 | 20755 | 5034 | 100.00% | 44.18% | SAMEA5043038 |
| T071UR | 80077 | 36770 | 29224 | 100.00% | 97.68% | SAMEA5043007 |
| T072UR | 73469 | 29671 | 19787 | 100.00% | 87.68% | SAMEA5042992 |
| T073UR | 73167 | 17577 | 3914 | 100.00% | 54.09% | SAMEA5042989 |
| T074UR | 59084 | 23906 | 21347 | 100.00% | 89.11% | SAMEA5042973 |
| T075UR | 60263 | 17726 | 15250 | 100.00% | 70.37% | SAMEA5042976 |
| T076UR | 37428 | 809 | 514 | 100.00% | 63.42% | SAMEA5042960 |
| T078UR | 76834 | 17034 | 4220 | 100.00% | 66.75% | SAMEA5042958 |
| T080UR | 12172 | 609 | 61 | 100.00% | 52.46% | SAMEA5042932 |
| T081UR | 63432 | 14841 | 6915 | 100.00% | 86.49% | SAMEA5043029 |
| T082UR | 26941 | 693 | 609 | 100.00% | 2.96% | SAMEA5043047 |
| T083UR | 69149 | 34307 | 30270 | 100.00% | 95.45% | SAMEA5043055 |
| T084UR | 59304 | 25863 | 22866 | 100.00% | 89.34% | SAMEA5042928 |
| T085UR | 65565 | 20426 | 1344 | 100.00% | 78.57% | SAMEA5043030 |
| T086UR | 66605 | 23828 | 21243 | 100.00% | 95.16% | SAMEA5043025 |
| T087UR | 62480 | 16656 | 6414 | 100.00% | 76.55% | SAMEA5043057 |
| T088UR | 82733 | 32538 | 3794 | 100.00% | 71.09% | SAMEA5042994 |
| T089UR | 110227 | 27223 | 11761 | 100.00% | 24.49% | SAMEA5042944 |
| T090UR | 70526 | 29296 | 1917 | 100.00% | 71.62% | SAMEA5043020 |
| T091UR | 27973 | 913 | 739 | 100.00% | 27.74% | SAMEA5042952 |
| T092UR | 69694 | 10825 | 7894 | 100.00% | 55.26% | SAMEA5043050 |
| T093UR | 58492 | 14656 | 7272 | 100.00% | 84.27% | SAMEA5042980 |
| T094UR | 194564 | 59268 | 35224 | 100.00% | 37.26% | SAMEA5042971 |
| T095UR | 42681 | 1009 | 560 | 100.00% | 45.00% | SAMEA5042974 |
Due to anatomical structures, voided urine samples from women were considered to be easily contaminated by microbiota from the surrounding vulvovaginal region [22]. Most vaginal communities (88%) in this cohort were dominated by one genus with >50% relative abundance within data from individuals. In contrast, the urinary microbiota in this study showed more heterogeneity. 56.93% of the cohort harboured a diverse type represented significantly by bacteria, including Streptococcus, Lactobacillus, Pseudomonas, Staphylococcus, Acinetobacter, and Vagococcus, though none of these species were dominant, i.e. reached >50% relative abundance (Figure 2). In addition, 22.63% of the women harboured >50% Streptococcus, and 13.87% of the women harboured >50% Lactobacillus (Figure 2A, B). Rare subtypes such as Enterococcus (2.19%), Bifidobacteriaceae (1.46%), Prevotella (0.73%), Enterobacteriaceae (0.73%), Coriobacteriaceae (0.73%), and Veillonella (0.73%) were also detected in this cohort (Figure 2A, B). Notably, the median relative abundances of Lactobacillus, Pseudomonas, and Acinetobacter in the urine samples were more similar to the uterus samples (Figure 2C) [2]. At the phylum level, urinary microbiota were dominated by Firmicutes and Proteobacteria (Figure 2C).
Figure 2.
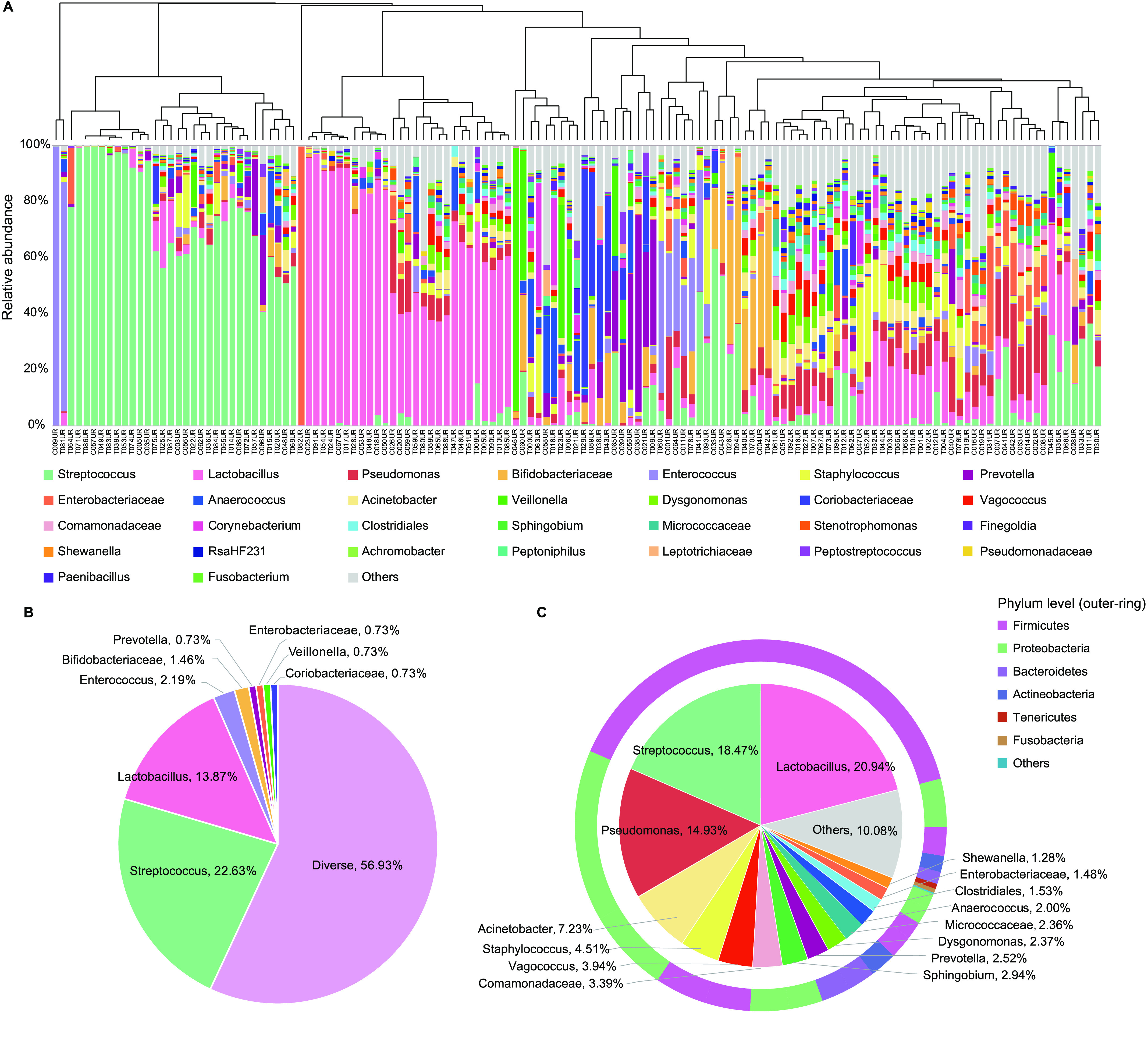
Urinary microbiota of the initial cohort of 137 Chinese reproductive-age women. (A) The relative abundances of genera detected in each individual are shown in the bar chart. The dendrogram is a result of a centroid linkage hierarchical clustering based on Euclidean distances between the microbial composition proportion of urinary bacterial communities. (B) The ratio of different urinary microbiota types. The genus whose relative abundance accounted for >50% in an individual was selected as an identified type. The genera that accounted for <50% of the microbiota in an individual were identified as diverse type. (C) Pie chart for the urinary microbial genera according to their median relative abundance. Genera that took up less than 1% of the microbiota are labelled together as ‘others’. The outer ring indicates the distribution of microbiota at the phylum level.
Cultivation of live bacteria from transurethral catheterized urine
The question of whether bacterial DNA signals have originated from live bacteria or fragments in the urine samples has been a subject of much debate [22]. To demonstrate the utility of the data for addressing this question, we performed a validation study using live bacteria cultures from urine samples provided by an additional cohort of 10 women.
We tried to culture and isolate bacterial colonies from freshly collected urine samples. Urine samples were serial diluted and spread on three different kinds of agar plates and incubated under both aerobic and anaerobic conditions. Six different positive isolates belonging to 5 genera, including Lactobacillus, Staphylococcus, Clostridium, Enterococcus, and Propionibacterium were obtained from 3 out of 10 subjects (Table 2). The 5 genera were also found as dominant in our 16S rRNA gene amplicon sequencing data and consistent with previous cultivation results of published papers [23–26] (Table 2). Reassuringly, no isolates were detected from the negative controls (sterile saline and ultrapure water). Therefore, these data verified the existence of live bacteria in the urine by obtaining isolates using conventional culturing methods.
Table 2.
Identification of cultured microbial isolates from urine of the 10 additional women by sequencing of partial 16S rRNA gene.
| Sample ID | Condition | Medium | 16S rRNA gene-PCR Identification | Accessions | Identity (%) | Supported by previous cultivation |
|---|---|---|---|---|---|---|
| S001U | Anaerobic, 37 °C | EG | Clostridium cochlearium | LR761333.1 | 99.26 | Meijer-Severs et al. [24] |
| S001U | Anaerobic, 37 °C | 104 | Streptococcus sp. (S. tigurinus/S. mitis) | LR761334.1 | 99.72 | Hilt et al. [23] |
| S003U | Anaerobic, 37 °C | BHI | Enterococcus faecalis | LR761335.1 | 99.91 | Hilt et al. [23], Guzmàn et al. [25], Fraimow et al. [26], |
| S003U | Anaerobic, 37 °C | 104 | Lactobacillus crispatus | LR761337.1 | 99.82 | Hilt et al. [23] |
| S003U | Anaerobic, 37 °C | 104 | Propionibacterium granulosum | LR761336.1 | 99.02 | Ormerod et al. [27] |
| S008U | Anaerobic, 37 °C | 104, BHI, EG | Streptococcus agalactiae | LR761340.1, LR761339.1, LR761338.1 | 99.65, 99.35, 99.52 | Hilt et al. [23] |
Considerable bacterial biomass revealed by qPCR
To provide additional evidence of the bacterial communities in the urine, a species-specific quantitative real-time PCR method was utilized to focus on the four common vaginal Lactobacillus species, i.e. L. crispatus, L. iners, L. jensenii and L. gasseri (QPCR Lactobacillus.csv [8]). The Lactobacillus species we examined presented a similar distribution and abundance along the female reproductive tract, and the corresponding urinary Lactobacillus ranged between the upper and lower reproductive tracts (Figure 3A). Among them, L. iners occurred most frequently (59%) in the urine samples, while L. crispatus only occurred in 26% of women sampled (Figure 3B). L. iners was reported far less protective against bacterial and viral infections compared to L. crispatus [28]. 80% of the cohort was detected to harbour at least one of these four Lactobacillus species (Figure 3B). The occurrence rate of Lactobacillus in the genus level of 16S rRNA gene amplicon sequencing data was 94% (Figure 2A). The total bacterial biomass is approximated by the ratio of the copy number from the result of qPCR to the relative abundance according to the result of 16S rRNA gene sequencing of the same sample (QPCR bacterial_biomass.csv [8]). The result gave an estimation of 107 copies/sample, placing the urinary bacterial biomass between the vaginal-cervical sites (1010–1011 copies/sample) and the endometrium (ET) samples (106–107 copies/sample) [2] (Figure 3A), all of which were orders of magnitude above potential background noise [29]. These results were interestingly consistent with a weakly acidic pH of the urine, in comparison to pH < 4.5 in the vagina or pH ∼ 8 in the peritoneal fluid [30].
Figure 3.
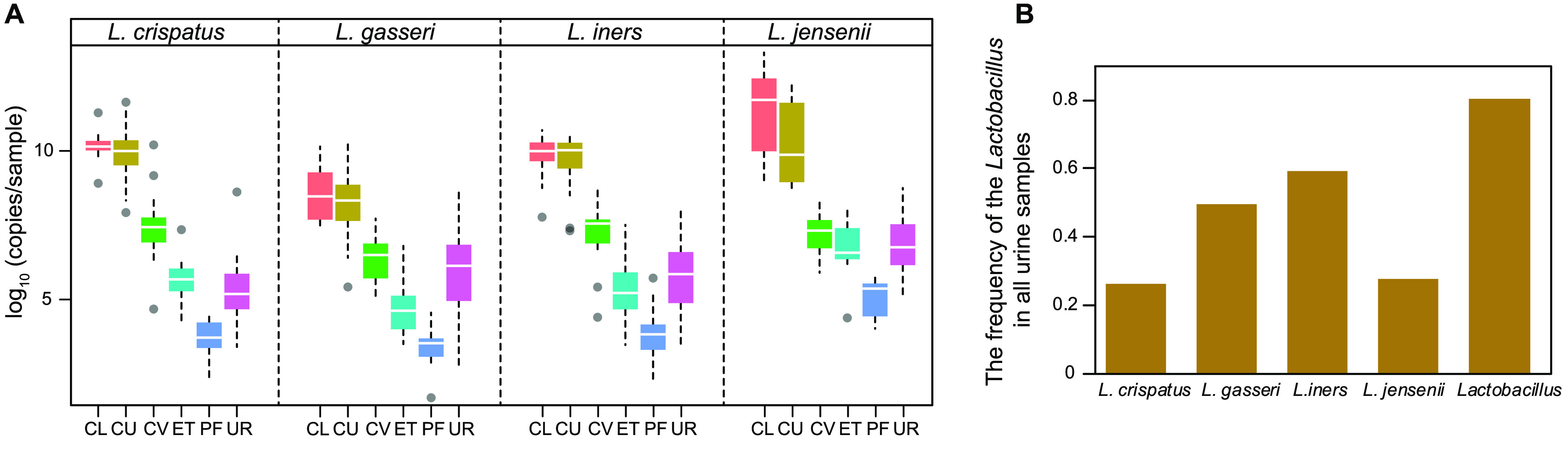
The concentrations of the dominant Lactobacillus species at urine and the reproductive tract. Samples derive from the initial cohort of 137 Chinese reproductive-age women. (A) The abundance of L. iners, L. jensenii, L. crispatus and L. gasseri calculated by qPCR results in different samples. Boxes denote the interquartile range (IQR) between the first and third quartiles (25th and 75th percentiles, respectively), and the lines inside the boxes denote the median. The whiskers denote the lowest and highest values within 1.5 times the IQR from the first and third quartiles, respectively. (B) The frequency of the respective Lactobacillus detected in all urine sample.
Intra-individual similarity in the urine-reproductive tract microbiota
To further assess the microbiota relationship between the urine and the six positions of the female reproductive tract, we computed intra-individual correlation between the microbial profiles in the urine and those found in different sites of the reproductive tract, and then clustered the individuals into 4 groups (Spearman’s correlation coefficient, Figure 4A, relative_abundance_correlation.csv [8]). Interestingly, the microbiota of group 3, which accounted for 41% of the cohort, showed significant correlation between the urine samples and the female reproductive tract samples, of which the coefficient increased gradually along the anatomical site from CL to CV, ET, and PF (Figure 4B). In contrast, 9% of women in group 1 presented a reverse trend. In group 2 (22%) and group 4 (27%), there appeared to be a weak relationship between the microbiota of the urine and female reproductive tract. Taken together, we observed the most similar distribution of microbiota between urine and CV/ET (Figure 4A). The principal coordinate analyses (PCoA) of the weighted and unweighted intra-individual UniFrac distance further corroborated our conclusion that there is an intra-individual similarity of the microbiota between the urine and the upper sites of female reproductive tract, especially the junction sites (CV and ET) (Figure 5).
Figure 4.
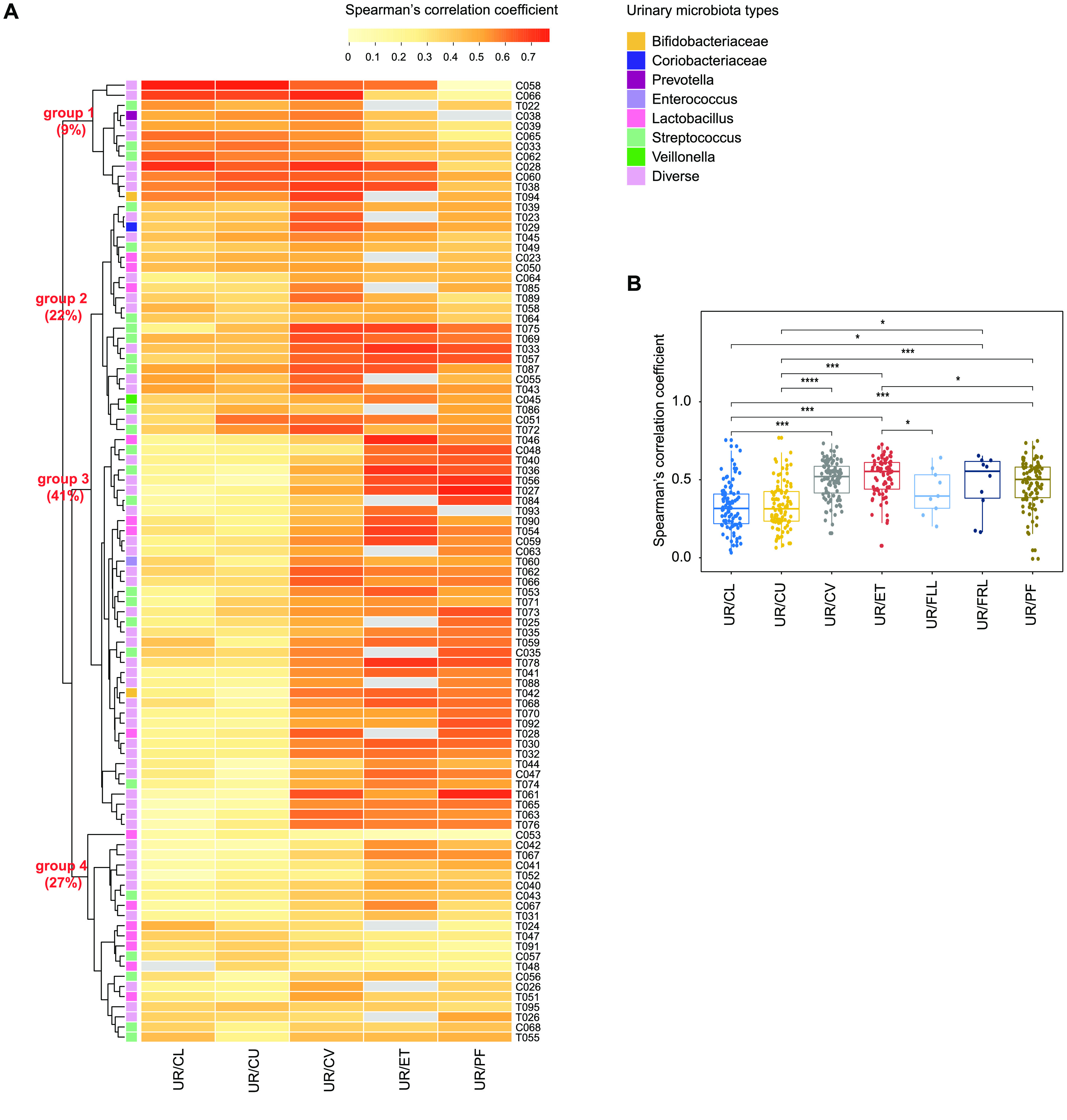
Similarity of the urine-reproductive tract microbiota within individuals. (A) Heatmap for the intra-individual Spearman’s correlation coefficient between microbiota identified in the urine and at different sites in the reproductive tract (relative_abundance_correlation.csv [8]). Samples derived from the initial cohort of 95 Chinese reproductive-age women, who collected both the urine and reproductive tract samples. As the number of samples from fallopian tubes (FLL, FRL) is too small, the correlation between microbiota in the urine and those in fallopian tubes are not shown. The dendrogram is a result of a centroid linkage hierarchical clustering based on Euclidean distances between the intra-individual Spearman’s correlation coefficient of different body sites. The colored squares illustrate the subtypes found within the urinary microbiome. (B) Spearman’s correlation coefficient between microbiota found in the urine and those from different sites of the reproductive tract. The Wilcoxon ranked sum test was used to calculate the difference. Boxes denote the interquartile range (IQR) between the first and third quartiles (25th and 75th percentiles, respectively), and the line inside the boxes denote the median. The whiskers denote the lowest and highest values within 1.5 times the IQR from the first and third quartiles, respectively. An asterisk denotes p <0.05, two asterisks denote p <0.01, three asterisks denote p <0.001.
Figure 5.
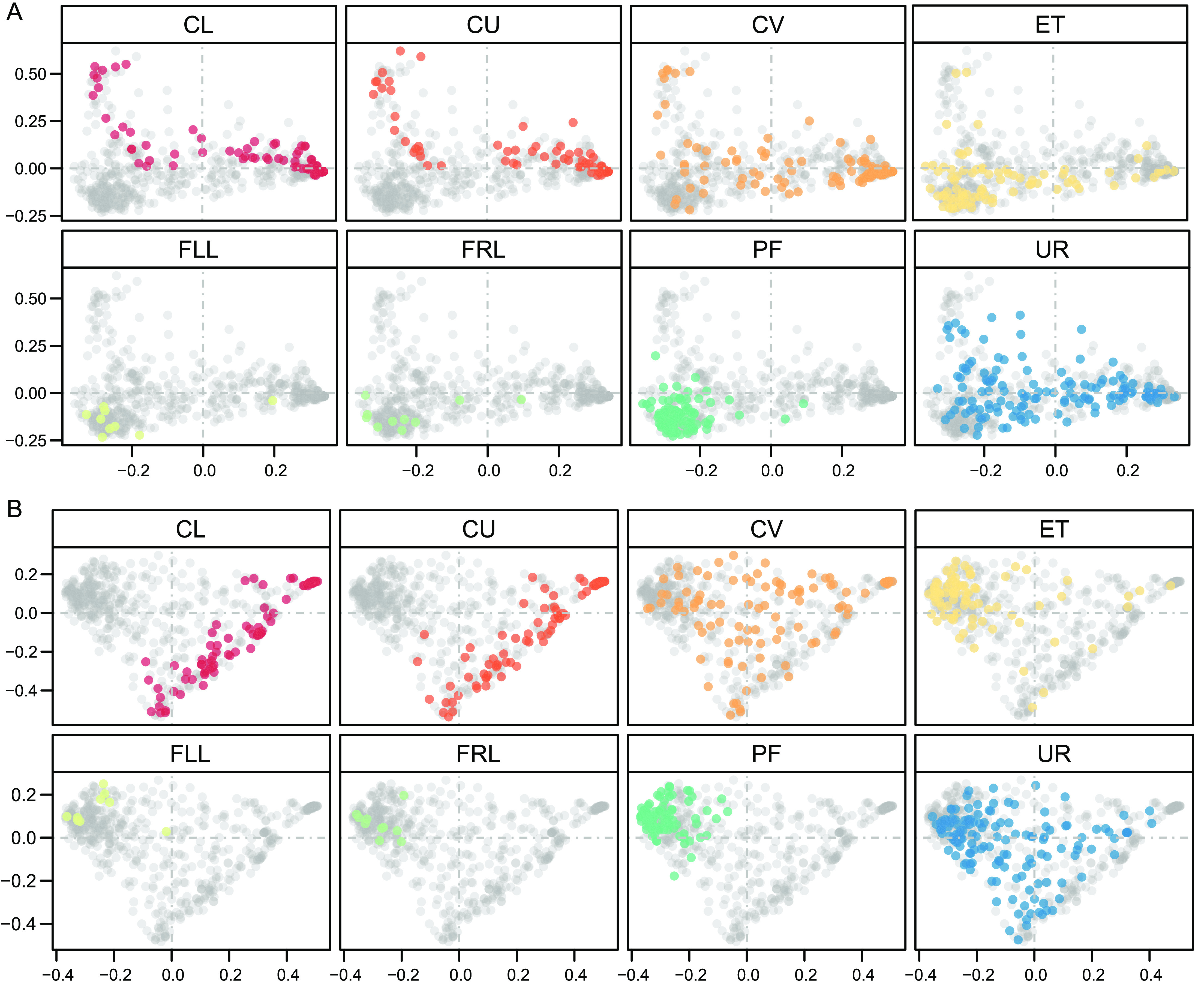
PCoA on the samples based on Unweighted-UniFrac (A) and Weighted-UniFrac (B) distances. Samples were taken from UR, CL, CU, and CV before operation, and from ET and PF during operation. Samples were derived from the initial cohort of 137 Chinese reproductive-age women. Each dot represents one sample (n =94 CL, 95 CU, 95 CV, 80 ET, 93 PF, 9 FLL, 10 FRL, and 137 UR).
Lifestyle and clinical factors influencing the urinary microbiota
The human microbiome is dynamic and highly affected by its host environment. Age, menstrual cycle, benign conditions such as adenomyosis, and infertility due to endometriosis have previously been reported to shape the vagino-uterine microbiota [2]. With our comprehensive collection of demographic and baseline clinical characteristics from women of reproductive age (sample_metadata.csv [8]), such variations in the urinary microbiota can be explored in this dataset. Urinary microbial composition was significantly associated with these factors, such as age, surgical history, abortion, vaginal deliveries, experience of given birth (multipara vs. nullipara), infertility due to endometriosis, and hysteromyoma (PERMANOVA, P <0.05, q <0.05, Table 3). Although the urinary microbiota also correlated with some other factors, such as menstrual phase, contraception, endometriosis, pelvic adhesiolysis, and anemia, statistical significance was not achieved after controlling for multiple testing (PERMANOVA, P <0.05 but q >0.05, Table 3). The initial results here indicate a close link between the urinary microbiota and the general and diseased physiological conditions, and this link could be further understood by exploring this data more deeply.
Table 3.
PERMANOVA for the influence of phenotypes on the urinary microbiota.
| Phenotype | Bray-Curtis | Unweighted-UniFrac | Weighted-UniFrac | ||||||
|---|---|---|---|---|---|---|---|---|---|
| R2 | P value | Fdr | R2 | P value | Fdr | R2 | P value | Fdr | |
| Age | 0.018 | 0.005 | 0.049 | 0.010 | 0.178 | 0.541 | 0.019 | 0.026 | 0.419 |
| Age-2 groups | 0.013 | 0.050 | 0.236 | 0.011 | 0.116 | 0.429 | 0.016 | 0.042 | 0.452 |
| Age-3 groups | 0.032 | 0.026 | 0.150 | 0.025 | 0.228 | 0.577 | 0.030 | 0.135 | 0.539 |
| Pulses | 0.015 | 0.019 | 0.131 | 0.010 | 0.159 | 0.505 | 0.015 | 0.072 | 0.456 |
| Frequent colds | 0.011 | 0.080 | 0.270 | 0.011 | 0.108 | 0.429 | 0.021 | 0.017 | 0.419 |
| Antibiotics | 0.014 | 0.036 | 0.194 | 0.012 | 0.108 | 0.429 | 0.007 | 0.525 | 0.782 |
| Constipation | 0.011 | 0.114 | 0.325 | 0.014 | 0.039 | 0.400 | 0.018 | 0.033 | 0.419 |
| Surgical history | 0.018 | 0.005 | 0.049 | 0.018 | 0.006 | 0.172 | 0.034 | 0.001 | 0.091 |
| Abdominal surgical history | 0.010 | 0.187 | 0.418 | 0.007 | 0.466 | 0.755 | 0.019 | 0.030 | 0.419 |
| Menstrual cycle | 0.009 | 0.200 | 0.421 | 0.018 | 0.005 | 0.172 | 0.015 | 0.065 | 0.455 |
| Menstrual phase (lower) | 0.018 | 0.260 | 0.468 | 0.024 | 0.048 | 0.408 | 0.020 | 0.207 | 0.623 |
| Menstrual phase (upper) | 0.018 | 0.006 | 0.056 | 0.018 | 0.009 | 0.172 | 0.014 | 0.096 | 0.456 |
| Contraception | 0.044 | 0.010 | 0.086 | 0.038 | 0.098 | 0.429 | 0.029 | 0.470 | 0.782 |
| Vaginal deliveries | 0.018 | 0.003 | 0.049 | 0.016 | 0.014 | 0.172 | 0.016 | 0.051 | 0.455 |
| Abortion | 0.028 | 0.004 | 0.049 | 0.014 | 0.239 | 0.585 | 0.016 | 0.198 | 0.623 |
| Multipara / nullipara | 0.019 | 0.003 | 0.049 | 0.017 | 0.014 | 0.172 | 0.013 | 0.091 | 0.456 |
| Infertility due to endometriosis | 0.045 | 0.000 | 0.008 | 0.029 | 0.013 | 0.172 | 0.019 | 0.181 | 0.599 |
| Endometriosis | 0.014 | 0.022 | 0.141 | 0.011 | 0.095 | 0.429 | 0.005 | 0.644 | 0.857 |
| Pelvic adhesiolysis | 0.008 | 0.346 | 0.572 | 0.013 | 0.042 | 0.400 | 0.006 | 0.489 | 0.782 |
| Anemia | 0.016 | 0.012 | 0.090 | 0.008 | 0.354 | 0.740 | 0.006 | 0.511 | 0.782 |
| Hysteromyoma | 0.018 | 0.003 | 0.049 | 0.012 | 0.057 | 0.429 | 0.021 | 0.014 | 0.419 |
Potential uses
As a large-scale cohort for studying the female urinary microbiota, our data provide a useful baseline and reference dataset in women of reproductive age. We also explored the association between the composition of urinary microbiota and that of the female reproductive tract microbiota. It is valuable to note that a higher intra-individual compositional similarity was observed between the microbiota of the urine and those of the cervical canal/uterus than between the microbiota of the urine and those of the vagina. This finding indicates that sampling of midstream urine (the least invasive and the easiest way) could be potentially used to survey the micro-environment of the cervical canal and uterus in the general population. This is relevant to the demonstrated associations between the urinary microbiota and various uterine-related diseases, such as hysteromyoma and infertility due to endometriosis. Our data provide a reference for clinical diagnosis and warrants further detailed exploration.
There are three limitations for this study. Firstly, as it was not possible to directly sample the upper reproductive tract of perfectly healthy women, we have included women who underwent minimally invasive laparoscopy or laparotomy for conditions that are not known to involve infection. This was the best proxy for sampling the upper reproductive tract in healthy women. Nevertheless, the relevance of the urinary microbiota between healthy women and women in our cohort would require further comparison. Secondly, for the low bacterial biomass of urine samples, a more comprehensive sampling process should be taken into consideration in subsequent studies, such as disinfection of the urethra and vulvovaginal region with 75% alcohol before urine self-collection, including a sample of sterile saline with the self-collection kit as a negative control and asking participants to fill another vial with it immediately following urine collection. A comparison of the microbial composition between the catheter-collected and self-collected specimens in the same individual would also require further inspection. Together, we hope that this dataset helps promote a new round of accelerated discoveries, including a novel scientific explanation for uterine-related diseases via longitudinal studies on the microbiota of the urinary and reproductive tracts.
Acknowledgements
The study was supported by the Shenzhen Municipal Government (No. SZXK027 and No. SZSM202011016), Shenzhen Peacock Plan (No. KQTD20150330171505310), and the Medical Scientific Research Foundation of Guangdong (No. A2019035). The authors really appreciate colleagues at BGI-Shenzhen for DNA extraction, library construction, and sequencing.
Funding Statement
The study was supported by the Shenzhen Municipal Government (No. SZXK027 and No. SZSM202011016), Shenzhen Peacock Plan (No. KQTD20150330171505310), and the Medical Scientific Research Foundation of Guangdong (No. A2019035).
Declarations
Ethics approval and consent to participate
The study was approved by the Institutional Review Board of BGI-Shenzhen (No. BGI-IRB 17219) and Peking University Shenzhen Hospital (Version 1.0.20140301). All participants gave written informed consent prior to their recruitment into the study.
Data availability
The sequence reads generated by 16S rRNA gene amplicon sequencing have been deposited in both the European Nucleotide Archive with the accession number PRJEB29341 and the CNSA (https://db.cngb.org/cnsa/) of CNGB database with accession code CNP0000166. Additional data, result and a STORMS (Strengthening The Organizing and Reporting of Microbiome Studies) checklist are available from the GigaScience GigaDB repository [8]. The sequences of bacterial isolates have been deposited in the European Nucleotide Archive with the accession number PRJEB36743.
Author contributions
H.J. and R.W. organized this study. W.W., J.D., H.D., L.Z., H.T., T.W., and R.W. performed the sample collection, and phenotypic information collection. F.L., L.S., C.C., and J.L. performed the molecular biology experiments. C.C., L.H., and F.L. performed the bioinformatic analyses. C.C., X.Z., F.L., and H.J., wrote the manuscript.
Competing interests
There were no competing financial interests.
References
- 1.Whiteside Sa, Razvi H, Dave S, Reid G, Burton JP, . The microbiome of the urinary tract — A role beyond infection. Nat. Rev. Urol., 2015; 81–90. [DOI] [PubMed] [Google Scholar]
- 2.Chen C, Song X, Wei W, Zhong H, Dai J, Lan Z et al. The microbiota continuum along the female reproductive tract and its relation to uterine-related diseases. Nat. Commun., 2017; 8: 875. doi:10.1038/s41467-017-00901-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siddiqui H, Nederbragt AJ, Lagesen K, Jeansson SL, Jakobsen KS., . Assessing diversity of the female urine microbiota by high throughput sequencing of 16S rDNA amplicons. BMC Microbiol., 2011; 11: 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wolfe AJ, Toh E, Shibata N, Rong R, Kenton K, FitzGerald MP et al. Evidence of uncultivated bacteria in the adult female bladder. J. Clin. Microbiol., 2012; 50: 1376–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thomas-White K, Forster SC, Kumar N, Van Kuiken M, Putonti C, Stares MD et al. Culturing of female bladder bacteria reveals an interconnected urogenital microbiota. Nat Commun., 2018; 9: 1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gottschick C, Deng ZL, Vital M, Masur C, Abels C, Pieper DH et al. The urinary microbiota of men and women and its changes in women during bacterial vaginosis and antibiotic treatment. Microbiome, 2017; 5: 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hao L, . Protocols for “The female urinary microbiota in relation to the reproductive tract microbiota”. 2020; 10.17504/protocols.io.bpyumpww. [DOI] [PMC free article] [PubMed]
- 8.Chen C, Hao L, Wei W, Li F, Song X, Zhang X et al. Data from the female urinary microbiota. 2020, GigaScience Database; 10.5524/100754. [DOI]
- 9.Hao L, . DNA extraction for human microbe samples. protocols.io 2020; 10.17504/protocols.io.bcmriu56. [DOI]
- 10.Prepare Amplicon Libraries without Fragmentation Using the Ion Plus Fragment Library Kit. https://assets.thermofisher.com/TFS-Assets/LSG/manuals/MAN0006846_PrepAmpliconLibr_using_IonPlusFragLibraryKit_UB.pdf.
- 11.Ion Personal Genome Machine™ (PGM™) System REFERENCE GUIDE. https://tools.thermofisher.com/content/sfs/manuals/MAN0009783_IonPGM_RefGuide.pdf.
- 12.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol., 2009; 75: 7537–7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods, 2010; 7: 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hao L, . A Bioinformatics Analysis workflow for 16S rRNA Amplicon Sequencing data. protocols.io 2020; 10.17504/protocols.io.bntpmemn. [DOI]
- 15.Anderson MJ, . A new method for non-parametric multivariate analysis of variance. Aust. Ecol., 2001; 26: 32–46. [Google Scholar]
- 16.Feng Q, Liang S, Jia H, Stadlmayr A, Tang L, Lan Z et al. Gut microbiome development along the colorectal adenoma-carcinoma sequence. Nat. Commun., 2015; 6: 6528. [DOI] [PubMed] [Google Scholar]
- 17.Zapala MA, Schork NJ, . Multivariate regression analysis of distance matrices for testing associations between gene expression patterns and related variables. Proc. Natl Acad. Sci. USA, 2006; 103: 19430–19435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Backer E, Verhelst R, Verstraelen H, Alqumber M a, Burton JP, Tagg JR et al. Quantitative determination by real-time PCR of four vaginal Lactobacillus species, Gardnerella vaginalis and Atopobium vaginae indicates an inverse relationship between L. gasseri and L. iners. BMC Microbiol., 2007; 7: 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen C, . Quantitative real-time PCR for the four Lactobacillus species. protocols.io 2020; 10.17504/protocols.io.bps2mnge. [DOI]
- 20.Augustinos AA, Kyritsis GA, Papadopoulos NT, Abd-Alla AMM, Cáceres C, Bourtzis K, . Exploitation of the medfly gut microbiota for the enhancement of sterile insect technique: Use of Enterobacter sp. in larval diet-based probiotic applications. PLoS One, 2015; 10: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.EzBioCloud. https://www.ezbiocloud.net/.
- 22.Karstens L, Asquith M, Caruso V, Rosenbaum JT, Fair DA, Braun J et al. Community profiling of the urinary microbiota: considerations for low-biomass samples. Nat. Rev. Urol., 2018; 15: 735–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hilt EE, McKinley K, Pearce MM, Rosenfeld AB, Zilliox MJ, Mueller ER et al. Urine is not sterile: Use of enhanced urine culture techniques to detect resident bacterial flora in the adult female bladder. J. Clin. Microbiol., 2014; 52: 871–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meijer-Severs GJ, Aarnoudse JG, Mensink WFA, Dankert J., . The presence of antibody-coated anaerobic bacteria in asymptomatic bacteriuria during pregnancy. J. Infect. Dis., 1979; 140: 653–658. [DOI] [PubMed] [Google Scholar]
- 25.Guzmàn CA, Pruzzo C, LiPira G, Calegari L, . Role of adherence in pathogenesis of Enterococcus faecalis urinary tract infection and endocarditis. Infect. Immun., 1989; 57: 1834–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fraimow HS, Jungkind DL, Lander DW, Delso DR, Dean JL, . Urinary tract infection with an Enterococcus faecalis isolate that requires vancomycin for growth. Ann. Intern. Med., 1994; 121: 22–26. [DOI] [PubMed] [Google Scholar]
- 27.Ormerod AD, Petersen J, Hussey JK et al. Immune complex glomerulonephritis and chronic anaerobic urinary infection–complications of filariasis. Postgrad. Med. J., 1983; 59: 730–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Petricevic L, Domig KJ, Nierscher FJ, Sandhofer MJ, Fidesser M, Krondorfer I et al. Characterisation of the vaginal Lactobacillus microbiota associated with preterm delivery. 2014;1–6. [DOI] [PMC free article] [PubMed]
- 29.Salter SJ, Cox MJ, Turek EM, Calus ST, Cookson WO, Moffatt MF et al. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol., 2014; 12: 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O’Hanlon DE, Come RA, Moench TR, . Vaginal pH measured in vivo: lactobacilli determine pH and lactic acid concentration. BMC Microbiol., 2019; 19: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]