

Abstract
Neonatal Marfan syndrome (nMFS) is a rare and severe form of Marfan syndrome (MFS) with a poor prognosis, that presents with a highly variable phenotype, particularly regarding skeletal, ocular, and cardiovascular manifestations. Mutations in the fibrillin-1 (FBN1) gene are known as the principal cause of MFS and MFS-related syndromes. Here, we report on a full-term female neonate with postnatal characteristics suggestive of nMFS, including severe cardiovascular disease resulting in cardiorespiratory failure and death by 4 mo of age. We identified a novel large genomic in-frame deletion of FBN1 exons 42–45, c.(5065 + 1_5066 − 1)_(5545 + 1_5546 − 1)del. Large FBN1 in-frame deletions between exons 24 and 53 have been associated with severe MFS. The deletion in our patient differs from the FBN1 region associated with the majority of nMFS cases, exons 24–32.
Keywords: aortic aneurysm, arachnodactyly, cardiomegaly, hyperextensible thumb, microretrognathia, pulmonary hypertension, pulmonary valve defects, tricuspid regurgitation
INTRODUCTION
Marfan syndrome (MFS; OMIM #154700), first described by Antoine Marfan in 1896, is a rare but severe and potentially life-threatening genetic disorder of fibrous connective tissue, with pronounced pleiotropism and a wide spectrum of clinical phenotypes. The cardinal symptoms of MFS occur in the skeletal, ocular, and cardiovascular systems, and include aortic root enlargement and lens luxation (Loeys et al. 2010). Neonatal Marfan syndrome (nMFS) is characterized by additional clinical features including facial dysmorphism (megalocornea, iridodonesis, ectopia lentis, crumpled ears, and loose skin with a senile facial appearance), joint flexion contractures, pulmonary emphysema, progressive cardiovascular disease with aortic dilatation, severe mitral or tricuspid valve insufficiency, and skeletal abnormalities including arachnodactyly, dolichostenomelia, and pectus deformities (Booms et al. 1999). Gastrointestinal symptoms are rare in nMFS. Inguinal hernia is frequently observed, and hiatal, paraoesophageal hernia and intrathoracic stomach have also been described and are thought to be related to anomalies of gastric ligaments and diaphragm (Herman et al. 2013; Serradilla et al. 2018; Veiga-Fernández et al. 2020).
Marfan syndrome is caused by mutations of the fibrillin-1 (FBN1) gene on Chromosome 15q21.1 (Dietz et al. 1991; Hayward and Brock 1997; Comeglio et al. 2007). Fibrillin is the principal component of the extracellular microfibrils, which are widely distributed in connective tissue matrices. Tiecke et al. analyzed exons 24–40 of the FBN1 gene in 127 patients with Marfan syndrome or related disorders, and—similarly to others (Putnam et al. 1996; Booms et al. 1999; Comeglio et al. 2007)—observed a significant clustering of mutations in exons 24–32. All mutations were associated with nMFS and most of the mutations had been related to atypical severe MFS located in this region. However, they found mutations associated with classical MFS to be localized to this region as well. These findings led the authors to conclude that the presence of a mutation in exons 24–32 is not predictive for a classic, atypically severe or neonatal MFS (Tiecke et al. 2001). Moreover, nMFS has also been associated with mutations outside this FBN1 region (Eayrs et al. 2013; Šípek et al. 2014; Nazarali et al. 2017).
The diagnosis of MFS is based on a set of clinical criteria known as the Ghent nosology (Loeys et al. 2010). In the latest revision of these criteria by an international expert panel, the cardiovascular manifestations were given more weight, and aortic root aneurysm and ectopia lentis were designated as principal clinical features (Loeys et al. 2010).
In this case report, we describe a neonate with a severe form of nMFS, who suffered fatal cardiac failure caused by mitral and tricuspid valve insufficiency and aortic intimal dissection. Genetic evaluation revealed a novel large FBN1 in-frame deletion.
RESULTS
A female neonate was born at 39 wk of gestation after an uneventful pregnancy and uncomplicated vaginal delivery. She was the first child of nonconsanguineous parents of Asian origin (mother was aged 34, father was aged 40). There was no family history of inherited disorders or congenital heart disease. The APGAR scores were 9/10/10, and immediate postnatal adaptation was uneventful. The birth biometrics were 3226 g (43th percentile), 52 cm for length (65th percentile), and 35 cm head circumference (78th percentile). In the first hours of life, hypothermia and a heart murmur were noted, and the baby was admitted to the neonatal intensive care unit. Empirical treatment with ampicillin/sulbactam was started for clinically suspected neonatal sepsis, but discontinued after 3 d as there was neither microbiological nor other laboratory evidence for sepsis. Arachnodactyly, thumb hyperextension and micro- and retrognathia were noted (Fig. 1). Ophthalmologic examination revealed bilateral lens subluxation. Inguinal and/or umbilical herniation were not present. Echocardiography showed signs of pulmonary hypertension, patent ductus arteriosus, atrial septal defect, atrioventricular valve prolapse, severe tricuspid, and mitral valve regurgitation as well as aortic ectasia. Chest computed tomography (CT) angiography confirmed a descending thoracic aortic aneurysm, aortic root dilatation with a maximum diameter of 19 mm (z-score = +4.8), and aortic intimal dissection (Fig. 2). Conservative treatment with lisinopril and bisoprolol was started. Until day 15 of life, respiratory assistance was necessary with a nasal flow cannula and intermittent short-term oxygen supplementation. On day 17, the baby was released from hospital in good clinical condition with spontaneous room-air breathing and exclusive breastfeeding. She was followed up at regular intervals by the pediatric cardiology team.
Figure 1.

Clinical features of neonate with neonatal Marfan syndrome (nMFS) at the age of 16 d, showing microretrognathia (A) and arachnodactyly of fingers and toes (B,C).
Figure 2.

Computed tomography (CT) scan of the patient at the age of 7 d. Aortic dilatation is shown in the frontal (A) and axial (B) plane, with a maximal diameter of 20 × 19 mm at the ventricle level.
By the age of 3 mo the baby developed tachypnea, cough, breathlessness, and sweating, particularly during breastfeeding. Because of progressive deterioration with congestive heart failure and hepatomegaly, diuretics were added to the treatment.
A multidisciplinary team concluded that cardiosurgical intervention could not be expected to improve prognosis. The severe nMFS phenotype and the likelihood of high postoperative morbidity were the main concerns. The parents sought a second opinion, but an independent cardiology/cardiosurgery team considered mitral and tricuspid valve repair a nonviable option because of the aortic intimal dissection. Following extensive consultation with the baby's family, the decision was made for the child to receive palliative home care.
At 4 mo of age, the baby was readmitted with signs of progressive cardiac and respiratory decompensation (Fig. 3), and the infant died soon thereafter while receiving symptomatic cardiorespiratory therapy.
Figure 3.
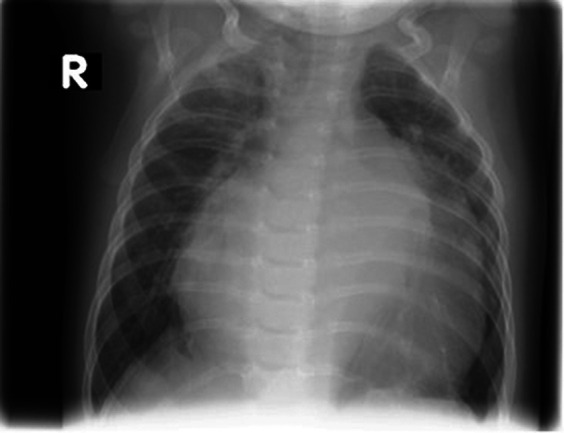
Chest X-ray at the age 3 mo, showing cardiomegaly at the time of worsening cardiac failure (R, right side).
In light of the dysmorphic features and the cardiovascular anomalies, comprehensive genetic studies were carried out on suspicion of nMFS. Next-generation sequencing (NGS) revealed no pathogenic sequence variants of the FBN1, SMAD3, TGFBR1, and TGFBR2 genes known to be associated with Marfan or Marfan-like syndrome. Subsequent multiplex ligation-dependent probe amplification (MLPA) analysis of FBN1 identified a previously undescribed pathogenic in-frame deletion of exons 42-45 (c.(5065 + 1_5066 − 1)_(5545 + 1_5546 − 1)del) in hemizygous state with position +1 corresponding to the A of the ATG start codon (in exon 2 of altogether 66 exons) of the mRNA reference sequence (Ensembl ENST00000316623.10, GenBank NM_000138.3) at the cDNA (c) level (Fig. 4A,B; Tab). Additional copy-number variation (CNV) analysis of the NGS data also detected the deletion of these exons, but additionally indicated a questionable deletion of the 5′-adjacent exon 41 as well as a deletion of the 3′-adjacent exon 46 (minimum sequencing coverage of 48-fold for exon 41, 46-fold for exon 42, 15-fold for exon 43, 61-fold for exon 44, 85-fold for exon 45, and 78-fold for exon 46) (Fig. 4C). Neither parent carried this deletion, indicating that the mutation had occurred de novo.
Figure 4.

Results of (A,B) semiquantitative multiplex ligation-dependent probe amplification (MLPA)- and (C) next-generation sequencing (NGS)-based analyses of the patient for copy-number variations (CNVs) in the FBN1 gene. Bars in the upper and lower histograms represent MLPA probes (A,B) and exons covered by NGS data (C), respectively. Upper histogram: blue bars, mean relative peak area (RPA; A,B) and mean relative product coverage (RPC; C) of the reference DNAs with standard deviations; green bars, RPA (A,B) and RPC (C) of patient DNA. Numbers below the bars in A and B: amplicon length (bp) of each MLPA probe. Lower histogram with the bar for each probe/exon indicating the ratio RPA (A,B) and ratio RPC (C), respectively (RPA/RPC of the patient divided by the RPA/RPC of the reference DNAs). Light blue bars: ratio RPAs (A,B) and RPCs (C) ranging within the limits from 75% to 125% (red lines). Dark blue bars: ratio RPAs (A,B) and RPCs (C) below the limit of 75% indicating a deletion; deletions were detected for FBN1 exons 42–45 by MLPA analysis (SALSA MLPA Kits P065-C1 in A and P066-C1 in B, respectively) and for exons 42–46 with an additional questionable deletion of exon 41 by NGS analysis (C).
Meanwhile, the mother has given birth to a male infant who is clinically healthy and has no anomalies on echocardiographic and ophthalmological examination.
DISCUSSION
Because of severe cardiac anomalies and secondary pulmonary manifestations, nMFS is associated with higher morbidity and mortality than classic MFS (Tekin et al. 2007). As nMFS patients suffer from severe mitral and/or tricuspid regurgitation and aortic root dilatation (Stuart and Williams 2007), the clinical course is characterized by rapidly congestive heart failure, and death often occurs within the first year of life (Booms et al. 1999).
Our patient was clinically diagnosed with nMFS soon after birth. Previously reported prenatal ultrasound findings associated with nMFS include cardiomegaly, dilatation of the aortic and pulmonary roots, diaphragmatic hernia or eventration, and overgrowth of the femoral or humeral length (Veiga-Fernández et al. 2020). None of these symptoms was described in our patient prenatally.
Early recognition of nMFS is critical because it allows for a timely assessment of the cardiac anomalies and prognosis. The treatment requires a multidisciplinary approach, and carefully planned specialist follow-up influences the prognosis. The pharmacological management of the cardiovascular abnormalities in nMFS is controversial. Evidence exists to indicate that β-blockers, calcium channel blockers, angiotensin-converting enzyme inhibitors, and angiotensin II receptor blockers may reduce the progression of aortic dilatation. However, whether this has a beneficial effect on mortality is unclear (Stuart and Williams 2007; Thakur et al. 2013). Early cardiosurgical intervention may be considered, and cases have been reported in whom corrective cardiac surgery resulted in improved survival (Amado et al. 2014; Kitahara et al. 2016; Carande et al. 2017; Tognato et al. 2019). However, the overall effectiveness of cardiac surgical intervention is undetermined and should be evaluated individually. Two independent multidisciplinary teams evaluated our patient for cardiac surgery. However, both considered cardiosurgical intervention inappropriate because of the extended aortic intimal dissection in this baby.
The Ghent nosology for the diagnosis of MFS initially consisted of a set of clinical criteria. With the advent of MLPA and NGS, however, mutation analysis of the FBN1 gene has become widely available, and the most recent revised diagnostic Ghent nosology criteria now include the identification of a pathogenic FBN1 variant (Loeys et al. 2010).
Dietz et al. were the first to identify FBN1 mutations in association with MFS (Dietz et al. 1991). To our knowledge, a deletion of FBN1 exons 42–45 (c.(5065 + 1_5066 − 1)_(5545 + 1_5546 − 1)del) has not been described in the MFS literature or in disease-specific variant databases: ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/), Human Gene Mutation Database (HGMD; http://www.hgmd.cf.ac.uk/ac/index.php), Leiden Open Variation Database (LOVD; https://www.lovd.nl/), and Genome Aggregation Database (gnomAD; https://gnomad.broadinstitute.org/). This large genomic in-frame deletion is predicted to lead to a functional impairment of the FBN1 protein, and to cause nMFS in our patient. Large genomic FBN1 deletions were recently described to result in variable MFS phenotypes (Li et al. 2017), and interestingly, in-frame deletions in the region spanning exons 24–53 are frequently associated with severe MFS. An international study by Faivre et al. investigated FBN1 genotype–phenotype correlations and designated the region spanning exons 24–32 as the “neonatal region” of FBN1, with mutations in this region being associated with a high risk for cardiac manifestations and severe prognosis through a dominant-negative effect (Faivre et al. 2007). A further recent study revisited FBN1 genotype–phenotype correlations and found in-frame mutations, but not haploinsufficiency-linked protein-truncating mutations in the neonatal region to be associated with severe clinical courses. Particularly missense mutations substituting a cystein for another amino acid had more adverse effects notably regarding cardiovascular manifestations that required surgical treatments more often and at much earlier age (Arnaud et al. 2021). It should be emphasized, however, that not only mutations in this so-called neonatal region are associated with severe phenotypes.
In our CNV analysis of the NGS data, we confirmed the deletion of FBN1 exons 42–45, but additionally detected a questionable deletion of the 5′-adjacent exon 41 as well as a deletion of the 3′-adjacent exon 46. In this context, it has to be mentioned that CNV calling from NGS data is less reliable and sometimes controversial compared to MLPA. Although the putative deletions of exons 41 and 46 very likely reflect the shortcomings of quantitative analyses of NGS data, we cannot exclude that the discrepancy between the NGS and MLPA data here results from extension of the deletion to the 3′ region of exon 41 and to the 5′ region of exon 46, with breakpoints before the central regions where the MLPA hybridization probes of these exons each bind. Consequently, we cannot completely rule out that the deletion leads to an out-of-frame instead of the predicted in-frame rearrangement of the FBN1 coding sequence.
It is further important to note that some MFS cases harboring biallelic homozygous or compound heterozygous FBN1 mutations have been reported with early age of onset and a very severe clinical course (Karttunen et al. 1994; de Vries et al. 2007; Van Dijk et al. 2009; Hilhorst-Hofstee et al. 2010; Hogue et al. 2013; Khan et al. 2014; Nayak et al. 2021). On the other hand, biallelic FBN1 mutations have been also described in families exhibiting classical or mild clinical MFS signs with later ages at diagnosis (Arnaud et al. 2017). Though, it is still possible that compound heterozygosity of the deletion detected in this study and a second FBN1 mutation not detectable with the methods used here (e.g., in deep intronic regions of the FBN1 gene) caused the severe neonatal phenotype of our patient.
Several reports have documented familial transmission of FBN1 mutations in association with nMFS (Tekin et al. 2007; Elshershari and Harris 2014; Šípek et al. 2014; Le Gloan et al. 2016). Two of these studies described families with low level of somatic mosaicism for the causal mutation in one of the patients’ parents who displayed no or only very incomplete (unilateral lens ectopy) clinical signs of MFS (Tekin et al. 2007; Šípek et al. 2014). The other two studies reported families with the causal mutation and no evidence for somatic mosaicism in one of the patients’ parents who exhibited the rather classical MFS phenotype (Elshershari and Harris 2014; Le Gloan et al. 2016). Appropriate genetic counseling of the parents of an infant with nMFS therefore requires both the identification of the causal mutation and a parental segregation analysis. Neither parent of our patient carried the FBN1 exons 42–45, c.(5065 + 1_5066 − 1)_(5545 + 1_5546 − 1) deletion, indicating a de novo germline mutation. However, a 3%–5% residual recurrence risk remains in case of negative parental testing because of the possibility of parental germline mosaicism (Sutherell et al. 2007; Tekin et al. 2007). Following genetic counseling, the parents of our patient decided to continue with active family planning and recently the mother gave birth to a healthy boy.
METHODS
Genomic DNA was extracted from peripheral blood lymphocytes of the affected girl and her parents. The patient´s sample was subjected to targeted NGS of the FBN1, TGFBR1, TGFBR2, and SMAD3 genes as described previously (Preising et al. 2019) with library preparation using the KAPA HyperPlus kit (Kapa Biosystems), enrichment using the IDT xGen inherited Diseases Panel v1.0 (IDT Integrated Technologies) paired-end sequencing on an Illumina NextSeq500 system (Illumina) and data analysis using the SeqNext module of the SeqPilot software (JSI Medical Systems).
MLPA analyses of the samples of the patient and her parents were performed using the SALSA MLPA Kits P065-C1 and P066-C1 (MRC Holland), capillary electrophoresis with an Applied Biosystems 3130xl Genetic Analyzer (Applied Biosystems, and data analysis using the MLPA module of the SeqPilot software (JSI Medical Systems).
ADDITIONAL INFORMATION
Data Deposition and Access
The authors confirm that all data supporting the conclusions of this study are available within the manuscript. The interpreted variant has been deposited in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and can be found under accession number SCV002577329 (Table 1).
Table 1.
Variant table
| Gene | Chromosome | HGVS DNA reference | HGVS protein reference | Variant type | Predicted effect | dbSNP/dbVar ID | Genotype | ClinVar ID | Parent of origin | Observed effect | Comments |
|---|---|---|---|---|---|---|---|---|---|---|---|
| FBN1 | 15 | c.(5065 + 1_5066 − 1)_(5545 + 1_5546 − 1)del | p.? | Deletion | In-frame deletion | N/A | Heterozygous | (Most probably) de novo | N/A | N/A | N/A |
Ethics Statement
The authors obtained permission from the patient's parents to publish the data within this report, as well as the clinical pictures. University Hospital Frankfurt/Main does not require IRB approval for case reports.
Acknowledgments
The authors are grateful to the patient's parents, who allowed reporting this case study, including their daughter's pictures. We thank all medical, nursing, and supportive staff at the Department of Neonatology, Clinic for Children and Adolescents, University Hospital Frankfurt/Main, Germany, for their competent patient care, as well as all colleagues involved in patient care. Medical writing support, in the form of editorial services, was provided by An Billiau, M.D. Ph.D., Celsus Medical Writing.
Funding
The research of H.J.B. is supported by funding from the Dr. Senckenbergische Stiftung, Frankfurt, Germany.
Competing Interest Statement
The authors have declared no competing interest.
Referees
Peter N Robinson
David Liang
REFERENCES
- Amado M, Calado MA, Ferreira R, Lourenco T. 2014. Neonatal Marfan syndrome: a successful early multidisciplinary approach. BMJ Case Rep 2014: bcr2013202438. 10.1136/bcr-2013-202438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaud P, Hanna N, Aubart M, Leheup B, Dupuis-Girod S, Naudion S, Lacombe D, Milleron O, Odent S, Faivre L, et al. 2017. Homozygous and compound heterozygous mutations in the FBN1 gene: unexpected findings in molecular diagnosis of Marfan syndrome. J Med Genet 54: 100–103. doi: 10.1136/jmedgenet-2016-103996 [DOI] [PubMed] [Google Scholar]
- Arnaud P, Milleron O, Hanna N, Ropers J, Ould Ouali N, Affoune A, Langeois M, Eliahou L, Arnoult F, Renard P, et al. 2021. Clinical relevance of genotype–phenotype correlations beyond vascular events in a cohort study of 1500 Marfan syndrome patients with FBN1 pathogenic variants. Genet Med 23: 1296–1304. 10.1038/s41436-021-01132-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booms P, Cisler J, Mathews KR, Godfrey M, Tiecke F, Kaufmann UC, Vetter U, Hagemeier C, Robinson PN. 1999. Novel exon skipping mutation in the fibrillin-1 gene: two ‘hot spots’ for the neonatal Marfan syndrome. Clin Genet 55: 110–117. 10.1034/j.1399-0004.1999.550207.x [DOI] [PubMed] [Google Scholar]
- Carande EJ, Bilton SJ, Adwani S. 2017. A case of neonatal Marfan syndrome: a management conundrum and the role of a multidisciplinary team. Case Rep Pediatr 2017: 8952428. 10.1155/2017/8952428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comeglio P, Johnson P, Arno G, Brice G, Evans A, Aragon-Martin J, da Silva FP, Kiotsekoglou A, Child A. 2007. The importance of mutation detection in Marfan syndrome and Marfan-related disorders: report of 193 FBN1 mutations. Hum Mutat 28: 928. 10.1002/humu.9505 [DOI] [PubMed] [Google Scholar]
- de Vries BB, Pals G, Odink R, Hamel BC. 2007. Homozygosity for a FBN1 missense mutation: clinical and molecular evidence for recessive Marfan syndrome. Eur J Hum Genet 15: 930–935. 10.1038/sj.ejhg.5201865 [DOI] [PubMed] [Google Scholar]
- Dietz HC, Cutting GR, Pyeritz RE, Maslen CL, Sakai LY, Corson GM, Puffenberger EG, Hamosh A, Nanthakumar EJ, Curristin SM, et al. 1991. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature 352: 337–339. 10.1038/352337a0 [DOI] [PubMed] [Google Scholar]
- Eayrs K, Shettihalli N, Adwani S. 2013. Down syndrome masked by Marfan syndrome in a neonate. BMJ Case Rep 2013: bcr2013008807. 10.1136/bcr-2013-008807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elshershari H, Harris C. 2014. Paternal fibrillin-1 mutation transmitted to an affected son with neonatal Marfan syndrome: the importance of early recognition. Cardiol Young 24: 735–738. 10.1017/S1047951113001029 [DOI] [PubMed] [Google Scholar]
- Faivre L, Collod-Beroud G, Loeys BL, Child A, Binquet C, Gautier E, Callewaert B, Arbustini E, Mayer K, Arslan-Kirchner M, et al. 2007. Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am J Hum Genet 81: 454–466. 10.1086/520125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward C, Brock DJ. 1997. Fibrillin-1 mutations in Marfan syndrome and other type-1 fibrillinopathies. Hum Mutat 10: 415–423. [DOI] [PubMed] [Google Scholar]
- Herman TE, Siegel MJ, Mathur A, Vachharajani A. 2013. Neonatal Marfan syndrome with hiatus hernia and intrathoracic stomach. J Perinatol 33: 652–653. 10.1038/jp.2013.15 [DOI] [PubMed] [Google Scholar]
- Hilhorst-Hofstee Y, Rijlaarsdam MEB, Scholte AJHA, Swart-van de Berg M, Versteegh MIM, van der Schoot-van Velzen I, Schäbitz HJ, Bijlsma EK, Baars MJ, Kerstjens-Frederikse WS, et al. 2010. The clinical spectrum of missense mutations of the first aspartic acid of cbEGF-like domains in fibrillin-1 including a recessive family. Hum Mutat 31: E1915–E1927. 10.1002/humu.21372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogue J, Lee C, Jelin A, Strecker MN, Cox VA, Slavotinek AM. 2013. Homozygosity for a FBN1 missense mutation causes a severe Marfan syndrome phenotype. Clin Genet 84: 392–393. doi: 10.1111/cge.12073 [DOI] [PubMed] [Google Scholar]
- Karttunen L, Raghunath M, Lönnqvist L, Peltonen L. 1994. A compound-heterozygous Marfan patient: two defective fibrillin alleles result in a lethal phenotype. Am J Hum Genet 55: 1083–1091. [PMC free article] [PubMed] [Google Scholar]
- Khan AO, Bolz HJ, Bergmann C. 2014. Results of fibrillin-1 gene analysis in children from inbred families with lens subluxation. J Aapos 18: 134–139. 10.1016/j.jaapos.2013.11.012 [DOI] [PubMed] [Google Scholar]
- Kitahara H, Aeba R, Takaki H, Shimizu H. 2016. Palliative mitral valve repair during infancy for neonatal Marfan syndrome. Ann Thorac Surg 2016: 1987–1988. 10.1016/j.athoracsur.2015.06.115 [DOI] [PubMed] [Google Scholar]
- Le Gloan L, Hauet Q, David A, Hanna N, Arfeuille C, Arnaud P, Boileau C, Romefort B, Benbrik N, Gournay V, et al. 2016. Neonatal Marfan syndrome: report of a case with an inherited splicing mutation outside the neonatal domain. Mol Syndromol 6: 281–286. 10.1159/000443867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Wu W, Lu C, Liu Y, Wang R, Si N, Liu F, Zhou J, Zhang S, Zhang X. 2017. Gross deletions in FBN1 results in variable phenotypes of Marfan syndrome. Clin Chim Acta 474: 54–59. 10.1016/j.cca.2017.08.023 [DOI] [PubMed] [Google Scholar]
- Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, Devereux RB, Hilhorst-Hofstee Y, Jondeau G, Faivre L, Milewicz DM, et al. 2010. The revised Ghent nosology for the Marfan syndrome. J Med Genet 47: 476–485. 10.1136/jmg.2009.072785 [DOI] [PubMed] [Google Scholar]
- Nayak SS, Schneeberger PE, Patil SJ, Arun KM, Suresh PV, Kiran VS, Siddaiah S, Maiya S, Venkatachalagupta SK, Kausthubham N, et al. 2021. Clinically relevant variants in a large cohort of Indian patients with Marfan syndrome and related disorders identified by next-generation sequencing. Sci Rep 11: 764. 10.1038/s41598-020-80755-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazarali S, Nazarali SA, Antoniuk A, Greve M, Damji KF. 2017. Childhood glaucoma in neonatal Marfan syndrome resulting from a novel FBN1 deletion. Can J Ophthalmol 52: e171–e173. 10.1016/j.jcjo.2017.03.008 [DOI] [PubMed] [Google Scholar]
- Preising MN, Görg B, Friedburg C, Qvartskhava N, Budde BS, Bonus M, Toliat MR, Pfleger C, Altmüller J, Herebian D, et al. 2019. Biallelic mutation of human SLC6A6 encoding the taurine transporter TAUT is linked to early retinal degeneration. FASEB J 33: 11507–11527. 10.1096/fj.201900914RR [DOI] [PubMed] [Google Scholar]
- Putnam EA, Cho M, Zinn AB, Towbin JA, Byers PH, Milewicz DM. 1996. Delineation of the Marfan phenotype associated with mutations in exons 23–32 of the FBN1 gene. Am J Med Genet 62: 233–242. [DOI] [PubMed] [Google Scholar]
- Serradilla J, Bueno A, De La Torre C, Gamarra EA, Romo MM, Nava Hurtado de Saracho FB, Barrial MA, Cervantes MG, Santamaria ML. 2018. Neonatal intrathoracic gastric volvulus in Marfan's syndrome. European J Pediatr Surg Rep 6: e48–e51. 10.1055/s-0038-1666795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šípek A, Grodecká L, Baxová A, Cibulková P, Dvořáková M, Mazurová S, Magner M, Zeman J, Honzik T, Freiberger T. 2014. Novel FBN1 gene mutation and maternal germinal mosaicism as the cause of neonatal form of Marfan syndrome. Am J Med Genet A 164A: 1559–1564. 10.1002/ajmg.a.36480 [DOI] [PubMed] [Google Scholar]
- Stuart AG, Williams A. 2007. Marfan's syndrome and the heart. Arch Dis Child 92: 351–356. 10.1136/adc.2006.097469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherell J, Zarate Y, Tinkle BT, Markham LW, Cripe LH, Hyland JC, Witte D, Hopkin RJ, Hinton RB. 2007. Novel fibrillin 1 mutation in a case of neonatal Marfan syndrome: the increasing importance of early recognition. Congenit Heart Dis 2: 342–346. 10.1111/j.1747-0803.2007.00123.x [DOI] [PubMed] [Google Scholar]
- Tekin M, Cengiz FB, Ayberkin E, Kendirli T, Fitoz S, Tutar E, Ciftci E, Conba A. 2007. Familial neonatal Marfan syndrome due to parental mosaicism of a missense mutation in the FBN1 gene. Am J Med Genet A 143A: 875–880. 10.1002/ajmg.a.31660 [DOI] [PubMed] [Google Scholar]
- Thakur V, Rankin KN, Hartling L, Mackie AS. 2013. A systematic review of the pharmacological management of aortic root dilation in Marfan syndrome. Cardiol Young 23: 568–581. 10.1017/S1047951112001412 [DOI] [PubMed] [Google Scholar]
- Tiecke F, Katzke S, Booms P, Robinson PN, Neumann L, Godfrey M, Mathews KR, Scheuner M, Hinkel GK, Brenner RE, et al. 2001. Classic, atypically severe and neonatal Marfan syndrome: twelve mutations and genotype–phenotype correlations in FBN1 exons 24-40. Eur J Hum Genet 9: 13–21. 10.1038/sj.ejhg.5200582 [DOI] [PubMed] [Google Scholar]
- Tognato E, Perona A, Aronica A, Bertola A, Cimminelli L, De Vecchi S, Eshraghy MR, Loperfido B, Vivenza C, Manzoni P. 2019. Neonatal Marfan syndrome. Am J Perinatol 36: S74–S76. 10.1055/s-0039-1691770 [DOI] [PubMed] [Google Scholar]
- Van Dijk FS, Hamel BC, Hilhorst-Hofstee Y, Mulder BJM, Timmermans J, Pals G, Cobben JM. 2009. Compound-heterozygous Marfan syndrome. Eur J Med Genet 52: 1–5. 10.1016/j.ejmg.2008.11.004 [DOI] [PubMed] [Google Scholar]
- Veiga-Fernández A, Joigneau Prieto L, Álvarez T, Ruiz Y, Pérez R, Gámez F, Ortega Abad V, Yllana F, De León-Luis J. 2020. Perinatal diagnosis and management of early-onset Marfan syndrome: case report and systematic review. J Matern Fetal Neonatal Med 33: 2493–2504. 10.1080/14767058.2018.1552935 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that all data supporting the conclusions of this study are available within the manuscript. The interpreted variant has been deposited in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and can be found under accession number SCV002577329 (Table 1).
Table 1.
Variant table
| Gene | Chromosome | HGVS DNA reference | HGVS protein reference | Variant type | Predicted effect | dbSNP/dbVar ID | Genotype | ClinVar ID | Parent of origin | Observed effect | Comments |
|---|---|---|---|---|---|---|---|---|---|---|---|
| FBN1 | 15 | c.(5065 + 1_5066 − 1)_(5545 + 1_5546 − 1)del | p.? | Deletion | In-frame deletion | N/A | Heterozygous | (Most probably) de novo | N/A | N/A | N/A |