

Abstract
Long-read single-molecule sequencing has revolutionized de novo genome assembly and enabled the automated reconstruction of reference-quality genomes. It has also been widely used to study structural variants, phase haplotypes and more. Here, we introduce the assembler SMARTdenovo, a single-molecule sequencing (SMS) assembler that follows the overlap-layout-consensus (OLC) paradigm. SMARTdenovo (RRID: SCR_017622) was designed to be a rapid assembler, which, unlike contemporaneous SMS assemblers, does not require highly accurate raw reads for error correction. It has performed well in the evaluation of congeneric assemblers and has been successfully users for various assembly projects. It is compatible with Canu for assembling high-quality genomes, and several of the assembly strategies in this program have been incorporated into subsequent popular assemblers. The assembler has been in use since 2015; here we provide information on the development of SMARTdenovo and how to implement its algorithms into current projects.
Introduction
The development of high-throughput sequencing provides the means to deliver fast, inexpensive, and accurate information for assembling whole genomes. As a result, there has been rapid growth in the number of whole-genome sequencing projects [1–3]. Single-molecule sequencing (SMS) technologies, such as Pacific Biosciences (PacBio) and Oxford Nanopore, which generate sequencing reads of >20 kb in length, are now widely used in whole genome projects. These long reads are advantageous because they span polymorphic regions, repeats, and transposable elements, and because they provide long-range information for assemblies that are usually too complex to be resolved by short reads alone.
The huge demand for long-range DNA sequencing and mapping technologies has catalysed a renaissance of the development of high-quality SMS assemblers, such as PBcR [4, 5], Falcon (RRID: SCR_016089) [6], Canu (RRID: SCR_015880) [7], Miniasm [8], Ra [9], Wtdbg2 (RRID: SCR_017225) [10], Flye (RRID: SCR_017016) [11], Shasta [12], and ABruijn [13]. In fact, highly available SMS assemblers have always been essential for improving the quality of genome assemblies.
SMARTdenovo (RRID: SCR_017622) is a long-read SMS assembler that follows the overlap-layout-consensus (OLC) paradigm. It assembles genomes following four steps: overlapping, trimming, layout, and consensus. The source code for SMARTdenovo was released in GitHub in 2015 [14]. Assessment by others has shown that it performs well compared with other congeneric assemblers [15], and it has been widely used for generating highly accurate contigs in many genome assemblies [16–18]. For datasets from both PacBio and Oxford Nanopore, such as 20–30× 2D reads of different varieties of yeast, SMARTdenovo assembled more accurate and more highly contiguous sequences than other assemblers [15, 19]. SMARTdenovo was also used successfully for datasets from the wild tomato Solanum pennellii (∼1.2 Gb) and Sorghum bicolor (∼732 Mb) using Oxford Nanopore reads [18, 20], and for the long-read datasets for Taraxacum kok-saghyz (∼1.04 Gb) and the woody plant, Rhizophora apiculata (∼274 Mb) using PacBio RSII reads [16, 21]. Here, we explain how we developed SMARTdenovo and provide use cases to show its ability.
Implementation
The SMARTdenovo algorithm
SMARTdenovo uses four steps for assembly: overlapping, trimming, layout, and consensus. We used homopolymer-compressed (HPC) k-mers for seed-indexing and identifying collinear seeds. HPCs have been widely adopted in Minimap2, Wtdbg2 and Shasta [10, 12, 22]. We then trimmed low-quality regions and chimeric reads based on the overlapping reads. We applied the Best-Overlap-Graph [23] to generate the layout of the reads and the PBDAG-Con algorithm [24] to generate a consensus.
Overlapper
The algorithm for alignment follows a typical seed-chain-align procedure that is used by most full-genome aligners. In this step, we constructed each read by contracting homopolymer reads to a single base, called HPC strings. An HPC k-mer, a 1000-bp substring of an HPC string, was treated as a seed (“wtzmo -k 16 by default”). For HPC-based k-mer-indexing, we scanned all the reads and counted k-mers in a hashtable with a 64-bit key to store a k-mer, and a 64-bit value to store its count. If a k-mer was present more than 500 times, it was filtered out (“wtzmo -K 500”). A seed array was also created and filled with the “read_id” and the orientation of the remaining seeds. To manage the cost of memory for k-mer-indexing, we used two different parameters: (1) we kept only the index with smaller values between the k-mer and its reverse complement; (2) we randomly selected k-mers according to the hash code (one quarter was set as the default). All queried k-mers were indexed against the hashtable and seed array to identify candidate reads. We sorted the seeds by the “read_id” and “strand” and calculated the coverage length of the overlaps. If the coverage was longer than 300 bp (“wtzmo -d 300”), the candidate was kept. The top 500 candidates were chosen for each query (“wtzmo –A 500”).
To refine the collinearity relationship between query and candidate, we further built a similar but shorter HPC-based index called a “z-mer” on queries for seed chaining. Each z-mer was recorded with its offset and strand. Pairs of matched seeds between candidate and query were obtained. However, because the high rate of insertions and deletions (indels) made the distance between two nearby z-mers highly variable, wtzmo was used to identify the synteny between query and candidate using sliding windows (“wtzmo –y 800”) on query instead of using the whole overlapping region. We first filtered z-mer windows using a minimum match length of 200 bp. We then developed a scoring algorithm to filter excessive matches. The adjacent matched pairs of seeds (pi, pi+1) were scored based on the relationship of z-mers on candidates: Si+1 = Si + Li+1 − Distancei−(i+1), S, L, D represented the sort of seeds, length of seeds, and the distance between the adjacent seeds, respectively. If Si+1 was larger than Li+1, pairs of pi, pi+1 were considered to be a syntenic block, and the block would be extended until Sj was smaller than Lj. At that point, the block ended, Sj recovered to Lj and the next round of syntenic block identification began. The z-mer-block containing the highest value of S was the block chosen as the best syntenic overlap between the two windows. If the coverage was longer than 100 bp, the matched windows were retained. Seed windows were also scored using the same method as above for searching the best colinear pairs. Finally, candidates with the best colinear window-block that covered more than 300 bp were retained for pairwise alignment.
To speed calculation time and reduce unnecessary computation, we developed a weighting algorithm that negatively ranked a repetitive region based on its depth in the alignment process. “Wtzmo –q 100” was set so that the weights ranged between 1 and 0 if the depth ranged from 10–100. Large numbers of false candidates containing similar repeats were eliminated with queries. The pairwise alignment procedure based on the collinearity relationship was split into four steps: (1) first, the bases of the z-mers were decompressed and gaps were added between the matched z-mers; (2) global alignment was conducted between two adjacent z-mers within a z-mer window; (3) the two adjacent z-mer windows were aligned using the banded global Smith–Waterman algorithm, with a dynamic bandwidth that increased according to the length of the gaps (“wtzmo –w 50 ∼ 3200”); (4) the two ends were extended by global alignment with the band width set at 800 bp (Figure 1).
Figure 1.
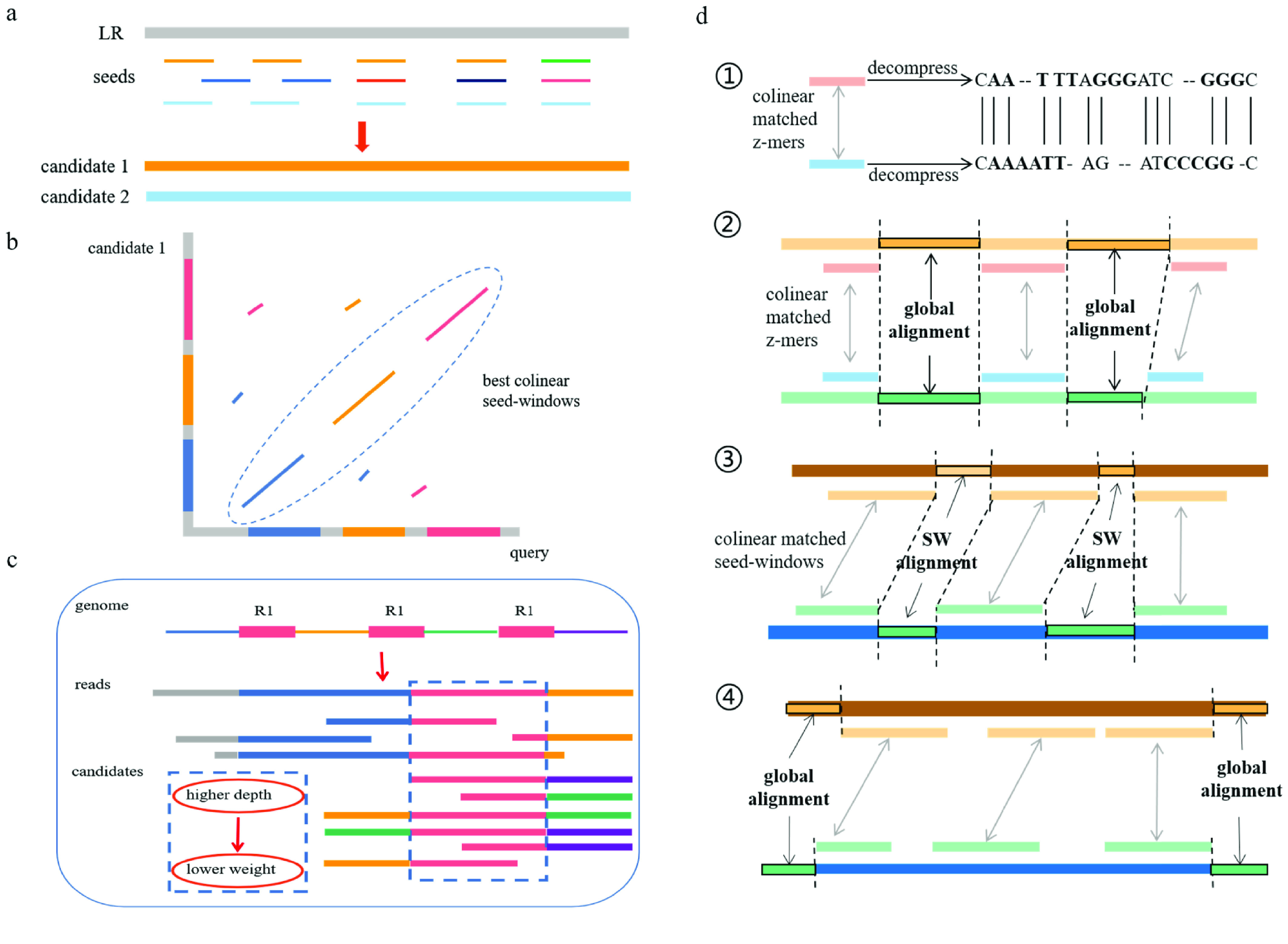
The algorithm for processing overlaps. (a) k-mer indexing and identification of candidate reads based on overlap coverage. LR, long reads. (b) Seed chaining based on sliding windows. (c) Weighting algorithm negatively ranking the repetitive regions according to their depth. R, repeat; red blocks represent reads and candidates represent the repeat region; and these bases of high depth would be marked with lower weight. (d) Pairwise alignment with four steps: (1) decompress the matched z-mers; (2) global alignment between two adjacent matched z-mers within a z-mer window; (3) global dynamic SW-alignment between the two adjacent paired z-mer windows; (4) extend the two ends by global alignment.
Trimming
Wtclp trimmed or discarded reads to a maximum total length of the valid overlaps. It took one read as a reference, and tiled all reads containing overlaps. A functional model, “call_legal_overlaps_wtclp”, calculated the length of valid overlaps. First, it clipped the ends that had high error rates. Then, it detected chimeras and trimmed them according to their depths. Structures containing partially aligned reads were called “spurs”. We counted the depth of reads crossing the “spurs” as “m” and counted the number of reads with partial alignments as “n”. If the m of a read was less than half of the average depth, or if n was larger than the average depth, or if n was larger than “m∕2”, the read was considered chimeric and discarded. Otherwise, it was considered to be a sequencing error and the maximum region of reads was retained. Errors in the structure were corrected based on the graph. If a single read connected two subgraphs, it was considered a chimera. We then used wtclp to check for any alternative path formed by valid overlaps of tiled reads.
Layout
Wtlay was used to achieve the Best-Overlap-Graph [23] to generate a layout of reads following the OLC paradigm. In general, if an overlap was not end-to-end, leaving n (no greater than 100) bp unaligned, it would be treated as true (“wtlay –w 100”). Owing to the high indel rate, wtlay identified the best overlap with an alignment score ≥ 0.95, instead of picking out the longest one (“wtlay –r 0.95”). Using this process would keep bubbles from being merged, and instead find one appropriate path. The wtlay script also filtered out each unitig sharing more than 40% identity with another unitig to avoid islands. The output included uncorrected unitigs and all the parameters needed by the consensus caller.
Consensus
We used the wtcns command to implement the PBDAG-Con algorithm described in HGAP to generate consensus [24]. Because an alignment algorithm is integrated into wtcns, it required no other alignment tools. Wtcns took the layout file as input and output the consensus sequences in fasta format.
Results
Assembling the genome of the fruit fly and evaluating accuracy of the assembly
We benchmarked SMARTdenovo against the SMS assemblers Flye, Canu, and Ra using the dataset of the fruit fly, Drosophila melanogaster, and calculated the accuracy of the assembly by aligning it with the reference genome. A total of 29.3 Gb PacBio reads from the National Center for Biotechnology Information Sequence Read Archive database (SRX499318) [25] was assembled with the command line “smartdenovo.pl -c 1 -t 16 reads.fa > wtasm.mak && make -f wtasm.mak”. The length of the genome sequence was 146 Mb, with an N50 value of 11.59 Mb. We also tested three other SMS assemblers (Canu, Flye, Ra) using this dataset. SMARTdenovo was superior to Flye and Ra in both total length and contig N50, but was inferior to Canu (Table 1).
Table 1.
Evaluation of long-read assemblies on the fruit fly genome (PacBio datasets).
| Parameter | SMS assembler | |||
|---|---|---|---|---|
| Canu | Flye | Ra | SMARTdenovo | |
| Total length, Mb | 161.98 | 137.02 | 139.27 | 146.29 |
| Count of contigs | 633 | 970 | 424 | 242 |
| Total length (≥50,000 bp), Mb | 151.43 | 131.37 | 135.14 | 143.15 |
| Largest contig, Mb | 25.87 | 25.74 | 5.82 | 23.29 |
| N50, Mb | 21.40 | 9.57 | 1.02 | 11.59 |
| L50, n | 4 | 4 | 37 | 4 |
| Misassemblies, n | 3,296 | 783 | 242 | 1,382 |
| Mismatches per 100 kb, n | 158.82 | 36.93 | 63.60 | 103.44 |
| CPU hours | 2810.14 | 348.47 | 226.28 | 830.95 |
| peak MEM, Gb | 15.70 | 134.66 | 92.50 | 24.02 |
The genome size of D. melanogaster is usually estimated to be 180 Mb. Of this, 60 Mb of the genome comprises centric heterochromatin, making it intractable for assembly [26]. Compared with the released reference [27], SMARTdenovo and Canu were able to create longer assemblies: ∼146.29 Mb and ∼161.97 Mb, respectively. SMARTdenovo and Canu performed better than the other two SMS assemblers, not only by creating longer assembly lengths, but also having higher coverage when aligned to the reference genome.
Assembling the genome of the wild tomato
We also compared the performance of SMARTdenovo with that of three other SMS assemblers: Flye, Canu, and Ra using the dataset for the wild tomato Solanum pennellii. A total of 27.5 Gb Oxford Nanopore reads was downloaded from the European Nucleotide Archive database (PRJEB19787) [28]. A k-mer analysis of this dataset indicated that this accession of S. pennellii (LYC1722) has a genome size between 1 and 1.2 Gb [20]. We assembled a 30-fold Oxford Nanopore dataset and achieved an assembly of 902.96 Mb, with an N50 value of 339 kb (Table 2). We also tried Flye and Ra on this dataset: Flye obtained the longest genome sequence (1.27 Gb) and a higher N50 value (429 kb). Ra was unable to achieve the same sequence length as SMARTdenovo. When taking into account the computation time, SMARTdenovo required 651 central processing unit (CPU) hours, which was 70 hours faster than Flye (Table 2).
Table 2.
Comparison of different assemblers on the wild tomato genome (Oxford Nanopore datasets).
| Parameter | SMS assembler | |||
|---|---|---|---|---|
| Canu | Flye | Ra | SMARTdenovo | |
| Total length, Mb | 801.62 | 1265.09 | 815.60 | 902.99 |
| Count of contigs, n | 14,286 | 10,323 | 6490 | 4395 |
| Total length (≥50,000 bp), Mb | 606.49 | 1,070.26 | 767.88 | 860.21 |
| Largest contig, Mb | 2.25 | 4.54 | 2.98 | 3.34 |
| N50, kb | 114.88 | 429.25 | 161.11 | 399.45 |
| L50, n | 1728 | 720 | 1,292 | 633 |
| Misassemblies, n | 97 | 3539 | 1096 | 136 |
| Mismatches per 100 kb, n | 881.26 | 2656.12 | 2250.65 | 931.47 |
| CPU hours | 11,885.30 | 723.91 | 507.97 | 651.72 |
| peak MEM, Gb | 19.09 | 135.57 | 135.50 | 27.78 |
Discussion
SMARTdenovo is an accurate and efficient SMS assembler compatible with data formats output of both PacBio and Oxford Nanopore technologies. It comprises several command line tools: wtzmo, to overlap reads; wtclp, to trim low-quality regions and chimeras: wtlay, to generate the assembly graph layout; and wtcns to calculate the consensus. Based on the results of tests on the wild tomato dataset, we found that SMARTdenovo was more memory-intensive than the other SMS assemblers, but its performance was comparable, and it was faster. SMARTdenovo has been successfully used to assemble data from various species such as plasmids [29], protists [17, 30], fungi [19, 31, 32], microorganisms [33], and complex plants [18, 20, 21].
In addition to its solid performance, SMARTdenovo includes multiple algorithms that can be—and have been—useful for improving other programs. These algorithms have had a positive impact on popular SMS assemblers. For example, in developing SMARTdenovo, we introduced the first algorithm to use HPC-based k-mers, and this has now been incorporated into many other assemblers [10, 12, 22]. SMARTdenovo has had a more extensive impact on our development of the assembler Wtdbg2, as it includes several of its algorithms for handling long reads, including those for indexing, seed chaining, trimming, consensus, and some of the data formation.
There are several other algorithms within SMARTdenovo that have not yet been taken advantage of for use in other programs. An example includes its weighting algorithm for handling repeat regions, which significantly improves both its speed and the accuracy of the alignment. At this point, no other long read assemblers have this feature.
SMARTdenovo has been available on GitHub since 2015, but its performance not only remains comparable with current assemblers, it also has several advantages as described. Furthermore, given its excellent performance for use on corrected long sequence reads, it continues to be widely used in for genome assembly projects today [34–43].
Acknowledgements
Not applicable.
Funding Statement
This study was supported by the National Key R&D Program of China (2019YFA0707003), the Natural Science Foundation of China (31822029).
Availability of source code and requirements
Project name: SMARTdenovo
Project home page: https://github.com/ruanjue/smartdenovo
Operating systems: 64-bit Linux
Programming language: C 93.3%, C++ 4.6%, Perl 1.5%, other 0.6%
Other requirements: None
License: GNU GPL-3.0
RRID: RRID:SCR_017622
Data availability
A Code Ocean capsule to execute SMARTdenovo is available (Figure 2) [44]. Supporting data are available in the GigaScience GigaDB repository [45].
Figure 2.
Code Ocean capsule to execute SMARTdenovo [44]. https://doi.org/10.24433/CO.4665826.v1
Declarations
List of abbreviations
HPC, homopolymer compressed; indel, insertion and deletion; OLC, overlap-layout-consensus; PacBio, Pacific Biosciences; SMS, single-molecule sequencing.
Ethical approval
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Funding
This study was supported by the National Key R&D Program of China (2019YFA0707003), the Natural Science Foundation of China (31822029).
Authors’ contributions
J.R. initiated the program, coordinated the project. H.L.L. and J.R. wrote the manuscript. S.G.W., A.L.L and H.L.L conducted the software testing. J.R. and H.L.L revised the manuscript. All authors read and approved the final manuscript.
References
- 1.Metzker ML, . Sequencing technologies - the next generation. Nat. Rev. Genet., 2010; 11: 31–46. [DOI] [PubMed] [Google Scholar]
- 2.Huang SW, et al. The genome of the cucumber, Cucumis sativus L. Nat. Genet., 2009; 41: 1275–1281. [DOI] [PubMed] [Google Scholar]
- 3.Li RQ, et al. The sequence and de novo assembly of the giant panda genome. Nature, 2010; 463: 311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koren S, et al. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat. Biotechnol., 2012; 30: 693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berlin K, Koren S, Chin CS, Drake JP, Landolin JM, AM Phillippy, . Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat. Biotechnol., 2015; 33: 623–630. [DOI] [PubMed] [Google Scholar]
- 6.Chin CS, et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods, 2016; 13: 1050–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM, . Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res., 2017; 27: 722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li H, . Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics, 2016; 32: 2103–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Laboratory for Bioinformatics and Computational Biology, University of Zagreb, Faculty of Electrical Engineering and Computing. Ra (v0.2.1), 2019; https://github.com/lbcb-sci/ra.
- 10.Ruan J, Li H, . Fast and accurate long-read assembly with wtdbg2. Nat. Methods, 2020; 17: 155–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kolmogorov M, Yuan J, Lin Y, Pevzner PA, . Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol., 2019; 37: 540–546. [DOI] [PubMed] [Google Scholar]
- 12.Shafin K, et al. Nanopore sequencing and the Shasta toolkit enable efficient de novo assembly of eleven human genomes. Nat. Biotechnol., 2020; 38: 1044–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin Y, Yuan J, Kolmogorov M, Shen MW, Chaisson M, Pevzner PA, . Assembly of long error-prone reads using de Bruijn graphs. Proc. Natl Acad. Sci. USA, 2016; 113: E8396–E8405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ruan J., . SMARTdenovo. 2015; https://github.com/ruanjue/smartdenovo.
- 15.Istace B, et al. De novo assembly and population genomic survey of natural yeast isolates with the Oxford Nanopore MinION sequencer. Gigascience, 2017; 6: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu SH, et al. The origin, diversification and adaptation of a major mangrove clade (Rhizophoreae) revealed by whole-genome sequencing. Natl Sci. Rev., 2017; 4: 721–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Belkhelfa S, et al. Complete genome sequence of the facultative methylotroph Methylobacterium extorquens TK 0001 isolated from soil in Poland. Genome Announc., 2018; 6: e00018–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deschamps S, et al. A chromosome-scale assembly of the Sorghum genome using nanopore sequencing and optical mapping. Nat. Commun., 2018; 9: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fournier T, et al. High-quality de novo genome assembly of the Dekkera bruxellensis yeast using nanopore MinION sequencing. G3 (Bethesda), 2017; 7: 3243–3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schmidt MH-W, et al. De novo assembly of a new Solanum pennellii accession using nanopore sequencing. Plant Cell, 2017; 29: 2336–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin T, et al. Genome analysis of Taraxacum kok-saghyz Rodin provides new insights into rubber biosynthesis. Natl Sci. Rev., 2018; 5: 78–87. [Google Scholar]
- 22.Li H., . Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics, 2018; 34: 3094–4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller JR, et al. Aggressive assembly of pyrosequencing reads with mates. Bioinformatics, 2008; 24: 2818–2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chin CS, et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods, 2013; 10: 563–569. [DOI] [PubMed] [Google Scholar]
- 25.Pacific Biosciences. SRX499318: Pacific Biosciences model organism genome sequencing – Drosophila melanogaster. NCBI Sequence Read Archive; https://www.ncbi.nlm.nih.gov/sra/SRX499318.
- 26.Adams MD, et al. The genome sequence of Drosophila melanogaster. 2000; 287: 2185–2195. [DOI] [PubMed] [Google Scholar]
- 27.Hoskins RA, et al. The Release 6 reference sequence of the Drosophila melanogaster genome. Genome Res., 2015; 25: 445–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.RWTH Aachen. Sequencing the gigabase plant genome of the wild tomato species Solanum pennellii using Oxford Nanopore single molecule sequencing. European Nucleotide Archive; https://www.ebi.ac.uk/ena/browser/view/PRJEB19787.
- 29.Fang H, et al. Sequencing of pT5282-CTXM, p13190-KPC and p30860-NR, and comparative genomics analysis of IncX8 plasmids. Int. J. Antimicrob. Agents, 2018; 52: 210–217. [DOI] [PubMed] [Google Scholar]
- 30.Pollo SM, et al. MinION re-sequencing of Giardia genomes and de novo assembly of a new Giardia isolate. BioRxiv. 2018; 10.1101/343541. [DOI]
- 31.Wang X, et al. Genome sequencing illustrates the genetic basis of the pharmacological properties of Gloeostereum incarnatum. Genes (Basel)., 2019; 10: 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sossah FL, et al. Genome sequencing of Cladobotryum protrusum provides insights into the evolution and pathogenic mechanisms of the cobweb disease pathogen on cultivated mushroom. Genes (Basel), 2019; 10: 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shin SC, et al. Nanopore sequencing reads improve assembly and gene annotation of the Parochlus steinenii genome. Sci. Rep., 2019; 9: 5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perumal S, et al. High contiguity long read assembly of Brassica nigra allows localization of active centromeres and provides insights into the ancestral Brassica genome. BioRxiv. 2020; 10.1101/2020.02.03.932665. [DOI]
- 35.Xu Z, et al. Comparative genome analysis of Scutellaria baicalensis and Scutellaria barbata reveals the evolution of active flavonoid biosynthesis. Genomics Proteomics Bioinformatics, 2020; 18(3): . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dussert Y, et al. Identification of the first oomycete mating-type locus sequence in the grapevine downy mildew pathogen, Plasmopara viticola. Curr. Biol., 2020; 30: 3897–3907, e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Adams TM, et al. Genomic investigation of the strawberry pathogen Phytophthora fragariae indicates pathogenicity is associated with transcriptional variation in three key races. Front. Microbiol., 2020; 11: 490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang S, et al. Chromosome-scale genome assembly provides insights into speciation of allotetraploid and massive biomass accumulation of elephant grass (Pennisetum purpureum Schum.). BioRxiv. 2020; 10.1101/2020.02.28.970749. [DOI] [PubMed]
- 39.Fang Y, et al. Long transposon-rich centromeres in an oomycete reveal divergence of centromere features in Stramenopila-Alveolata-Rhizaria lineages. PLoS Genet., 2020; 16: e1008646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takehana Y, et al. Genome sequence of the Euryhaline javafish Medaka, Oryzias javanicus: a small aquarium fish model for studies on adaptation to salinity. G3 (Bethesda), 2020; 10: 907–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Armitage AD, et al. Genomics evolutionary history and diagnostics of the Alternaria alternata species group including apple and asian pear pathotypes. Front. Microbiol., 2019; 10: 3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Feron R, et al. Characterization of a Y-specific duplication/insertion of the anti-Mullerian hormone type II receptor gene based on a chromosome-scale genome assembly of yellow perch, Perca flavescens. Mol. Ecol. Resour., 2020; 20: 531–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Large CR, et al. Genomic stability and adaptation of beer brewing yeasts during serial repitching in the brewery. BioRxiv. 2020; 10.1101/2020.06.26.166157. [DOI]
- 44.Ruan J., . SMARTdenovo: a de novo assembler using long noisy reads [Source Code]. Code Ocean. 2020; 10.24433/CO.4665826.v1. [DOI] [PMC free article] [PubMed]
- 45.Liu H, Wu s, Li A, Ruan J, . Supporting data for “SMARTdenovo: a de novo assembler using long noisy reads”. 2021, GigaScience Database; 10.5524/100881. [DOI] [PMC free article] [PubMed]