

Abstract
Objective:
To identify genetic variants associated with severity of post-burn hypertrophic scarring (HTS) using a genome-wide approach.
Background:
Risk of severe post-burn HTS is known to depend on race, but the genetic determinants of HTS are unknown.
Methods:
We conducted a genome-wide association study (GWAS) in a prospective cohort of adults admitted with deep-partial-thickness burns from 2007 through 2014. Scar severity was assessed over time using the Vancouver Scar Scale (VSS), and DNA was genotyped with a >500,000-marker array. We performed association testing of SNPs with minor allele frequency (MAF) >0.01 using linear regression of VSS height score on genotype adjusted for patient- and injury characteristics as well as population genetic structure. Array-wide significance was based on Bonferroni correction for multiple testing.
Results:
Of 538 patients (median age 40 years, median burn size 6.0% body surface area), 71% were male and 76% were white. The mean VSS height score was 1.2 (range: 0–3). Of 289,639 SNPs tested, a variant in the CUB and Sushi multiple domains 1 (CSMD1) gene (rs11136645; MAF = 0.49), was significantly associated with decreased scar height (regression coefficient = −0.23, p = 7.9×10−8).
Conclusions:
In the first published GWAS of HTS, we report that a common intronic variant in the CSMD1 gene is associated with reduced severity of post-burn HTS. If this association is confirmed in an independent cohort, investigating the potential role of CSMD1 in wound healing may elucidate HTS pathophysiology.
Mini-abstract
Severity of post-burn hypertrophic scarring (HTS) is known to depend on race, but the genetic determinants of HTS are unknown. In the first genome-wide association study of HTS, we report that a common variant in the CUB and Sushi multiple domains 1 (CSMD1) gene is associated with reduced HTS severity.
Introduction
Approximately 40,000 people are hospitalized for burn injuries each year in the U.S.,1 and over 70% of these patients are estimated to develop hypertrophic scarring (HTS).2,3 This exuberant response to injury results in thick, contracted, dyspigmented scar raised above the level of the surrounding skin but remaining within the boundaries of the original wound.4 In addition to being disfiguring and stigmatizing, HTS often causes pain, pruritus, and functional impairment, disabling conditions that impede return to work5 and contribute to decreased quality of life.6 Despite years of research, HTS pathophysiology remains unknown7 with no reliable methods available to prevent or treat HTS.8
The long-standing observation that individuals of dark-skinned race are at increased risk of HTS3,9,10 strongly implies a genetic mechanism. However, thus far there have been only a few candidate-gene association studies of HTS in burned patients.10,11 These studies have been limited by their reliance on a priori etiologic hypotheses and consideration of only a small number of targeted genetic variants. Thus, the genetic determinants of HTS are largely unknown.
Genome-wide association studies (GWAS) test thousands of genetic markers, predominantly single-nucleotide-polymorphisms (SNPs), for association with complex phenotypes. Following the first successful GWAS in 2005, the number of published GWAS projects has risen exponentially; currently about 2,000 published GWASs12 identify over 2,000 robust genetic associations with hundreds of human traits and diseases.13 Unlike candidate-gene studies, GWASs do not rely on existing knowledge and thus are valuable for generating novel biological hypotheses. They have been effective in identifying risk loci in genes not previously associated with a particular phenotype, risk loci shared by diseases not previously known to have etiologic overlap, and associations with gene-poor chromosomal regions.14 GWAS findings are being translated to clinical use in risk prediction, disease classification, and drug development.13 As there have been no published GWASs of HTS to date, the objective of our study was to identify genetic variants associated with severity of post-burn HTS using a genome-wide approach.
METHODS
Study design, population, and setting
We conducted a GWAS in a prospective cohort of patients admitted to the UW Medicine Regional Burn Center. This study was approved by the University of Washington Institutional Review Board, and a National Institutes of Health Federal Certificate of Confidentiality was obtained. From 2007 through 2014, we enrolled adults (age ≥18) with deep-partial-thickness burns or burns that showed delayed healing (≥2 weeks), characteristics known to be associated with increased risk of HTS.15 Subjects provided blood samples for genotyping, and age, sex, burn size, number of operations, and self-identified race and ethnicity were obtained from the electronic medical record. Scars were assessed in clinic by research nurses using the Vancouver Scar Scale (VSS)16. Although we sought to evaluate each subject at both early (1–6 months post-burn) and late (6–12 months post-burn) time points, we included in our analysis any subject with at least one completed scar assessment.
Genotyping and quality control
Genomic DNA was isolated from venous whole-blood samples using a QIAamp DNA Mini Kit (Qiagen, Valencia, CA) and quantified using a Quant-iT dsDNA Broad Range Assay Kit (Life Technologies, Carlsbad, CA). Sex was verified using a Taqman GTXpress kit on a Viia 7 real-time PCR system (both from Life Technologies, Carlsbad, CA). Samples were genotyped at the University of Washington Center for Clinical Genomics on HumanCoreExome-12 v1.1 BeadChips containing 542,585 genome-wide markers (Illumina, San Diego, CA) using a Freedom EVO robotic platform (Tecan Systems, San Jose, CA) and an iScan array scanner (Illumina, San Diego, CA). Samples with <3% missing genotypes were retained for further analysis. Markers with minor allele frequency (MAF) >1%, <10% missingness, and Hardy-Weinberg disequilibrium P>1×10−6 were included in association testing.
Statistical analysis
Subject characteristics are summarized as number (percent) for categorical variables and median (interquartile range [IQR]) for continuous variables. Population genetic structure was assessed by principal component analysis (PCA). Testing for difference in SNP frequency according to self-identified race was performed using the χ2 test. Association testing was based on multivariate linear regression with VSS height score (corresponding to scar height above the level of the surrounding skin) as the dependent variable. We used VSS height score alone as our outcome since raised height is the defining feature of hypertrophic scars4 and since pigmentation, vascularity, and pliability assessments are inherently more subjective, potentially limiting reproducibility of our findings. Height score was coded as follows: 0 mm (0), <2 mm (1), 2–5 mm (2), or >5 mm (3). For subjects with more than one scar assessment, the highest height score was used. In each model, the independent variable of interest was marker genotype, with biallelic markers modeled assuming an additive model of inheritance. The following covariates were included for adjustment: age in years, sex, percent body surface area burned, number of operations, and, to correct for population genetic structure, the first two global principal components.17 For each marker, we estimated the adjusted regression coefficient (β) with its 95% confidence interval (CI) and asymptotic P-value. To account for the number of markers tested, the array-wide significance level was adjusted by the Bonferroni method. Quality control, PCA, association testing, and linkage-disequilibrium analysis were carried out using PLINK18 version 1.90. R version 3.1.2 was used to summarize the clinical data, calculate the genomic inflation factor, and generate Manhattan and quantile-quantile (Q-Q) plots.
Study power
Based on a previous analysis of scar outcomes at our institution10, we anticipated a mean VSS height score of approximately 1 with standard deviation of 0.75 among our study subjects. Using these estimates, we calculated the number of subjects required for 80% power to detect β values of 0.1–0.5 for markers with MAF from 0.1–0.5 with an array-wide type-I error rate of 0.05/289,639 ≈ 2×10−7 (two-sided) assuming an additive model of inheritance (Table 1). Given that >500 subjects were included in our analysis, we concluded that our study was adequately powered to detect moderate to large effect sizes associated with common markers (eg, β ≥ 0.35 for a marker with MAF of 0.20). Sample-size calculations were performed using Quanto19 version 1.2.4 (University of Southern California Biostatistics, Los Angeles, CA).
TABLE 1.
Estimated number of subjects required for detection* of various effect sizes (β#) according to marker minor allele frequency.
| Regression coefficient β# | ||||||
|---|---|---|---|---|---|---|
| 0.10 | 0.20 | 0.30 | 0.40 | 0.50 | ||
| Minor allele frequency | 0.10 | 11386 | 2833 | 1249 | 694 | 438 |
| 0.20 | 6397 | 1585 | 694 | 382 | 238 | |
| 0.30 | 4869 | 1204 | 525 | 287 | 177 | |
| 0.40 | 4258 | 1051 | 457 | 249 | 152 | |
| 0.50 | 4087 | 1008 | 438 | 238 | 145 | |
With 80% power and a type-I error rate of 2×10−7, assuming an additive model of inheritance.
Linear regression coefficient corresponding to average difference in Vancouver Scar Scale height score (range: 0–3) associated with each additional rare allele.
RESULTS
Of 638 subjects enrolled, 25 were lost to follow-up prior to providing a blood sample and/or undergoing any scar assessment, leaving 613 samples for genotyping. Of these, 15 failed post-genotyping quality control, and 60 were either lost to follow-up or had not yet had a scar assessment by the time of the analysis and were thus excluded, leaving 538 subjects with complete genotypic and clinical data available for association testing (Table 2). Subjects were predominantly non-Hispanic White males, consistent with demographics of patients treated at our burn center. The majority of the study subjects underwent at least one burn operation, reflecting enrollment based on deep or slow-healing burn wounds. At a median follow-up of 6.4 months (IQR 3.7–7.7 months), the majority (85%) of subjects had raised scars, indicating some degree of HTS (Table 3).
TABLE 2.
Characteristics* of 538 subjects.
| Age# (years) | 40 | (28–53) |
| Sex | ||
| Male | 382 | (71%) |
| Female | 156 | (29%) |
| Ethnicity† | ||
| Non-Hispanic | 72 | (13%) |
| Hispanic | 448 | (83%) |
| Race‡ | ||
| White | 408 | (76%) |
| Asian | 26 | (5%) |
| Black/AA | 19 | (4%) |
| Native American | 11 | (2%) |
| Other/multiple | 57 | (11%) |
| Burn size# (%TBSA) | 6 | (2–14) |
| Number of operations | ||
| None | 201 | (37%) |
| At Least One | 337 | (63%) |
Data presented as number (%), except where indicated.
Reported as median (interquartile range).
Missing or reported as unknown for 18 subjects.
Missing or reported as unknown for 17 subjects. AA, African American; %TBSA, percent total body surface area burned.
TABLE 3.
Scar outcomes for 538 subjects.
| Scar height (mm) |
VSS height score |
N | (%) |
|---|---|---|---|
| 0 | 0 | 83 | (15.4) |
| <2 | 1 | 302 | (56.1) |
| 2–5 | 2 | 131 | (24.4) |
| >5 | 3 | 22 | (4.1) |
After quality control and filtering, 289,639 markers were retained for subsequent analysis. PCA demonstrated considerable population genetic structure, with SNP frequencies varying according to self-identified race (Fig. 1A) but not ethnicity (Fig. 1B), consistent with a previous report.20 This analysis also revealed substantial genetic substructure within racial groups, particularly among those self-identifying as White (Fig. 1A). Following association testing with adjustment for the first two principal components, a QQ plot of the obtained P-values showed no systematic deviation of the observed distribution from the expected null distribution (Fig. 2). Accordingly, the genomic inflation factor, which quantifies the extent of test-statistic inflation and excess false-positive rate (with a value of 1 signifying no inflation), was estimated to be 1.005, indicating adequate correction for population genetic structure in our genetically admixed study population.
FIGURE 1.
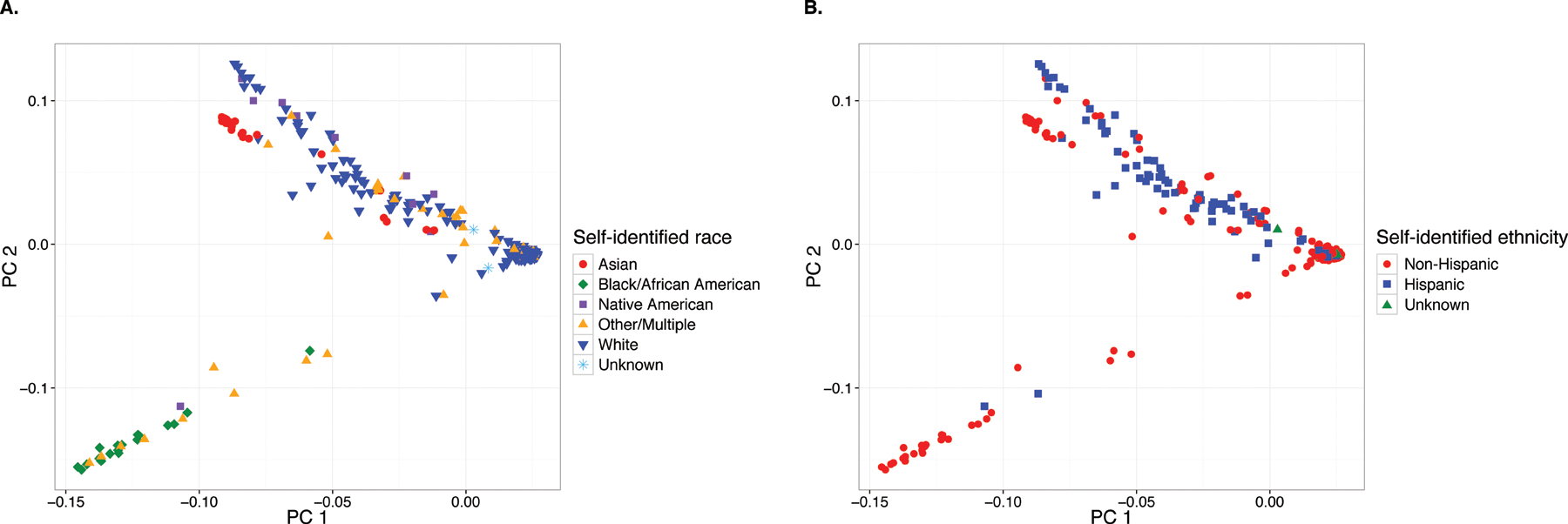
Principal component analysis (PCA) biplots for 538 study subjects based on 289,639 genome-wide markers. (A) Subjects are identified based on self-identified race, illustrating both clustering in accordance with self-identified race as well as considerable genetic substructure, particularly among Whites. (B) Subjects are identified based on self-identified ethnicity, illustrating lack of separation between Hispanics from Non-Hispanics.
FIGURE 2.
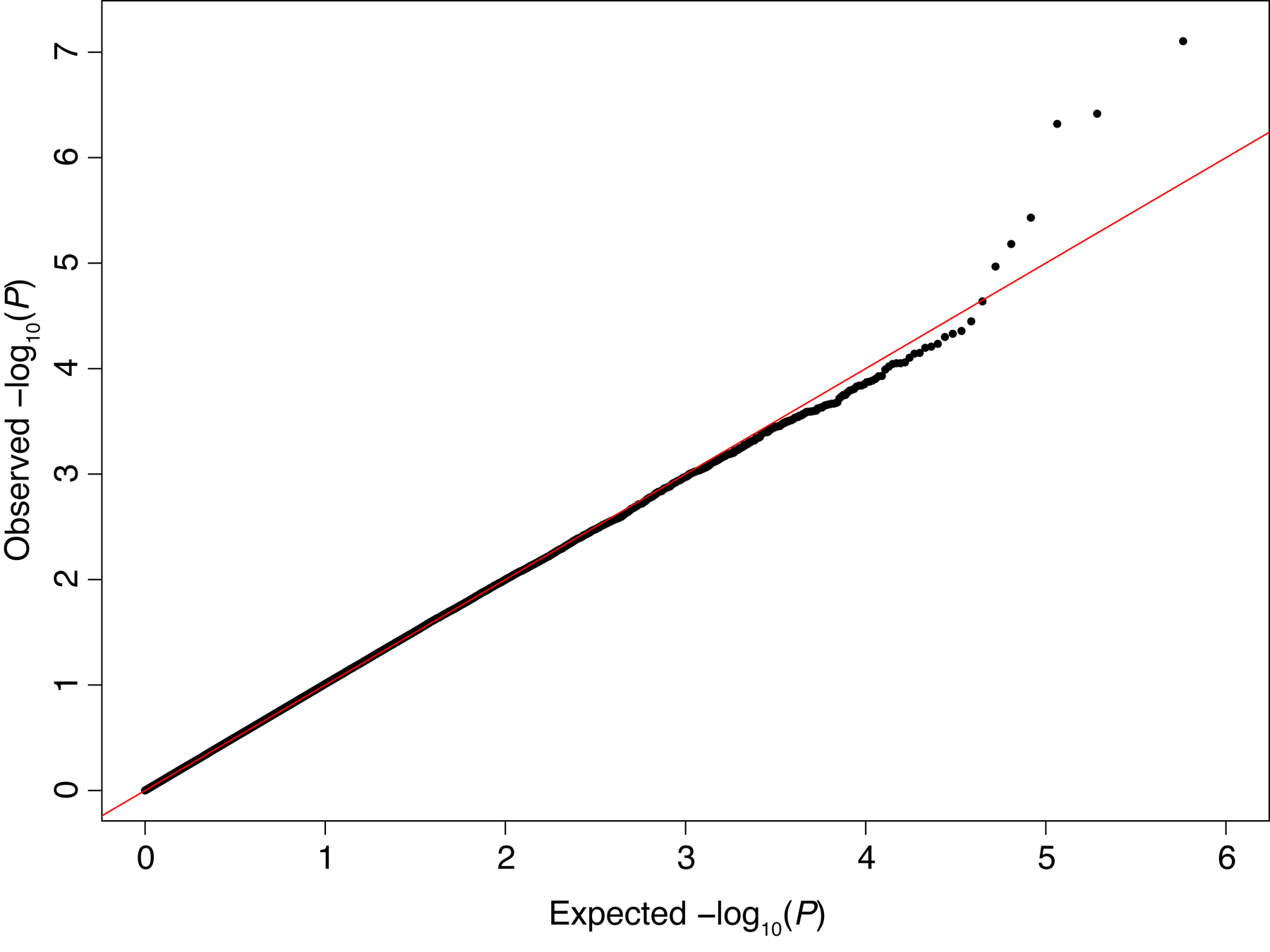
Quantile-quantile (QQ) plot of P values from association testing of genome-wide markers with post-burn scar height. Close correspondence of the observed P-values to the expected null distribution (solid line) indicates adequate correction for population genetic structure.
Using an array-wide significance threshold of P < 2×10−7, one SNP (rs11136645) was significantly associated with HTS (β = −0.23, P = 7.9×10−8; Fig. 3). This common (overall MAF 0.49) intronic variant in the CUB and Sushi multiple domains 1 (CSMD1) gene was associated with decreased scar height; specifically, each additional rs11136645 minor allele was associated with a 0.23 point lower height score (95% CI: −0.32, −0.15) after adjusting for age, sex, burn size, number of operations, and population genetic structure (by principal components). Two other CSMD1 SNPs approached array-wide significance and were similarly associated with decreased scar height: rs2449199 (β = −0.22; 95% CI: −0.31, −0.14; P = 3.8×10−7) and rs13279667 (β = −0.22; 95% CI: −0.30, −0.13; P = 4.8×10−7). Both of these common SNPs were in near-complete linkage disequilibrium with rs11136645 (MAF = 0.49, r2 = 0.82, and D’ = 0.91 for rs2449199; MAF = 0.49, r2 = 0.96, and D’ = 0.98 for rs13279667).
FIGURE 3.
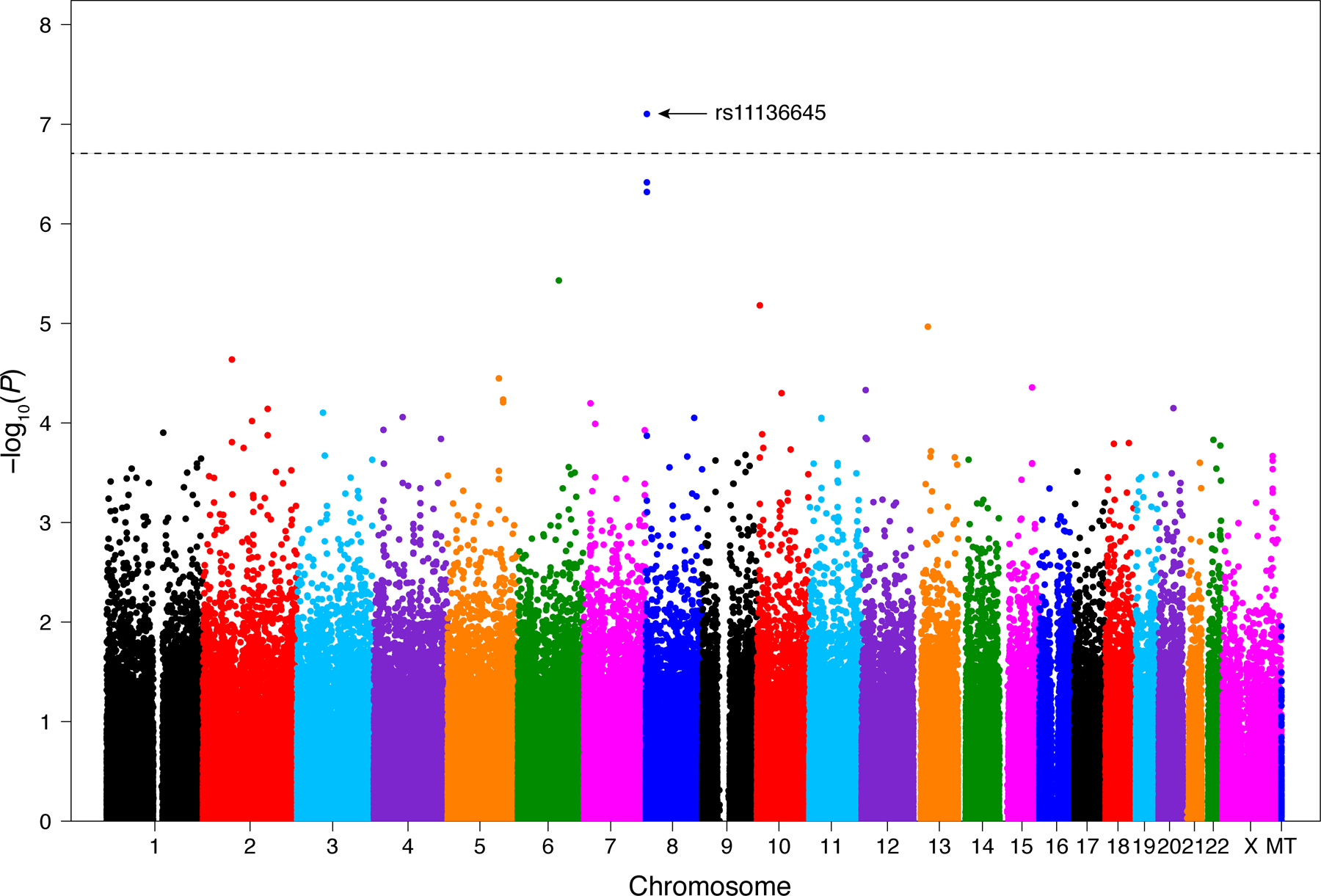
Manhattan plot of P values for genome-wide SNP association testing with post-burn scar height demonstrates a single SNP reaching array-wide significance (P = 2×10−7;dashed line). MT, mitochondrial DNA.
Despite considerable population genetic structure among our study population (Fig. 1), the frequency of rs11136645 did not vary significantly according to self-identified race (P = 0.66). However, given the potential for effect modification by genetic or environmental factors associated with race, we examined the association between rs11136645 with scar height among the White subjects only (N = 408). Similar to our results based on the entire study population, we found that among Whites, each additional rs11136645 minor allele was associated with a 0.20 point lower height score (95% CI: −0.29, −0.11; P = 4.5×10−5) after adjusting for age, sex, burn size, number of operations, and population substructure (by principal components). We conclude that the relatively small proportion of non-White subjects included in the analysis did not drive the strong association between rs11136645 and decreased scar height.
DISCUSSION
In the first published GWAS of hypertrophic scarring, we report that rs11136645, a common variant in the CSMD1 gene, is significantly associated with decreased scar height. Our results demonstrate the feasibility of applying the GWAS approach to the study of HTS and support further studies in populations at increased risk of HTS. Because ours is the only published GWAS of HTS to date, our results have not yet been confirmed in an independent clinical cohort. Due to the possibility of false-positive associations, replication of primary GWAS results is critically important prior to clinical translation of findings.21 If our findings are replicated, understanding the biological mechanism underlying the association between rs11136645 and HTS severity may help elucidate the pathogenesis of HTS.
Given that rs11136645 is located in an intronic region of the CSMD1 gene, this variant allele would not result directly in an amino-acid substitution. However, intronic SNPs can alter function of their gene products through a variety of mechanisms, including by affecting mRNA splicing or translation, among others.22 Although one or more ungenotyped variant(s) in linkage disequilibrium with rs11136645 max explain the association between rs11136645 and decreased scar height, a functional effect on CSMD1 is also possible. Because our results implicate CSMD1 in wound healing for the first time, examining the spatial and temporal pattern of CSMD1 expression during normal and pathologic wound healing will be critical.
The CSMD1 gene was discovered in an effort to characterize 8p23 chromosomal deletions associated with multiple types of cancer including head and neck squamous cell carcinoma and prostatic adenocarcinoma.23 The name reflects the structure of the ~3,500 amino-acid transmembrane protein23 it encodes, which consists of 14 CUB and 27 Sushi domains.23 CUB domains are found on a variety of proteins involved in developmental processes and were named for the first three identified proteins of this family (complement subcomponents C1r/C1s, epidermal growth factor related sea urchin protein, and bone morphogenetic protein 1).24 Sushi domains, also known as short consensus repeats (SCRs) or complement control protein (CCP) modules, are present in the majority of complement inhibitors.25 We propose three novel mechanistic hypotheses that could potentially explain an association between CSMD1 and post-burn scar height.
Consistent with its characteristic protein structure, CSMD1 inhibits the classical complement pathway in rats26 and both the classical and lectin complement pathways in human cells.27 Reconciling this function with the role of CSMD1 as a tumor suppressor, Escudero-Esparza and colleagues hypothesize that CSMD1 loss of function promotes tumorigenesis by increasing inflammation through loss of complement inhibition.27 Given the known role of inflammation in the pathogenesis of fibroproliferative cutaneous scarring,28 we propose that one potential mechanism linking CSMD1 to HTS might be through regulation of complement-induced inflammation. Consistent with this hypothesis, mice deficient in the complement inhibitor CD59 demonstrate increased renal interstitial fibrosis characterized by increased myofibroblasts and extracellular matrix,29 key histologic features of hypertrophic burn scars.30 If the rs11136645 variant were associated with increased complement inhibition by CSMD1, it may attenuate the inflammatory response to burn injury, leading ultimately to decreased fibrogenesis and scarring.
A second potential mechanistic link between CSMD1 and post-burn HTS derives from the observation that CSMD1 is highly expressed in developing rat26 and human27 brain tissue and localizes to neuronal growth cones.26 These are specialized structures located at the tips of axons and dendrites involved in neuronal growth during development and, in peripheral nerves, regeneration after injury.31 We and others have reported increased nerve fibers in hypertrophic scar samples,32 and we have shown that hypertrophic burn scars have elevated levels of substance P,33 a neuroinflammatory mediator known to be released from peripheral nerves after injury. Thus, dysregulated neuronal modulation of cutaneous wound repair may contribute to HTS formation.32 Hence, it is possible that CSMD1 could affect HTS formation by influencing epidermal nerve fiber growth or signaling during burn wound repair. Specifically, the association between the rs11136645 variant and reduced HTS formation could be explained by decreased CSMD1-mediated neuroinflammation following cutaneous burn injury.
A third putative mechanism linking CSMD1 to HTS comes from its clinical associations with disease processes resulting from aberrant morphogenesis or morphostasis.34 Since its initial characterization as a putative tumor suppressor in head and neck squamous cell carcinoma, CSMD1 has been linked to other cancers, including colorectal,35,36 breast,37,38 liver,39 and skin cancers.38,40 Consistent with experimental evidence suggesting a role for CSMD1 in neurodevelopment, genetic association studies have implicated CSMD1 variants in various neuropsychiatric disorders including multiple sclerosis,41 Alzheimer’s disease,42,43 schizophrenia,44 bipolar disorder,45 and autism.46 We report an association between CSMD1 and pathologic scarring. Thus, CSMD1 has been implicated in abnormal development, tumorigenesis, and dysfunctional wound healing, suggesting that it may regulate cellular mechanisms involved in morphogenesis and morphostasis.34 For instance, the transforming growth factor beta (TGF-β) proteins have well-studied roles in development,47 tumorigenesis,48 and wound healing.49 Tang and colleagues studied the interaction of CSMD1 with TGF-β mediated SMAD signaling in melanoma cells and found that CSMD1 increases expression and activation of SMADs involved in TGF-β signal transduction.40 Given that overactive TGF-β1/SMAD signaling in hypertrophic-scar fibroblasts is believed to be a central component of HTS pathogenesis,30 CSMD1 may influence post-burn HTS by modulating SMAD signaling. If the rs11136645 variant were associated with diminished CSMD1 function, it may decrease TGF-β1/SMAD signaling and attenuate post-burn scarring. Each of these proposed potential mechanisms warrants investigation in the laboratory setting pending confirmation of our findings in an independent study.
This study has several limitations. Due to demographics of our burn-center catchment area, our study subjects were predominantly non-Hispanic Whites; thus, we were unable to detect variants potentially explaining the increased scarring propensity in individuals of dark-skinned races.3,9,10 Based on our desire to use a simple, objective outcome measure to facilitate reproducibility of our analysis as well as our finding that VSS height score alone had the best accuracy in distinguishing hypertrophic from normotrophic burn scars, we used scar height alone as an indicator of scar severity in our analysis. However, this approach may have underrepresented scar severity in some subjects (for instance in the case of a stiff, erythematous, dyspigmented scar that is only slightly raised). In addition, despite enrolling the largest prospective cohort of burned patients with genotype- and scar-outcome data to date and detecting an association with the common (MAF 0.49) rs11136645 variant, our study was underpowered to detect associations with less common variants or variants with relatively small effect sizes (Table 1). Whereas a case-control design comparing burned subjects with any degree of raised scarring to a large number of uninjured healthy might confer greater statistical power, wound-healing phenotypes of the uninjured controls would be unknown. Given the >70% incidence of post-burn HTS,2 this approach would likely result in an unacceptably high degree of differential misclassification, predisposing to false-positive associations and/or over-estimated effect sizes. Instead, we sought to maximize power within the confines of a prospective-cohort design by enrolling subjects at increased risk of scarring and testing for linear association with scar height. Finally, our findings await validation in an independent clinical cohort.
In summary, taking a genome-wide approach, we have identified a common variant in the CSMD1 gene that is associated with decreased scar height, suggesting a novel role for CSMD1 in wound healing. CSMD1 could potentially influence post-burn HTS through regulation of complement activation, neuronal regeneration, and/or TGF-β1/SMAD signaling. Understanding its role in wound healing may advance our knowledge of HTS pathophysiology. Our results support further genome-wide association studies of post-burn scarring both to confirm our findings and to identify risk loci in high-risk racial groups.
DISCUSSANTS.
Dr. Ronald G. Tompkins (Boston, MA):
I have no disclosures to make.
One feature of genome-wide association studies is that polymorphisms are more often related to race than diseases, making this a little bit problematic. Do the SNPs correlate with demographics in this patient population? I understand the small numbers of nonwhites. Specifically, given the large white male population, is there a correlation between CMNDI SNPs, hypertrophic scars if you restrict the analysis to only those non-Hispanic white cohort?
Also, whereas your numbers of the racially diverse groups is rather small, do you have any evidence that CSMDI SNPs are more common in different races, particularly those races that you mentioned that have a predilection for hypertrophic scarring?
Regarding the measurement of height in scars, this is 1 mm. One could argue the degree to which that is actually hypertrophic scarring, but is height enough or sufficient to measure abnormal scar formation given the manifestations of abnormal scar formation that are rather late (and there are many of those)?
Last question, what are the next steps to study and define the biological role that this SNP might play, and CSMDI, in wound healing in hypertrophic scars?
I'm particularly interested because this is an intronic SNP, and understanding exactly how biologically this is functioning would be of a great deal of interest.
Response from R.F. Sood:
With regard to your first question about whether, in our study, SNP frequencies were associated with self-identified race, they very much were, which was illustrated by that scatter plot from the PCA. Thaťs why it was so critically important to adjust for the differences in SNP frequencies according to race or essentially the population substructure, the overall genetic similarities due to common ancestry.
In regard to your question about repeating the analysis limited to whites only, when we did that, there end up being about 350 subjects included in the analysis, so there is a significant loss of power, but we did see that CSMDI SNP was still strongly associated with scar height. The effect size is in the same range. The regression coefficient was about minus 0.2, and the P value was about 2 times 10 to the minus 4, so there was still a strong association, but, as would be expected given the reduction in power, nothing quite reached genome-wide significance.
You asked whether the frequency of this SNP differed according to race. It was actually very common across races. It was slightly less common among blacks or African Americans, with a minor allele frequency of about 40%, but there was no significant difference in minor allele frequency between races.
You asked a question about whether scar height is enough to measure scar severity. I think thaťs an important question that I donť have a definitive answer to, because I donť think we have sufficiently answered that question simply because there havenť been enough studies examining how to take information from scar scales and translate them to variables to be used in epidemiologic studies.
I can explain, though, why we chose height alone. A main reason is that replication of findings from genome-wide association studies is critically important. A lot of the variables used to assess and track scar severity have a lot of inherent subjectivity to them, like pliability, pigmentation, vascularity, itching, pain, and functional impairment. We wanted to pick the one that was most subjective in order to hopefully facilitate replication of our findings.
Another piece of data supporting our decision to choose the height score alone is we conducted a different study in which we asked burn care providers to rate scar photographs according to the Vancouver Scar Scale. We tested each sub-score of the Vancouver Scar Scale as well as combinations of scores to see which performed best in diagnosing hypertrophic scarring. It turned out that height score alone did better than any of the other combinations. Those were the reasons for us selecting height score.
Finally, regarding the next steps with research: as you pointed out, this is an intronic SNP. It very well may not be impacting CSMD1 function in the ways that I suggested, but I think, especially if our findings are replicated, iťs warranted to start looking not specifically at the SNP but at CSMDI in general to see: is it expressed in burn wounds? Is it expressed in burn scars? Then we can take the next step by manipulating CSMD1 expression in in vitro models of fibroproliferative scarring. If we see promise there, we could then go on to examine the functional effect of individual SNPs.
Dr. Clive Callender (Washington, D.C.):
I donť have a question, but I have a comment. For example, the title is genome-wide association study. As we all know, the Human Genome Project in 2000-plus identified that we have one race. Thaťs homo sapiens.
Why you continue to divide us into races when we are different ethic groups and not races, I take issue with, and would request that you recognize that and make that change in your paper.
Response from R.F. Sood:
Thank you for the comments. I talk about race simply because there are differences in ancestry, even though, as you point out, we are all Homo sapiens. As a result, there are systematic differences in the frequency of different mutations that are inherent to certain groups. My point was to highlight those differences.
Dr. Clive Callender (Washington, D.C.):
I hope you're not saying that there are different races, though. We are all one race, is that not correct? We are all Homo sapiens. Thaťs a sociological determination by race. We all know we are Homo sapiens, so thaťs a characterization that I think is inappropriate and somewhat harmful.
Dr. Paul Kuo (Maywood, IL):
This is a bit of a tangential question, and please excuse me for this. When you look at your PCA, I'm curious as to what the primary variables are that had the highest loadings and what were the scores? How much of the variability was accounted for with those two principal components?
Response from R.F. Sood:
We have not examined the factor loadings, in part because there is no option to do so in PLINK, the software package we used for PCA and association analysis. Regarding the first two principal components, although they each explained far more variability than any of the other individual principal components, combined they accounted for only about 10% of the total variability, which is not surprising given that there were nearly 300,000 SNP variables included in the analysis. However, we felt comfortable that adjusting for the first two would likely be sufficient to account for the population substructure. The appearance of our QQ plot and our estimate of the genomic inflation factor support that.
Dr. Scott Lemaire (Houston, TX):
I have no disclosures to make.
You mentioned the possibility that this SNP is just in linkage disequilibrium with other variants in adjacent genes. What are the other genes in the region that might also be of interest to this particular process?
Response from R.F. Sood:
In a preliminary effort to try to answer that question, I've plugged in the top few SNPs that came up in our GWAS into an online tool that relies on data from the HapMap Project to see what other SNPs are in linkage disequilibrium. In the preliminary results, all of the SNPs that came up are all actually within CSMD1. So there certainly might be others, but we will have to do more analysis to see what those might be.
Acknowledgements
We would like to thank Dr. Deborah Nickerson and the members of her laboratory for genotyping the samples. We also gratefully acknowledge funding support from the National Institutes of Health (R01 GM089704 and T32 GM007037), the Jack Ruppelt Endowed Fellowship, and the David and Nancy Auth—Washington Research Foundation Endowed Chair for Restorative Burn Surgery.
Funding support:
NIH R01 GM089704
NIH T32 GM007037
David and Nancy Auth—Washington Research Foundation Endowed Chair for Restorative Burn Surgery
Jack Ruppelt Endowed Fellowship
REFERENCES
- 1.ABA. Burn Incidence and Treatment in the United States: 2013 Fact Sheet 2013 Available at: http://www.ameriburn.org/resources_factsheet.php. Accessed 22 Feb, 2015.
- 2.Gangemi EN, Gregori D, Berchialla P, et al. Epidemiology and risk factors for pathologic scarring after burn wounds. Arch Facial Plast Surg 2008;10:93–102. [DOI] [PubMed] [Google Scholar]
- 3.Bombaro KM, Engrav LH, Carrougher GJ, et al. What is the prevalence of hypertrophic scarring following burns? Burns 2003;29:299–302. [DOI] [PubMed] [Google Scholar]
- 4.Atiyeh BS, Costagliola M, Hayek SN. Keloid or hypertrophic scar: the controversy: review of the literature. Ann Plast Surg 2005;54:676–80. [DOI] [PubMed] [Google Scholar]
- 5.Mason ST, Esselman P, Fraser R, et al. Return to work after burn injury: a systematic review. J Burn Care Res 2012;33:101–9. [DOI] [PubMed] [Google Scholar]
- 6.Rumsey N, Clarke A, White P. Exploring the psychosocial concerns of outpatients with disfiguring conditions. J Wound Care 2003;12:247–52. [DOI] [PubMed] [Google Scholar]
- 7.van der Veer WM, Bloemen MC, Ulrich MM, et al. Potential cellular and molecular causes of hypertrophic scar formation. Burns 2009;35:15–29. [DOI] [PubMed] [Google Scholar]
- 8.Friedstat JS, Hultman CS. Hypertrophic burn scar management: what does the evidence show? A systematic review of randomized controlled trials. Ann Plast Surg 2014;72:S198–201. [DOI] [PubMed] [Google Scholar]
- 9.Deitch EA, Wheelahan TM, Rose MP, et al. Hypertrophic burn scars: analysis of variables. J Trauma 1983;23:895–898. [PubMed] [Google Scholar]
- 10.Thompson CM, Hocking AM, Honari S, et al. Genetic risk factors for hypertrophic scar development. J Burn Care Res 2013;34:477–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Castagnoli C, Peruccio D, Stella M, et al. The HLA-DR beta 16 allogenotype constitutes a risk factor for hypertrophic scarring. Hum Immunol 1990;29:229–232. [DOI] [PubMed] [Google Scholar]
- 12.Hindorff LA, MacArthur J, Morales J, et al. A Catalog of Published Genome-Wide Association Studies Available at: http://www.genome.gov/gwastudies. Accessed 22 Feb, 2015.
- 13.Manolio TA. Bringing genome-wide association findings into clinical use. Nat Rev Genet 2013;14:549–558. [DOI] [PubMed] [Google Scholar]
- 14.Hindorff LA, Sethupathy P, Junkins HA, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A 2009;106:9362–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lawrence JW, Mason ST, Schomer K, et al. Epidemiology and impact of scarring after burn injury: a systematic review of the literature. J Burn Care Res 2012;33:136–146. [DOI] [PubMed] [Google Scholar]
- 16.Baryza MJ, Baryza GA. The Vancouver Scar Scale: an administration tool and its interrater reliability. J Burn Care Rehabil 1995;16:535–538. [DOI] [PubMed] [Google Scholar]
- 17.Price AL, Patterson NJ, Plenge RM, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 2006;38:904–909. [DOI] [PubMed] [Google Scholar]
- 18.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007;81:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.QUANTO 1.1: A computer program for power and sample size calculations for genetic-epidemiology studies, http://hydra.usc.edu/gxe [computer program]. 2006. [Google Scholar]
- 20.Aalami OO, Nacamuli RP, Lenton KA, et al. Applications of a mouse model of calvarial healing: differences in regenerative abilities of juveniles and adults. Plast Reconstr Surg 2004;114:713–720. [DOI] [PubMed] [Google Scholar]
- 21.McCarthy MI, Abecasis GR, Cardon LR, et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet 2008;9:356–369. [DOI] [PubMed] [Google Scholar]
- 22.Cooper DN. Functional intronic polymorphisms: Buried treasure awaiting discovery within our genes. Hum Genomics 2010;4:284–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun PC, Uppaluri R, Schmidt AP, et al. Transcript map of the 8p23 putative tumor suppressor region. Genomics 2001;75:17–25. [DOI] [PubMed] [Google Scholar]
- 24.Bork P, Beckmann G. The CUB domain. A widespread module in developmentally regulated proteins. J Mol Biol 1993;231:539–545. [DOI] [PubMed] [Google Scholar]
- 25.Holmquist E, Okroj M, Nodin B, et al. Sushi domain-containing protein 4 (SUSD4) inhibits complement by disrupting the formation of the classical C3 convertase. FASEB J 2013;27:2355–2366. [DOI] [PubMed] [Google Scholar]
- 26.Kraus DM, Elliott GS, Chute H, et al. CSMD1 is a novel multiple domain complement-regulatory protein highly expressed in the central nervous system and epithelial tissues. J Immunol 2006;176:4419–4430. [DOI] [PubMed] [Google Scholar]
- 27.Escudero-Esparza A, Kalchishkova N, Kurbasic E, et al. The novel complement inhibitor human CUB and Sushi multiple domains 1 (CSMD1) protein promotes factor I-mediated degradation of C4b and C3b and inhibits the membrane attack complex assembly. FASEB J 2013;27:5083–5093. [DOI] [PubMed] [Google Scholar]
- 28.Stramer BM, Mori R, Martin P. The inflammation-fibrosis link? A Jekyll and Hyde role for blood cells during wound repair. J Invest Dermatol 2007;127:1009–1017. [DOI] [PubMed] [Google Scholar]
- 29.Turnberg D, Lewis M, Moss J, et al. Complement activation contributes to both glomerular and tubulointerstitial damage in adriamycin nephropathy in mice. J Immunol 2006;177:4094–4102. [DOI] [PubMed] [Google Scholar]
- 30.Zhu Z, Ding J, Shankowsky HA, et al. The molecular mechanism of hypertrophic scar. J Cell Commun Signal 2013;7:239–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kadomatsu K, Sakamoto K. Mechanisms of axon regeneration and its inhibition: roles of sulfated glycans. Arch Biochem Biophys 2014;558:36–41. [DOI] [PubMed] [Google Scholar]
- 32.Scott JR, Muangman P, Gibran NS. Making sense of hypertrophic scar: a role for nerves. Wound Repair Regen 2007;15 Suppl 1:S27–31. [DOI] [PubMed] [Google Scholar]
- 33.Scott JR, Muangman PR, Tamura RN, et al. Substance P levels and neutral endopeptidase activity in acute burn wounds and hypertrophic scar. Plast Reconstr Surg 2005;115:1095–1102. [DOI] [PubMed] [Google Scholar]
- 34.Oviedo NJ, Beane WS. Regeneration: The origin of cancer or a possible cure? Semin Cell Dev Biol 2009;20:557–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Farrell C, Crimm H, Meeh P, et al. Somatic mutations to CSMD1 in colorectal adenocarcinomas. Cancer Biol Ther 2008;7:609–613. [DOI] [PubMed] [Google Scholar]
- 36.Sheffer M, Bacolod MD, Zuk O, et al. Association of survival and disease progression with chromosomal instability: a genomic exploration of colorectal cancer. Proc Natl Acad Sci U S A 2009;106:7131–7136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kamal M, Shaaban AM, Zhang L, et al. Loss of CSMD1 expression is associated with high tumour grade and poor survival in invasive ductal breast carcinoma. Breast Cancer Res Treat 2010;121:555–563. [DOI] [PubMed] [Google Scholar]
- 38.Ma C, Quesnelle KM, Sparano A, et al. Characterization CSMD1 in a large set of primary lung, head and neck, breast and skin cancer tissues. Cancer Biol Ther 2009;8:907–916. [DOI] [PubMed] [Google Scholar]
- 39.Midorikawa Y, Yamamoto S, Tsuji S, et al. Allelic imbalances and homozygous deletion on 8p23.2 for stepwise progression of hepatocarcinogenesis. Hepatology 2009;49:513–522. [DOI] [PubMed] [Google Scholar]
- 40.Tang MR, Wang YX, Guo S, et al. CSMD1 exhibits antitumor activity in A375 melanoma cells through activation of the Smad pathway. Apoptosis 2012;17:927–937. [DOI] [PubMed] [Google Scholar]
- 41.Baranzini SE, Wang J, Gibson RA, et al. Genome-wide association analysis of susceptibility and clinical phenotype in multiple sclerosis. Hum Mol Genet 2009;18:767–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Swaminathan S, Kim S, Shen L, et al. Genomic Copy Number Analysis in Alzheimer’s Disease and Mild Cognitive Impairment: An ADNI Study. Int J Alzheimers Dis 2011;2011:729478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parcerisas A, Rubio SE, Muhaisen A, et al. Somatic signature of brain-specific single nucleotide variations in sporadic Alzheimer’s disease. J Alzheimers Dis 2014;42:1357–1382. [DOI] [PubMed] [Google Scholar]
- 44.Havik B, Le Hellard S, Rietschel M, et al. The complement control-related genes CSMD1 and CSMD2 associate to schizophrenia. Biol Psychiatry 2011;70:35–42. [DOI] [PubMed] [Google Scholar]
- 45.Xu W, Cohen-Woods S, Chen Q, et al. Genome-wide association study of bipolar disorder in Canadian and UK populations corroborates disease loci including SYNE1 and CSMD1. BMC Med Genet 2014;15:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cukier HN, Dueker ND, Slifer SH, et al. Exome sequencing of extended families with autism reveals genes shared across neurodevelopmental and neuropsychiatric disorders. Mol Autism 2014;5:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pera MF, Tam PP. Extrinsic regulation of pluripotent stem cells. Nature 2010;465:713–720. [DOI] [PubMed] [Google Scholar]
- 48.Ikushima H, Miyazono K. TGFbeta signalling: a complex web in cancer progression. Nat Rev Cancer 2010;10:415–424. [DOI] [PubMed] [Google Scholar]
- 49.Penn JW, Grobbelaar AO, Rolfe KJ. The role of the TGF-beta family in wound healing, burns and scarring: a review. Int J Burns Trauma 2012;2:18–28. [PMC free article] [PubMed] [Google Scholar]