

Summary
Apical-basal polarity is an essential epithelial trait controlled by the evolutionarily conserved PAR-aPKC polarity network. Dysregulation of polarity proteins disrupts tissue organization during development and in disease, but the underlying mechanisms are unclear due to the broad implications of polarity loss. Here, we uncover how Drosophila aPKC maintains epithelial architecture by directly observing tissue disorganization after fast optogenetic inactivation in living adult flies and ovaries cultured ex vivo. We show that fast aPKC perturbation in the proliferative follicular epithelium produces large epithelial gaps that result from increased apical constriction, rather than loss of apical-basal polarity. Accordingly, we can modulate the incidence of epithelial gaps by increasing and decreasing actomyosin-driven contractility. We traced the origin of these large epithelial gaps to tissue rupture next to dividing cells. Live imaging shows that aPKC perturbation induces apical constriction in non-mitotic cells within minutes, producing pulling forces that ultimately detach dividing and neighboring cells. We further demonstrate that epithelial rupture requires a global increase of apical constriction, as it is prevented by the presence of non-constricting cells. Conversely, a global induction of apical tension through light-induced recruitment of RhoGEF2 to the apical side is sufficient to produce tissue rupture. Hence, our work reveals that the roles of aPKC in polarity and actomyosin regulation are separable and provides the first in vivo evidence that excessive tissue stress can break the epithelial barrier during proliferation.
Keywords: aPKC, epithelial tissue, cell division, Drosophila, cell polarity, actomyosin contractility, RhoGEF2, tissue integrity, optogenetics, apical constriction
Graphical abstract
Highlights
-
•
Optogenetic clustering disrupts aPKC in vivo and ex vivo with high temporal control
-
•
aPKC downregulates apical contractility in multiple Drosophila tissues
-
•
Rapid aPKC inactivation causes tissue rupture next to dividing follicle cells
-
•
Increased apical constriction can break the epithelial barrier during proliferation
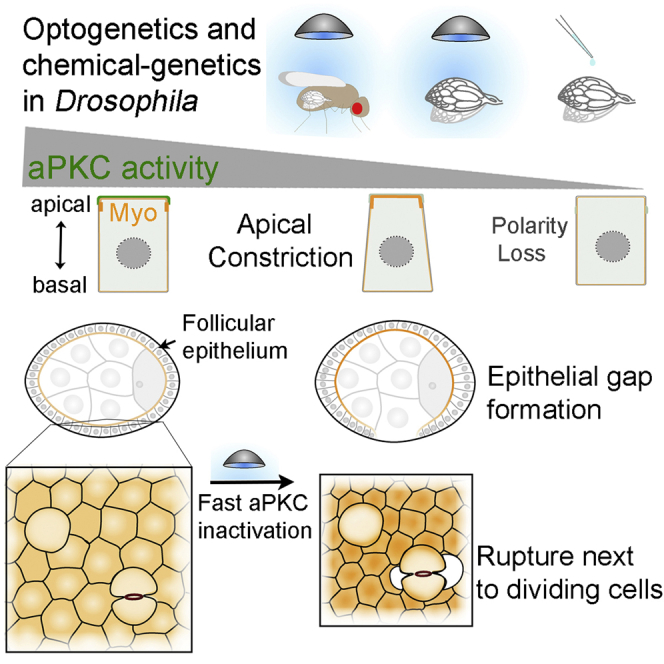
Osswald et al. use optogenetics and chemical genetics to rapidly modulate aPKC activity in Drosophila tissues and untangle its role as an inhibitor of apical actomyosin networks from its function in cell polarity. They show that acute induction of apical constriction rapidly leads to epithelial tissue rupture next to dividing cells in the fly ovary.
Introduction
Cell polarity is a defining feature of epithelial architecture and function. Apical-basal polarity ensures the asymmetric localization of intercellular junctions that maintain tissue cohesion and thereby preserve the epithelial barrier. Epithelial architecture is also regulated by the distribution of actomyosin-driven forces at the apical, basal, and junctional level.1 It is thus not surprising that polarity disruption induces epithelial disorganization during animal development or disease.2, 3, 4, 5 This raises the importance of spatial cues provided by polarity regulators to build and support the three-dimensional structure of an organ. However, because polarity proteins are involved in many different processes that can ultimately affect tissue shape, how these proteins maintain epithelial architecture remains a critical, longstanding question.
Interfering with polarity regulators in monolayered epithelia leads to different defects that disrupt epithelial integrity. These include the formation of multilayered tissue,3,4,6 lumen defects,4,7 and appearance of gaps.5,6,8 Extensive characterization using loss- or gain-of-function perturbations linked these defects to junctional disorganization, misoriented cell division, defective control of proliferation, or mis-differentiation. However, direct observation of how an epithelium becomes disorganized upon disruption of polarity regulators is still missing, which prevents a clear understanding of how defects arise.
Atypical protein kinase C (aPKC) is part of the apical PAR complex (Cdc42-Par6-aPKC) and is a central regulator of animal cell polarity9; it generates apical-basal asymmetry through phosphorylation of several polarity proteins, including Baz/Par3, Lgl, Yurt, and Crb. Their phosphorylation regulates local cortical binding through modulation of multivalent protein interactions, homo-oligomerization, or simply by reducing electrostatic interactions with plasma membrane phospholipids.10, 11, 12, 13, 14, 15 Apical-basal polarization ultimately positions belt-like adherens junctions (AJs) at the apical-lateral border where they mechanically link neighboring cells.
In addition to its well-studied role in polarity regulation, aPKC regulates cell fate, epithelial-to-mesenchymal transition, cell-cycle length, cell division orientation, actomyosin contractility, and microtubule dynamics.7,16, 17, 18, 19, 20, 21, 22, 23 In fact, some aPKC targets are not polarity proteins. Phosphorylation of Rho-associated coiled-coil-containing kinase (ROCK) limits the localization of this myosin activator to apical junctions and thereby inhibits apical constriction in mammalian cells.22,24 Moreover, aPKC can both regulate and respond to actomyosin networks and acts in a negative feedback loop to regulate pulsatile apical constriction in the Drosophila amnioserosa.24, 25, 26 aPKC function is also linked to actomyosin reorganization during cell division in fly tissues, which is consistent with its mitotic redistribution along the lateral cortex in mouse and sea anemone blastomeres.27, 28, 29 Thus, aPKC may ensure epithelial architecture through different functional outputs, demanding the separation of its contribution toward actomyosin and apical-basal polarity regulation.
Here, we combined optogenetic with chemical-genetic approaches to fine-tune aPKC inactivation with high temporal control. This allowed us to disentangle the functions of aPKC in the regulation of actomyosin contractility and polarity. The monolayered follicular epithelium that encloses the Drosophila germline is a powerful system to explore the regulation of epithelial architecture in vivo. Through acute perturbation we show that epithelial gaps form during proliferative stages, arising from tissue rupture next to dividing follicle cells. This phenotype stems from the role of aPKC as an inhibitor of apical actomyosin networks in non-mitotic cells, which become hypercontractile after aPKC downregulation and pull on dividing cells until detachment occurs. Our work reveals the importance of keeping apical contractility in check during proliferation-mediated growth to maintain epithelial integrity.
Results
Optogenetic clustering inactivates aPKC
To explore how apical polarity maintains epithelial architecture, we developed an approach to inactivate aPKC with high temporal control in the Drosophila follicular epithelium with an optogenetic clustering tool—light-activated reversible inhibition by assembled trap (LARIAT).30 When exposed to blue light, LARIAT components—CRY2 fused to a GFP nanobody (VHH) and CIBN fused to a multimerization domain—interact with each other and cluster. To target and sequester aPKC, flies co-expressed endogenously GFP-tagged aPKC and GAL4-driven UAS-LARIAT (UAS-VHH::CRY2-P2A-CIBN::MP) (Figure 1A), which enabled high temporal control of LARIAT expression specifically in the follicular epithelium by temperature shift from 18°C to 29°C.
Figure 1.

Optogenetic clustering inactivates aPKC and disrupts tissue architecture in vivo
(A) Schematic representation of optogenetic aPKC inactivation strategy using LARIAT (VHH::CRY2 and CIBN::MP). GFP::aPKC is targeted by CRY2 fused with a GFP nanobody (VHH). Exposing flies to blue light triggers CRY2 binding to CIBN fused with a multimerization domain (MP).
(B) Living flies were exposed to blue light for 48 h to cluster GFP::aPKC or kept in the dark (control) before egg chambers were stained for F-actin and DNA. Flies were either homozygous or heterozygous for endogenously tagged GFP::aPKC. Arrows point to epithelial gap (yellow) and multilayering (cyan).
(C) Frequency of epithelial defects in egg chambers (stages 3–8) from flies with the indicated combinations of wild-type, GFP::aPKC, or apkck06403 null allele after 24 h blue-light exposure (n, number of egg chambers). LARIAT was expressed in the follicular epithelium when indicated. Control flies were kept in the dark. Fisher’s exact test compared the incidence of defects between different samples (ns, not significant).
(D) Midsagittal sections of control and LARIAT egg chambers from flies expressing GFP::aPKC and Lgl::mCherry exposed to blue light for 24 h. Arrows point to apical Lgl::mCherry. Yellow boxes define inset region.
(E) Ratio of apical/lateral mean pixel intensity of Lgl::mCherry in control (n = 684 cells, 19 egg chambers) and LARIAT (n = 447 cells, 23 egg chambers). Graphs show mean ± SD (unpaired t test); gray points represent average for individual egg chambers. Scale bars, 10 μm.
See also Figure S1.
Expression of the UAS-LARIAT system in homozygous GFP::aPKC flies that remained in the dark did not interfere with aPKC localization or protein levels, nor did it produce defects in epithelial organization (Figures 1B and S1B). This shows that GFP::aPKC is fully functional in the presence of the LARIAT components. We then exposed female flies to blue light continuously for at least 24 h to test whether optogenetic clustering reproduced the aPKC mutant phenotypes described for the follicular epithelium.31,32 GFP::aPKC clustered in puncta and led to the anticipated defects in epithelial architecture, namely epithelial gaps (regions where the germline is not covered by epithelial tissue), and multilayering (layers of cells piling on top of each other) (Figures 1B and 1C). A similar frequency of tissue defects was also visible after clustering heterozygous GFP::aPKC in the presence of an apkc mutant allele but not in presence of the untagged wild-type allele (Figures 1B and 1C), which suggests that clustering inactivates GFP::aPKC. Optogenetic clustering of GFP-tagged proteins can induce co-recruitment of its binding partners.33 Par6 is indeed co-sequestered in GFP::aPKC clusters, whereas Cdc42 was not incorporated in the clusters (Figure S1A). This further suggests that the clustered aPKC-Par6 complex is inactive since it lacks Par-6 binding to Cdc42, which is necessary for full aPKC activation.34 Furthermore, as predicted for aPKC inactivation, its substrate Lgl mislocalized to the apical domain upon aPKC optogenetic clustering in follicle cells (Figures 1D and 1E). Taken together, these results show that illuminating living flies is sufficient to trigger CRY2-CIBN heterodimerization and perturb aPKC in vivo. We further evaluated the impact of optogenetic aPKC clustering on the asymmetric distribution of Miranda during Drosophila neural stem cell division, where it is a relevant aPKC substrate.9 aPKC clustering prevented Miranda’s release from the apical domain of dividing larval neuroblasts (Figures S1C and S1D). Thus, LARIAT-mediated clustering is applicable to study aPKC in distinct contexts of cell polarity.
Optogenetic aPKC inactivation leads to fast tissue disorganization
We took advantage of optogenetic perturbation in vivo to monitor the progression of tissue disorganization in flies exposed to light for specific periods of time (Figures 2A and 2B). We narrowed the analysis to stages 4–6 of egg chamber development to determine the impact of aPKC perturbation on epithelial architecture prior to major morphogenetic changes. Multilayering was the most prevalent phenotype in tissue from flies exposed to light for longer periods of time, whereas gaps were the most frequent defect upon 2 h of GFP::aPKC clustering in vivo (2 h, ∼50% egg chambers with gaps and ∼30% with multilayering; 4 h, ∼25% gaps and ∼85% multilayering; Figures 2A and 2B). The two phenotypes were not mutually exclusive (Figure 1C) and were commonly observed in different positions of the egg chamber (Figure 2C). Epithelial gaps appeared almost exclusively at the dorsal/ventral region, whereas multilayering was largely restricted to egg chamber poles, suggesting a distinct basis for the two phenotypes.
Figure 2.

aPKC inactivation leads to fast tissue disorganization
(A) Midsagittal sections and quantification of epithelial defects in GFP::aPKC homozygous egg chambers in proliferative stages (4–6) expressing LARIAT and stained for F-actin and DNA. Flies were exposed to light for the indicated time before ovary fixation.
(B) Frequency (mean ± SD) of epithelial gaps (yellow arrow in A) and multilayering (cyan arrows in A).
(C) Data in (B) re-analyzed to show the spatial distribution of defects. n = total amount of gaps or multilayering events.
(D) Time-lapse of an egg chamber expressing LARIAT in the follicular epithelium, GFP::aPKC, and H2A::RFP shows epithelial gaps (yellow arrows) and multilayering (cyan arrows). Imaging with 488 nm laser triggered LARIAT clustering from min 0 onward.
(E) Midsagittal images and quantification of epithelial gaps (yellow arrow) and multilayering (cyan arrow) in proliferative aPKCas4 egg chambers treated ex vivo for 2 h with the indicated concentrations of 1NA-PP1 and stained for F-actin and DNA.
(F) Graph shows frequency (mean ± SD) of epithelial defects.
Gray data points (graphs in B and F) represent independent experiments; n, number of egg chambers. Scale bars, 10 μm.
See also Video S1.
Intriguingly, gap frequency declined with increasing duration of light exposure (Figures 2A and 2B), suggesting that epithelial gaps appear specifically during the initial phase of aPKC clustering and before the formation of multilayered tissue. We confirmed these results by live imaging using fluorescent markers of the nucleus (H2A::RFP) and the plasma membrane in egg chambers cultured ex vivo. GFP::aPKC formed large clusters at the apical domain within minutes of exposure to blue light (Figure 2D; Video S1). Epithelial gaps formed within 30 min of light exposure and earlier than multilayering (Figure 2D; Video S1).
Live imaging of egg chamber (midsagittal view) expressing GFP::aPKC (green), LARIAT, H2A::RFP (magenta; right panel), and stained with membrane marker (magenta; right panel). Imaging with 488 nm laser triggered GFP::aPKC clustering from time 0 onward. Epithelial gaps appear at the dorsal and ventral side and multilayering at the posterior side.
aPKC is likely only partially inactive during the initial clustering period due to the time necessary to completely cluster and mislocalize aPKC. To test whether the predominance of distinct defects was associated with the extent of aPKC inactivation, we treated ovaries mutant for an aPKC ATP-analog-sensitive allele (aPKCas4) for 2 h with a range of 1NA-PP1 inhibitor concentrations. Treatment with 1 μM 1NA-PP1, which in vitro reduces aPKC activity to ∼15%,35 led predominantly to epithelial gaps (∼50% of egg chambers with gaps versus 10% with multilayering), whereas increasing drug concentrations led predominantly to multilayering (Figures 2E and 2F). Thus, epithelial gaps are associated with partial aPKC inhibition, whereas multilayering arises upon strong loss of aPKC function.
aPKC antagonizes apical constriction in multiple Drosophila tissues
Our results show that gap formation is the earliest defect in epithelial architecture after aPKC inactivation. To identify the primary cellular effect underlying epithelial gaps, we clustered aPKC and analyzed the immediate impact on polarity, adhesion, and the actomyosin cytoskeleton. Par6::mCherry is trapped together with aPKC from the onset of light exposure (Figure S2A). However, in contrast to long-term clustering, which mislocalized Lgl (Figure 1D), apical-basal polarity is unaffected before gap formation, as both Lgl::mCherry and E-cad::mKate2 remained enriched at the lateral membrane and apical junctions, respectively (Figures 3A and S2B). These results could be explained by partial aPKC inhibition during the initial period of clustering. Strikingly, we observed that the fluorescence of Sqh::mKate2, a tagged version of non-muscle myosin II regulatory light chain (MyoII-RLC), increased rapidly at the apical side of the epithelium within minutes of light exposure and prior to gap formation (Figures 3B and 3C; Video S2). Furthermore, apical myosin increase was accompanied by an increase in apical circularity, measured as the extent to which the shape of a cross-section of the whole apical surface of the epithelium approximates a circle (Figures 3B and 3D). This tissue deformation could result from alterations in the apical area of individual cells. Accordingly, live imaging of mosaic epithelia with clonal expression of UAS-LARIAT showed that optogenetic aPKC clustering induced constriction of the apical area of LARIAT-expressing cells (Figures 3E and 3F; Video S3). Recent work showed that aPKC prevents Yurt mislocalization to the apical membrane of follicle cells, where Yurt binding to Crumbs induces apical constriction.36 We therefore tested Yurt and Crumbs response in tissues clonally expressing UAS-LARIAT and exposed to light for 40 min. Crumbs largely maintained its apical localization, despite partial co-localization with the GFP::aPKC clusters (Figures S2C and S2D). More importantly, Yurt became enriched at the apical level (Figures S2E–S2G). Thus, induction of apical constriction is the earliest effect upon optogenetic aPKC clustering and may be linked to Yurt mislocalization.
Figure 3.

aPKC antagonizes apical constriction in Drosophila tissues
(A and B) Time-lapse of egg chambers expressing LARIAT, GFP::aPKC, and either Lgl-mCherry (A, top), E-cad::mKate2 (A, bottom), or Sqh::mKate2 (B). Imaging with 488 nm laser triggered aPKC clustering from min 0 onward. Yellow arrows in (A) indicate epithelial gaps.
(C) Sqh accumulation at the apical surface after aPKC clustering (mean ± SD) measured at the anterior/posterior (A/P) and dorsal/ventral (D/V) regions, corrected for cytoplasm intensity and normalized to its initial value (n ≥ 96 A/P and ≥ 96 D/V cells, 12 egg chambers).
(D) Egg chamber circularity (mean ± SD) measured at the apical surface of the follicular epithelium (n = 10 egg chambers).
(E) Time-lapse images of GFP::aPKC follicular epithelium cells with mosaic LARIAT expression (marked by 2xnls::RFP). Imaging with 488 nm laser triggered aPKC clustering and apical domain constriction in LARIAT-expressing cells.
(F) Apical cell area (mean ± SD) within LARIAT-expressing clones before and after aPKC clustering. Connected points (blue line indicates mean value) represent average apical area change within each measured clone (n = 161 cells, 12 clones, ANOVA for paired samples).
(G) Contraction of aPKCas4::mScarlet larval brain neuroepithelium following the addition of 10 μM 1NA-PP1 at min 0. Blue shading depicts the segmented region used for quantification.
(H) Graph shows the apical area (mean ± SD, normalized to its initial value) of aPKCas4::mScarlet larval brain neuroepithelial tissue after addition of 10 μM 1NA-PP1 (n = 10 neuroepithelia with an average of 157 cells, 5 independent experiments).
(I) Close up of the neuroepithelium of an aPKCas4::mScarlet larvae expressing YFP-tagged non-muscle myosin II heavy chain (Zip::YFP) following addition of 10 μM 1NA-PP1.
(J) Zip::YFP intensity (mean ± SD) at the apical junctions and apicomedial region and the corresponding change in junctional perimeter (mean ± SD; n = 3 neuroepithelia, 54 cells).
(K) Zip::YFP integrated density (mean ± SD) for the dataset in (J). Scale bars, 5 μm.
Time-lapse video shows that Sqh::mKate2x3 (magenta; right panel) increases at the apical membrane during the initial period of GFP::aPKC (green) clustering. Imaging with 488 nm laser triggered GFP::aPKC clustering from time 0 onward.
Live imaging of GFP::aPKC (green) follicular epithelial cells (surface view) with mosaic expression of LARIAT (marked by the presence of two copies of nlsRFP (bright magenta) and aPKC clustering). Imaging with 488 nm laser triggered GFP::aPKC clustering from time 0 onward. Right panel shows GFP::aPKC.
We further tested whether apical myosin increase was the first consequence of aPKC inactivation using chemical genetics (Figures S2H and S2I). Live imaging showed that treatment of aPKCas4 egg chambers with 1 μM 1NA-PP1 led to a quick increase of apical Sqh::mKate2, which persisted for 50 min in regions of the follicular epithelium without gaps. A higher inhibitor concentration (10 μM 1NA-PP1) also increased apical Sqh::mKate2 initially. However, this effect was transient, possibly due to the quicker loss of apical-basal polarity, which occurs around 20 min after addition of 10 μM 1NA-PP1 in the follicular epithelium.35 Perturbation of aPKC activity with high temporal control therefore demonstrates that aPKC regulates apical contractility prior to polarity loss.
We analyzed the effect of aPKC inhibition on the neuroepithelium of the developing fly brain to determine if downregulation of apical myosin intensity is a general function of aPKC. Previous genetic perturbation in neuroepithelial cells indicated that aPKC could instead promote apical contractility by maintaining the polarized myosin distribution.17 Hence, fast and controlled inactivation could also be necessary to separate the roles of aPKC in contractility and polarity in this tissue. We performed live imaging of aPKCas4::mScarlet larval brains to follow the initial impact of aPKC inhibition on apical shape and myosin accumulation. aPKC inhibition led to a dramatic constriction of the neuroepithelium (Figures 3G and 3H) that was initiated within 10 min of inhibitor addition, and which was associated with an increase in junctional and apicomedial myosin II intensity (Figures 3I and 3J). This was due to myosin becoming concentrated in a smaller area, since the total amount of myosin was slightly reduced (Figure 3K). Hence, aPKC inhibits apical constriction to control the shape of distinct epithelial tissues.
Epithelial gaps result from increased apical contractility
To determine whether increased actomyosin contractility is necessary to generate epithelial gaps in the follicular epithelium, we first disrupted the actin cytoskeleton with Latrunculin A (Lat-A). Time-lapse imaging with E-cad::mKate2 to measure the apical area at the AJ level showed that treatment with Lat-A before light exposure blocks apical constriction during optogenetic clustering of aPKC (Figures 4A and 4B). Moreover, disruption of the actin cytoskeleton prior to aPKC clustering prevented the formation of epithelial gaps in tissue exposed ex vivo to light for 2 h (Figures 4C and 4D). To further test whether increased MyoII activity promotes gap formation, we modulated actomyosin contractility and evaluated the presence of epithelial gaps after in vivo aPKC clustering for 2 h. Overexpressed unphosphorylatable (SqhAA) or phosphomimetic (SqhEE) versions of myosin-RLC respectively reduce and increase contractility when they form bipolar filaments with wild-type myosin-RLC.37,38 Upon optogenetic aPKC inactivation, SqhAA overexpression restored epithelial integrity, whereas SqhEE overexpression increased epithelial-gap frequency (Figure 4E). Together, these results indicate that actomyosin-dependent cell contractility promotes and is necessary for the formation of epithelial gaps after aPKC inactivation.
Figure 4.

Increased apical contractility underlies gap formation after aPKC optogenetic clustering
(A) Live imaging of GFP::aPKC in control or LARIAT egg chambers co-expressing E-cad::mKate2 (apical view) and treated with DMSO or Lat-A prior to aPKC clustering (triggered by imaging with the 488 nm laser). Scale bars, 5 μm.
(B) Apical cell surface area (mean ± SD) normalized to the area before clustering (n ≥ 7 egg chambers per condition).
(C) Representative midsagittal images and quantification of epithelial gaps (arrow) in proliferative follicular epithelium expressing GFP::aPKC and LARIAT and stained for F-actin and DNA. Ovaries were exposed to blue light ex vivo before fixation. Scale bars, 10 μm.
(D) Gap frequency (mean ± SD) scored in the presence (+) or absence of LARIAT (−) in egg chambers treated with DMSO or Lat-A.
(E) Gap frequency (mean ± SD) upon overexpression of mCherry (control), SqhE20E21 (SqhEE), or SqhA20A21 (SqhAA) in the follicular epithelium of GFP::aPKC LARIAT flies. Flies were exposed to blue light (+) or kept in the dark (−) for 2 h prior to fixation.
Gray data points represent independent experiments; n, number of egg chambers scored; Fisher’s exact test (ns, not significant).
Epithelial gaps form by tissue rupture next to dividing cells
During live imaging of Sqh::mKate2 in midsagittal egg chamber sections, we noticed that epithelial gaps frequently formed next to dividing cells (Figure S3A; Video S4). To test whether epithelial gaps appear specifically in proliferative tissue, we analyzed stage 8 egg chambers, which are non-proliferative during the 2 h period of light exposure. Epithelial gaps were almost absent in these egg chambers (Figure S3B), suggesting that cell division challenges cell attachment in the follicular epithelium. To address how cell division contributes to loss of tissue integrity, we imaged epithelial gaps forming in the follicular epithelium marked with a membrane marker. Strikingly, the majority of epithelial gaps initiated as ruptures between cells undergoing cytokinesis and their neighbors (Figures 5A and 5B; Video S4)
Figure 5.

Cell division challenges tissue cohesion upon aPKC inactivation
(A) Time-lapse images of an egg chamber (surface view) expressing LARIAT and GFP::aPKC and stained with membrane marker. Imaging with 488 nm laser clustered aPKC from min 0 onward. Epithelial gaps (arrows) form adjacent to dividing cells (colored).
(B) Quantification of epithelial-gap origin according to cell division stage of neighboring cells; n, number of gaps scored.
(C and D) Gap frequency in the presence or absence of LARIAT in egg chambers treated with Binucleine-2 (Bin-2) to inhibit AurB (C), or colchicine (Colch) to depolymerize microtubules (D), before light exposure for 2 h ex vivo.
(E) Frequency of epithelial gaps scored in control and TumRNAi egg chambers from flies expressing GFP::aPKC LARIAT and exposed (+) or not (−) to blue light for 2 h.
Graphs in (C)–(E) show mean ± SD; gray data points represent independent experiments; n, number of egg chambers scored; Fisher’s exact test (ns, not significant).
(F and H) Time-lapse images of the follicular epithelium (surface views) expressing LARIAT, GFP::aPKC, and H2A::RFP (chromatin) and stained with membrane marker. Bin-2 (F) or Colch (H) was added at least 15 min prior to clustering from min 0 onward. Epithelial gaps (arrows) form adjacent to dividing cells (colored) despite cytokinesis failure (F, chromatin decondenses without chromosome separation) or mitotic arrest (H, condensed chromatin throughout the video).
(G and I) Epithelial-gap origin according to cell division stage of neighboring cells in LARIAT egg chambers treated with Bin-2 (G) or Colch (I); n, number of gaps scored; scale bars, 10 μm.
Egg chamber expressing GFP::aPKC (green), LARIAT and Sqh::mKate2x3 (magenta, right panel) and imaged in midsagittal view is shown on top, whereas an egg chamber expressing GFP::aPKC (green), LARIAT, stained with membrane marker (magenta, right panel) and imaged at the surface is shown at the bottom. Imaging with 488 nm laser triggered GFP::aPKC clustering from time 0 onward.
Cytokinetic ring constriction could promote epithelial rupture in the context of aPKC downregulation by exerting an opposite pulling force on neighboring cells undergoing apical constriction. To test whether cytokinesis was necessary for cell detachment, we blocked cytokinesis either by disrupting contractile ring assembly with the Aurora B inhibitor Binucleine 2 (Bin2) or by blocking cells in prometaphase with the microtubule-depolymerizing drug colchicine (Colch) (Figure S3C). We also depleted Tumbleweed/RacGAP50C (Tum; Figure S3D), a component of the centralspindlin complex that regulates contractile ring assembly. None of these treatments induced epithelial gaps on their own, but they also did not prevent gap formation upon aPKC clustering (Figures 5C–5E). To determine how gaps were formed in the absence of cytokinesis, we performed live imaging of egg chambers treated with Colch or Bin2. These treatments allowed mitotic rounding and did not change apical cell area on their own (Figures S3E–S3J), but optogenetic aPKC clustering still led to recurrent tissue rupture next to rounded mitotic cells (Figures 5G and 5H). We conclude that although tissue rupture upon aPKC perturbation is commonly observed next to dividing cells, it does not require cytokinesis. Thus, other aspects of cell division likely provide an additional challenge to epithelial integrity upon increased apical constriction.
Dividing cells are stretched by hypercontractile neighbors after aPKC inactivation
To understand why epithelial gaps form between dividing and neighboring cells, we characterized the impact of aPKC on MyoII distribution in mitotic and non-mitotic cells. During interphase, MyoII is enriched at junctions and at the apicomedial surface of follicle cells, where it drives pulses of apical constriction.39 Live imaging showed that GFP::aPKC accumulated at the apical contacts and displayed a smaller dynamic apicomedial pool, which formed puncta during constriction of interphasic cells. aPKC intensity at the apicomedial surface correlated with cycles of apicomedial MyoII accumulation and inversely correlated with apical area, even though GFP::aPKC puncta did not fully overlap with the apicomedial actomyosin (Figures 6A–6C; Video S5). During mitosis, both MyoII and aPKC extended along the lateral cortex (Figure 6D, blue arrow; Video S6). Thus, we evaluated the effect of aPKC inactivation on the distinct MyoII pools. Upon optogenetic or chemical aPKC inhibition, MyoII accumulated rapidly at the apicomedial level in interphasic cells (orange inset; Figures 6D, 6E, and S4; Video S6), whereas a similar MyoII accumulation was not observed in mitotic cells (Figure 6F). In fact, aPKC inactivation still enabled lateral myosin extension as well as the reduction in apicomedial myosin intensity even when cells entered mitosis after aPKC inactivation (yellow inset; Figures 6D and 6G). This suggests that aPKC is required to antagonize apicomedial actomyosin, specifically during interphase.
Figure 6.

aPKC inactivation leads to excessive pulling forces on dividing cells
(A) Live imaging of GFP::aPKC and Sqh::mKate2 at the apical surface shows dynamic accumulation at the apicomedial region (arrows).
(B) Mean pixel intensity (normalized to first frame) of GFP::aPKC and Sqh::mKate2 in the apicomedial region of the cell shown in (A). The corresponding variation in apical area is also shown.
(C) Correlation coefficient between the mean intensity curves of GFP::aPKC and Sqh::mKate2 at the apical surface, and between apical area and the intensity of GFP::aPKC or Shq::mKate2 (n = 41 cells, 20 egg chambers). Graph shows mean ± SD. Gray points represent value for individual cells.
(D) Live imaging of GFP::aPKC in control or LARIAT egg chambers co-expressing Sqh::mKate2 (apical and lateral projections). Note the redistribution of GFP::aPKC and Sqh::mKate2 to the lateral cortex of the mitotic cell (blue arrow). Imaging with the 488 nm laser triggered aPKC clustering from min 0 in LARIAT egg chambers. After GFP::aPKC clustering, Sqh::mKate2 accumulates at the apicomedial region in interphase cells (orange dashed squares) but not in mitotic cells (yellow dashed square). Red arrows depict ring canals.
(E and F) Apicomedial Sqh intensity (normalized to the mean intensity before blue-light [488 nm] exposure) in (E) interphasic cells in control (n = 42 cells, 14 egg chambers) and LARIAT (n = 51 cells, 17 egg chambers), and (F) in mitotic cells in control (n = 14 cells, 9 egg chambers) and LARIAT (n = 12 cells, 8 egg chambers). Time is shown in respect of the onset of blue-light exposure.
(G) Apicomedial Sqh intensity (normalized to the mean intensity of interphasic control cells before blue-light exposure) in control (n = 18 cells, 9 egg chambers) and LARIAT (n = 17 cells, 9 egg chambers) cells that entered mitosis 5–25 min after the onset of blue-light exposure. Time is shown in respect of mitotic entry.
(H) Live imaging of GFP::aPKC and E-cad::mKate2 in the follicular epithelium (apical projection). aPKC clustering was triggered up to 5 min before mitotic entry. Cells enter anaphase at min 0.
(I) Apical surface area in dividing cells measured at the junction level until anaphase onset (n = 27 cells from 6 control egg chambers and 12 cells from 8 LARIAT egg chambers).
Graphs (E–G and I) show mean ± SD; scale bars, 5 μm.
Live imaging of GFP::aPKC (green, middle panel) and MyoII(sqh)::mKate2 (magenta, right panel) in the follicular epithelium (surface view).
Live imaging of GFP::aPKC (green; right panel) and Sqh::mKate2 (magenta; middle panel) in the follicular epithelium (surface view) of control and LARIAT egg chambers. Imaging with 488 nm laser triggered GFP::aPKC clustering in the presence of LARIAT from time 0 onward.
We hypothesized that increased apical constriction in neighboring non-mitotic cells could produce excessive pulling on dividing cells, which would be unable to sustain this force due to the decrease in apical myosin. To test this hypothesis, we quantified the effect of optogenetic aPKC inactivation on the apical surface area of mitotic cells labeled with E-cad::mKate2. In contrast to interphasic cells, mitotic cells did not contract but rather expanded their apical domain upon mitotic entry and later detached from constricting neighbors (Video S7). Quantification of the apical area of cells that were mitotic during the initial period of light exposure showed that clustering increased the expansion of the apical domain (Figures 6H and 6I). Hence, excessive apical contractility in non-mitotic cells induces stretching of dividing cells and promotes tissue disruption.
Live imaging of a GFP::aPKC (green; right panel) and E-cad::mKate2 (magenta) in the follicular epithelium (surface view). Imaging with the 488 nm laser triggered GFP::aPKC clustering from time point 0 onwards. Unlike interphase cells (blue asterisks) constrict, mitotic cells (yellow asterisks) still expand after aPKC clustering.
Global increase in apical contractility at the tissue level induces epithelial gaps
To address whether rupture of the follicular epithelium was produced by a global or local increase in apical contractility, we analyzed proliferative tissue with clonal expression of UAS-LARIAT. In contrast to egg chambers expressing UAS-LARIAT in all follicle cells, there was no rupture in mosaic egg chambers containing cells insensitive to light (Figures 7A and 7B), neither when LARIAT cells divided adjacent to wild-type cells (n = 16) nor within LARIAT clones (n = 28). This suggests that the apical side of wild-type cells may stretch to accommodate the constriction of neighboring tissue and prevent rupture. Accordingly, wild-type cells neighboring UAS-LARIAT clones expanded their apical area during light exposure (Figure 7C). Whereas the closed connection of the epithelial monolayer around the egg chamber restrains simultaneous constriction of follicle cells, the larval neuroepithelium, a pseudostratified epithelium that is free to constrict at the borders, is likely to be more deformable. To determine whether aPKC inhibition induces tissue rupture in the neuroepithelium, we performed live imaging of aPKCas4::mScarlet brains expressing E-cad::GFP to label AJs (Figures S5A–S5C) or Utrophin::GFP to label the whole-cell membrane (Figure S5D). Interestingly, apical contacts are lost specifically in post-mitotic cells after aPKC inhibition, as new daughter cells fail to accumulate aPKC apically and E-cad at newly formed AJs (Figures S5A–S5C). Nevertheless, this is insufficient to cause neuroepithelial rupture, because orthogonal views show these unpolarized cells remain integrated with their surrounding tissue while it contracts (Figures S5B and S5D). Thus, whereas fast aPKC inactivation disrupts tissue shape, epithelial rupture only occurs if there are mechanical constraints that prevent cell deformations from accommodating increased apical constriction.
Figure 7.

Global increase of apical contractility induces epithelial gaps
(A) Time-lapse images (surface of egg chamber) show that epithelial rupture does not occur in GFP::aPKC follicular epithelia with mosaic LARIAT expression (marked by 2xnls::RFP in magenta) despite multiple dividing cells (asterisks).
(B) Frequency of gaps in tissue with mosaic LARIAT expression (approximate clone area [% of total visible egg chamber surface] = 41% ± 26%) compared with whole-tissue expression of LARIAT (data from Figure 5B re-analyzed, Fisher’s exact test).
(C) Variation in wild-type apical cell area in control (fully wild-type tissue [n = 60 cells, 2 egg chambers]) and mosaic (wild-type cells adjacent to LARIAT-expressing cells [n = 82 cells, 13 egg chambers]) immediately upon blue-light exposure (min 0) and 15 min after. Gray points represent individual cells, and blue points represent mean apical cells. ANOVA for paired samples (ns, not significant).
(D) Live imaging of egg chamber expressing PatJ::CIBN::pmGFP and RhoGEF2::CRY2::mCherry (optoGEF) in the follicular epithelium (midsagittal view). Imaging with the 488 nm laser targeted RhoGEF2 to the apical domain from min 0 onward. Yellow arrows denote gap formation in a region with a dividing cell.
(E) Time-lapse images of the follicular epithelium expressing PatJ::CIBN::pmGFP, RhoGEF2::CRY2, and Sqh::mKate2 (midsagittal view). Pseudo-colored panel shows increased Sqh::mKate2 intensity at the apical surface after RhoGEF2 apical recruitment.
(F) Time-lapse images of the follicular epithelium expressing PatJ::CIBN::pmGFP and RhoGEF2::CRY2::mCherry showing epithelial gaps (arrows) forming adjacent to dividing cells (colored).
(G) Frequency of gap formation upon optoGEF activation ex vivo.
(H) Epithelial-gap origin according to the cell division stage of neighboring cells; n, number of gaps scored.
(I) Representative midsagittal images and epithelial-gap quantification (yellow arrow) in egg chambers (stages 4–6) from flies expressing PatJ::CIBN::pmGFP and RhoGEF2::CRY2::mCherry exposed to blue light for 2 h prior to fixation and staining for DNA. Control flies were left in the dark. Graphs show mean ± SD; gray data points represent independent experiments; n, total amount of egg chambers; Fisher’s exact test; scale bars, 5 μm.
(J) Model depicting how downregulation of aPKC activity increases apical constriction in the non-mitotic cells of a polarized epithelium, generating excessive pulling forces on dividing cells that induce detachment and, ultimately, epithelial gaps.
To test whether the global increase in apical contractility is sufficient to drive tissue rupture in the follicular epithelium, we used an optogenetic tool to stimulate apical constriction by light-dependent recruitment of the RhoGTPase activator RhoGEF2 (RhoGEF2::CRY2::mCherry) to the apically enriched PatJ::CIBN::GFP::CAAX fusion (optoGEF2).40 Live imaging showed cytoplasmic RhoGEF2::CRY2::mCherry was quickly recruited to the apical domain after light exposure (Figure 7D). Apical recruitment of RhoGEF2::CRY2::mCherry induced apical MyoII accumulation and produced gaps, which often formed adjacent to dividing cells (Figures 7D–7H; Video S8). Moreover, in vivo exposure of flies expressing optoGEF2 to 2 h of light led to a high frequency of egg chambers with epithelial gaps (Figure 7I). Thus, a global increase of apical contractility is sufficient to disrupt the integrity of an epithelium, further supporting the idea that aPKC protects epithelial integrity through regulation of apical actomyosin.
Imaging with 488 nm laser triggered RhoGEF2::CRY2::mCherry (magenta; right panel) recruitment to apical PatJ::CIBN::pmGFP (green).
Discussion
Apical-basal polarity provides positional information at the cellular level that is essential for tissue architecture; however, it remains ill-defined how loss of polarity regulators leads to epithelial architecture defects. Even though genetic approaches have yielded substantial insight, the inherent temporal constraints preclude direct visualization of the underlying events. Here, we used fast aPKC perturbation approaches in Drosophila epithelia to shed light on how aPKC regulates epithelial architecture. We show that epithelial gaps form prior to loss of apical-basal polarity and within minutes of aPKC optogenetic perturbation in the follicular epithelium. aPKC inactivation increases apical contractility in non-mitotic cells. This increase pulls dividing and neighboring cells apart, causing epithelial ruptures. We propose that downregulation of apical contractility by aPKC prevents the build-up of excessive forces that can compromise epithelial integrity (Figure 7J).
We have developed a strategy to quickly inactivate aPKC in epithelial and neural stem cells by employing optogenetic clustering in the abdomen of living flies or ex vivo in intact organs. By complementing optogenetic clustering with the ability to modulate aPKC activity with chemical genetics, we show that immediately after clustering, aPKC is only partially inactive. Strikingly, this decrease in aPKC activity does not disrupt apical-basal polarity and leads instead to an increase in apical contractility that underlies the formation of gaps in the follicular epithelium. This recapitulates the phenotype of hypomorphic aPKC alleles that produce gaps but do not disrupt apical-basal polarity.8,31 Our results therefore indicate that a high threshold of aPKC inactivation is required to disrupt apical-basal polarity, which suggests that polarized epithelia can withstand fluctuations in aPKC activity. In turn, the higher sensitivity of the apical actomyosin cytoskeleton likely enables aPKC-dependent regulation of contractility without compromising apical-basal polarity.
aPKC is essential for apical-basal polarity and thereby provides spatial cues that position the actomyosin network apically. Other apical polarity proteins can also act as positive regulators of apical constriction, such as Crumbs associated with Yurt36 or Cysts,41 and Cdc42, which activates MyoII through MRCK.24,42 Thus, it is anticipated that whenever a genetic perturbation of aPKC disrupts apical-basal polarity, it would also prevent apical constriction. Although this could indirectly place aPKC as a positive regulator of apical contractility, our work highlights the integration of aPKC function on apical-basal polarity with a role as a negative regulator of apical constriction. aPKC has been reported to antagonize apical actomyosin networks during different morphogenetic events in Drosophila and mammalian embryos.25,43,44 Interestingly, we found that whereas inhibition of aPKC induces myosin accumulation at the apicomedial surface in follicle cells, myosin becomes more concentrated at junctions in neuroepithelial tissue. Thus, the role of aPKC as an inhibitor of actomyosin contractility must be locally modulated for different morphogenetic and homeostatic purposes. The molecular nature of these functions is yet to be fully understood. Phosphorylation of ROCK by aPKC induces its cortical dissociation to downregulate junctional contractility in mammalian cells22 but aPKC does not regulate equivalent sites in Drosophila Rok.45 Alternatively, aPKC may target other actomyosin regulators or function through other apical polarity proteins implicated in the regulation of apical actomyosin contractility, such as Crumbs and Lulu2/Yurt.24,41,45, 46, 47, 48 In line with recent work showing aPKC prevents apical constriction by repressing apical Yurt accumulation,36 we observed Yurt apical enrichment after optogenetic inactivation of aPKC .
We propose that aPKC inhibits apical contractility to balance forces within proliferating epithelia to maintain epithelial integrity. Additionally, our findings suggest that the physical constraints of different tissues define the phenotypic outcome of apical constriction. For instance, in egg chambers, epithelial gaps form almost exclusively at the dorsal/ventral regions where tension at the apical cortex is higher.49 Moreover, the follicular epithelium is physically constrained at the basal side by a stiff basement membrane and at the apical side by the growing germline, which may keep the epithelium stretched.50,51 Thus, egg chamber organization likely opposes the shape changes necessary to accommodate global apical constriction, leading to an increase in tension and tissue rupture upon aPKC inactivation. In contrast, the presence of deformable wild-type cells during aPKC inactivation in mosaic tissue, or the less constrained mechanical context of the pseudostratified neuroepithelium, enables constricting cells to freely reduce their apical area, which likely releases tension and enables constriction without rupture. Hence, on top of possible differences in the local actomyosin response, our findings stress the importance of physical boundaries, tissue geometry, and mechanical context in the outcome of increased apical contractility in different tissues.
We also provide direct evidence that large epithelial rupture can arise by intercellular detachment during cell division, which provides a weak spot primed for disruption upon increased mechanical stress. This is consistent with previous observations that reinforcement of junctional attachment to the cytoskeleton prevents detachment during cell division in the Drosophila embryonic epithelium and in mammalian cell culture.52,53 Dividing cells do not generate gaps upon aPKC inactivation in a mosaic tissue, showing that a direct effect in mitotic cells is not responsible for gap formation by itself. Then, why are mitotic cells prone to separate from the hypercontractile surrounding tissue? Mitotic cells downregulate apicomedial actomyosin and revert apical constriction, which makes them more susceptible to extrinsic forces.54,55 We show that increased pulling forces exerted by the constricting non-mitotic tissue indeed further expand the apical surface of mitotic cells upon aPKC clustering. These forces could amplify outward pulling forces at the poles of dividing cells56 and spatially oppose pulling forces by the contractile ring on cell adhesion during cytokinesis. We observed that ruptures generally occur next to the equatorial region during cytokinesis, which is consistent with opposing forces overcoming cell adhesion in this region (Figure 7J). Furthermore, local remodeling of cell adhesion during mitosis28,57 and cytokinesis58, 59, 60, 61 may favor detachment next to dividing cells.
Different cellular events must be integrated at the tissue level to drive concerted shape changes during morphogenesis. Apical constriction is frequently used to bend or fold epithelia during development. Cell division actively contributes to tissue morphogenesis by controlling tissue material properties62,63 and driving shape change64, 65, 66 or cellular rearrangements.67 However, the cell-intrinsic mitotic remodeling of the cytoskeleton can disrupt morphogenetic processes that require apical constriction.54,68, 69, 70 Our results now show that forces produced by apical constriction challenge cohesion at the dividing neighboring cell interface and thereby disrupt epithelial integrity. Hence, this study shows the importance of a strict control over apical constriction in proliferative tissues to enable growth and morphogenesis without compromising epithelial integrity.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-Miranda (1:2000) | Betschinger et al.71 | N/A |
| Mouse monoclonal anti-phospho-Histone H3 (pH3) Ser 10 (1:1000) | Cell Signalling | Cat# 9706; RRID: AB_331748 |
| Rabbit anti-phospho-Histone H3 (pH3) Ser10 (1:2000) | Merck Millipore | Cat# 06-570; RRID: AB_310177 |
| Rabbit anti-aPKC (c20, 1:2000) | Santa Cruz Biotechnology | Cat# sc-208; RRID: AB_2168668 |
| Mouse anti-Crumbs (1:50) | Developmental Studies Hybridoma Bank | Cat# cq4; RRID: AB_528181 |
| Rabbit anti-Yurt (1:2000) | Biehler et al.36 | RRID: AB_2568494 |
| Mouse anti-α-Tubulin (DM1A, 1:10 000) | Santa Cruz Biotechnology | Cat# sc-32293; RRID: AB_628412 |
| Alexa Fluor 568-conjugated goat anti-rabbit | Thermo Fisher Scientific | Cat# A-11036; RRID: AB_10563566 |
| Alexa Fluor 647-conjugated goat anti-rabbit | Thermo Fisher Scientific | Cat# A-31573; RRID: AB_2536183 |
| Alexa Fluor 647-conjugated goat anti-mouse | Thermo Fisher Scientific | Cat# A-31571; RRID: AB_162542 |
| Chemicals, peptides, and recombinant proteins | ||
| Insulin solution from bovine pancreas | Sigma-Aldrich | Cat# I0516 |
| Schneider’s Insect Medium | Sigma-Aldrich | Cat# S0146 |
| Phalloidin-FITC | Sigma-Aldrich | Cat# P5282 |
| Phalloidin-TRITC | Sigma-Aldrich | Cat# P1951 |
| Phalloidin-CruzFluor647 | Santa Cruz Biotechnology | Cat# sc-363797 |
| Cell Mask Orange Plasma Membrane Stain | Thermo Fisher | Cat# C10045 |
| Latrunculin A | Sigma-Aldrich | Cat# 428021 |
| Colchicine | Sigma-Aldrich | Cat# C9754 |
| Binucleine-2 | Sigma-Aldrich | Cat# B1186 |
| Thrombin | Sigma-Aldrich | Cat# T7513 |
| Glucose | Sigma-Aldrich | Cat# G7021 |
| Fibrinogen | Sigma-Aldrich | Cat# F8630 |
| Experimental models: Organisms/strains | ||
| D. melanogaster: Par6::mCherry:;; UAS-Par6mCherry | Doerflinger et al.72 | N/A |
| D. melanogaster: GFP::aPKC:;aPKC>GFP::aPKC; | Chen et al.73 | N/A |
| D. melanogaster: Lgl::mCherry:;lgl>lgl::mCherry; | Dong et al.15 | N/A |
| D. melanogaster: E-cad::GFP:;shg>shg::GFP; | Huang et al.74 | N/A |
| D. melanogaster: E-cad::mKate2:;shg>shg::3xmKate2/CyoGFP; | Pinheiro et al.75 | N/A |
| D. melanogaster: E-cad::3xGFP:;shg>shg::3xGFP/Cyo; | Pinheiro et al.75 | N/A |
| D. melanogaster: Zip::YFP:;PBac{544.SVS-1}zipCPTI100036/SM6a; | Kyoto Stock Center | RRID: DGGR_115082 |
| D. melanogaster: Sqh::mKate2:;P{w+, sqh>sqh::3xmKate2}/CyO; | Pinheiro et al.75 | N/A |
| D. melanogaster: Sqh::mKate2:;;P{w+, sqh>sqh::3xmKate2}/TM6B,Tb | Pinheiro et al.75 | N/A |
| D. melanogaster: H2A::RFP:;;P{w+, his2Av>his2Av::mRFP}/MKRS | Pandey et al.76 | N/A |
| D. melanogaster: mCherry::Cdc42:;;Sqh-mCherry::Cdc42 | Bloomington Drosophila Stock Center | RRID: BDSC_42237 |
| D. melanogaster: UAS-LARIAT:;P{w+, UASt>VHH(GFP)::SNAP::CRY2(PHR)::P2A::CIBN::MP}/CyO; | Lee et al.30 | N/A |
| D. melanogaster: UAS-LARIAT:;;P{w+, UASt>VHH(GFP)::SNAP::CRY2(PHR)::P2A::CIBN::MP}/TM6B,Tb | Qin et al.77 | N/A |
| D. melanogaster: PatJ::CIBN::GFPpm:;P{w+, UASp>PatJ::CIBN::pmGFP}/CyO; | Krueger et al.40 | N/A |
| D. melanogaster: RhoGEF::CRY2::mCherry:;;P{w+, UASp>RhoGEF2::CRY2::mCherry}/TM3,Ser | Izquierdo et al.78 | N/A |
| D. melanogaster: RhoGEF::CRY2:;;P{w+, UASp> RhoGEF2::CRY2}/TM3,Ser | Izquierdo et al.78 | N/A |
| D. melanogaster: mCherry:;;P{y+ v+, UAS>mCherry.VALIUM10}attP2 | Bloomington Drosophila Stock Center | RRID: BDSC_35787 |
| D. melanogaster: Tum RNAi:;;P{y+ v+, UAS>tum RNAi.VALIUM10}attP2 | Bloomington Drosophila Stock Center | RRID: BDSC_28982 |
| D. melanogaster: SqhE20E21:;;P{w+, UASp> UASp>SqhE20E21} | Bloomington Drosophila Stock Center | RRID: BDSC_64411 |
| D. melanogaster: SqhA20A21:;;P{w+, UASp> UASp>SqhA20A21} | Bloomington Drosophila Stock Center | RRID: BDSC_64114 |
| D. melanogaster: tj>Gal4:;P{w+=GawB}NP1624/CyO; | Kyoto Stock Center | RRID: DGGR_104055 |
| D. melanogaster: GR1>Gal4:;;P{w+=GawB}GR1/TM6B, Tb | Gupta and Schüpbach79 | N/A |
| D. melanogaster: pnt>Gal4: | Vienna Drosophila Resource Center | VDRC_VT212057 (discarded) |
| D. melanogaster: Gal80ts:;;P{w+, tubP>Gal80ts} | Bloomington Drosophila Stock Center | RRID: BDSC_7018 |
| D. melanogaster: nlsRFP hs-Flp FRT19A: P{w+, ubi>nlsRFP} w∗P{ry+, hs>Flp}12 P{ry+, neoFRT}19A;; | Bloomington Drosophila Stock Center | RRID: BDSC_31418 |
| D. melanogaster: hs-Flp Gal80 FRT19A: P{ry+, hs>Flp}1 P{w+, tubP>Gal80} w∗P{ry+, neoFRT}19A;; | Bloomington Drosophila Stock Center | RRID: BDSC_5133 |
| D. melanogaster: aPKC-:;P{lacW}aPKCk06403/CyO; | Wodarz et al.80 | N/A |
| D. melanogaster: aPKCas4:;aPKCas4; | Hannaford et al.35 | N/A |
| D. melanogaster: aPKCas4::mScarlet:;aPKCas4::mScarlet; | This paper | N/A |
| Software and algorithms | ||
| FIJI | Schindelin et al.81 | N/A |
| GraphPad Prism 6 and 8 | GraphPad Software (La Jolla, CA, USA) | N/A |
| Other | ||
| Blue light LED bulb (474nm) | SuperBrightLEDs | Cat# MR16-B48SMD |
| Amber light LED bulb (593nm) | SuperBrightLEDs | Cat# MR16-Y48SMD |
| Laser Scanning Confocal Microscope TCS SP5 II | Leica Microsystems | N/A |
| Laser Scanning Confocal Microscope TCS SP8 | Leica Microsystems | N/A |
| Zeiss Axio Imager Z1 microscope | Carl Zeiss | N/A |
| Zeiss Axio Imager Z1 Apotome microscope | Carl Zeiss | N/A |
| Zeiss LSM880 Confocal | Carl Zeiss | N/A |
| Zeiss LSM710 Confocal | Carl Zeiss | N/A |
| Spinning disk confocal microscope Andor Revolution XD | Andor Technology | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Eurico Morais-de-Sá (eurico.sa@ibmc.up.pt).
Materials availability
All reagents generated in this study will be shared upon request by the lead contact without any restrictions.
Experimental model and subject details
Drosophila melanogaster lines and maintenance
We performed all experiments using Drosophila melanogaster. We raised fly lines on standard fly food (cornmeal/agar/molasses/yeast) at 18°C or 25°C with 60% humidity and 12h/12h dark light cycle, except when otherwise indicated in the method details section. The molecular details of the used fly lines are listed in the method details and the key resources table. Fly genotypes for each experiment can be found in Table S1.
Method details
Drosophila strains
The following fly lines were used:
-
•
Under regulation of the respective endogenous promoters: H2A::RFP76 and Sqh::mKate2x3 – Drosophila non-muscle myosin II regulatory light chain tagged with three tandem mKate2 and inserted in chromosomes II and III (Pinheiro et al.,75 gift from Yohanns Bellaïche, Institut Curie, Paris);
-
•
Tagged in the respective endogenous locus: GFP::aPKC (Chen et al.,73 gift from Daniel St Johnston, The Gurdon Institute, Cambridge), Lgl::mCherry15 and E-cad::GFP74 (both gifts from Yang Hong, University of Pittsburgh), E-cad::mKate2x3 and E-cad::GFPx3 (tagged with three tandem mKate2 or GFP,75 gift from Yohanns Bellaïche, Institut Curie, Paris) and Zip::YFP – Drosophila non-muscle myosin II heavy chain82;
-
•
mCherry::Cdc42 under regulation of the Sqh promoter (BDSC_42237) and UAS-Par6::mCherry (Doerflinger et al.72 gift from Daniel St Johnston, The Gurdon Institute, Cambridge);
-
•
UAS-LARIAT inserted in chromosomes II and III: CRY2 PHR domain fused to SNAPtag and a GFP nanobody (VHH) and N-terminal CIB domain (residues 1-170) fused to the CaMKIIα multimerization domain (MP), both expressed from a single construct with the help of a P2A self-cleaving peptide (Qin et al.,77 gift from Xiabo Wang, CBI, Université de Toulouse);
-
•
For optogenetic RhoGEF2 recruitment to the apical membrane: PatJ::CIBN::pmGFP, for UAS-driven expression of PatJ fused to N-terminal CIB domain (residues 1–170) and membrane targeted GFP fused to human Kras4B CAAX,40 RhoGEF2::CRY2 and RhoGEF2::CRY2::mCherry, for UAS-driven expression of tagged and untagged catalytic DHPH domain of RhoGEF2 fused to CRY2 PHR,78 gifts from Stefano de Renzis, EMBL, Heidelberg;
-
•
To drive Gal4-UAS mediated construct expression: GR1-Gal4, an enhancer trap line where Gal4 is under regulation of unknown regulatory sequences that drive Gal4 expression in the follicular epithelium79 and tj-Gal4, an enhancer trap line where Gal4 is under regulation of traffic jam regulatory sequences (DGGR_104055); pnt-Gal4 (VDRC_VT212057, discarded)
-
•
Gal80ts, temperature-sensitive Gal80 under regulation of the αTub84B promotor (BDSC_7018);
-
•
nlsRFP hs-Flp FRT19A (BDSC_31418) and hs-Flp Gal80 FRT19A (BDSC_5133), for FLP/FRT-mediated generation of Gal80 clones.
-
•
aPKCk06403, an aPKC null allele obtained by insertional mutagenesis of a P-element construct80;
-
•
aPKCas4 is an ATP analog-sensitive aPKC allele (I342A and T405A mutations introduced in the endogenous locus through CRISPR/Cas9)35;
-
•
aPKC::mScarletas4 was made by scar-less (inDroso co-CRISPR approach) CrispR gene editing. The mScarlet-I sequence, preceded and followed by short two amino acid (VAL GLY) linkers, was inserted into the genome of the apkcas4 line using the AATGGATCCTCCGGTGGCGGTGG guide RNA. The insert position was the same as previously published for GFP83: the mScarlet-I amino acid sequence, including the ATG and framed by the linkers, was inserted after amino acid 228 of aPKC-PA.
-
•
UAS-mCherry (BDSC_35787), UAS-Tum RNAi (BDSC_28982) and phosphomimetic and nonphosphorylatable Sqh - UAS-SqhE20E21 (BDSC_64411) and UAS-SqhA20A21 (BDSC_64114).
For optogenetic experiments where flies were exposed to blue light, female offspring of the same cross with the same genotype were randomly assigned to experimental groups (dark vs light). For each independent ex vivo experiment with drug treatment of egg chambers, we dissected ovaries from all flies of the same genotype, mixed them together, separated their ovarioles and then randomly distributed them by the experimental groups. For live imaging of egg chambers, we imaged 2-3 egg chambers per fly.
Optogenetics in the follicular epithelium
To inactivate apical polarity with optogenetics, we combined aPKC tagged with GFP in its endogenous locus73 with UAS-LARIAT.77 We used tj-Gal4 or GR1-Gal4 to drive UAS constructs expression in the follicular epithelium. To minimize premature UAS construct expression, crosses were kept at 18°C. 1-3 days after eclosion, adult offspring were transferred to 29°C to drive expression of UAS constructs (1 day at 29°C for all UAS constructs, except for optogenetic RhoGEF2 membrane recruitment, which was induced for 2-3 days at 29°C). To avoid unintended optogenetic system activation by light, we kept fly vials inside cardboard boxes or wrapped in aluminum foil and handled them in a dark room under a 593nm LED light source (SuperBrightLEDs) from this point forward. For co-expression of LARIAT with other UAS constructs (except UAS-Par6::mCherry), we used temperature-sensitive Gal80ts to fully suppress premature Gal4-UAS driven transcription prior to temperature shift to 29°C. To express LARIAT in clones, we generated Gal80 clones through FLP/FRT-mediated recombination.84 These crosses were kept at 18°C, protected from light and heat shocked at 37°C for 2 hours 3-5 times. LARIAT-expressing cells were marked by the presence of 2 nlsRFP copies, while wild-type cells had either 1 or no nlsRFP copies. Alternatively, in Figure S2B, LARIAT-expressing cells were identified by the presence of GFP::aPKC clusters.
For in vivo optogenetic experiments, flies were exposed continuously to blue light for the indicated periods of time (in Table S1 and figure legends) by placing vials at approximately 8 cm from a 472nm LED bulb (SuperBrightLEDs) at room temperature. Afterwards, we dissected their ovaries and fixed them. For each independent experiment, control flies from each genotype were kept in the dark and dissected in a dark room to avoid triggering CIBN-CRY2 interaction. To control for potential blue light toxicity, we also exposed flies without optogenetic constructs to blue light using the same setup (data included in Figure 1).
For ex vivo optogenetic experiments, ovaries were dissected in a dark room in ex vivo culture medium (Schneider’s medium (Sigma-Aldrich) supplemented with 10% FBS (fetal bovine serum, heat inactivated; Thermo Fisher) and 200 μg/mL insulin (Sigma-Aldrich)). Afterwards, the dissected ovaries were transferred to new ex vivo culture medium and the ovarioles were partially separated by pipetting up and down gently. The separated ovarioles were exposed to blue light for the indicated periods of time (in Table S1 and figure legends), in 24-well-plates using the same setup as for whole flies, and then they were fixed and stained. When indicated in the figures and figure legends, specific drugs (or DMSO or ethanol for control samples) were added 20 minutes before exposure to blue light: Colchicine (Sigma-Aldrich; 30 μM; prepared in ethanol) to depolymerize microtubules and block cells in mitosis; Binucleine-2 (Sigma-Aldrich; 40 μM; prepared in DMSO) to inhibit Aurora B85; and Latrunculin A to disrupt the actin cytoskeleton (Sigma Aldrich; 5 μg/mL; prepared in DMSO). To confirm that Binucleine-2 blocked cytokinesis and Colchicine blocked cells in mitosis in the follicular epithelium, ovarioles were treated with these drugs for 30 minutes and then fixed (without exposing them to blue light).
For live imaging, ovaries were dissected in a dark room and CIBN-CRY2 interaction was only triggered with the 488 nm laser used for GFP-tagged protein imaging.
Fixation and staining of egg chambers
Drosophila ovaries were dissected in Schneider’s medium (Sigma-Aldrich) supplemented with 10% FBS (fetal bovine serum, heat inactivated; Thermo Fisher), washed once with PBT (PBS + 0.05% Tween 20 (Sigma-Aldrich)) and fixed in 4% paraformaldehyde solution (prepared in PBS with 0.2% Tween 20 (Sigma-Aldrich)) for 20 min. After washing three times for 10 min with PBT, samples were mounted with Vectashield with DAPI (Vector Laboratories). Alternatively, for antibody staining, after the post-fixation washes, egg chambers were blocked for 2 hours at room temperature with 10% FBS (prepared in PBS + 0.2% Tween 20) and incubated overnight at 4°C with the primary antibody diluted in PBT + 1% FBS. Afterwards, the samples were washed four times with PBT + 1% FBS for 30 minutes and incubated again for at least two hours at room temperature with the secondary antibody diluted in PBT + 0.1% FBS. After washing them three times for 10 minutes with PBT, the samples were finally mounted with Vectashield with DAPI (Vector Laboratories). For F-actin staining, we added Phalloidin-FITC (Sigma-Aldrich), Phalloidin-TRITC (Sigma-Aldrich) or Phalloidin-CruzFluor647 (Santa Cruz Biotechnology) to the fixative solution and increased the incubation time to 30 min. Alternatively, for F-actin staining of antibody-stained samples, egg chambers were incubated for 30 minutes with Phalloidin diluted in PBT and washed three times for 10 minutes with PBT before mounting. We used the following primary and secondary antibodies: rabbit anti-phospho-Histone H3 (pH3) Ser10 (1:2000; Upstate Biotechnology), mouse anti-Crumbs (1:50; DSHB), rabbit anti-Yurt (1:2000; Biehler et al., 36 gift from Patrick Laprise, Centre de Recherche sur le Cancer, Québec ), Alexa Fluor 568-conjugated goat anti-rabbit (Invitrogen; 1:300), Alexa Fluor 647-conjugated anti-rabbit (Invitrogen) and Alexa Fluor 647-conjugated anti-mouse (Invitrogen). For Crumbs staining, fixed egg chambers were denatured with guanidine hydrochloride after the post-fixation washes with PBT: we rinsed samples twice in 50 mM Tris-HCl, pH 7.5, before incubating for 20 min in 6M guanidine hydrochloride prepared in 50 mM, pH 7.5, washing three times for 10 min with PBT and proceeding to block and stain with the antibodies as described.
Neuroblast immunofluorescence and optogenetics
We used pnt-Gal4 to drive UAS-LARIAT expression in type II neuroblasts. Following a 12h egg-laying period, control and LARIAT embryos were kept in the dark until wandering L3 larvae (wL3) stage. wL3 of both conditions were exposed to light for 1h. Brains were then dissected in PBS 1x, fixed in 4% paraformaldehyde for 20 min at room temperature and washed three times with PBST (0.1% Triton X-100 in 1x PBS). Brains were blocked with 1% normal goat serum in 0.1% PBST for at least 20 min at room temperature and incubated overnight at 4°C with rabbit anti-Miranda (1:2000, Betschinger et al.,71 gift from Juergen A. Knoblich) and mouse monoclonal anti-phospho-Histone H3 (pH3) Ser10 (1:1000, Cell Signalling, 9706), diluted in blocking solution. Afterwards, brains were washed three times, blocked for 20 min and incubated for 2h at room temperature with secondary antibodies Alexa Fluor 647-conjugated goat anti-mouse and Alexa Fluor 568-conjugated goat anti-rabbit (Invitrogen), used at 1:1000. Finally, brains were mounted in Aqua Polymount (Polysciences).
Imaging of fixed tissues
Images of fixed Drosophila egg chambers were collected with a 1.1 NA/40x water or 1.30 NA/63x glycerine objectives on an inverted laser scanning confocal microscope Leica TCS SP5 II (Leica Microsystems) or 1.30 NA/63x glycerol objective on an inverted laser scanning confocal microscope Leica SP8 (Leica Microsystems). To score epithelial defects and evaluate mitotic progression, images for egg chamber staging were collected with a 10x objective on a Zeiss Axio Imager Z1 microscope (Carl Zeiss, Germany) or a Zeiss Axio Imager Z1 Apotome microscope (Carl Zeiss, Germany). To evaluate epithelial architecture defects (epithelial gaps and/or multilayering), midsagittal cross-sections of egg chambers were inspected with a 20x or 40x Oil objective. To evaluate mitotic progression, images from the follicular epithelium at the surface of egg chambers were acquired with a 40x Oil objective on a Zeiss Axio Imager Z1 microscope (Carl Zeiss, Germany). Images from Drosophila larvae brains were acquired with a Zeiss LSM880 confocal microscope (Zeiss) using a LD LCI Plan-Apochromat 40x/1.2 Imm Corr DIC M27 water objective.
Live imaging
For live imaging of Drosophila egg chambers, individual ovarioles were dissected in ex vivo culture medium (Schneider’s medium (Sigma-Aldrich) supplemented with 10% FBS (fetal bovine serum, heat inactivated; Thermo Fisher) and 200 μg/uL insulin (Sigma-Aldrich)) and the enveloping muscle removed. Ovarioles were transferred to new culture medium and imaged on uncoated coverslips or glass bottom dishes (MatTek; No 1.5; P35G-1.5-7-C) with an Andor XD Revolution Spinning Disk Confocal system equipped with two solid state lasers – 488nm and 561nm -, an iXonEM+ DU-897 EMCCD camera and a Yokogawa CSU-22 unit built on an inverted Olympus IX81 microscope with a PLAPON 60x/NA 1.42 or a UPLSAPO 100x/NA 1.40 objective using iQ software (Andor). On average 2 egg chambers were imaged per fly. When indicated in the figures, to mark the cell membrane, ovarioles were stained with CellMask Orange Plasma membrane Stain (ThermoFisher; C10045; diluted 1:10 000 in culture medium) for 15 minutes and washed twice with ex vivo culture medium before imaging. Live imaging was performed at 25°C. When indicated in the figures and figure legends, Colchicine (Sigma-Aldrich; 30 μM; prepared in ethanol), Binucleine-2 (Sigma-Aldrich; 40 μM; prepared in DMSO) or Latrunculin A (Sigma Aldrich; 5 μg/mL; prepared in DMSO) were added at least 15 minutes before imaging. Midsagittal egg chamber cross-sections were used to image the follicular epithelium along the apical-basal axis and z-stacks at the surface of the egg chamber to cross-section the follicular epithelium along the apical-basal axis.
For live imaging of larval brain neuroepithelia, brains from L3 larvae were dissected in Schneider's medium supplemented with glucose (1 mg/ml, Sigma-Aldrich, G7021) and insulin (0.2 mg/ml) and transferred to a 10 μl drop of the same medium supplemented with Fibrinogen (0.2 mg/ml) on a 25 mm glass-bottom dish. Brains were oriented on their side and the Fibrinogen (Sigma-Aldrich, F8630) was clotted using thrombin (100 U/ml, Sigma-Aldrich, T7513). After 3 min, 190 μl Schneider's medium supplemented with glucose and insulin was pipetted on top of the clot. The neuroepithelia were imaged for 15 minutes on a Zeiss 710 Spinning Disk microscope using a 63x Plan-Apochromat 1.4 NA objective. 200 μl Schneider's medium supplemented with glucose, insulin and 1NA-PP1 (20 μM) was then added for a final concentration of 10 μM 1NA-PP1, after which imaging was resumed.
Protein extracts and Western blot
To confirm endogenous and GFP::aPKC levels in the different genotypes used for optogenetic aPKC inactivation (Figure S1B), we prepared protein extracts from Drosophila ovaries (at least 15 flies per genotype) dissected in a dark room. Dissected Drosophila ovaries were transferred to lysis buffer (150mM KCl, 75mM HEPES pH 7.5, 1.5 mM EGTA, 1.5mM MgCl2, 15% glycerol, 0.1% NP-40, 1x protease inhibitors cocktail (Roche) and 1x phosphatase inhibitors cocktail 3 (Sigma-Aldrich)), frozen in liquid nitrogen, thawed and then disrupted through sonication. We clarified lysates through two consecutive centrifugations at 14000 rpm for 10 min at 4°C. Protein concentration was determined with NanoDrop 1000 Spectrophotometer (Thermo Fisher). Samples were then resolved through SDS-PAGE and transferred to a nitrocellulose membrane using the iBlot Dry Blotting System (Invitrogen) for Western blotting. Protein transfer was confirmed by Ponceau staining (0.25% Ponceau S in 40% methanol and 15% acetic acid). The membranes were blocked for two hours at room temperature with 5% dry milk prepared in PBT and incubated overnight at 4°C with the primary antibodies (rabbit anti-aPKC 1:2000 (c-20, Santa Cruz Biotechnology) and mouse anti-α-Tubulin 1:10 000 (DM1A, Santa Cruz Biotechnology)) diluted in PBT + 1% dry milk. After washing three times for 10 min with PBT, membranes were incubated with the secondary antibodies anti-mouse and anti-rabbit conjugated with horseradish peroxidase diluted in PBT + 1% dry milk for one hour at room temperature. After washing again three times for 10 min with PBT, blots were developed with ECL Chemiluminescent Detection System (Amersham) according to the manufacturer’s instructions and revealed with a ChemiDoc XRS+ (BioRad).
aPKCas4 allele inactivation
For epithelial defect analysis in the follicular epithelium, Drosophila ovaries from aPKCas4 flies (prepared as previously described in the optogenetic experiments section) were cultured ex vivo for 2 hours in the presence of the ATP analog 1NA-PP1 (Calbiochem; prepared in DMSO; at the concentrations indicated in Figures 2E and 2F) before fixation. DMSO was added to control samples. For live imaging of egg chambers and larval neuroepithelium, 1NA-PP1 (at the concentration indicated in figure legends) or DMSO was added to the culture medium at the indicated timing.
Quantification and statistical analysis
Image processing and quantifications were done with FIJI.81 Statistical analysis and graphs were done in GraphPad Prism 8 (GraphPad Software, La Jolla, CA, USA), except when otherwise indicated.
Epithelial defects analysis
To evaluate epithelial architecture, we scored the amount of egg chambers at specific developmental stages with one or more epithelial gaps, one or more multilayering events or both in midsagittal cross-sections. As egg chambers develop, they grow in size. Thus, we determined the developmental stage of egg chambers by measuring their area in midsagittal cross-sections, as a proxy for size. To define the area intervals corresponding to each developmental stage, we staged control egg chambers from GFP::aPKC flies according to phenotypic characteristics, as in Jia et al.,86 and correlated their stage with their size. We scored epithelial defects (epithelial gaps and/or multilayering) and their position (anterior, posterior, dorsal-ventral) by inspecting midsagittal cross-sections of egg chambers: for LARIAT and aPKCas4 experiments, egg chambers were stained with DAPI (DNA) and Phalloidin (F-actin); for optogenetic RhoGEF2 membrane recruitment, egg chambers were stained with DAPI (DNA) and PatJ-CIBN-pmGFP and RhoGEF2-CRY2-mCherry fluorescence was used. For the initial analysis of aPKC inactivation with LARIAT (Figure 1C), results from 3 independent experiments (≥ 8 flies per condition per experiment) were summed up in a single contingency table and the graph shows the relative amount of egg chambers (stages 3 to 8) with each type of defect found. For statistical analysis, epithelial gaps and/or multilayering were grouped in a single defect category and Fisher’s exact test with Bonferroni correction for multiple comparison was used. For other experiments, graphs show mean percentage of egg chambers with the indicated type of defect ± standard deviation (SD). The percentages of defective egg chambers obtained for each independent experiment for each condition (≥ 8 flies per condition per independent experiment) are represented as individual data points in the graphs. The total amount of egg chambers scored in each analysis is indicated in the respective graph as n. To ensure consistent LARIAT expression levels, only proliferative stages 4 to 6 were included in analyses (except in Figures 1 and S3B). To test for statistical significance, we built contingency tables comparing the sum of egg chambers from all replicates with one or more epithelial gap vs no gap and used Fisher’s exact test, with Bonferroni correction for multiple comparisons when necessary. To compare the frequency of epithelial gap and multilayering formation at the anterior, posterior and dorsal-ventral regions, we analyzed how many of the epithelial gaps and multilayering events detected upon aPKC clustering in proliferative egg chambers were present at these different regions irrespective of how long the samples had been exposed to blue light (data in Figure 2C).
Epithelial gap analysis live
To evaluate whether and where gaps formed in the follicular epithelium, 4D stacks of surface cross-sections from egg chambers stained with membrane marker were analyzed. Gaps were inspected to verify if they span the whole length of the apical-basal axis and were only included in the analysis when all neighbor cells were in sight, so as to be able to determine whether any of them were in mitosis. The number of independent gaps detected in the 13 control, 8 Binucleine-2-treated and 6 Colchicine-treated egg chambers is indicated as n in Figures 5B, 5G, and 5I, respectively.
Egg chamber circularity
To assess egg chamber deformation after aPKC inactivation, we measured egg chamber circularity in single plane midsagittal cross-sections of Sqh::mKate2x3 egg chambers acquired during live imaging. The apical surface of the follicular epithelium was manually segmented and circularity (4π(area/perimeter2)) was measured. Egg chamber circularity was only quantified while no epithelial defect appeared.
Mitotic progression
To confirm the effect of Binucleine-2 and Colchicine, we analyzed mitotic progression in control and drug treated egg chambers. Mitotic cells were identified through positive staining with pH3 (number of cells counted indicated as n in Figures S3C ). DAPI staining was used to verify whether sister chromatids had separated and group cells into early mitosis (prophase, prometaphse, metaphase) and late mitosis (anaphase, telophase) or cytokinesis. Actin staining was used to verify whether cells had elongated, confirming anaphase onset, and whether they had assembled a cytokinetic ring.
Apical area in the follicular epithelium
To evaluate apical constriction in interphase cells and pulling forces on mitotic cells upon aPKC inactivation, we measured epithelial cell area in cross-sections at the junctional level of the follicular epithelium acquired during live imaging. For each egg chamber, we quantified the mean apical cell area (average of at least 3 interphase cells per egg chamber, individually and manually segmented using E-cad::mKate2). Surface area was normalized to the initial mean value, obtained by averaging the corresponding cell area for the 3 frames before aPKC clustering (from min -2 to 0). A similar procedure was used to segment cells that entered mitosis up to 5 minutes after aPKC clustering was initiated. Anaphase onset was defined as the first frame of cell elongation (determined through E-cad::mKate2 signal at the lateral cortex). Mitotic entry was defined as the first frame of visible mitotic rounding in a lateral cortex cross-section.
To verify whether Bin2 or Colch by themselves promote apical constriction, we performed a similar analysis of epithelial cell area in cross-sections acquired at the junctional level of Ecad::GFP follicle cells. Surface area was normalized to the initial mean value.
To evaluate apical constriction in clones of LARIAT-expressing cells, we measured the epithelial cell area at the apical surface of the follicular epithelium acquired during live imaging. For each clone, we quantified the mean apical cell area by manually segmenting the entire clone or the visible fraction in 4D stacks using GFP::aPKC and a plasma membrane marker and dividing the area by the number of cells inside (clones included in this analysis had at least 6 cells within the region quantified). To measure the apical area of individual wild-type cells adjacent to LARIAT-expressing cells, they were individually segmented in 4D stacks using GFP::aPKC and a plasma membrane marker (Figure 7C).
Lgl, Crb and Yurt localization
The ratio of apical over lateral Lgl and Yurt fluorescence intensities was used to analyze how aPKC clustering affects their respective asymmetric distributions along the apical-basal axis in the follicular epithelium. For each egg chamber, the lateral and apical cortex of all epithelial cells was manually segmented, average fluorescence intensity was extracted and corrected for average Lgl intensity in the cytoplasm or Yurt background intensity, respectively. Each point in Figures 1E and S2F represents the apical/lateral Lgl or Yurt ratio, respectively, for an individual egg chamber. Additionally, we also analyzed the distribution of Yurt and Crb along the lateral epithelial cell cortex (Figures S2D and S2G). We segmented lateral cell-cell interfaces from apical to basal manually for all cells in each egg chamber, measured Yurt and Crb fluorescence intensities along them using the Plot Profile function from FIJI and corrected these values for the respective background intensities. To average the profiles from cell-cell interfaces with different apical-basal lengths, we normalized the length of each individual interface to 100%. Fluorescence intensities for each control or LARIAT-expressing cell interface were normalized to the average fluorescence intensity of all control cell interfaces in the same egg chamber. Parts of the epithelium presenting multilayering were excluded from these analyses.
Apical myosin II in the follicular epithelium
To quantify apical accumulation of myosin II after aPKC inactivation, we measured Sqh::mKate2x3 fluorescence intensity at the apical domain of follicular epithelial cells in single plane midsagittal cross-sections of egg chambers acquired during live imaging. For each egg chamber, 4 regions of interest (ROIs), each one encompassing the apical domain of at least 4 cells in a different region (dorsal, ventral, anterior or posterior), were manually defined and tracked through time. Mean apical fluorescence intensity (AFI) for each timepoint was extracted from raw video datasets, corrected for mean cytoplasm fluorescence intensity (CFI; average value measured for each timepoint in 3 follicular epithelial cells) and normalized to the corrected fluorescence intensity before aPKC inactivation (AFIinitial-CFIinitial) as follows:
For aPKC LARIAT experiments, AFIinitial-CFIinitial corresponds to the AFI-CFI value at min 0 (when optogenetic clustering is triggered). For aPKCas4 experiments AFIinitial-CFIinitial was obtained by averaging AFI-CFI for the 5 frames before aPKC inactivation with 1NA-PP1 at min 0. Whenever, an epithelial gap started forming at a particular region, quantification at that same region was stopped.
Apicomedial myosin II in follicle cells
To quantify the effect of aPKC optogenetic clustering on apicomedial myosin II during interphase and mitosis, we measured apicomedial Sqh::mKate2x3 fluorescence intensity in interphase cells (three cells per egg chamber), dividing cells that were already in mitosis when blue light was turned on, and dividing cells that entered mitosis 5-25 min after blue light was turned on. We acquired z-stacks (Δz = 0.5 μm) of the surface of egg chambers by live imaging. We identified and tracked individual cells through time with the help of Sqh::mKate2x3 accumulation at cell-cell junctions and ring canals. We measured mean Sqh::mKate2x3 fluorescence intensity inside a 4.4 μm2 circular ROI placed in the middle of the apicomedial region of individual cells in maximum intensity projections of the z-stacks. Mean apicomedial fluorescence intensity for each timepoint was corrected for mean background fluorescence intensity (average value measured in three ROIs placed outside the egg chamber). For Figures 6E and 6F, Sqh::mKate2x3 fluorescence intensity for interphase and mitotic cells was normalized to the average Sqh::mKate2x3 fluorescence intensity in the frames prior to stimulation with blue light. For Figure 6G, Sqh::mKate2x3 fluorescence intensity in control and LARIAT samples was normalized to the Sqh::mKate2x3 fluorescence intensity from control cells, which was obtained by averaging Sqh::mKate2x3 fluorescence intensity at the apicomedial region for all measured control interphase cells (n = 42, 14 egg chambers) in the 5 frames prior to stimulation with blue light. We determined the timing of mitotic entry as the first frame of visible mitotic rounding with Sqh::mKate2x3 accumulation in a lateral cortex cross-section.
Apical area, aPKC and myosin II correlation
To analyze how myosin II and aPKC levels change at the apicomedial surface through time relative to each other and to the apical cell area, we measured these three factors in z-stacks (Δz = 0.5 μm) of the surface of egg chambers acquired during live imaging (Δt = 5 s). The maximum intensity of Sqh::mKate2 and aPKC::GFP from 3 z-planes centered around the apical surface was projected into a 2D image with the FIJI plugin LocalZProjector.87 To measure apical cell area, we segmented individual cells manually using GFP::aPKC signal (up to 3 cells per egg chamber). To measure fluorescence intensities at the apicomedial surface, the segmented ROIs encompassing the whole apical cell area were reduced by 0.66 μm to exclude junctional Sqh::mKate2x3 and GFP::aPKC signal. For each individual cell analyzed, we tested whether there was a temporal correlation between GFP::aPKC and Sqh::mKate2x3 levels, as well as between either of these variables and apical cell area, with Excel (Microsoft Corporation). The correlation coefficient for each individual cell is shown as an individual point in the graph in Figure 6C.
Apical area and myosin in the neuroepithelium
To measure apical surface contraction in the neuroepithelium upon aPKCas4 inactivation with 1NA-PP1, the ROI edges were manually tracked as dots using aPKCas-mScarlet signal to detect apical cell edges. These dots were then connected using a steerable filter for line detection. The resulting shape was then filled and dilated (blue mask in Figure 3G) to approximate the area to measure. Apical area was normalized to its initial value at min 0. To measure myosin II intensity at the apicomedial region and apical junctions after aPKCas4 inactivation with 1NA-PP1, the apical junctions of individual cells were segmented using aPKCas-mScarlet signal. The aPKC signal to noise ratio was increased by a steerable filter detecting lines and a junctional mask was generated by thresholding. The apicomedial regions of 54 individual cells were tracked and segmented based on this junctional mask. Individual junctional masks were generated by making a 10 pixels wide band around the resulting individual apicomedial masks. Myosin II intensity was obtained by extracting average Zip::YFP intensity and Zip::YFP integrated density with these masks and normalizing to average intensity and integrated density at junctions before aPKC inactivation at min 0. The junctional perimeter was obtained using the junctional masks of individual cells and normalized to the average perimeter before aPKC inactivation at min 0.
Miranda asymmetry in neuroblasts
The distribution of Miranda along the membrane of dividing NBs (pH3 positive) was analyzed. To extract Miranda intensity profile, that is, the intensity of Miranda (IM) along the length of the cell membrane (L), we proceeded as in Rodriguez et al.,88 with minor changes: we used a 30-pixel wide stripe to delineate the membrane; profile extraction was initiated in the basal membrane section, so that L<50% correspond to basal Miranda intensity values. In the end, Miranda intensity (IM) was plotted as a function of percentage of membrane length (L). To obtain the asymmetry index (ASI) for Miranda, Basal (B) and Apical (A) intensities were calculated as the area under the Miranda intensity plot (IM) for the basal (L ≤50%) and apical (L>50%) sections of the membrane. Absolute ASI values were calculated as in Hannaford et al.35 with the following formula:
ASI values were then normalized relative to control mean so that, an ASI of 1 represents normal asymmetry and lower values (∼0) indicate loss of asymmetry. As pnt-GAL4 only drives LARIAT expression in type II neuroblasts, only this subtype was considered in all calculations. Statistical analysis was performed using Prism 6 (GraphPad Software, La Jolla, CA, USA), mean ± SD are depicted and individual ASI values represented (normalized to control mean). Statistical significance of the difference of means was calculated using unpaired t test and considered significant when p < 0.05.
Image preparation
Representative images were processed and prepared using FIJI. Representative midsagittal images from egg chambers are from a single optical section or 2-5 planes maximum intensity projection. Surface images from egg chambers are maximum intensity projections of all optical sections covering the epithelial domain of interest. When necessary, videos were registered with the FIJI plugin StackReg (EPFL; Biomedical Imaging Group), to correct for whole egg chamber movement during live imaging; and a Gaussian Blur or Gaussian Blur 3D filter was applied to remove random noise.
Acknowledgments
We thank Daniel St Johnston, Juergen Knoblich, Patrick Laprise, Stefano de Renzis, Xiaobo Wang, Yohanns Bellaiche, and the Bloomington and Kyoto Drosophila Stock Centers for reagents. We also thank Yohanns Bellaiche, Ivo Telley, and Romain Levayer for insightful comments on the manuscript. This work is funded by National Funds through FCT—Fundação para a Ciência e a Tecnologia, I.P., under the project PTDC/BIA-CEL/1511/2021. E.M.-d.-S.'s salary is funded by the “FCT Scientific Employment Stimulus” program. M.O.,A.B.-C., and A.M.C. were supported by PhD fellowships from FCT. M.O.'s salary was also supported by the Maria de Sousa Award Research in the J.J. lab was supported by Wellcome Trust, the Royal Society, and BBSRC (BB/V001353/1). The authors acknowledge the i3S Scientific Platform ALM, member of the national infrastructure Portuguese Platform of Bioimaging, and the Dundee Imaging Facility for excellent support.
Author contributions
E.M.-d.-S. and M.O. conceptualized the study and wrote the original manuscript draft. M.O., A.B.-C., A.M.C., and E.M.-d.-S. performed all experiments in follicular epithelium, apart from Figure S4, which was performed by N.L. N.L. performed experiments in larval neuroepithelium and P.C.G. performed neuroblast experiments. Data analysis and interpretation, M.O., A.B.-C., A.M.C., N.L., P.C.G., C.C.F.H., J.J., and E.M.-d.-S.; supervision, C.E.S., C.C.F.H., J.J., and E.M.-d.-S.; funding acquisition, C.C.F.H., J.J., and E.M.-d.-S. All authors reviewed the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: September 15, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.cub.2022.08.063.
Supplemental information
Data and code availability
-
•
All original microscopy data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Munjal A., Lecuit T. Actomyosin networks and tissue morphogenesis. Development. 2014;141:1789–1793. doi: 10.1242/dev.091645. [DOI] [PubMed] [Google Scholar]
- 2.Halaoui R., Rejon C., Chatterjee S.J., Szymborski J., Meterissian S., Muller W.J., Omeroglu A., McCaffrey L. Progressive polarity loss and luminal collapse disrupt tissue organization in carcinoma. Genes Dev. 2017;31:1573–1587. doi: 10.1101/gad.300566.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bilder D., Li M., Perrimon N. Cooperative regulation of cell polarity and growth by Drosophila tumor suppressors. Science. 2000;289:113–116. doi: 10.1126/science.289.5476.113. [DOI] [PubMed] [Google Scholar]
- 4.Horne-Badovinac S., Lin D., Waldron S., Schwarz M., Mbamalu G., Pawson T., Jan Y., Stainier D.Y., Abdelilah-Seyfried S. Positional cloning of heart and soul reveals multiple roles for PKC lambda in zebrafish organogenesis. Curr. Biol. 2001;11:1492–1502. doi: 10.1016/s0960-9822(01)00458-4. [DOI] [PubMed] [Google Scholar]
- 5.Imai F., Hirai S., Akimoto K., Koyama H., Miyata T., Ogawa M., Noguchi S., Sasaoka T., Noda T., Ohno S. Inactivation of aPKClambda results in the loss of adherens junctions in neuroepithelial cells without affecting neurogenesis in mouse neocortex. Development. 2006;133:1735–1744. doi: 10.1242/dev.02330. [DOI] [PubMed] [Google Scholar]
- 6.Tanentzapf G., Smith C., McGlade J., Tepass U. Apical, lateral, and basal polarization cues contribute to the development of the follicular epithelium during Drosophila oogenesis. J. Cell Biol. 2000;151:891–904. doi: 10.1083/jcb.151.4.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sallee M.D., Pickett M.A., Feldman J.L. Apical PAR complex proteins protect against programmed epithelial assaults to create a continuous and functional intestinal lumen. eLife. 2021;10 doi: 10.7554/eLife.64437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guilgur L.G., Prudêncio P., Ferreira T., Pimenta-Marques A.R., Martinho R.G. Drosophila aPKC is required for mitotic spindle orientation during symmetric division of epithelial cells. Development. 2012;139:503–513. doi: 10.1242/dev.071027. [DOI] [PubMed] [Google Scholar]
- 9.Hong Y. aPKC: the kinase that phosphorylates cell polarity. F1000Res. 2018;7:7. doi: 10.12688/f1000research.14427.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Soriano E.V., Ivanova M.E., Fletcher G., Riou P., Knowles P.P., Barnouin K., Purkiss A., Kostelecky B., Saiu P., Linch M., et al. aPKC inhibition by Par3 CR3 flanking regions controls substrate access and underpins apical-junctional polarization. Dev. Cell. 2016;38:384–398. doi: 10.1016/j.devcel.2016.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morais-de-Sá E., Mirouse V., St Johnston D. aPKC phosphorylation of Bazooka defines the apical/lateral border in Drosophila epithelial cells. Cell. 2010;141:509–523. doi: 10.1016/j.cell.2010.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krahn M.P., Bückers J., Kastrup L., Wodarz A. Formation of a Bazooka-Stardust complex is essential for plasma membrane polarity in epithelia. J. Cell Biol. 2010;190:751–760. doi: 10.1083/jcb.201006029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gamblin C.L., Parent-Prévost F., Jacquet K., Biehler C., Jetté A., Laprise P. Oligomerization of the FERM-FA protein Yurt controls epithelial cell polarity. J. Cell Biol. 2018;217:3853–3862. doi: 10.1083/jcb.201803099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bailey M.J., Prehoda K.E. Establishment of par-polarized cortical domains via phosphoregulated membrane motifs. Dev. Cell. 2015;35:199–210. doi: 10.1016/j.devcel.2015.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dong W., Zhang X., Liu W., Chen Y.J., Huang J., Austin E., Celotto A.M., Jiang W.Z., Palladino M.J., Jiang Y., et al. A conserved polybasic domain mediates plasma membrane targeting of Lgl and its regulation by hypoxia. J. Cell Biol. 2015;211:273–286. doi: 10.1083/jcb.201503067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Niessen M.T., Scott J., Zielinski J.G., Vorhagen S., Sotiropoulou P.A., Blanpain C., Leitges M., Niessen C.M. aPKClambda controls epidermal homeostasis and stem cell fate through regulation of division orientation. J. Cell Biol. 2013;202:887–900. doi: 10.1083/jcb.201307001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Franco M., Carmena A. Eph signaling controls mitotic spindle orientation and cell proliferation in neuroepithelial cells. J. Cell Biol. 2019;218:1200–1217. doi: 10.1083/jcb.201807157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jung H.Y., Fattet L., Tsai J.H., Kajimoto T., Chang Q., Newton A.C., Yang J. Apical-basal polarity inhibits epithelial-mesenchymal transition and tumour metastasis by PAR-complex-mediated SNAI1 degradation. Nat. Cell Biol. 2019;21:359–371. doi: 10.1038/s41556-019-0291-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sabherwal N., Thuret R., Lea R., Stanley P., Papalopulu N. aPKC phosphorylates p27Xic1, providing a mechanistic link between apicobasal polarity and cell-cycle control. Dev. Cell. 2014;31:559–571. doi: 10.1016/j.devcel.2014.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hao Y., Du Q., Chen X., Zheng Z., Balsbaugh J.L., Maitra S., Shabanowitz J., Hunt D.F., Macara I.G. Par3 controls epithelial spindle orientation by aPKC-mediated phosphorylation of apical Pins. Curr. Biol. 2010;20:1809–1818. doi: 10.1016/j.cub.2010.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dias Gomes M., Letzian S., Saynisch M., Iden S. Polarity signaling ensures epidermal homeostasis by coupling cellular mechanics and genomic integrity. Nat. Commun. 2019;10:3362. doi: 10.1038/s41467-019-11325-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ishiuchi T., Takeichi M. Willin and Par3 cooperatively regulate epithelial apical constriction through aPKC-mediated ROCK phosphorylation. Nat. Cell Biol. 2011;13:860–866. doi: 10.1038/ncb2274. [DOI] [PubMed] [Google Scholar]
- 23.Harris T.J., Peifer M. aPKC controls microtubule organization to balance adherens junction symmetry and planar polarity during development. Dev. Cell. 2007;12:727–738. doi: 10.1016/j.devcel.2007.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zihni C., Vlassaks E., Terry S., Carlton J., Leung T.K.C., Olson M., Pichaud F., Balda M.S., Matter K. An apical MRCK-driven morphogenetic pathway controls epithelial polarity. Nat. Cell Biol. 2017;19:1049–1060. doi: 10.1038/ncb3592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.David D.J., Tishkina A., Harris T.J. The PAR complex regulates pulsed actomyosin contractions during amnioserosa apical constriction in Drosophila. Development. 2010;137:1645–1655. doi: 10.1242/dev.044107. [DOI] [PubMed] [Google Scholar]
- 26.Durney C.H., Harris T.J.C., Feng J.J. Dynamics of PAR proteins explain the oscillation and ratcheting mechanisms in dorsal closure. Biophys. J. 2018;115:2230–2241. doi: 10.1016/j.bpj.2018.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosa A., Vlassaks E., Pichaud F., Baum B. Ect2/Pbl acts via Rho and polarity proteins to direct the assembly of an isotropic actomyosin cortex upon mitotic entry. Dev. Cell. 2015;32:604–616. doi: 10.1016/j.devcel.2015.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ragkousi K., Marr K., McKinney S., Ellington L., Gibson M.C. Cell-cycle-coupled oscillations in apical polarity and intercellular contact maintain order in embryonic epithelia. Curr. Biol. 2017;27:1381–1386. doi: 10.1016/j.cub.2017.03.064. [DOI] [PubMed] [Google Scholar]
- 29.Zenker J., White M.D., Gasnier M., Alvarez Y.D., Lim H.Y.G., Bissiere S., Biro M., Plachta N. Expanding actin rings zipper the mouse embryo for blastocyst formation. Cell. 2018;173:776–791.e17. doi: 10.1016/j.cell.2018.02.035. [DOI] [PubMed] [Google Scholar]
- 30.Lee S., Park H., Kyung T., Kim N.Y., Kim S., Kim J., Heo W.D. Reversible protein inactivation by optogenetic trapping in cells. Nat. Methods. 2014;11:633–636. doi: 10.1038/nmeth.2940. [DOI] [PubMed] [Google Scholar]
- 31.Kim S., Gailite I., Moussian B., Luschnig S., Goette M., Fricke K., Honemann-Capito M., Grubmüller H., Wodarz A. Kinase-activity-independent functions of atypical protein kinase C in Drosophila. J. Cell Sci. 2009;122:3759–3771. doi: 10.1242/jcs.052514. [DOI] [PubMed] [Google Scholar]
- 32.Franz A., Riechmann V. Stepwise polarisation of the Drosophila follicular epithelium. Dev. Biol. 2010;338:136–147. doi: 10.1016/j.ydbio.2009.11.027. [DOI] [PubMed] [Google Scholar]
- 33.Ventura G., Moreira S., Barros-Carvalho A., Osswald M., Morais-de-Sá E. Lgl cortical dynamics are independent of binding to the Scrib-Dlg complex but require Dlg-dependent restriction of aPKC. Development. 2020;147:dev186593. doi: 10.1242/dev.186593. [DOI] [PubMed] [Google Scholar]
- 34.Pichaud F., Walther R.F., Nunes de Almeida F. Regulation of Cdc42 and its effectors in epithelial morphogenesis. J. Cell Sci. 2019;132:jcs217869. doi: 10.1242/jcs.217869. [DOI] [PubMed] [Google Scholar]
- 35.Hannaford M., Loyer N., Tonelli F., Zoltner M., Januschke J. A chemical-genetics approach to study the role of atypical protein kinase C in Drosophila. Development. 2019;146:dev170589. doi: 10.1242/dev.170589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Biehler C., Rothenberg K.E., Jette A., Gaude H.M., Fernandez-Gonzalez R., Laprise P. Pak1 and PP2A antagonize aPKC function to support cortical tension induced by the Crumbs-Yurt complex. eLife. 2021;10 doi: 10.7554/eLife.67999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vasquez C.G., Heissler S.M., Billington N., Sellers J.R., Martin A.C. Drosophila non-muscle myosin II motor activity determines the rate of tissue folding. eLife. 2016;5 doi: 10.7554/eLife.20828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kasza K.E., Farrell D.L., Zallen J.A. Spatiotemporal control of epithelial remodeling by regulated myosin phosphorylation. Proc. Natl. Acad. Sci. USA. 2014;111:11732–11737. doi: 10.1073/pnas.1400520111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alégot H., Pouchin P., Bardot O., Mirouse V. Jak-Stat pathway induces Drosophila follicle elongation by a gradient of apical contractility. eLife. 2018;7 doi: 10.7554/eLife.32943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krueger D., Tardivo P., Nguyen C., De Renzis S. Downregulation of basal myosin-II is required for cell shape changes and tissue invagination. EMBO J. 2018;37 doi: 10.15252/embj.2018100170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Silver J.T., Wirtz-Peitz F., Simões S., Pellikka M., Yan D., Binari R., Nishimura T., Li Y., Harris T.J.C., Perrimon N., Tepass U. Apical polarity proteins recruit the RhoGEF cysts to promote junctional myosin assembly. J. Cell Biol. 2019;218:3397–3414. doi: 10.1083/jcb.201807106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marston D.J., Higgins C.D., Peters K.A., Cupp T.D., Dickinson D.J., Pani A.M., Moore R.P., Cox A.H., Kiehart D.P., Goldstein B. MRCK-1 drives apical constriction in C. elegans by linking developmental patterning to force generation. Curr. Biol. 2016;26:2079–2089. doi: 10.1016/j.cub.2016.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maître J.L., Turlier H., Illukkumbura R., Eismann B., Niwayama R., Nédélec F., Hiiragi T. Asymmetric division of contractile domains couples cell positioning and fate specification. Nature. 2016;536:344–348. doi: 10.1038/nature18958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Röper K. Anisotropy of Crumbs and aPKC drives myosin cable assembly during tube formation. Dev. Cell. 2012;23:939–953. doi: 10.1016/j.devcel.2012.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sidor C., Stevens T.J., Jin L., Boulanger J., Röper K. Rho-kinase planar polarization at tissue boundaries depends on phospho-regulation of membrane residence time. Dev. Cell. 2020;52:364–378.e7. doi: 10.1016/j.devcel.2019.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Flores-Benitez D., Knust E. Crumbs is an essential regulator of cytoskeletal dynamics and cell-cell adhesion during dorsal closure in Drosophila. eLife. 2015;4 doi: 10.7554/eLife.07398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Letizia A., Sotillos S., Campuzano S., Llimargas M. Regulated Crb accumulation controls apical constriction and invagination in Drosophila tracheal cells. J. Cell Sci. 2011;124:240–251. doi: 10.1242/jcs.073601. [DOI] [PubMed] [Google Scholar]
- 48.Nakajima H., Tanoue T. Lulu2 regulates the circumferential actomyosin tensile system in epithelial cells through p114RhoGEF. J. Cell Biol. 2011;195:245–261. doi: 10.1083/jcb.201104118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Finegan T.M., Na D., Cammarota C., Skeeters A.V., Nádasi T.J., Dawney N.S., Fletcher A.G., Oakes P.W., Bergstralh D.T. Tissue tension and not interphase cell shape determines cell division orientation in the Drosophila follicular epithelium. EMBO J. 2019;38 doi: 10.15252/embj.2018100072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Balaji R., Weichselberger V., Classen A.K. Response of Drosophila epithelial cell and tissue shape to external forces in vivo. Development. 2019;146:dev171256. doi: 10.1242/dev.171256. [DOI] [PubMed] [Google Scholar]
- 51.Crest J., Diz-Muñoz A., Chen D.Y., Fletcher D.A., Bilder D. Organ sculpting by patterned extracellular matrix stiffness. eLife. 2017;6 doi: 10.7554/eLife.24958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Manning L.A., Perez-Vale K.Z., Schaefer K.N., Sewell M.T., Peifer M. The Drosophila Afadin and ZO-1 homologues Canoe and Polychaetoid act in parallel to maintain epithelial integrity when challenged by adherens junction remodeling. Mol. Biol. Cell. 2019;30:1938–1960. doi: 10.1091/mbc.E19-04-0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Monster J.L., Donker L., Vliem M.J., Win Z., Matthews H.K., Cheah J.S., Yamada S., de Rooij J., Baum B., Gloerich M. An asymmetric junctional mechanoresponse coordinates mitotic rounding with epithelial integrity. J. Cell Biol. 2021;220 doi: 10.1083/jcb.202001042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ko C.S., Kalakuntla P., Martin A.C. Apical constriction reversal upon mitotic entry underlies different morphogenetic outcomes of cell division. Mol. Biol. Cell. 2020;31:1663–1674. doi: 10.1091/mbc.E19-12-0673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Godard B.G., Dumollard R., Munro E., Chenevert J., Hebras C., McDougall A., Heisenberg C.P. Apical relaxation during mitotic rounding promotes tension-oriented cell division. Dev. Cell. 2020;55:695–706.e4. doi: 10.1016/j.devcel.2020.10.016. [DOI] [PubMed] [Google Scholar]
- 56.Gupta V.K., Nam S., Yim D., Camuglia J., Martin J.L., Sanders E.N., O'Brien L.E., Martin A.C., Kim T., Chaudhuri O. The nature of cell division forces in epithelial monolayers. J. Cell Biol. 2021;220 doi: 10.1083/jcb.202011106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aguilar-Aragon M., Bonello T.T., Bell G.P., Fletcher G.C., Thompson B.J. Adherens junction remodelling during mitotic rounding of pseudostratified epithelial cells. EMBO Rep. 2020;21 doi: 10.15252/embr.201949700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guillot C., Lecuit T. Adhesion disengagement uncouples intrinsic and extrinsic forces to drive cytokinesis in epithelial tissues. Dev. Cell. 2013;24:227–241. doi: 10.1016/j.devcel.2013.01.010. [DOI] [PubMed] [Google Scholar]
- 59.Higashi T., Arnold T.R., Stephenson R.E., Dinshaw K.M., Miller A.L. Maintenance of the epithelial barrier and remodeling of cell-cell junctions during cytokinesis. Curr. Biol. 2016;26:1829–1842. doi: 10.1016/j.cub.2016.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Herszterg S., Leibfried A., Bosveld F., Martin C., Bellaiche Y. Interplay between the dividing cell and its neighbors regulates adherens junction formation during cytokinesis in epithelial tissue. Dev. Cell. 2013;24:256–270. doi: 10.1016/j.devcel.2012.11.019. [DOI] [PubMed] [Google Scholar]
- 61.Founounou N., Loyer N., Le Borgne R. Septins regulate the contractility of the actomyosin ring to enable adherens junction remodeling during cytokinesis of epithelial cells. Dev. Cell. 2013;24:242–255. doi: 10.1016/j.devcel.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 62.Petridou N.I., Corominas-Murtra B., Heisenberg C.P., Hannezo E. Rigidity percolation uncovers a structural basis for embryonic tissue phase transitions. Cell. 2021;184:1914–1928.e19. doi: 10.1016/j.cell.2021.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Saadaoui M., Rocancourt D., Roussel J., Corson F., Gros J. A tensile ring drives tissue flows to shape the gastrulating amniote embryo. Science. 2020;367:453–458. doi: 10.1126/science.aaw1965. [DOI] [PubMed] [Google Scholar]
- 64.Hoijman E., Rubbini D., Colombelli J., Alsina B. Mitotic cell rounding and epithelial thinning regulate lumen growth and shape. Nat. Commun. 2015;6:7355. doi: 10.1038/ncomms8355. [DOI] [PubMed] [Google Scholar]
- 65.Kondo T., Hayashi S. Mitotic cell rounding accelerates epithelial invagination. Nature. 2013;494:125–129. doi: 10.1038/nature11792. [DOI] [PubMed] [Google Scholar]
- 66.Guirao B., Rigaud S.U., Bosveld F., Bailles A., López-Gay J., Ishihara S., Sugimura K., Graner F., Bellaïche Y. Unified quantitative characterization of epithelial tissue development. eLife. 2015;4 doi: 10.7554/eLife.08519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Firmino J., Rocancourt D., Saadaoui M., Moreau C., Gros J. Cell division drives epithelial cell rearrangements during gastrulation in chick. Dev. Cell. 2016;36:249–261. doi: 10.1016/j.devcel.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mata J., Curado S., Ephrussi A., Rørth P. Tribbles coordinates mitosis and morphogenesis in Drosophila by regulating string/CDC25 proteolysis. Cell. 2000;101:511–522. doi: 10.1016/s0092-8674(00)80861-2. [DOI] [PubMed] [Google Scholar]
- 69.Seher T.C., Leptin M. Tribbles, a cell-cycle brake that coordinates proliferation and morphogenesis during Drosophila gastrulation. Curr. Biol. 2000;10:623–629. doi: 10.1016/s0960-9822(00)00502-9. [DOI] [PubMed] [Google Scholar]
- 70.Großhans J., Wieschaus E. A genetic link between morphogenesis and cell division during formation of the ventral furrow in Drosophila. Cell. 2000;101:523–531. doi: 10.1016/S0092-8674(00)80862-4. [DOI] [PubMed] [Google Scholar]
- 71.Betschinger J., Mechtler K., Knoblich J.A. Asymmetric segregation of the tumor suppressor brat regulates self-renewal in Drosophila neural stem cells. Cell. 2006;124:1241–1253. doi: 10.1016/j.cell.2006.01.038. [DOI] [PubMed] [Google Scholar]
- 72.Doerflinger H., Vogt N., Torres I.L., Mirouse V., Koch I., Nüsslein-Volhard C., St Johnston D. Bazooka is required for polarisation of the Drosophila anterior-posterior axis. Development. 2010;137:1765–1773. doi: 10.1242/dev.045807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen J., Sayadian A.C., Lowe N., Lovegrove H.E., St Johnston D. An alternative mode of epithelial polarity in the Drosophila midgut. PLoS Biol. 2018;16 doi: 10.1371/journal.pbio.3000041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Huang J., Zhou W., Dong W., Watson A.M., Hong Y. From the cover: directed, efficient, and versatile modifications of the Drosophila genome by genomic engineering. Proc. Natl. Acad. Sci. USA. 2009;106:8284–8289. doi: 10.1073/pnas.0900641106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pinheiro D., Hannezo E., Herszterg S., Bosveld F., Gaugue I., Balakireva M., Wang Z., Cristo I., Rigaud S.U., Markova O., Bellaïche Y. Transmission of cytokinesis forces via E-cadherin dilution and actomyosin flows. Nature. 2017;545:103–107. doi: 10.1038/nature22041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pandey R., Heidmann S., Lehner C.F. Epithelial re-organization and dynamics of progression through mitosis in Drosophila separase complex mutants. J. Cell Sci. 2005;118:733–742. doi: 10.1242/jcs.01663. [DOI] [PubMed] [Google Scholar]
- 77.Qin X., Park B.O., Liu J., Chen B., Choesmel-Cadamuro V., Belguise K., Heo W.D., Wang X. Cell-matrix adhesion and cell-cell adhesion differentially control basal myosin oscillation and Drosophila egg chamber elongation. Nat. Commun. 2017;8 doi: 10.1038/ncomms14708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Izquierdo E., Quinkler T., De Renzis S. Guided morphogenesis through optogenetic activation of Rho signalling during early Drosophila embryogenesis. Nat. Commun. 2018;9:2366. doi: 10.1038/s41467-018-04754-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gupta T., Schüpbach T. Cct1, a phosphatidylcholine biosynthesis enzyme, is required for Drosophila oogenesis and ovarian morphogenesis. Development. 2003;130:6075–6087. doi: 10.1242/dev.00817. [DOI] [PubMed] [Google Scholar]
- 80.Wodarz A., Ramrath A., Grimm A., Knust E. Drosophila atypical protein kinase C associates with Bazooka and controls polarity of epithelia and neuroblasts. J. Cell Biol. 2000;150:1361–1374. doi: 10.1083/jcb.150.6.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lowe N., Rees J.S., Roote J., Ryder E., Armean I.M., Johnson G., Drummond E., Spriggs H., Drummond J., Magbanua J.P., et al. Analysis of the expression patterns, subcellular localisations and interaction partners of Drosophila proteins using a pigP protein trap library. Development. 2014;141:3994–4005. doi: 10.1242/dev.111054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Besson C., Bernard F., Corson F., Rouault H., Reynaud E., Keder A., Mazouni K., Schweisguth F. Planar cell polarity breaks the symmetry of PAR protein distribution prior to mitosis in Drosophila sensory organ precursor cells. Curr. Biol. 2015;25:1104–1110. doi: 10.1016/j.cub.2015.02.073. [DOI] [PubMed] [Google Scholar]
- 84.Xu T., Rubin G.M. Analysis of genetic mosaics in developing and adult Drosophila tissues. Development. 1993;117:1223–1237. doi: 10.1242/dev.117.4.1223. [DOI] [PubMed] [Google Scholar]
- 85.Smurnyy Y., Toms A.V., Hickson G.R., Eck M.J., Eggert U.S. Binucleine 2, an isoform-specific inhibitor of Drosophila Aurora B kinase, provides insights into the mechanism of cytokinesis. ACS Chem. Biol. 2010;5:1015–1020. doi: 10.1021/cb1001685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jia D., Xu Q., Xie Q., Mio W., Deng W.M. Automatic stage identification of Drosophila egg chamber based on DAPI images. Sci. Rep. 2016;6 doi: 10.1038/srep18850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Herbert S., Valon L., Mancini L., Dray N., Caldarelli P., Gros J., Esposito E., Shorte S.L., Bally-Cuif L., Aulner N., et al. LocalZProjector and DeProj: a toolbox for local 2D projection and accurate morphometrics of large 3D microscopy images. BMC Biol. 2021;19:136. doi: 10.1186/s12915-021-01037-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rodriguez J., Peglion F., Martin J., Hubatsch L., Reich J., Hirani N., et al. aPKC cycles between functionally distinct PAR protein assemblies to drive cell polarity. Dev. Cell. 2017;42:400–415.e409. doi: 10.1016/j.devcel.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Live imaging of egg chamber (midsagittal view) expressing GFP::aPKC (green), LARIAT, H2A::RFP (magenta; right panel), and stained with membrane marker (magenta; right panel). Imaging with 488 nm laser triggered GFP::aPKC clustering from time 0 onward. Epithelial gaps appear at the dorsal and ventral side and multilayering at the posterior side.
Time-lapse video shows that Sqh::mKate2x3 (magenta; right panel) increases at the apical membrane during the initial period of GFP::aPKC (green) clustering. Imaging with 488 nm laser triggered GFP::aPKC clustering from time 0 onward.
Live imaging of GFP::aPKC (green) follicular epithelial cells (surface view) with mosaic expression of LARIAT (marked by the presence of two copies of nlsRFP (bright magenta) and aPKC clustering). Imaging with 488 nm laser triggered GFP::aPKC clustering from time 0 onward. Right panel shows GFP::aPKC.
Egg chamber expressing GFP::aPKC (green), LARIAT and Sqh::mKate2x3 (magenta, right panel) and imaged in midsagittal view is shown on top, whereas an egg chamber expressing GFP::aPKC (green), LARIAT, stained with membrane marker (magenta, right panel) and imaged at the surface is shown at the bottom. Imaging with 488 nm laser triggered GFP::aPKC clustering from time 0 onward.
Live imaging of GFP::aPKC (green, middle panel) and MyoII(sqh)::mKate2 (magenta, right panel) in the follicular epithelium (surface view).
Live imaging of GFP::aPKC (green; right panel) and Sqh::mKate2 (magenta; middle panel) in the follicular epithelium (surface view) of control and LARIAT egg chambers. Imaging with 488 nm laser triggered GFP::aPKC clustering in the presence of LARIAT from time 0 onward.
Live imaging of a GFP::aPKC (green; right panel) and E-cad::mKate2 (magenta) in the follicular epithelium (surface view). Imaging with the 488 nm laser triggered GFP::aPKC clustering from time point 0 onwards. Unlike interphase cells (blue asterisks) constrict, mitotic cells (yellow asterisks) still expand after aPKC clustering.
Imaging with 488 nm laser triggered RhoGEF2::CRY2::mCherry (magenta; right panel) recruitment to apical PatJ::CIBN::pmGFP (green).
Data Availability Statement
-
•
All original microscopy data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.