

Abstract
Aziridines—three-membered nitrogen-containing cyclic molecules—are important synthetic targets. Their substantial ring strain and resultant proclivity towards ring-opening reactions makes them versatile precursors of diverse amine products1–3, and, in some cases, the aziridine functional group itself imbues important biological (for example, anti-tumour) activity4–6. Transformation of ubiquitous alkenes into aziridines is an attractive synthetic strategy, but is typically accomplished using electrophilic nitrogen sources rather than widely available amine nucleophiles. Here we show that unactivated alkenes can be electrochemically transformed into a metastable, dicationic intermediate that undergoes aziridination with primary amines under basic conditions. This new approach expands the scope of readily accessible N-alkyl aziridine products relative to those obtained through existing state-of-the-art methods. A key strategic advantage of this approach is that oxidative alkene activation is decoupled from the aziridination step, enabling a wide range of commercially available but oxidatively sensitive7 amines to act as coupling partners for this strain-inducing transformation. More broadly, our work lays the foundations for a diverse array of difunctionalization reactions using this dication pool approach.
Despite tremendous progress in aziridine synthesis over the past decade, diverse N-substituted aziridine products remain difficult to access efficiently. A deceptively straightforward strategy for obtaining N-alkyl aziridine products would be the coupling of an alkene and a primary amine, liberating H2 as a by-product (Fig. 1a). Unfortunately, this strain-inducing oxidative reaction is thermodynamically uphill (with a ΔG0 value greater than 30 kcal mol−1), and thus this idealized transformation is not feasible. In theory, the aziridine formation could be coupled to the quenching of a chemical oxidant rather than formation of H2. However, oxidants that are strong enough to render this aziridination favourable are incompatible with most amine coupling partners as well as many functional groups7. Instead, established alkene aziridination methodologies use a high-energy electrophilic nitrogen reagent (for example, iminoiodinane) or nitrene precursor (such as organoazide) that serves both as the stoichiometric oxidant and as the nitrogen source1,8–10. These reagents typically bear electron-withdrawing N-substituents to strike the requisite balance of reactivity and reagent stability (Fig. 1b). As a consequence, there is a paucity of general strategies to transform alkenes into aziridines bearing N-alkyl substituents, despite the utility of these aziridines in constructing diverse aliphatic amine products and their presence in biologically active molecules. A recently reported intramolecular aziridination methodology11 provides access to a broader range of N-alkyl-substituted aziridine products via copper hydride chemistry; however, multistep synthesis to access the appropriately activated allylic amine substrate is required. Pioneering advances have introduced electrophilic reagents to furnish N–H aziridines12–15, but analogous reagents to access diverse N-alkyl aziridines through intermolecular reaction with alkenes have been slow to follow. Indeed, despite substantial effort, there is only a handful of electrophilic nitrogen reagents that are capable of delivering N-alkyl aziridines or even the corresponding vicinal aminohalide precursors from simple alkenes (fewer than ten unique N-substituents). Furthermore, each new reagent requires multistep de novo synthesis16,17, and there are only isolated examples of in situ generation of electrophilic nitrogen reagents to form N-alkyl aziridine products18.
Fig. 1 |. Development of an oxidative coupling strategy for aziridine synthesis.
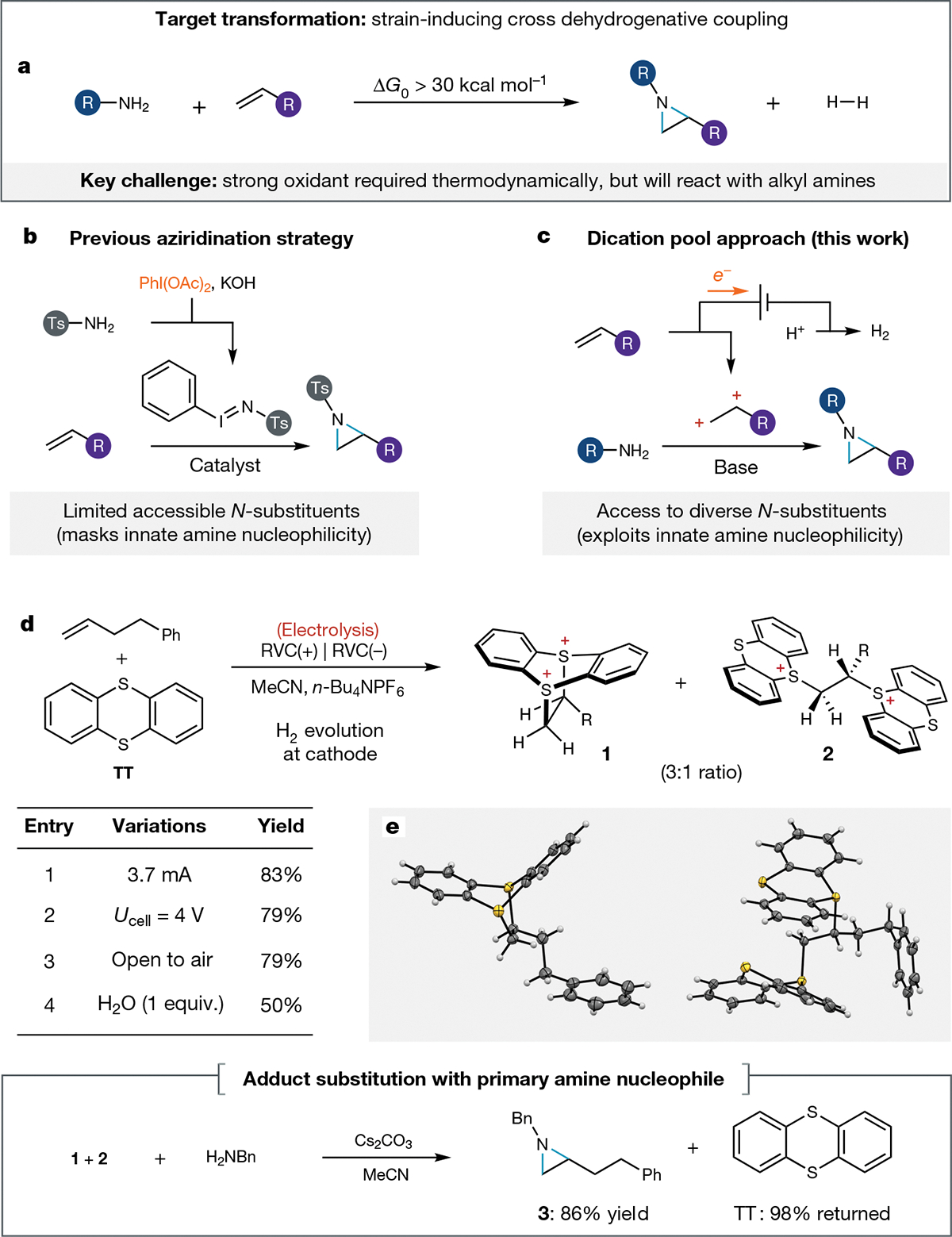
a, Thermodynamic challenges associated with ideal aziridine synthesis. b, Representative example of previous strategies for alkene aziridination rely on electrophilic nitrogen reagents that limit the accessible N-substituents. c, Our proposed electrochemical aziridine synthesis, involving the coupling of structurally diverse amines and alkenes through a strategic dicationic intermediate. d, Development of our electrochemical reaction. Bottom, aziridination with a model primary amine. e, Adducts characterized by X-ray crystallography. All yields were determined by 1H-NMR. Ph, phenyl; R, (CH2)2Ph; RVC, reticulated vitreous carbon; Ts, toluenesulfonyl.
In principle, transformation of an alkene into a dielectrophile—reversing the conventional strategy of pre-oxidizing the amine—could leverage intrinsic amine nucleophilicity, rather than masking it. Given that there are more than one million commercially available primary amines (commercial availability of primary amines: 1,054,553)19, this complementary approach would dramatically expand the scope of readily accessible N-alkyl aziridines. Dibromination is a well established reaction for transforming alkenes into dielectrophiles; however, the vast majority of vicinal dihalides do not undergo substitution with primary amines, and undesired elimination reactions are observed instead (see Supplementary Table 1). As a result, initial alkene oxygenation (epoxidation or dihydroxylation) followed by iterative stoichiometric activation and substitution steps remains the most common synthetic workaround to accomplish the net coupling of alkenes and primary amines to form N-alkyl aziridines20–22.
We envisaged that electrochemistry could enable the clean, efficient and scalable transformation of alkenes into a metastable dielectrophile of sufficient reactivity to be leveraged for aziridine synthesis (Fig. 1c). Pioneering work in electroorganic synthesis has illustrated that electrochemistry can replace chemical redox agents with safer and more environmentally benign conditions and, in some cases, enable reactivity that would be impractical to otherwise access23–27. Unfortunately, despite recent progress in other electrochemical alkene oxidation reactions28,29, electrochemical aziridinations still depend on direct oxidation of the coupling partners and, consequently, remain limited to electron-rich styrene derivatives, specific nucleophiles, or both30–33.
To address the implicit limitations of prior approaches, we drew inspiration from the stabilized cation pool strategy, wherein a potent cationic electrophile is anodically generated from a hydrocarbon and subsequently treated with an oxidatively sensitive nucleophile in a one-pot process34,35. Unfortunately, extension of this strategy to alkene functionalization has proven difficult. Therefore, instead of direct oxidation of the alkene substrate, we decided to target the anodic activation of a safe and inexpensive reagent in situ that would transform an alkene into a potent dielectrophile. To this end, we were attracted to the dicationic adducts first prepared through the reaction of thianthrene radical cations (TT+•) with alkenes36,37. Although we suspected that these dicationic adducts might be potent dielectrophiles, little is known of their reactivity with organic nucleophiles38. In recent years, the immense synthetic value of C(sp2) thianthrenium salts, generated from C(sp2)–H bonds, has been illustrated in a wide range of cross-coupling reactions39–41. However, electrochemical methods to access thianthrenium salts from hydrocarbons are underdeveloped42, and examples that generate C(sp3) products through hydrocarbon thianthrenation are rare43. We hypothesized that dicationic thianthrenenium alkene adducts could be cleanly prepared electrochemically and would undergo efficient reaction with amines to furnish N-alkyl aziridines. Given the expansive pool of commercially available primary amines—as well as the fact that thianthrene (TT) is both safe44 and inexpensive (costing less than 0.15 USD mmol−1)—this approach would dramatically expand the scope of readily accessible N-alkyl aziridine products. Herein we confirm that this new strategy allows us to take a large step towards an ideal N-alkyl aziridine synthesis, by coupling unactivated terminal alkenes and aliphatic primary amines using electricity to circumvent the need for a conventional chemical oxidant.
First, we probed the viability of electrochemical generation of the key electrophilic adduct between an alkene and thianthrene. Both constant current and constant cell potential electrolysis of a solution of 4-phenyl-1-butene, thianthrene and electrolyte delivered a 3:1 mixture of dicationic adducts 1 and 2 in excellent combined yield (Fig. 1d, entries 1 and 2). Despite the presumably high reactivity of these species, we characterized both adducts 1 and 2 unambiguously through X-ray crystallography. We found that the electrochemical generation of these dicationic adducts is robust; the yield of adducts 1 and 2 is insensitive to exposure to air (entry 3) and even tolerated intentional addition of exogenous water (entry 4). Having established that electrochemistry can promote the oxidative coupling of thianthrene and an unactivated terminal alkene, we next evaluated the substitution chemistry of this presumably reactive intermediate. To our delight, exposure of the mixture of adducts 1 and 2 to benzylamine and a heterogeneous base fully consumed both adducts and delivered the desired aziridine (3) in excellent yield (86%). As expected, thianthrene was returned after substitution (98% returned). The aziridination of 1 and 2 could proceed either through direct iterative nucleophilic substitution or via elimination to form a transient vinyl thianthrenium salt that undergoes subsequent substitution45. The overall transformation uses trifluoroacetic acid (TFA) as the formal oxidant, producing H2 and trifluoroacetate as stoichiometric by-products. We translated these preliminary data into a simple one-pot, two-step procedure that could furnish synthetically useful yields of N-alkyl aziridine products using either limiting amine (68%) or limiting alkene (80%), with a modest excess (three to four equivalents) of the other coupling partner (see Supplementary Fig. 1). This reaction requires no expensive specialized electrochemical equipment; it can be conducted using a commercially available DC power supply (costing less than 100 USD) paired with cheap carbon electrodes in a simple glass divided cell.
We next probed the scope of the amines amenable to this process using 1-octene as a representative unactivated alkene. These experiments show aziridine products to be accessible from a wide range of amine nucleophiles, dramatically expanding the limited range of viable aziridine N-substituents accessible from alkenes using conventional electrophilic nitrogen reagents (Fig. 2). Primary amines bearing diverse steric profiles (4–8) were well tolerated. Amines containing an alkene (7) and an alkyne (8) incorporated handles for further functionalization that would be difficult to install using conventional electrophilic nitrogen approaches. Diverse heterocycles are broadly represented in biologically active molecules, yet many of these species are oxidatively sensitive and/or poison the transition-metal catalysts required for conventional aziridination reactions. In stark contrast, using the metal-free dication pool strategy outlined herein, aziridines bearing a range of both heteroaromatic and saturated heterocyclic groups could be efficiently accessed from inexpensive commercially available amines (9–18). We anticipate that efficient access to these versatile heterocyclic building blocks will be of particularly high value in medicinal chemistry efforts. We were delighted to find that primary amines bearing potential competing nucleophiles, such as tryptamine, primaquine and ethanolamine, were each selectively transformed into aziridine products (17–19). Of note, each of the primary amines evaluated here was commercially available and inexpensive, illustrating the substantial benefit of this aziridination approach relative to multistep synthesis of new electrophilic reagents for each distinct N-substituent.
Fig. 2 |. Scope of the aziridination reaction.
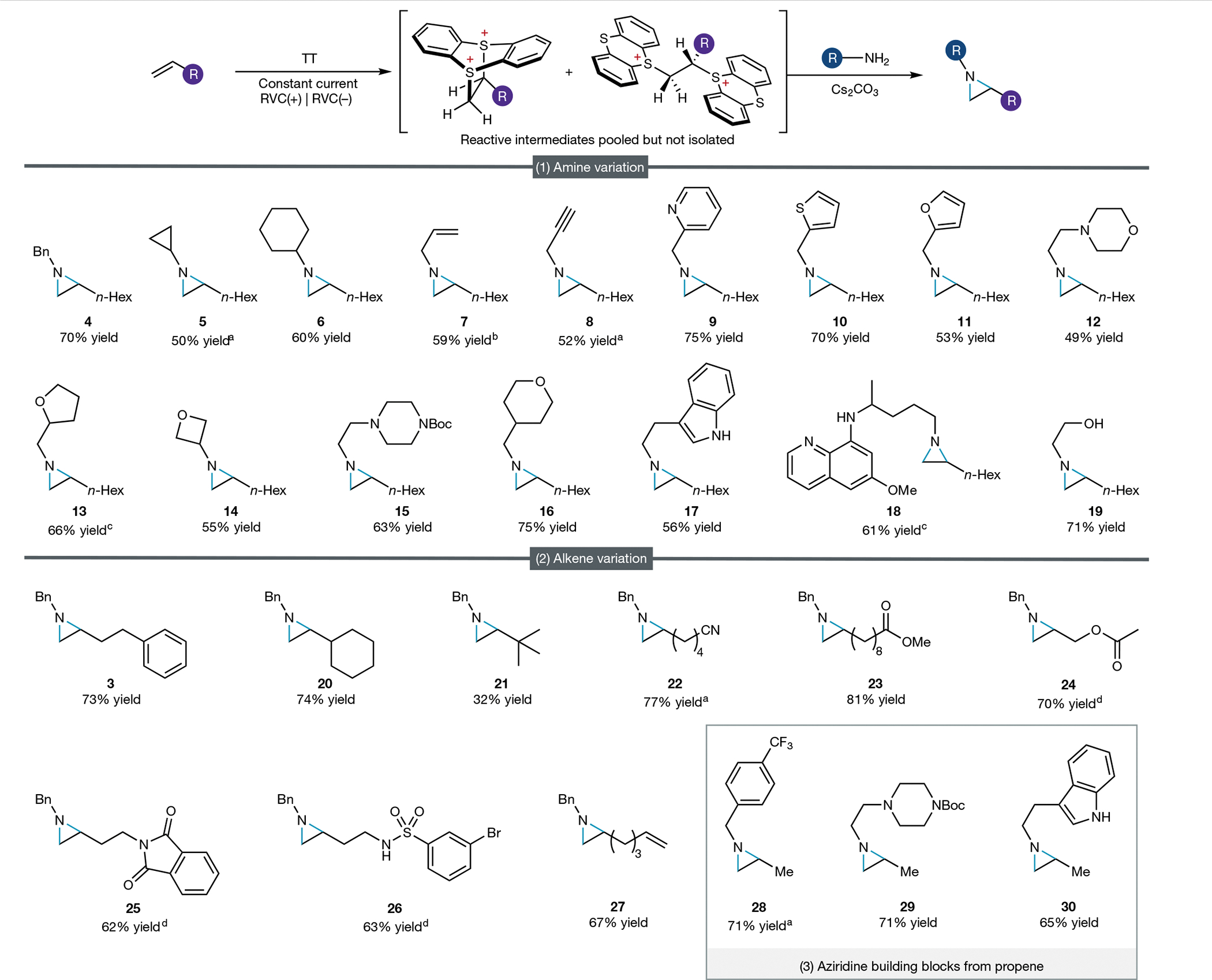
All yields, unless otherwise noted, are for isolated aziridine products. (1) Limiting amine conditions: 1-octene (1.2 mmol), thianthrene (1.8 mmol), 4 ml MeCN (0.2 M n-Bu4NPF6), I = 11.1 mA, 7 h (2.5 F mol−1 alkene); then amine (0.4 mmol) Cs2CO3 (3.2 mmol), 16 h. (2) Limiting alkene conditions: alkene (0.4 mmol), thianthrene (0.6 mmol), trifluoroacetic acid (0.8 mmol), 8 ml MeCN (0.2 M n-Bu4NPF6), I = 3.7 mA, 7 h (2.5 F mol−1 alkene); then benzylamine (1.6 mmol), Cs2CO3 (2.4 mmol), 16 h. (3) Propene conditions: thianthrene (1.8 mmol), 4 ml MeCN (0.2 M n-Bu4NPF6) under propene (1 atm), I = 11.1 mA, 7 h (2.5 F mol−1 alkene); then amine (0.4 mmol), Cs2CO3 (3.2 mmol), 16 h. aYield determined by NMR spectroscopy. bYield obtained using limiting alkene conditions (2) instead of limiting amine conditions (1). cIsolated in 1:1 diastereomeric ratio. dI = 12.0 mA, 2 h. See Supplementary Information for further experimental details.
Next, we turned our attention to the scope of alkene substrates. Under limiting alkene conditions, we found that modest steric hindrance about the alkene had a minimal impact on aziridine yield (19, 20), and even an alkene bearing a hindered allylic quaternary carbon was transformed into the corresponding aziridine, albeit in diminished yield (21). However, disubstituted alkenes were not compatible with this aziridination methodology. Although 1,2-disubstituted thianthrenium adducts could be formed, attempted substitution delivered either vinyl thianthrenium salts or intractable mixtures (Supplementary Fig. 2). Our initial efforts have focused on simple unactivated aliphatic alkenes, but alkene substrates bearing functional groups such as esters, nitriles, sulfonamides, aryl halides and phthalimides in both distal and proximal positions each furnished the desired aziridine products (23–26). Notably, an unconjugated diene underwent selective monofunctionalization to provide a useful building block that contains two orthogonal sites for further functionalization (27). Finally, this oxidative coupling strategy is also amenable to functionalization of gaseous feedstock alkenes using primary amine nucleophiles. Electrolysis of thianthrene under one atmosphere of propene, followed by addition of base and amine, furnished a range of synthetically attractive but previously unknown aziridine building blocks from exceptionally inexpensive starting materials (28–30).
We envisaged that this newly realized oxidative aziridination methodology would streamline the synthesis of important complex amines—a class of compounds broadly represented in medicinally relevant molecules. Consistent with this proposal, we were able to prepare aziridine 31—an intermediate in the synthesis of a collection of patented agonists of the dopamine D2 and 5-hydroxytryptamine (5-HT) 1B and 2A receptors (Pfizer; central nervous system indications)—from a commercially available primary amine and propene gas in high yield (Fig. 3a). By contrast, the previous route hinged on a low-yielding (17%) aziridine N-alkylation reaction46. Beyond offering a substantial improvement in synthetic efficiency, the new oxidative coupling route enables rapid synthesis of analogues (32, 33) because the alkene partner can be varied. Supporting its practical synthetic utility, we found this electrochemical transformation to be readily amenable to scaling up (Fig. 3b). We used a commercially available flow reactor to prepare 3.2 g (13.4 mmol) of aziridine 3, and recovery of thianthrene was straightforward (76% recovered). The cost of thianthrene on even this scale was trivial (3.60 USD), but we envisage that reisolation could be attractive for large-scale industrial processes.
Fig. 3 |. Synthetic applications of the dication pool strategy.
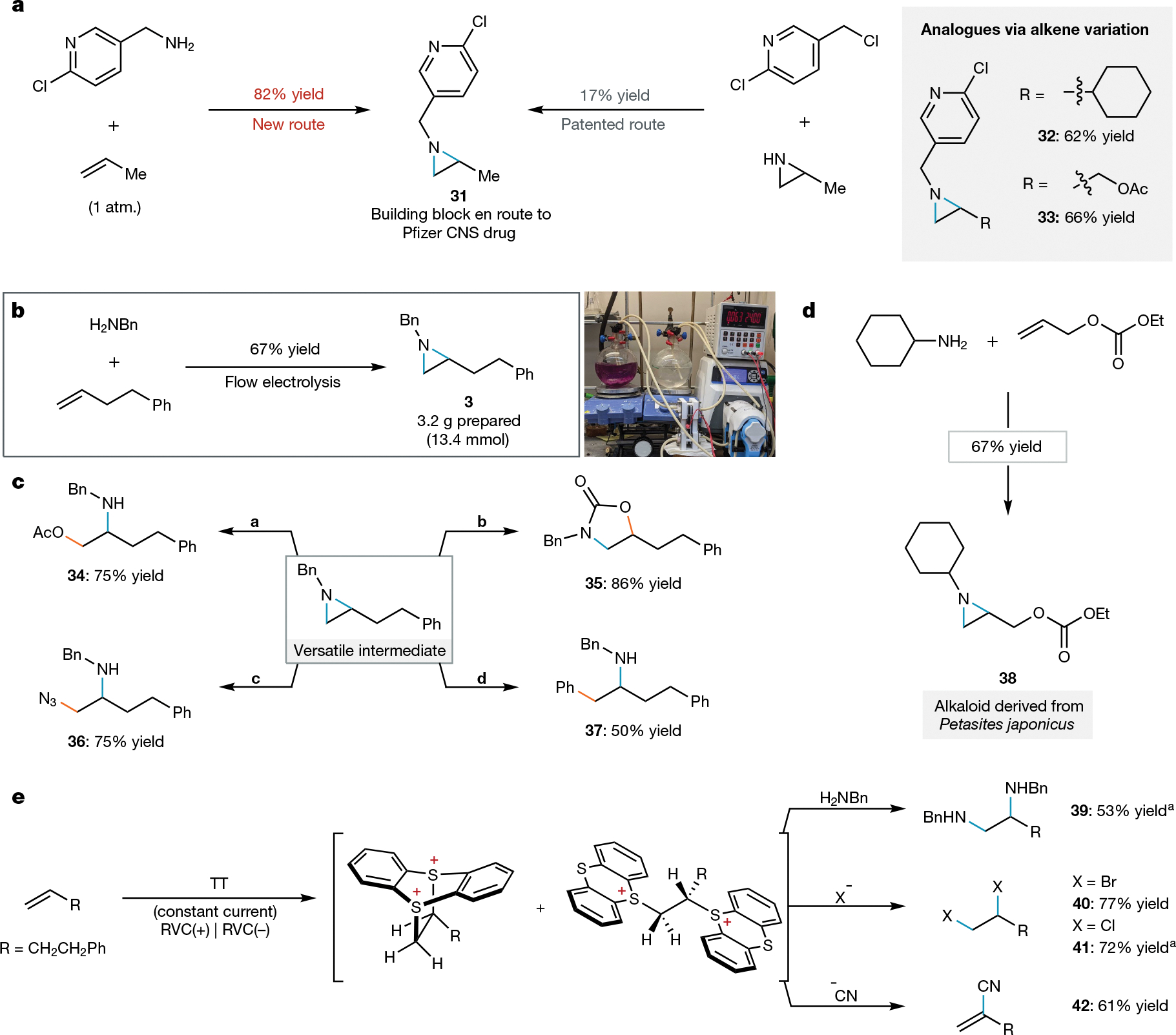
All yields are isolated products, and electrochemical alkene activation was conducted using standard conditions (2) from Fig. 2 unless otherwise noted. See Supplementary Information for experimental details. a, Comparison of our new dicationic adduct aziridination approach with the patented N–H aziridine alkylation route in the synthesis of a patented pharmaceutical intermediate and new analogues. CNS, central nervous system. b, Multigram aziridination via electrochemical formation of dicationic adducts using constant current flow electrolysis and subsequent substitution. c, Synthetic versatility of N-alkylaziridine products. Diversification conditions: a, acetic acid (AcOH), dichlormethane (DCM), 72 h; b, LiI, CO2 (1 atm.), tetrahydrofuran (THF), 44 h; c, trimethylsilyl azide (TMSN3), AcOH, DCM, 48 h; d, lithium diphenyl cuprate (Ph2CuLi), boron trifluoride etherate (BF3OEt2), THF, 4 h. d, Synthesis of an aziridine-containing natural product. e, Preliminary expansion of the dication pool strategy beyond aziridination. aYield determined by 1H-NMR. See Supplementary Information for experimental details.
Transformation of this aziridine provides easy access to diverse, valuable compounds through numerous selective ring-opening functionalization reactions (Fig. 3c). Derivatives of both vicinal amino alcohol isomers (34, 35), aminoazides (36) and medicinally relevant phenethylamines (37) are each smoothly generated in a single step from the N-alkyl aziridine product. In addition to serving as versatile synthetic precursors to diverse amines, N-alkyl aziridines themselves can possess attractive biological activity4. Using the electrochemical methodology reported herein, we prepared an N-cyclohexyl aziridine alkaloid natural product (38) from inexpensive starting materials (Fig. 3d). Finally, we probed whether this dication pool approach could be used outside of the context of aziridine synthesis. We found that omission of exogeneous base under our aziridination conditions resulted in direct diamination of alkenes (39); treatment of the adduct with halide salts resulted in dihalogenation (40, 41); and addition of potassium cyanide furnished the vinyl nitrile (42). Collectively, these data show that adding this electrochemical alkene activation strategy to the synthetic lexicon will dramatically accelerate the preparation of diverse compounds containing the aliphatic amine moiety and beyond.
We next investigated the mechanism underlying the electrochemical process that generates electrophilic adducts such as 1 and 2. In principle, either the thianthrene radical cation or its dication (TT2+) could react with an alkene to produce these dicationic adducts41,47. We found that standard constant current conditions operate slightly above the requisite voltage to oxidize TT (half-wave potential (E1/2) for TT = 0.8 V versus ferrocene (Fc/Fc+) for the majority of the reaction, and substantially below the voltage required to oxidize TT+• to TT2+ (E1/2 (TT+•) = 1.4 V versus Fc/Fc+) (Fig. 4a)41,48. However, we suspected that disproportionation of TT+• to provide the TT2+ in situ could be kinetically relevant (see Supplementary Fig. 4 for cyclic voltammetry experiments consistent with this hypothesis)47,49,50. Given that disproportionation will become increasingly favoured as TT is anodically consumed, we monitored the conversion of alkene over the course of a typical reaction. Following a brief induction period as TT+• is generated, alkene is consumed slowly in a pseudo 0th order fashion, but midway through the reaction the rate increases sharply. Electrochemical analysis reveals that this inflection point occurs when sufficient charge has been passed to oxidize all of the neutral thianthrene to its radical cation congener (1 F mol−1 TT); however, no notable increase in anodic voltage occurs in parallel with this rate change. Given that comproportionation processes will be minimized once TT has been anodically consumed, these data are consistent with the hypothesis that disproportionation to form TT2+ is responsible for the increased rate of adduct formation in this second regime. Furthermore, increasing the initial concentration of thianthrene but maintaining the same current delays the sharp rate increase but has no impact on the initial product-forming regime of the reaction (Fig. 4b). This is consistent with addition of TT+• to the alkene dominating the reaction until TT is consumed.
Fig. 4 |. Mechanistic insight into adduct formation.
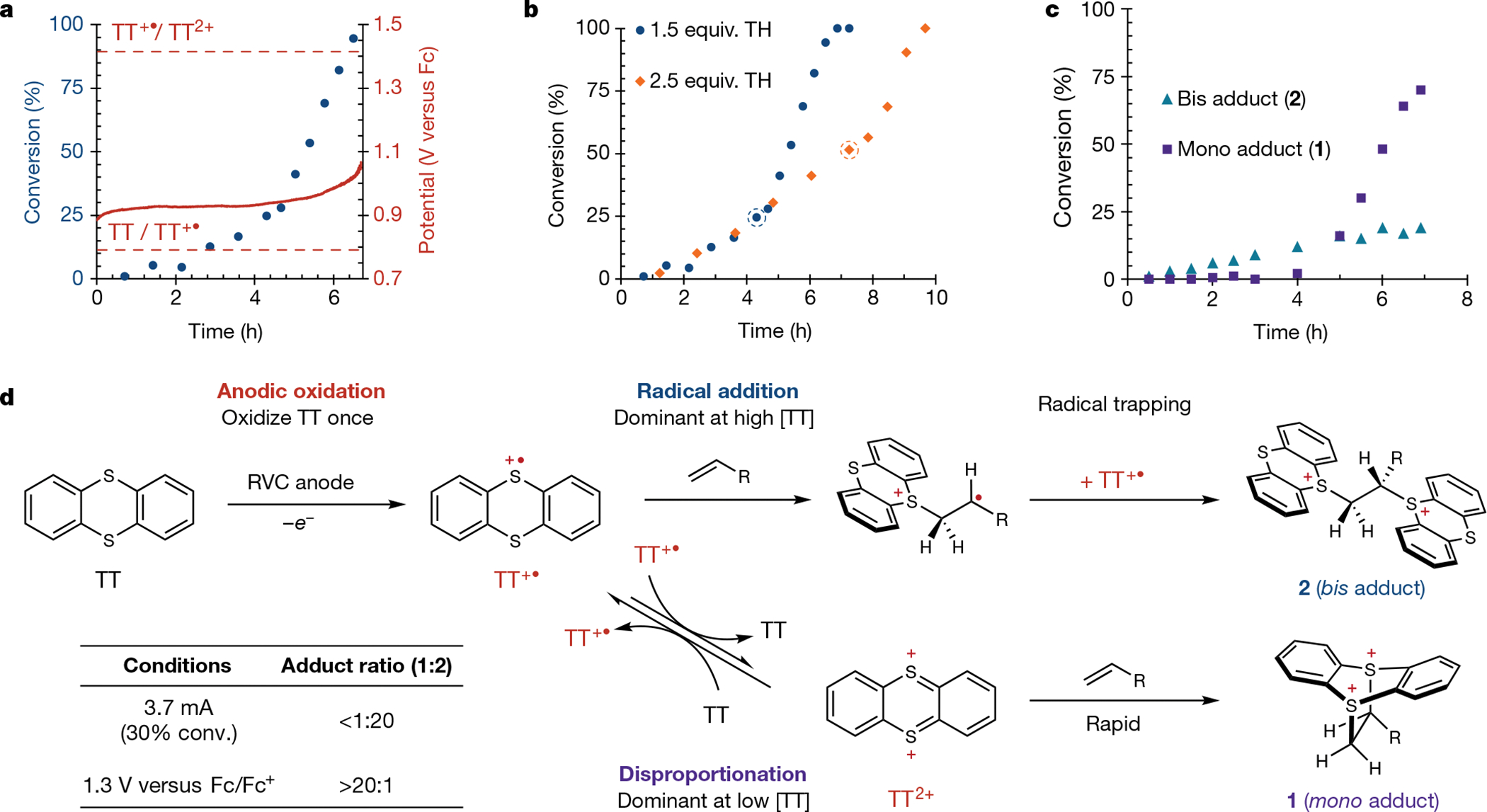
a, Reaction profile under conditions of standard constant current for alkene conversion (left y-axis, blue) as a function of time (x-axis) overlaid with anodic working potential (right y-axis, red). b, Impact of increased thianthrene equivalents on alkene conversion under constant current electrolysis (3.7 mA). The dotted circle indicates the point at which 1 F mol−1 thianthrene has been passed. c, Formation of adducts 1 and 2 as a function of time during constant current electrolysis (3.7 mA). d, Plausible routes for the formation of mono and bis adducts. See Supplementary Information for full experimental details. Fc, ferrocene; R, (CH2)2Ph. Percentage yield and percentage conversion are relative to the alkene starting material.
Given our prediction that these two phases of the reaction proceed through distinct mechanistic pathways, we evaluated the selectivity for mono (1) versus bis (2) adducts as a function of time. We found that, initially, bis adduct 2 is formed exclusively; this is followed by rapid generation of mono adduct 1, once the concentration of TT is low enough to render disproportionation kinetically relevant. This constitutes an unusual kinetic scenario, wherein two products are each formed in exceptionally high selectivity but at different stages of the reaction. To further evaluate the relevance of a transiently generated thianthrene dication, we compared our typical electrolysis conditions with an applied potential sufficient to anodically oxidize the radical cation to the dication without relying on disproportionation. These conditions resulted in an appreciable rate acceleration relative to standard constant current reaction conditions along with exclusive formation of mono adduct 1 (Fig. 4d; see Supplementary Information section 3). Given these preliminary data, we propose that the mono adduct 1 is formed through the following sequence of steps under standard reaction conditions: first, anodic oxidation of thianthrene to the radical cation; second, disproportionation to generate the thianthrenium dication; and third, formal cycloaddition with the alkene (Fig. 4d). The bis adduct 2 could be formed by direct radical addition of TT+• across the alkene followed by coupling between the transient distonic radical cation and the persistent thianthrene radical cation TT+•.
Overall, we have identified an electrochemical strategy by which to access a metastable, dicationic intermediate from alkenes. We found that, unlike conventional dielectrophiles (for example, 1,2-dihalides), these species were cleanly transformed into aziridine products by simple treatment with base and amine. This method represents a new approach to generating N-alkyl aziridines by coupling widely available primary amine nucleophiles and unactivated alkene substrates. This scalable procedure unlocks efficient access to diverse aziridine building blocks bearing sensitive functional groups that are challenging to access through more conventional approaches. Reaction-monitoring experiments revealed an unusual kinetic scenario wherein the mechanism of adduct formation changes sharply as TT is anodically consumed. We anticipate that this new electrochemical transformation will find immediate application in organic synthesis. Moreover, the results reported herein set the stage for the development of a wide range of alkene difunctionalization reactions that remain challenging to accomplish with more conventional approaches.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41586-021-03717-7.
Supplementary Material
Acknowledgements
We thank A. Wendlandt, M. Levin and. D. Weix for suggestions and manuscript proofreading. We also acknowledge A. Hoque and F. Wang in the Stahl laboratory for the use of, and technical assistance with, the electrochemical flow reactor. Additionally, we thank the Weix, Stahl, Yoon and Schomaker groups for sharing their chemical inventories. B. J. Thompson is acknowledged for his assistance with the design and fabrication of the power supply. T. Drier is acknowledged for the fabrication of electrochemical glassware. A. M. Wheaton is acknowledged for assistance with crystallographic studies. We also acknowledge support and suggestions from all members of the Wickens group throughout the investigation of this project. This work was supported financially by the Office of the Vice Chancellor for Research and Graduate Education at the University of Wisconsin–Madison, with funding from the Wisconsin Alumni Research Foundation. Spectroscopic instrumentation was supported by a gift from P. J. and M. M. Bender, by National Science Foundation (NSF) grant CHE-1048642, and by National Institutes of Health (NIH) grants 1S10OD020022–1 and S10 OD01225. This study also made use of the National Magnetic Resonance Facility at Madison, which is supported by NIH grants P41GM136463 (old number P41GM103399 (NIGMS)) and P41RR002301. Equipment was purchased with funds from the University of Wisconsin–Madison, the NIH (grants P41GM103399, S10RR02781, S10RR08438, S10RR023438, S10RR025062 and S10RR029220), the NSF (DMB-8415048, OIA-9977486 and BIR-9214394), and the US Department of Agriculture (USDA). The Bruker D8 VENTURE Photon III X-ray diffractometer was partially funded by NSF award CHE-1919350 to the University of Wisconsin–Madison Department of Chemistry.
Footnotes
Competing interests The authors declare no competing interests.
Additional information
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41586-021-03717-7.
Peer review information Nature thanks Louis-Charles Campeau and David Hickey for their contribution to the peer review of this work. Peer reviewer reports are available.
Reprints and permissions information is available at http://www.nature.com/reprints.
Data availability
Crystallographic data for compounds 1 and 2 can be obtained free of charge from the Cambridge Crystallographic Data Centre (https://www.ccdc.cam.ac.uk). All data supporting the findings of this paper are available within the paper and its Supplementary Information.
References
- 1.Sweeney JB Aziridines: epoxides’ ugly cousins? Chem. Soc. Rev. 31, 247–258 (2002). [DOI] [PubMed] [Google Scholar]
- 2.Stanković S et al. Regioselectivity in the ring opening of non-activated aziridines. Chem. Soc. Rev. 41, 643–665 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Botuha C, Chemla F, Ferreira F & Pérez-Luna A in Heterocycles in Natural Product Synthesis 1–39 (John Wiley, 2011). [Google Scholar]
- 4.Ismail FMD, Levitsky DO & Dembitsky VM Aziridine alkaloids as potential therapeutic agents. Eur. J. Med. Chem. 44, 3373–3387 (2009). [DOI] [PubMed] [Google Scholar]
- 5.Thibodeaux CJ, Chang W & Liu H Enzymatic chemistry of cyclopropane, epoxide, and aziridine biosynthesis. Chem. Rev. 112, 1681–1709 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lowden PAS in Aziridines and Epoxides in Organic Synthesis 399–442 (John Wiley, 2006). [Google Scholar]
- 7.Roth HG, Romero NA & Nicewicz DA Experimental and calculated electrochemical potentials of common organic molecules for applications to single-electron redox chemistry. Synlett 27, 714–723 (2016). [Google Scholar]
- 8.Evans DA, Faul MM & Bilodeau MT Copper-catalyzed aziridination of olefins by (N-(p-toluenesulfonyl)imino)phenyliodinane. J. Org. Chem. 56, 6744–6746 (1991). [Google Scholar]
- 9.Sweeney JB in Aziridines and Epoxides in Organic Synthesis 117–144 (John Wiley, 2006). [Google Scholar]
- 10.Osborn HMI & Sweeney J The asymmetric synthesis of aziridines. Tetrahedron Asymmetry 8, 1693–1715 (1997). [Google Scholar]
- 11.Wang H, Yang JC & Buchwald SL CuH-catalyzed regioselective intramolecular hydroamination for the synthesis of alkyl-substituted chiral aziridines. J. Am. Chem. Soc. 139, 8428–8431 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Legnani L, Prina-Cerai G, Delcaillau T, Willems S & Morandi B Efficient access to unprotected primary amines by iron-catalyzed aminochlorination of alkenes. Science 362, 434–439 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Jat JL et al. Direct stereospecific synthesis of unprotected N–H and N-Me aziridines from olefins. Science 343, 61–65 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma Z, Zhou Z & Kürti L Direct and stereospecific synthesis of N–H and N-alkyl aziridines from unactivated olefins using hydroxylamine-O-sulfonic acids. Angew. Chem. Int. Edn 56, 9886–9890 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng Q-Q et al. Organocatalytic nitrogen transfer to unactivated olefins via transient oxaziridines. Nat. Catal. 3, 386–392 (2020). [Google Scholar]
- 16.Falk E, Makai S, Delcaillau T, Gürtler L & Morandi B Design and scalable synthesis of N-alkylhydroxylamine reagents for the direct iron-catalyzed installation of medicinally relevant amines. Angew. Chem. Int. Edn 59, 21064–21071 (2020). [DOI] [PubMed] [Google Scholar]
- 17.Munnuri S, Anugu RR & Falck JR Cu(II)-mediated N–H and N-alkyl aryl amination and olefin aziridination. Org. Lett. 21, 1926–1929 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Govaerts S et al. Photoinduced olefin diamination with alkylamines. Angew. Chem. Int. Edn 59, 15021–15028 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.eMolecules (eMolecules Inc, accessed 20 January 2021); https://www.emolecules.com [Google Scholar]
- 20.Lohray BB, Gao Y & Sharpless KB One pot synthesis of homochiral aziridines and aminoalcohols from homochiral 1,2-cyclic sulfates. Tetrahedr. Lett. 30, 2623–2626 (1989). [Google Scholar]
- 21.Wenker H The preparation of ethylene imine from monoethanolamine. J. Am. Chem. Soc. 57, 2328 (1935). [Google Scholar]
- 22.Li X, Chen N & Xu J An improved and mild Wenker synthesis of aziridines. Synthesis 20, 3423–3428 (2010). [Google Scholar]
- 23.Yan M, Kawamata Y & Baran PS Synthetic organic electrochemical methods since 2000: on the verge of a renaissance. Chem. Rev. 117, 13230–13319 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wiebe A et al. Electrifying organic synthesis. Angew. Chem. Int. Edn 57, 5594–5619 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Francke R & Little RD Redox catalysis in organic electrosynthesis: basic principles and recent developments. Chem. Soc. Rev. 43, 2492–2521 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Moeller KD Using physical organic chemistry to shape the course of electrochemical reactions. Chem. Rev. 118, 4817–4833 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Yoshida J, Kataoka K, Horcajada R & Nagaki A Modern strategies in electroorganic synthesis. Chem. Rev. 108, 2265–2299 (2008). [DOI] [PubMed] [Google Scholar]
- 28.Sauer GS & Lin S An electrocatalytic approach to the radical difunctionalization of alkenes. ACS Catal. 8, 5175–5187 (2018). [Google Scholar]
- 29.Doobary S, Sedikides AT, Caldora HP, Poole DL & Lennox AJJ Electrochemical vicinal difluorination of alkenes: scalable and amenable to electron-rich substrates. Angew. Chem. Int. Edn 59, 1155–1160 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Siu T & Yudin AK Practical olefin aziridination with a broad substrate scope. J. Am. Chem. Soc. 124, 530–531 (2002). [DOI] [PubMed] [Google Scholar]
- 31.Oseka M et al. Electrochemical aziridination of internal alkenes with primary amines. Chem 7, 255–266 (2021). [Google Scholar]
- 32.Chen J et al. Electrocatalytic aziridination of alkenes mediated by n-Bu4NI: a radical pathway. Org. Lett. 17, 986–989 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Li J et al. Electrochemical aziridination by alkene activation using a sulfamate as the nitrogen source. Angew. Chem. Int. Edn 57, 5695–5698 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Yoshida J, Shimizu A & Hayashi R Electrogenerated cationic reactive intermediates: the pool method and further advances. Chem. Rev. 118, 4702–4730 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Hayashi R, Shimizu A & Yoshida J The stabilized cation pool method: metal- and oxidant-free benzylic C–H/aromatic C–H cross-coupling. J. Am. Chem. Soc. 138, 8400–8403 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Shine HJ, Bandlish BK, Mani SR & Padilla AG Ion radicals. 43. Addition of thianthrene and phenoxathiin cation radicals to alkenes and alkynes. J. Org. Chem. 44, 915–917 (1979). [Google Scholar]
- 37.Shine HJ et al. Adducts of phenoxathiin and thianthrene cation radicals with alkenes and cycloalkenes. J. Org. Chem. 68, 8910–8917 (2003). [DOI] [PubMed] [Google Scholar]
- 38.Rangappa P & Shine HJ An overview of some reactions of thianthrene cation radical. Products and mechanisms of their formation. J. Sulfur Chem. 27, 617–664 (2006). [Google Scholar]
- 39.Berger F et al. Site-selective and versatile aromatic C−H functionalization by thianthrenation. Nature 567, 223–228 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Engl PS et al. C–N cross-couplings for site-selective late-stage diversification via aryl sulfonium salts. J. Am. Chem. Soc. 141, 13346–13351 (2019). [DOI] [PubMed] [Google Scholar]
- 41.Chen J, Li J, Plutschack MB, Berger F & Ritter T Regio- and stereoselective thianthrenation of olefins to access versatile alkenyl electrophiles. Angew. Chem. Int. Edn 59, 5616–5620 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Houmam A, Shukla D, Kraatz H-B & Wayner DDM Electrosynthesis of mono- and bisthianthrenium salts. J. Org. Chem. 64, 3342–3345 (1999). [DOI] [PubMed] [Google Scholar]
- 43.Iwai K & Shine HJ Ion radicals. 46. Reactions of the adducts of thianthrene and phenoxathiin cation radicals and cyclohexene with nucleophiles. J. Org. Chem. 46, 271–276 (1981). [Google Scholar]
- 44.Mitchell SC & Waring RH Fate of thianthrene in biological systems. Xenobiotica 47, 731–740 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Kaiser D, Klose I, Oost R, Neuhaus J & Maulide N Bond-forming and -breaking reactions at sulfur(IV): sulfoxides, sulfonium salts, sulfur ylides, and sulfinate salts. Chem. Rev. 119, 8701–8780 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bright GM, Brodney MA & Wlodecki B Pyridyloxymethyl and benzisoxazole azabicyclic derivatives. International patent WO/2004/081007 (2004).
- 47.Qian D-Q, Shine HJ, Guzman-Jimenez IY, Thurston JH & Whitmire KH Mono- and bisadducts from the addition of thianthrene cation radical salts to cycloalkenes and alkenes. J. Org. Chem. 67, 4030–4039 (2002). [DOI] [PubMed] [Google Scholar]
- 48.Speer ME et al. Thianthrene-functionalized polynorbornenes as high-voltage materials for organic cathode-based dual-ion batteries. Chem. Commun. 51, 15261–15264 (2015). [DOI] [PubMed] [Google Scholar]
- 49.Murata Y & Shine HJ Ion radicals. XVIII. Reactions of thianthrenium perchlorate and thianthrenium trichlorodiiodide. J. Org. Chem. 34, 3368–3372 (1969). [Google Scholar]
- 50.Sandford C et al. A synthetic chemist’s guide to electroanalytical tools for studying reaction mechanisms. Chem. Sci. 10, 6404–6422 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Crystallographic data for compounds 1 and 2 can be obtained free of charge from the Cambridge Crystallographic Data Centre (https://www.ccdc.cam.ac.uk). All data supporting the findings of this paper are available within the paper and its Supplementary Information.