

Abstract
The pathogenesis of tissue damage in chronic Trypanosoma cruzi infection has been a subject of long-standing debate. Conventional staining methods reveal a paucity of parasites in tissues from chronically infected individuals, which has led to the theory that the pathologic findings may be primarily autoimmune in origin. Immunostaining for T. cruzi antigens or in situ PCR methods show evidence for parasite components in chronic tissues; however, these methods do not address whether the stained material represents parasite debris or live organisms. An improved method for detecting intact T. cruzi in tissues was developed by making a genetically engineered strain that expresses Escherichia coli β-galactosidase. The expression of this enzyme allows the detection of T. cruzi in tissues by using the histochemical stain 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal). The technique was used to monitor tissue parasitism and its relation to pathologic findings in the mouse model of Chagas’ disease. Parasites were easily visible as bright blue structures in skeletal muscle, heart, bladder, peripheral nerve, liver, spleen, adrenal gland, brain, and adipose tissue in acutely infected mice. The number of viable parasites diminished >100-fold when tissues from 3-week-infected mice were compared with those from 10-month-infected mice. However, even at the lower level, parasites were clearly recognizable in sections of skeletal muscle and bladder at the 10-month time point. Inflammation remained robust in skeletal muscle, bladder, and sciatic nerve despite the near disappearance of parasites, suggesting three possibilities: exuberant host reactions to the few remaining parasites, autoimmune inflammation, or reactions to retained parasite antigens in the tissues.
Trypanosoma cruzi is the etiologic agent of Chagas’ disease and infects a total of 16 million to 18 million people in Latin America (56). During the acute phase of the infection, the parasites replicate inside host cells and then are released into the bloodstream, where they are visible microscopically as trypomastigotes. The immune response appears to contain the infection in the vast majority of individuals, resulting in the absence of microscopically evident parasitemia (46). However, persons remain chronically infected, as evidenced by the ability to culture parasites from blood or by persistently positive serologic tests (29, 30). Approximately 10 to 30% of chronically infected individuals develop Chagas’ disease, manifested usually as cardiomyopathy, megaesophagus, or megacolon (56). The exact pathogenesis of the damage in the organs has been a subject of long-standing interest and debate (8, 10, 15, 21, 23, 25, 27, 28, 34, 51), primarily because of the difficulty in identifying parasites in tissues of diseased organs (6, 20). The paucity of parasites has led to the theory that most or all of the pathologic findings result from autoimmune mechanisms (23, 25), damage to autonomic nerves (41, 42), or microvascular disease (17). In support of the autoimmune mechanisms, it has been shown that the parasite and host share a number of antigenic determinants (1, 5, 13, 16, 18, 24, 26, 33, 43, 44, 49, 52, 54, 55). It is unproven, however, whether the host response to cross-reactive antigens mediates the tissue inflammation and clinical disease (15). An alternative explanation for the pathologic findings is that the inflammatory response is, in fact, directed at parasites in the tissues and that the methods of observing these parasites are inadequate (10, 34, 51).
Two general methods that go beyond microscopic examination have been used to test for the presence of parasites in tissues of patients or animals infected with T. cruzi. These are immunohistochemistry (6, 7, 20, 39, 50, 55) and DNA detection methods, i.e., in situ hybridization, whole-tissue PCR, and in situ PCR (10, 22, 51). These methods have successfully demonstrated the presence of parasite antigens or parasite DNA in tissues; however, they are unable to demonstrate the extent to which viable parasites are present in the tissues. This raises yet a third potential pathogenic mechanism, which is that residual parasite antigens retained in the tissues may be the stimulus for the persistent inflammatory reaction (3, 7). The distinction between these various pathogenic mechanisms is important because if the immune response appears to be wholly directed to self antigens or materials from nonviable parasites, the role of antiparasitic therapy is dubious and immune system-altering therapy may be warranted. On the other hand, if viable parasites are clearly detectable in the involved organs, antiparasitic therapy may be a more logical therapeutic approach.
This paper describes a method to better visualize T. cruzi in infected host tissues. The method utilizes a transfected T. cruzi strain that stably expresses Escherichia coli β-galactosidase, making the parasites visible in tissues as blue structures when stained with 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal). The method was applied in a mouse model to study acute and chronic tissue parasitism and its relation to pathologic findings.
MATERIALS AND METHODS
Parasites and culture procedures.
A clone of the Tulahuen strain of T. cruzi, designated MHOM/CH/00/Tulahuen C2, in accordance with the World Health Organization classification, was provided by S. Reed (Infectious Diseases Research Institute, Seattle, Wash.). Trypomastigotes and amastigotes were grown on monolayers of mouse NIH 3T3 fibroblasts in Dulbecco’s modified Eagle’s medium (BioWhittaker, Walkersville, Md.) supplemented with either 10% fetal calf serum (HyClone Laboratories, Inc., Logan, Utah) or 10% Cosmic Calf Serum (HyClone Laboratories) plus glutamine, penicillin, and streptomycin as previously described (54). The parasites used in these experiments were a clone that had been stably transfected with the E. coli β-galactosidase gene, lacZ (11, 12). This gene was integrated into the genome with linkage to the calmodulin-ubiquitin locus (2). The β-galactosidase-expressing parasites were unaltered in their in vitro growth characteristics (11). Parasites used for mouse infections had been recovered from the blood of infected mice and expanded in tissue culture. These were tested for β-galactosidase activity by reactivity with chlorophenolred-β-d-galactopyranoside (11) before use in mouse experiments. For mouse experiments, the parasites were washed and resuspended in Dulbecco’s modified Eagle’s medium.
Mouse infections.
Female C3H/He mice, 6 to 8 weeks old (B&K Universal, Kent, Wash.), were injected subcutaneously at the base of the tail with 250 tissue culture-derived trypomastigotes in a volume of 200 μl by using a 25-gauge needle. Parasitemia was quantified beginning on day 7 postinfection by examining 2 drops of tail blood under a coverslip at a magnification of ×400. The mice were monitored daily for mortality.
Preparation and staining of mouse tissues.
While the mice were under deep anesthesia with ketamine and xylazine, the chest was opened and the heart was exposed. An incision was made in the right atrium, and approximately 4 ml of warm 0.9% saline with heparin (25 U/ml) was perfused systemically by injection into the left ventricle. Next, a similar volume of freshly prepared 2% paraformaldehyde was perfused through the mouse. The paraformaldehyde was prepared in phosphate-buffered saline (PBS) (pH 7.2)–2 mM MgCl2. Immediately, the heart, quadriceps muscles, bladder, sciatic nerves, liver, spleen, colon, kidney, and brain were collected and placed in a tube with 2% paraformaldehyde. Unpaired organs were bisected so that half could be stained with X-Gal and half could be stained with hematoxylin-eosin. The tissues to be stained with X-Gal were fixed for exactly 1 h and then rinsed twice with PBS (pH 7.2). (Longer fixations diminished β-galactosidase activity in tissues. Similarly, paraffin embedding of tissues resulted in loss of β-galactosidase activity, probably because of the heating of tissues.) The tissues were transferred to a tube containing 30% sucrose in PBS (pH 7.2)–2 mM MgCl2 and placed at 4°C until they sank (usually overnight). Next, the tissues were embedded in Tissue-Tek O.C.T. compound (Miles, Inc., Elkhart, Ind.) and frozen until used for sectioning. Frozen sections 10 μm thick were prepared on a refrigerated microtome. The tissues were mounted on Superfrost Plus glass slides (Fisher Scientific, Pittsburgh, Pa.), and stored at −20°C until stained. Tissues to be stained with hematoxylin-eosin were left in 2% paraformaldehyde until they were embedded in paraffin.
The frozen sections were warmed to room temperature, and each section (two per slide) was circumscribed with a wax pencil to contain the stain. The slides were layed horizontally, and the tissues were rehydrated with PBS–2 mM MgCl2 for 5 min. The liquid was removed, and approximately 500 μl of X-Gal stain was applied to each tissue section. The X-Gal stain was made in PBS (pH 7.2)–2 mM MgCl2–4 mM potassium ferricyanide–4 mM potassium ferrocyanide–1 mg of X-Gal (Sigma Chemical Co., St. Louis, Mo.) (38). The X-Gal was dissolved in dimethyl sulfoxide before being added to the aqueous buffer. The slides were sealed in containers with wet paper towels to prevent drying and incubated for 16 to 18 h at 37°C. They were then rinsed in PBS and counterstained with nuclear fast red (Sigma Chemical Co.) for 5 min in staining jars. The nuclear fast red was prepared by dissolving 100 mg in 100 ml of 5% aluminum sulfate, boiling, and filtering. The slides were rinsed in PBS and then prepared for mounting by the following steps: dipping in 75% ethanol, dipping in 95% ethanol, setting in 100% ethanol for 2 min three times, setting in Americlear (Baxter Diagnostics, Inc., Deerfield, Ill.) for 2 min, and setting in xylene for 3 min twice. The slides were mounted with Permount (Fisher Scientific, Fair Lawn, N.J.).
Paraffin-embedded tissues were sectioned at 6-μm thick, stained with hematoxylin-eosin, and mounted by standard methods (53).
Analysis of tissues.
Tissues stained with X-Gal were scanned microscopically at ×100 for blue-staining parasites, which were confirmed by higher-power examination. Entire sections were scanned, and the total number of individual parasites was quantified. (Scoring was done by a single observer to ensure consistent quantification, particularly where dense parasite nests required some estimation.) The density of parasites was determined as a function of the measured area of the section examined. Inflammation was scored by quantifying the nuclei in the tissues under ×400 magnification. The quantity of inflammatory cells was inferred by the number of nuclei present in the tissues above the number of nuclei present in tissues from uninfected age-matched mice. This method allowed rapid quantification of polymorphic inflammatory cells in the tissues (polymorphonuclear leukocytes, eosinophils, lymphocytes, macrophages, and fibroblasts). The density of inflammatory cells was determined as a function of the measured area of the section examined. Thirty random fields from two sections were quantified for inflammation scores.
RESULTS
Parasitemia and mortality from T. cruzi infection.
C3H/He mice were selected for the experiments because preliminary studies showed that inoculation with 250 Tulahuen strain trypomastigotes resulted in moderate acute parasitemia and the majority of mice survived into the chronic phase of infection. This contrasted with BALB/c and C57BL/6 mice, of which >75% die within the first 6 weeks of infection with this dose of Tulahuen strain parasites (data not shown). A total of 45 mice were infected with the β-galactosidase-expressing T. cruzi strain, 20 mice were infected with untransfected T. cruzi, and 5 mice were left uninfected to act as age-matched controls for the assessment of tissue inflammation. The course and amplitude of parasitemia were essentially the same for the β-galactosidase-expressing T. cruzi (lacZ) and the wild-type parasites (Fig. 1a). Approximately 50% of the animals from both groups survived beyond 90 days (Fig. 1b). All animals surviving beyond 90 days continued to live until they were sacrificed at the 6- or 10-month time point.
FIG. 1.
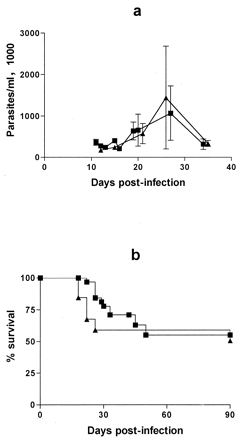
Parasitemia (a) and mortality (b) in C3H/He mice infected with Tulahuen strain T. cruzi expressing β-galactosidase (■) or untransfected (▴). Differences in parasitemia or mortality between the two groups were not statistically significant at any time points.
Tissue parasitism in acute infection.
Five mice were sacrificed after 3 weeks of infection, and frozen sections of their tissues were stained with X-Gal and counterstained with nuclear fast red. Tissue amastigotes were easily visible by light microscopy as round, blue-staining structures approximately 5 to 7 μm in diameter against a pink background (Fig. 2). The blue parasites were observed individually or in nests containing various numbers of amastigotes (in some cases more than 100). An occasional blue-flagellated trypomastigote was visible. The only blue-staining objects in the tissues were of the size and shapes of T. cruzi, leaving little doubt about their identity. The only exception was in sections of intestinal tissue, in which bacteria in the lumen stained blue. The shape and size of the structures were very clear, enabling an easy distinction between parasites and bacteria. Tissues from mice infected with untransfected T. cruzi had no blue-staining material in the tissues (except some bacteria in sections of the intestine).
FIG. 2.
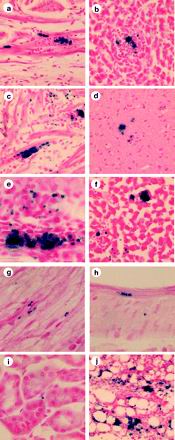
Photomicrographs of mouse tissues obtained 3 weeks postinfection. Tissues were stained with X-Gal and counterstained with nuclear fast red. The tissues shown are bladder (magnification ×200) (a), liver (×200) (b), heart (×200) (c), brain (×200) (d), skeletal muscle (×400) (e), adrenal gland (×200) (f), sciatic nerve (×400) (g), colon (×400) (h), kidney (×400) (i), and mesenteric fat (×100) (j).
All tissues examined 3 weeks after infection contained T. cruzi parasites. Large numbers of parasites were observed in skeletal muscle, heart, bladder, and connective tissues such as capsular tissues around visceral organs; several parasites were visible in most views at ×400 magnification. Smaller numbers of parasites were observed in sciatic nerve, liver, spleen, adrenal glands, and intestine. Rare parasites were observed in the kidney and brain (Fig. 2). The relative quantities of parasites in selected tissues are shown in Fig. 3a (see below).
FIG. 3.
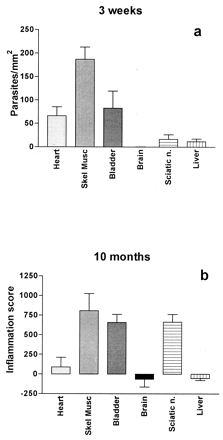
Quantification of tissue parasites from different tissues at 3 weeks postinfection (a) compared to tissue inflammation at 10 months postinfection (b).
Course of tissue parasitism and its relation to pathologic findings in tissue.
Mice were sacrificed at different time points following inoculation (five animals per group), and tissues were evaluated for the presence of parasites and inflammation. At 3 weeks after infection, tissue parasites were easily visible in most high-power (×400) views of skeletal muscle, heart, and bladder (as above). Individual parasites were frequently observed dispersed among inflammatory cells, which probably originated from a recently ruptured host cell. Intact pseudocysts were also readily visible. However, the numbers of parasites in tissues had decreased >30-fold by 6 weeks postinfection, and they decreased further at subsequent time points (Fig. 4a, for skeletal muscle). Most parasites observed at time points after 6 weeks were present in groups within pseudocysts, as opposed to being dispersed individually within inflammatory foci. Often, these pseudocysts had little immediate adjacent inflammation (Fig. 5i). At 6 weeks after infection and later, large areas of tissue had to be surveyed before any parasites could be observed. This made it difficult to quantify parasites except in tissues from which large sections could be obtained, such as skeletal muscle, in which 10 serial sections, each approximately 30 mm2, were examined. Rare but unequivocal tissue parasites were also detected in bladder tissue at 6 weeks and 3, 6, and 10 months. No parasites were observed in heart, sciatic nerve, brain, or liver after the 6-week time point. For the sciatic nerve, a relatively small total area was examined due to the small amount of this tissue. In the tissues collected after the 6-week time point, most inflammatory foci contained no identifiable parasites even though 10 serial sections, each 10 μm thick, were examined.
FIG. 4.
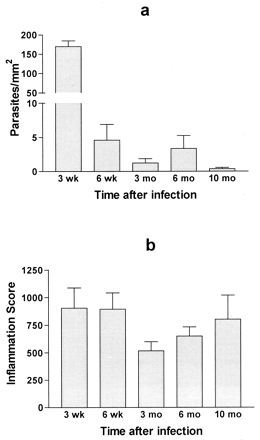
Quantification of tissue parasites (a) and tissue inflammation (b) in skeletal muscle at different time points following infection with T. cruzi.
FIG. 5.
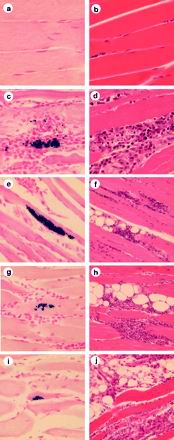
Photomicrographs of skeletal muscle tissue taken from uninfected mice (a and b) or T. cruzi-infected mice at 3 weeks (c and d), 6 weeks (e and f), 6 months (g and h), or 10 months (i and j) postinfection. Tissues were stained with X-Gal and nuclear fast red (a, c, e, g, and i) or with hematoxylin and eosin (b, d, f, h, and j). Magnifications, ×200 (a to d, g, and i) and ×100 (e, f, h, and j).
Inflammation in skeletal muscle was scored at the same time points that tissue parasitism was quantified (Fig. 4b). Despite the dramatic reduction in the number of tissue parasites by 6 weeks, inflammation persisted and in fact had intensified by the 10-month time point. At 3 weeks, intense interstitial inflammation was present in the muscle tissue with a mix of polymorphonuclear and mononuclear inflammatory cells (Fig. 5d). Myocyte necrosis was widespread. At 6 weeks and more so at 3 months, the inflammatory infiltrate consisted predominantly of mononuclear cells and myocyte regeneration was evident (Fig. 5f). At 6 and 10 months, intense mononuclear inflammation persisted in the intersititial and perivascular spaces within the muscle (Fig. 5h and j). Large areas within muscle fascicles were replaced by adipose and fibrous tissue, presumably at sites of myocyte necrosis (Fig. 5h and j). Tissues from mice infected with wild-type Tulahuen parasites were evaluated for inflammation at 6 and 10 months and were found to have a very similar pattern and intensity of inflammation to that found in the mice infected with the β-galactosidase-expressing parasites (data not shown).
Relation between acute tissue parasitism and chronic inflammation.
The most heavily parasitized tissues at the 3 week time point were skeletal muscle and bladder, and these tissues contained the highest degree of inflammation at 10 months (Fig. 3). Interestingly, the heart was heavily parasitized acutely yet showed very little inflammation during the late stage of the infection, when no parasites were demonstrable. In contrast, the sciatic nerve contained relatively few parasites during the acute infection but showed heavy inflammation later on. Finally, the brain and liver tissues contained relatively small numbers of parasites acutely and showed little inflammation at 10 months.
Stability of β-galactosidase gene expression in transfected parasites.
At the time of sacrifice, blood from infected mice was seeded onto NIH 3T3 fibroblasts to recover live parasites. Over 75% of cultures grew parasites. After expanding the T. cruzi in tissue culture, smears of the parasites were prepared and stained with X-Gal. Hundreds of individual parasites could be scanned by this method. Revertants that did not express β-galactosidase were never observed. Infected monolayers of NIH 3T3 fibroblasts were stained with X-Gal and showed uniform blue coloration of amastigotes and trypomastigotes, suggesting that the β-galactosidase was not variably expressed (data not shown).
DISCUSSION
A method was developed to better visualize tissue parasites in animal models of Chagas’ disease by using a T. cruzi strain that was genetically engineered to express E. coli β-galactosidase. The strength of the technique is that the presence and location of live parasites can easily be monitored in the tissues during the course of the infection. Standard tissue-staining techniques such as hematoxylin and eosin or trichrome do not permit easy visualization of tissue amastigotes, and special stains that significantly enhance the visibility of T. cruzi do not exist. Methods such as in situ PCR or immunohistochemistry demonstrate evidence of parasite DNA or antigens but do not necessarily show that live parasites are present. These methods are technically difficult and have potential problems with specificity and cross-reactivity. The β-galactosidase staining method enables the visualization of live parasites, since the staining results from enzymatic activity within a structure morphologically resembling T. cruzi. It seems highly improbable that β-galactosidase enzymatic activity could persist in host tissues after the parasites are destroyed. In fact, other investigators have shown that β-galactosidase activity in lacZ-transfected tumor cells disappears within hours as the cells die and are cleared by the immune response (36). The blue-staining material appears to be specific for T. cruzi, since the stain was associated only with round structures resembling amastigotes (with the exception of some bacteria found in the lumen of intestine) and no nonspecific staining was observed in tissues from uninfected mice. The specificity of the X-Gal stain for parasites and not host tissues is ascribed to the fact that E. coli β-galactosidase is active at pH 7 whereas mammalian β-galactosidase is active at pH 3 to 4.5 (53).
The β-galactosidase-expressing Tulahuen strain of T. cruzi appeared to be essentially the same as untransfected wild-type parasites in a number of ways. In a previous study, it was determined that the in vitro growth rates and ability to transform into the different life stages were no different from those of the wild-type parasites (11, 12). In this study, the in vivo effects of the parasites were compared and no significant differences in the ability to produce parasitemia or death were observed. In addition, the degree and appearance of the acute and chronic pathologic findings were not different between the β-galactosidase-expressing and untransfected T. cruzi. Although a potential concern was that the β-galactosidase protein expressed in the parasite might serve as an immunogen that could alter the host response to the parasite, this appeared unlikely based on previous immunological studies with this parasite strain. Specifically, murine antigen-presenting cells infected with the β-galactosidase-expressing T. cruzi strain did not present β-galactosidase antigen to an antigen-specific cytotoxic lymphocyte line (12).
It was important that the parasites maintain expression of the β-galactosidase gene in the absence of drug pressure. This was accomplished by targeting the lacZ gene into the parasite genome by homologous recombination, thus ensuring replication of the gene with each mitosis. Parasites recovered from blood of chronically infected mice were consistently positive for expression of β-galactosidase; hence, the possibility that many parasites were invisible because of loss of β-galactosidase expression is unlikely. An additional concern is that the stain failed to penetrate all parasites in the tissues. Although this is possible, it was evident that many more parasites were clearly identifiable with the X-Gal stain than with the hematoxylin and eosin stain (Fig. 5). Modifications that potentially could have enhanced the penetration of X-Gal, such as addition of the detergents deoxycholate or Triton X-100 to the staining buffer, did not improve the sensitivity of the method and appeared to diminish the quality of the tissue morphology.
This study resulted in some interesting observations about T. cruzi infection in a mouse model. First, parasites were visualized in all tissues examined during the acute infection, which included heart, skeletal muscle, bladder, intestine, spleen, liver, adrenal gland, brain, peripheral nerve, kidney, and fibrous and adipose connective tissue (Fig. 2). The observations confirm previous findings that T. cruzi has the potential to infect a wide range of host tissues (9, 14, 19, 32, 35, 45). The Tulahuen clone used in these experiments was myotropic, as has been previously reported (40). As seen in some other mouse models of Chagas’ disease, skeletal muscle was a site of heavier parasitism than heart muscle (9, 19). Infection of peripheral nerve tissue was observed to be relatively light in this system; however, attempts to specifically visualize autonomic nerves, which are believed to be important foci of infection (32, 41), were not made. Infection of the spleen and liver was relatively light, which indicates that the Tulahuen clone is not particularly reticulotropic, unlike some strains (4, 9, 14). Parasites were rare in the kidneys and brain, as previously reported (9, 32). Tissue tropism is known to vary among T. cruzi strains (4, 9, 31, 37); hence, the specific findings of tissue parasitism with this Tulahuen clone should not be generalized to all T. cruzi infections.
The relationship between acute tissue parasitism and chronic pathologic findings was investigated in this study (Fig. 3). Although the two most heavily infected tissues acutely (skeletal muscle and bladder) had the highest inflammation scores at 10 months, it cannot be said that this pattern was the rule. For example, cardiac tissue had relatively dense acute parasitism but showed minimal inflammation in the chronic stage. In contrast, peripheral nerve tissue had few parasites acutely but was heavily inflammed in the chronic stage. Hence, an assertion that chronic pathologic changes are merely extensions of acute inflammation would be an oversimplification.
Parasites were rare in the tissues of chronically infected animals compared to acutely infected animals (Fig. 4), which confirms the generally held belief there is a paucity of parasites in tissues of chronically infected animals (23, 25, 47). However, T. cruzi parasites were unquestionably present in skeletal muscle and bladder, demonstrating that the parasites are not completely cleared from the tissues. The inability to find parasites in other tissues may be due to the clearance of parasites (as is very possible with heart tissue and liver, where little inflammation was observed) or to the relatively small volume of tissue available for examination (as may be the case with peripheral nerve tissue, where a lot of inflammation was observed). Other methods for detecting parasites in chronically infected tissues have depended on assaying for parasite antigens or DNA. Specifically, Ben Younes-Chennoufi et al. (7) detected parasite antigens by immunohistochemical methods in 11% of inflammatory infiltrates, and Tarleton et al. (51) detected parasite kDNA by in situ PCR “in a diffuse staining pattern that is more intense in areas of heavy inflammation.” The quantity of blue-staining parasites in the tissues was unquestionably smaller than the amount of parasite antigen or DNA described in these papers. The differences may be due to the different parasite and mouse strains used, but they are more likely to be due to the detection of material from dead and disintegrating parasites by the immunocytochemical and PCR methods. This obviously raises the interesting issue of whether parasite antigens from destroyed T. cruzi organisms can remain in tissues for a long time and possibly provoke an ongoing inflammatory reaction (48). Clearly, a valuable future study would be to combine the methods of staining the tissues by the histochemical method described in this paper and the antigen- or DNA-detecting methods described by others, to relate the location and density of parasites with the presence of parasite antigen or DNA in the tissues.
The level of inflammation in skeletal muscle was dramatic from early in infection through the 10-month time point (Fig. 4 and 5), despite the near clearance of parasites from this tissue. One can argue either side of the autoimmunity debate with these data. On the one hand, the degree of inflammation seems to be out of proportion to the quantity of parasites observed in the tissues. The majority of inflammatory foci in muscle do not contain T. cruzi even when surveyed in three dimensions by looking at serial sections. This suggests (i) an autoimmune process directed at these neighboring cells, (ii) the presence on or in these cells of parasite antigens originating from disintegrating T. cruzi in the vicinity (7, 20), or (iii) tissue ischemia and subsequent inflammation (17). On the other hand, there were parasites in the tissues, and these may suffice to drive an ongoing exuberant host immune response targeted against T. cruzi. This implies that the degree of inflammatory response to each parasite increases with increasing time after infection (compare Fig. 4a and b).
The observations in the sciatic nerve are possibly explained by autoimmune mechanisms. T. cruzi was not visible in any sections from chronically infected mice at 3, 6, or 10 months despite the presence of dense inflammation in the nerve at these time points. Since considerably less nerve tissue than muscle tissue was available for the study, it may be that very few parasites were present and just were not detected. At the most, inflammation in the nerve occurred in the presence of very few T. cruzi parasites, and it possibly occurred in the presence of none.
The staining method described in this paper enabled a detailed observation of the natural history of T. cruzi in the mouse model of Chagas’ disease. The method may be useful with other parasite strain-mouse strain combinations or in other animals to more fully appreciate the spectrum of T. cruzi infection. The technique will be useful in monitoring tissue parasitism in animals undergoing interventions to alter infection, such as vaccination protocols, immunomodulatory therapy, or antiparasitic chemotherapy.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grant AI01258 to F.S.B. and American Heart Association grant-in-aid 95006700 to W.C.V.V.
We thank Emil Chi for services in preparing tissue sections, Shuzo Sumi for assistance in evaluating tissue sections, Lynn Barrett for technical assistance, and Peggy Sue O’Brien for secretarial assistance.
REFERENCES
- 1.Acosta A M, Santos-Buch C A. Autoimmune myocarditis induced by Trypanosoma cruzi. Circulation. 1984;71:1255–1261. doi: 10.1161/01.cir.71.6.1255. [DOI] [PubMed] [Google Scholar]
- 2.Ajioka J, Swindle J. The calmodulin-ubiquitin (CUB) genes of Trypanosoma cruzi are essential for parasite viability. Mol Biochem Parasitol. 1996;78:217–225. doi: 10.1016/s0166-6851(96)02627-8. [DOI] [PubMed] [Google Scholar]
- 3.Andrade S G, Magalhaes J B, Pontes A L. Evaluation of chemotherapy with benznidazole and nifurtimox in mice infected with Trypanosoma cruzi strains of different types. Bull W H O. 1985;63:721–726. [PMC free article] [PubMed] [Google Scholar]
- 4.Andrade V, Brodskyn C, Andrade S G. Correlation between isoenzyme patterns and biological behaviour of different strains of Trypanosoma cruzi. Trans R Soc Trop Med Hyg. 1983;77:796–799. doi: 10.1016/0035-9203(83)90292-4. [DOI] [PubMed] [Google Scholar]
- 5.Aznar C, Lopez-Bergami P, Brandariz S, Mariette C, Liegeard P, de Deus Alves M d C, Barreiro E L, Carrasco R, Lafon S, Kaplan D, Miguez H, Camacho C, Levitus G, Levin J M, Hontebeyrie M. Prevalence of anti-R-13 antibodies in human Trypanosoma cruzi infection. FEMS Immunol Med Microbiol. 1995;12:231–238. doi: 10.1111/j.1574-695X.1995.tb00197.x. [DOI] [PubMed] [Google Scholar]
- 6.Bellotti G, Bocchi E A, de Moraes A V, Higuchi M L, Barbero-Marcial M, Sosa E, Esteves-Filho A, Kalil R, Weiss R, Jatene A, Pileggi F. In vivo detection of Trypanosoma cruzi antigens in hearts of patients with chronic Chagas’ heart disease. Am Heart J. 1996;131:301–307. doi: 10.1016/s0002-8703(96)90358-0. [DOI] [PubMed] [Google Scholar]
- 7.Ben Younes-Chennoufi A, Hontebeyrie-Joskowicz M, Tricottet V, Eisen H, Reynes M, Said G. Persistence of Trypanosoma cruzi antigens in the inflammatory lesions of chronically infected mice. Trans R Soc Trop Med Hyg. 1988;82:77–83. doi: 10.1016/0035-9203(88)90269-6. [DOI] [PubMed] [Google Scholar]
- 8.Bestetti R B. Role of parasites in the pathogenesis of Chagas’ cardiomyopathy. Lancet. 1996;347:913–914. doi: 10.1016/s0140-6736(96)91403-8. [DOI] [PubMed] [Google Scholar]
- 9.Bice D E, Zeledon R. Comparison of infectivity of strains of Trypanosoma cruzi (Chagas, 1909) J Parasitol. 1970;56:663–670. [PubMed] [Google Scholar]
- 10.Brandariz S, Schijman A, Vigliano C, Arteman P, Viotti R, Beldjord C, Levin M J. Detection of parasite DNA in Chagas’ heart disease. Lancet. 1995;346:1370–1371. doi: 10.1016/s0140-6736(95)92388-8. [DOI] [PubMed] [Google Scholar]
- 11.Buckner F S, Verlinde C L M J, La Flamme A C, Van Voorhis W C. Efficient techniques for screening drugs for activity against Trypanosoma cruzi using parasites expressing β-galactosidase. Antimicrob Agents Chemother. 1996;40:2592–2597. doi: 10.1128/aac.40.11.2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buckner F S, Wipke B T, Van Voorhis W C. Trypanosoma cruzi infection does not impair major histocompatibility complex class I presentation of antigen to cytotoxic T lymphocytes. Eur J Immunol. 1997;27:2541–2548. doi: 10.1002/eji.1830271012. [DOI] [PubMed] [Google Scholar]
- 13.Cunha-Neto E, Coelho V, Guilherme L, Fiorelli A, Stolf N, Kalil J. Autoimmunity in Chagas’ disease. Identification of cardiac myosin-B13 Trypanosoma cruzi protein crossreactive T cell clones in heart lesions of a chronic Chagas’ disease cardiomyopathy patient. J Clin Invest. 1996;98:1709–1712. doi: 10.1172/JCI118969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Diego J A, del Rey J, Mayer R, Gamallo C. A comparative pathological study of three strains of Trypanosoma cruzi in an experimental model. Histol Histopathol. 1991;6:199–206. [PubMed] [Google Scholar]
- 15.Eisen H, Petry K, Van Voorhis W C. The origin of autoimmune pathology associated with Trypanosoma cruzi infection. In: Van Der Ploeg L H T, Cantor C R, Voegel H J, editors. Immune recognition and evasion: molecular aspects of host-parasite interaction. London, United Kingdom: Academic Press, Ltd.; 1990. pp. 91–103. [Google Scholar]
- 16.Elies R, Ferrari I, Wallukat G, Lebesgue D, Chiale P, Elizari M, Rosenbaum M, Hoebeke J, Levin M J. Structural and functional analysis of the B cell epitopes recognized by anti-receptor autoantibodies in patients with Chagas’ disease. J Immunol. 1996;157:4203–4211. [PubMed] [Google Scholar]
- 17.Factor S M, Cho S, Wittner M, Tanowitz H B. Abnormalities of the coronary microcirculation in acute murine Chagas’ disease. Am J Trop Med Hyg. 1985;34:246–253. doi: 10.4269/ajtmh.1985.34.246. [DOI] [PubMed] [Google Scholar]
- 18.Ferrari I, Levin M J, Wallukat G, Elies R, Lebesgue D, Chiale P, Elizari M, Rosenbaum M, Hoebeke J. Molecular mimicry between the immunodominant ribosomal protein P0 of Trypanosoma cruzi and a functional epitope on the human β1-adrenergic receptor. J Exp Med. 1995;182:59–65. doi: 10.1084/jem.182.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hanson W L, Roberson E L. Density of parasites in various organs and the relation to numbers of trypomastigotes in the blood during acute infections of Trypanosoma cruzi in mice. J Protozool. 1974;21:512–517. doi: 10.1111/j.1550-7408.1974.tb03689.x. [DOI] [PubMed] [Google Scholar]
- 20.Higuchi M L, De Brito T, Reis M M, Barbosa A, Bellotti G, Pereira-Barreto A C, Pilleggi F. Correlation between Trypanosoma cruzi parasitism and myocardial inflammation infiltrate in human chronic chagasic myocarditis: light microscopy and immunohistochemical findings. Cardiovasc Pathol. 1993;2:101–106. doi: 10.1016/1054-8807(93)90021-S. [DOI] [PubMed] [Google Scholar]
- 21.Hudson L. Autoimmune phenomena in chronic chagasic cardiopathy. Parasitol Today. 1985;1:6–9. doi: 10.1016/0169-4758(85)90099-7. [DOI] [PubMed] [Google Scholar]
- 22.Jones E M, Colley D G, Tostes S, Reis Lopes E, Vnencak-Jones C L, McCurley T L. Amplification of a Trypanosoma cruzi DNA sequence from inflammatory lesions in human Chagasic cardiomyopathy. Am J Trop Med Hyg. 1993;48:348–357. doi: 10.4269/ajtmh.1993.48.348. [DOI] [PubMed] [Google Scholar]
- 23.Kalil J, Cunha-Neto E. Autoimmunity in Chagas disease cardiomyopathy: fulfilling the criteria at last? Parasitol Today. 1996;12:396–399. doi: 10.1016/0169-4758(96)10058-2. [DOI] [PubMed] [Google Scholar]
- 24.Kaplan D, Ferrari I, Bergami P L, Mahler E, Levitus G, Chiale P, Hoebeke J, Van Regenmortel MHV, Levin MJ. Antibodies to ribosomal P proteins of Trypanosoma cruzi in Chagas disease possess functional autoreactivity with heart tissue and differ from anti-P autoantibodies in lupus. Proc Natl Acad Sci USA. 1997;94:10301–10306. doi: 10.1073/pnas.94.19.10301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khoury E L, Fields K L. Chagas’ disease and autoimmunity. Lancet. 1980;i:1088. doi: 10.1016/s0140-6736(80)91531-7. [DOI] [PubMed] [Google Scholar]
- 26.Khoury E L, Ritacco V, Cossio P M, Laguens R P, Szarfman A, Diez C, Arana R M. Circulating antibodies to peripheral nerve in American trypanosomiasis (Chagas’ disease) Clin Exp Immunol. 1979;36:8–15. [PMC free article] [PubMed] [Google Scholar]
- 27.Kierszenbaum F. Is there autoimmunity in Chagas disease? Parasitol Today. 1985;1:4–6. doi: 10.1016/0169-4758(85)90098-5. [DOI] [PubMed] [Google Scholar]
- 28.Kierszenbaum F. Chronic chagasic tissue lesions in the absence of Trypanosoma cruzi: a proposed mechanism. Parasitol Today. 1996;12:414–415. doi: 10.1016/0169-4758(96)20050-x. [DOI] [PubMed] [Google Scholar]
- 29.Koberle F. Chagas’ disease and Chagas’ syndromes: the pathology of American trypanosomiasis. Adv Parasitol. 1968;6:63–116. doi: 10.1016/s0065-308x(08)60472-8. [DOI] [PubMed] [Google Scholar]
- 30.Laranja F S, Dias E, Nobrega G, Miranda A. Chagas’ disease: a clinical, epidemiological, and pathologic study. Circulation. 1956;14:1035–1060. doi: 10.1161/01.cir.14.6.1035. [DOI] [PubMed] [Google Scholar]
- 31.Lauria-Pires L, Teixeira A R. Virulence and pathogenecity associated with diversity of Trypanosoma cruzi stocks and clones derived from Chagas’ disease patients. Am J Trop Med Hyg. 1996;55:304–310. doi: 10.4269/ajtmh.1996.55.304. [DOI] [PubMed] [Google Scholar]
- 32.Lenzi H L, Oliveira D N, Lima M T, Gattass C R. Trypanosoma cruzi: paninfectivity of CL strain during murine acute infection. Exp Parasitol. 1996;84:16–27. doi: 10.1006/expr.1996.0086. [DOI] [PubMed] [Google Scholar]
- 33.Levin M J. Molecular mimicry and Chagas’ heart disease: high anti-R-13 autoantibody levels are markers of severe Chagas heart complaint. Res Immunol. 1991;142:157–159. doi: 10.1016/0923-2494(91)90029-i. [DOI] [PubMed] [Google Scholar]
- 34.Levin M J. In chronic Chagas heart disease, don’t forget the parasite. Parasitol Today. 1996;12:415–416. doi: 10.1016/0169-4758(96)20051-1. [DOI] [PubMed] [Google Scholar]
- 35.Lima M T, Lenzi H L, Gattass C R. Negative tissue parasitism in mice injected with a noninfective clone of Trypanosoma cruzi. Parasitol Res. 1995;81:6–12. doi: 10.1007/BF00932410. [DOI] [PubMed] [Google Scholar]
- 36.Lin W-C, Culp L A. Altered establishment/clearance mechanisms during experimental micrometastasis with live and/or disabled bacterial lacZ-tagged tumor cells. Invasion Metastasis. 1992;12:197–209. [PubMed] [Google Scholar]
- 37.Melo R C, Brener Z. Tissue tropism of different Trypanosoma cruzi strains. J Parasitol. 1978;64:475–482. [PubMed] [Google Scholar]
- 38.Mendelsohn C, Ruberte E, LeMeur M, Morriss-Kay G, Chambon P. Developmental analysis of the retinoic acid-inducible RAR-β2 promoter in transgenic animals. Development. 1991;113:723–734. doi: 10.1242/dev.113.3.723. [DOI] [PubMed] [Google Scholar]
- 39.Mirkin G A, Jones M, Sanz O P, Rey R, Sica R E P, Gonzalez Cappa S M. Experimental Chagas’ disease: electrophysiology and cell composition of the neuromyopathic inflammatory lesions in mice infected with a myotropic and a pantropic strain of Trypanosoma cruzi. Clin Immunol Immunopathol. 1994;73:69–79. doi: 10.1006/clin.1994.1171. [DOI] [PubMed] [Google Scholar]
- 40.Morales M C, Cardoni R L, Rimoldi M T, Esteva M, Milei J. Heart damage comparing three strains of mice chronically infected with Trypanosoma cruzi. Medicina (Buenos Aires) 1987;47:493–499. [PubMed] [Google Scholar]
- 41.Mott K E, Hagstrom J W C. The pathologic lesions of the cardiac autonomic nervous system in chronic Chagas’ myocarditis. Circulation. 1965;31:273–286. doi: 10.1161/01.cir.31.2.273. [DOI] [PubMed] [Google Scholar]
- 42.Oliveira J S M, dos Santos M, Muccillo G, Ferreira A L. Increased capacity of the coronary arteries in chronic Chagas’ heart disease: further support for the neurogenic pathogenesis concept. Am Heart J. 1985;109:304–308. doi: 10.1016/0002-8703(85)90598-8. [DOI] [PubMed] [Google Scholar]
- 43.Petry K, Nudelman E, Eisen H, Hakomori S. Sulfated lipids represent common antigens on the surface of Trypanosoma cruzi and mammalian tissues. Mol Biochem Parasitol. 1988;30:113–122. doi: 10.1016/0166-6851(88)90104-1. [DOI] [PubMed] [Google Scholar]
- 44.Petry K, Voisin P, Baltz T. Complex lipids as common antigens to Trypanosoma cruzi, T. dionisii, T. vespertilionis and nervous tissue (astrocytes, neurons) Acta Trop. 1987;44:381–386. [PubMed] [Google Scholar]
- 45.Postan M, Cheever A W, Dvorak J A, McDaniel J P. A histopathological analysis of the course of myocarditis in C3H/He mice infected with Trypanosoma cruzi clone Sylvio-X10/4. Trans R Soc Trop Med Hyg. 1986;80:50–55. doi: 10.1016/0035-9203(86)90193-8. [DOI] [PubMed] [Google Scholar]
- 46.Prata A. Chagas’ disease. Infect Dis Clin North Am. 1994;8:61–76. [PubMed] [Google Scholar]
- 47.Rocken M, Shevach E M. Do parasitic infections break T-cell tolerance and trigger autoimmune disease. Parasitol Today. 1993;9:377–380. doi: 10.1016/0169-4758(93)90087-v. [DOI] [PubMed] [Google Scholar]
- 48.Said G, Joskowicz M, Barreira A A, Eisen H. Neuropathy associated with experimental Chagas’ disease. Ann Neurol. 1985;18:676–683. doi: 10.1002/ana.410180609. [DOI] [PubMed] [Google Scholar]
- 49.Santos-Buch C A, Acosta A M, Zweerink H J, Sadigursky M, Andersen O F, von Kreuter B F, Brodskyn C I, Sadigursky C, Cody R J. Primary muscle disease: definition of a 25-kDa polypeptide myopathic specific Chagas antigen. Clin Immunol Immunopathol. 1985;37:334–350. doi: 10.1016/0090-1229(85)90103-5. [DOI] [PubMed] [Google Scholar]
- 50.Snary D, Flint J E, Wood J N, Scott M T, Chapman M D, Dodd J, Jessell T M, Miles M A. A monoclonal antibody with specificity for Trypanosoma cruzi, central and peripheral neurones and glia. Clin Exp Immunol. 1983;54:617–624. [PMC free article] [PubMed] [Google Scholar]
- 51.Tarleton R L, Zhang L, Downs M O. “Autoimmune rejection” of neonatal heart transplants in experimental Chagas disease is a parasite-specific response to infected host tissue. Proc Natl Acad Sci USA. 1997;94:3932–3937. doi: 10.1073/pnas.94.8.3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tibbetts R S, McCormick T S, Rowland E C, Miller S D, Engman D M. Cardiac antigen-specific autoantibody production is associated with cardiomyopathy in Trypanosoma cruzi-infected mice. J Immunol. 1994;152:1493–1499. [PubMed] [Google Scholar]
- 53.Troyer H. Principles and techniques of histochemistry. Boston, Mass: Little, Brown & Co.; 1980. [Google Scholar]
- 54.Van Voorhis W C, Eisen H. FL-160: a surface antigen of Trypanosoma cruzi that mimics mammalian nervous tissue. J Exp Med. 1989;169:641–652. doi: 10.1084/jem.169.3.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wood J N, Hudson L, Jessell T M, Yamamoto M. A monoclonal antibody defining antigenic determinants on subpopulations of mammalian neurones and Trypanosoma cruzi parasites. Nature. 1982;296:34–38. doi: 10.1038/296034a0. [DOI] [PubMed] [Google Scholar]
- 56.World Health Organization. Control of Chagas’ disease. W H O Tech Rep Ser. 1991;811:1–93. [PubMed] [Google Scholar]