

Supplemental Digital Content is Available in the Text.
BACKGROUND
AbobotulinumtoxinA (aboBoNT-A) solution is a new ready-to-use formulation developed to reduce preparation time and improve reproducibility of injections.
OBJECTIVE
To further evaluate treatment of moderate-to-severe glabellar lines (GLs) using pooled data from 2 Phase III studies.
METHODS
Following double-blind treatment with 50 U aboBoNT-A solution (n = 251) or placebo (n = 123), GL severity was assessed by investigators (ILA) and subjects (SSA). Other assessments included subject-reported time to onset, subject satisfaction, FACE-Q, and adverse events.
RESULTS
One month after aboBoNT-A solution treatment, 88% had none-or-mild GLs at maximum frown and 93% had ≥1-grade improvement in ILA (similar for SSA), 24% to 27% remaining improved at Month 6. Glabellar lines responder rates remained higher than placebo throughout Month 6 (p < .001). Almost two-thirds of subjects reported onset within 3 days, nearly a quarter reporting effect by Day 1. Subject satisfaction with GL appearance, and FACE-Q satisfaction with facial appearance overall and psychological well-being were also improved over placebo throughout Month 6, p < .05. Treatment-related adverse events were nonserious and mild or moderate.
CONCLUSION
Pooled analysis confirmed a duration of effect on GLs of up to 6 months for aboBoNT-A solution, with onset starting within 24 hours, high subject satisfaction, and improved psychological well-being. The treatment was well tolerated.
AbobotulinumtoxinA (aboBoNT-A; Dysport, Ipsen Ltd, Slough, UK/Azzalure, Galderma SA, Lausanne, Switzerland) is a powder formulation neuromodulator widely used to improve the appearance of glabellar lines (GLs) with well-documented efficacy, subject satisfaction, and safety.1–4
A new ready-to-use formulation, aboBoNT-A solution for injection (Alluzience, Ipsen Ltd, Slough, UK/Galderma SA, Lausanne, Switzerland), was recently approved for marketing in several European countries, offering potential benefits over existing powder formulations, in convenience, consistency and precision of dosing, because there is no need for reconstitution. In addition, the formulation contains no human- or animal-derived excipients.
Previously published placebo-controlled data from 3 separate studies, including 1 Phase II and 2 Phase III studies, have demonstrated that 50 U of aboBoNT-A solution is efficacious and well tolerated when used to treat moderate-to-severe GL after single and repeated treatment.5–7
To further investigate the efficacy and duration of effect of ready-to-use aboBoNT-A solution in GLs, data from the double-blind, randomized, placebo-controlled cycle of the 2 multicenter Phase III studies mentioned above were pooled. This enabled analysis of the treatment effect in GLs more accurately in a larger population and includes the data supporting the marketing approvals.
Materials and Methods
Study Design
Data were pooled from the double-blind, randomized, placebo-controlled cycles of 2 multicenter Phase III studies (clinicaltrials.gov registration numbers NCT023538716 and NCT024939467, conducted in 2015 to 2016 in France, Germany, and the United Kingdom). Both studies were conducted in accordance with Good Clinical Practice and the Declaration of Helsinki, with appropriate approvals from independent ethics committees or institutional review boards and written informed consent from all subjects. Data pooling was justified because both studies had comparable populations, treatment, and endpoints.
In study NCT02353871, double-blinded follow-up was performed for all subjects up to Month 6, whereas in study NCT02493946, subjects could receive retreatment from Month 3 onward (if fulfilling eligibility criteria) and enter an open-label period (data not shown).
The present analysis aimed to thoroughly assess efficacy and safety of 50 U aboBoNT-A solution used for treatment of moderate-to-severe GLs, compared with placebo, in the pooled data set. The primary objective was identical in both studies, that is, to demonstrate superior efficacy of aboBoNT-A solution over placebo (none-or-mild ILA responder rates at Month 1) and these results are reported for each study in Ascher and colleagues6 and Kestemont and colleagues.7
Subjects and Treatment
To be eligible, subjects had to have moderate-to-severe (Grade 2 or 3) GL severity at maximum frown as assessed by investigators using the 4-graded Investigator's Live Assessment scale (ILA) and by subjects using the 4-graded Subject's Self-Assessment scale (SSA), and be dissatisfied or very dissatisfied (Grade 2 or 3) with their GL appearance before treatment and naïve to any serotype of botulinum toxin. Treatment assignment was randomized 2:1 to aboBoNT-A solution 50 U or placebo, injected at 5 injection points in the GL area, as 0.05 mL (10 U of aboBoNT-A or placebo) per injection.
Efficacy Assessments and Endpoints
Assessments included GL severity grading by investigators at maximum frown and at rest using a validated 4-graded ILA photographic scale8 from “none” (Grade 0) to “severe” (Grade 3) and by subjects at maximum frown using a corresponding 4-graded SSA. The primary endpoint for each study was identical: Response to treatment defined as improvement from a GL severity of moderate or severe to none or mild at maximum frown on the ILA scale at Month 1.
Subjects also reported onset of treatment response using a 7-day diary card, subject satisfaction with GL appearance using a 4-point scale from “very satisfied” (Grade 0), to “very dissatisfied” (Grade 3), and responded to 2 10-item FACE-Q scales, the Psychological Well-being scale and Satisfaction with Facial Appearance Overall scale, described in detail in previous publications.9,10
Safety Assessments
Treatment-emergent adverse events were collected throughout both individual studies.
Statistics
Statistical analyses of the pooled data were performed using SAS, version 9.4. Efficacy was analyzed on the modified intent-to-treat (mITT) population, defined as randomized subjects, treated in at least one injection site, with baseline and post-treatment data for ILA at maximum frown. Safety analysis was performed on randomized subjects, treated in at least one injection site.
For the pooled analyses of ILA, SSA, and subject satisfaction, a generalized linear model was used with a normal distribution and link identity function and gender, baseline ILA at maximum frown and study as fixed effects.
Responder rates were calculated based on the number of subjects in each treatment group with 95% confidence intervals (CIs). Subjects with missing data at a visit were considered nonresponders. Withdrawn subjects, and re-treated subjects (for study NCT02493946) were also considered nonresponders for subsequent visits.
Kaplan–Meier methods were used to estimate medians for time to onset of effect, time to loss of none or mild GL status and time to return to baseline GL severity.
For each FACE-Q scale, the subjects' scores for the 10 individual items were converted to a single Rasch transformed total score (0–100) as per the FACE-Q manual, with higher total scores indicating greater psychological well-being or subject satisfaction with facial appearance overall. Rasch transformed scores were analyzed using a general linear model with stratification factor and study as fixed effects.
Results
Subject Disposition and Demographics
The pooled data comprised 251 subjects treated with aboBoNT-A solution and 123 subjects treated with placebo. Of these, 250 subjects (aboBoNT-A solution) and 122 (placebo) were included in the mITT population. For details, see Supplemental Digital Content 1, subject disposition (Table S1, http://links.lww.com/DSS/B152). Baseline characteristics are summarized in Supplemental Digital Content 2, demographics and baseline characteristics (Table S2, http://links.lww.com/DSS/B153) and further detailed in Ascher and colleagues 6 and Kestemont and colleagues.7
Efficacy
Glabellar Line Severity Improvements at Maximum Frown
In the pooled data, peak responder rates of 88% (ILA) and 77% (SSA) were achieved at Month 1 at maximum frown in the aboBoNT-A solution group, using the none-or-mild GL severity responder definition, and 93% of subjects (ILA) and 90% (SSA) had ≥1-grade improvement. Responder rates were persistently significantly higher for aboBoNT-A solution than placebo throughout Month 6 (p-values are provided in Figure 1 and 2), with a retained none-or-mild response in 10% (ILA) and 15% of subjects (SSA), and a ≥1-grade improvement in 24% (ILA) and 27% of subjects (SSA) at Month 6 after treatment with aboBoNT-A solution.
Figure 1.
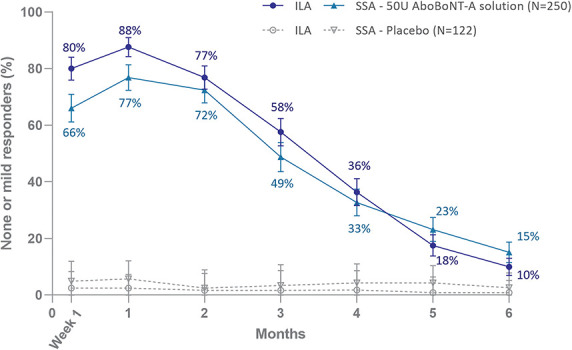
None-or-mild glabellar line severity responders at maximum frown (mITT population); a responder had improved from moderate (Grade 2) or severe (Grade 3) at baseline to none (Grade 0) or mild (Grade 1) post-treatment. Error bars show 95% confidence intervals. For ILA and SSA, the difference between aboBoNT-A solution and placebo was statistically significant at all time points, p < .0001 up to Month 5 and p < .001 at Month 6.
Figure 2.
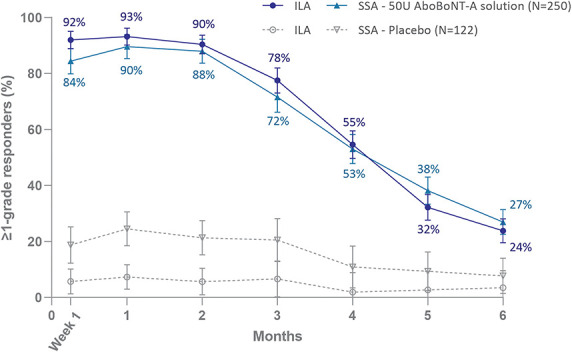
≥1-grade improvement from baseline in glabellar line severity at maximum frown (mITT population). Error bars show 95% confidence intervals. For ILA and SSA, the difference between aboBoNT-A solution and placebo was statistically significant at all time points, p < .0001.
Glabellar Line Severity Improvements at Rest
Investigator's Live Assessment responder rates at rest were also higher for aboBoNT-A solution than placebo throughout Month 6, p < .0001, peaking at Month 1 at 81% (none-or-mild response) and 87% (≥1-grade response). Improvement in GL severity at rest over time is shown in Supplemental Digital Content 3, none-or-mild glabellar line severity at rest (See Figure S1, http://links.lww.com/DSS/B148) and Supplemental Digital Content 4, ≥1-grade improvement from baseline in glabellar line severity at rest (See Figure S2, http://links.lww.com/DSS/B149).
Time to Onset
Time to onset of effect based on subject-reporting in a 7-day diary is shown in Supplemental Digital Content 5, subject-reported onset (see Table S3, http://links.lww.com/DSS/B154). In the aboBoNT-A solution group, 23% of subjects noted a treatment response on Day 1, which cumulated to 49% by Day 2, 64% by Day 3, with a median time to onset of 3.0 days (95% CI: 2.0–3.0). Among the 219 subjects in the aboBoNT-A solution group who were none-or-mild responders in ILA, maximum frown, at Month 1, that is fulfilled the responder definition of the primary endpoint, 24% noted a treatment response by Day 1, 53% by Day 2, and 68% by Day 3 and the median time to onset was 2.0 days (95% CI: 2.0–3.0).
Time to Loss of Response
The median time to loss of none-or-mild severity concomitantly on ILA and SSA scales was 127 days (95% CI: 112.0–134.0), that is 4.2 months, whereas the median time to return to baseline severity on both scales was 176 days (95% CI: 163.0–178.0), 5.9 months. Based on the ILA scale alone, the median time to loss of none-or-mild GL status at maximum frown in the aboBoNT-A solution group was 110 days (95% CI: 106.0–126.0), that is 3.7 months, and the median time to return to baseline was 141 days (95% CI: 135.0–143.0), 4.7 months.
Because of the small proportion of none-or-mild responders in the placebo group (3% for ILA, 6% for SSA), median time to loss of responder status or median time to return to baseline status for placebo were not evaluable.
Subject Satisfaction
The proportion of subjects who were satisfied with their GL appearance after treatment with aboBoNT-A solution peaked at Month 1 (85%) and was persistently higher than for placebo throughout Month 6, p < .0001, with 26% remaining satisfied at Month 6 in the aboBoNT-A solution group (Figure 3).
Figure 3.
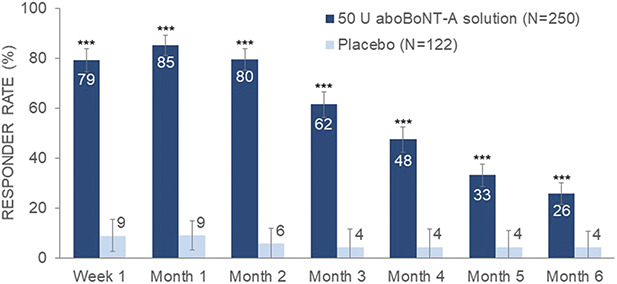
Subject satisfaction responder rates over time (mITT population). Responders were “satisfied” or “very satisfied” with the appearance of their glabellar lines post-baseline and “dissatisfied” or “very dissatisfied” at baseline. Error bars show 95% confidence intervals. The difference between aboBoNT-A solution and placebo was statistically significant at all time points, p < .0001.
Subject-Reported FACE-Q
In the subject-reported FACE-Q scales, assessing the more general impact of their GL treatment, psychological well-being and satisfaction with facial appearance overall were significantly improved from baseline in the aboBoNT-A solution group compared with placebo, throughout Month 6, p < .05. The changes from baseline over time for the total Rasch-transformed FACE-Q scores for each treatment group can be seen in Supplemental Digital Content 6, subjects' psychological well-being (See Figure S3, http://links.lww.com/DSS/B150), and Supplemental Digital Content 7, subject satisfaction with facial appearance overall (See Figure S4, http://links.lww.com/DSS/B151).
Safety
An overview of treatment-emergent adverse events (TEAEs) and the frequencies of the most common TEAEs judged as treatment-related from the pooled data analysis are summarized in Supplemental Digital Content 8, adverse events (See Table S4, http://links.lww.com/DSS/B155). In total, 17% of subjects in the aboBoNT-A solution group had TEAEs judged as treatment-related, versus 6% in the placebo group. The treatment-related TEAEs reported by >1% of subjects in the aboBoNT-A solution group were headache (6.4% vs 2.4% for placebo), injection-site pain (4.0%; 2.4%), hematoma (2.0%; 0%), eyelid edema (1.6%; 0%), and brow ptosis (1.2%; 0%).
Discussion
AboBoNT-A solution is the first liquid botulinum toxin type A formulation to be developed in the Western world and represents a new era in aesthetic botulinum toxin type A treatments. The aboBoNT-A solution has undergone thorough evaluation for the GL indication in Phase II and III trials. Pooling of the Phase III study data presented herein enabled analysis of a large double-blind placebo-controlled sample of subjects treated for GLs with aboBoNT-A solution, which was also used to support the recent marketing approvals in several European countries.
The results from this new pooled analysis confirmed the high level of efficacy in improving GL severity at maximum frown that was previously reported for each of the Phase III studies.6,7 Investigators and subjects reported peak responder rates of 77% to 93% and the aboBoNT-A solution achieved statistically significant higher responder rates for 6 months compared with placebo, both when using the none-or-mild definition (10%–15% responder rate) and ≥1-grade responder definitions (24%–27% responder rate). The pooled data analysis of the subject-reported diary also confirmed a rapid onset of effect (within 3 days) for almost two-thirds of subjects in the aboBoNT-A solution group (mITT population), with 23% reporting onset by Day 1, which is similar to the powder formulation.11
High subject satisfaction with post-treatment appearance of the GLs was also demonstrated. Peak subject satisfaction with GL appearance in the aboBoNT-A solution group coincided with the maximum observed effect on GL severity at Month 1, based on the ILA and SSA assessments. Satisfaction rates remained higher than for placebo up to Month 6, which was in line with the GL severity assessments.
Beyond the evaluations specific to the GLs, there was also clear evidence of a wider positive impact of the treatment. Even though only the GLs were treated, significantly improved psychological well-being and facial appearance overall was measured over placebo for 6 months after treatment in the 2 FACE-Q scales used.
All investigated efficacy parameters point to a long duration of action of aboBoNT-A solution with measurable effects up to 6 months in 1 in 4 treated subjects. This includes results from the ≥1-grade GL responder rate analysis at maximum frown, which have not been reported previously for the individual Phase III trials on aboBoNT-A solution.5 In the pooled analysis, a sustained ≥1-grade efficacy response up to 6 months after treatment was observed in 24% to 27% of subjects, as assessed by investigators and subjects. This severity improvement was closely matched by responder rates for subject satisfaction with GL appearance at Month 6, 26%, indicating that a ≥1-grade severity improvement corresponds well with a positive subject experience and is a clinically relevant response.
The pooled data reported herein suggest a duration and onset of effect for aboBoNT-A solution in the same range as for the currently available aboBoNT-A powder formulation.3,4 No new safety signals were identified in the pooled data analysis and a single treatment with aboBoNT-A solution was generally well tolerated. Treatment-related adverse events were mild or moderate in intensity and none were serious or indicative of a remote spread of effect. The incidence of injection-site pain was low and comparable to that reported for the powder aboBoNT-A formulation.1,12,13 Furthermore, in Phase III study NCT02493946, there was no evidence of development of BoNT-A neutralizing antibodies after repeat-treatment with aboBoNT-A solution.7
Altogether, the pooled data support that this ready-to-use formulation provides a high level of efficacy, long duration, and high subject satisfaction after aesthetic treatment, but with the added benefit of already being reconstituted and ready-to-use. The safety profile was as expected for aboBoNT-A treatment2,4,14 and in accordance with the aboBoNT-A labelling.
Further to reducing preparation time, the anticipated benefits of the ready-to-use aboBoNT-A solution include precise and consistent dose delivery, and this formulation has been shown to achieve consistent effect across treatment cycles.7
Conclusion
In conclusion, treatment with 50 U of ready-to-use aboBoNT-A solution was effective for improvement of moderate-to-severe GLs for up to 6 months, with 24% to 27% of subjects still showing improvement at Month 6. This was accompanied by high rates of subject satisfaction. Subject-reported onset of effect started within 24 hours, almost two-thirds of subjects reporting onset within 3 days. In support of a long duration of action, 26% of subjects remained satisfied with their GL appearance at Month 6 and a wider positive impact was shown for 6 months in the subject-reported Satisfaction with Facial Appearance Overall and Psychological Well-being scales. The treatment was well tolerated, with a safety profile consistent with powder formulations of aboBoNT-A.
Supplementary Material
Acknowledgments
The authors thank all patients, investigators, and staff who contributed to both studies, and Dr. Carolina Edwartz for contributions to manuscript preparation on behalf of Galderma.
Footnotes
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.dermatologicsurgery.org).
Ipsen (Paris, France) sponsored the studies described herein and provided funding to the investigational centers involved. S. Hilton, P. Kestemont, G. Sattler, C. Thompson, and B. Ascher are consultants for Ipsen and/or Galderma. M. Volteau is an employee of Ipsen. B. Andriopoulos, I. Prygova and A.-K. Berg are employees of Galderma.
Contributor Information
Gerhard Sattler, Email: gerhard.sattler@rosenparkklinik.de.
Magali Volteau, Email: magali.volteau@ipsen.com.
Catherine Thompson, Email: catherine.thompson@junepharma.com.
Bill Andriopoulos, Email: bill.andriopoulos@galderma.com.
Inna Prygova, Email: inna.prygova@galderma.com.
Anna-Karin Berg, Email: anna-karin.berg@galderma.com.
Benjamin Ascher, Email: benjaminascher@wanadoo.fr.
References
- 1.Monheit GD, Baumann L, Maas C, Rand R, et al. Efficacy, safety, and subject satisfaction after AbobotulinumtoxinA treatment for moderate to severe glabellar lines. Dermatol Surg 2020;46:61–9. [DOI] [PubMed] [Google Scholar]
- 2.Cohen JL, Scuderi N. Safety and patient satisfaction of AbobotulinumtoxinA for aesthetic use: a systematic review. Aesthet Surg J 2017;37:S32–S44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rzany B, Ascher B, Monheit G. Treatment of glabellar lines with botulinum toxin type A (Speywood Unit): a clinical overview. J Eur Acad Dermatol Venereol 2010;24(Suppl 1):1–14. [DOI] [PubMed] [Google Scholar]
- 4.Schlessinger J, Cohen JL, Shamban A, Jacob C, et al. A multicenter study to evaluate subject satisfaction with two treatments of AbobotulinumtoxinA a year in the glabellar lines. Dermatol Surg 2021;47:504–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ascher B, Kestemont P, Boineau D, Bodokh I, et al. Liquid formulation of AbobotulinumtoxinA exhibits a favorable efficacy and safety profile in moderate to severe glabellar lines: a randomized, double-blind, placebo- and active comparator-controlled trial. Aesthet Surg J 2018;38:183–91. [DOI] [PubMed] [Google Scholar]
- 6.Ascher B, Rzany B, Kestemont P, Hilton S, et al. Liquid formulation of AbobotulinumtoxinA: a 6-month, Phase 3, double-blind, randomized, placebo-controlled study of a single treatment, ready-to-use toxin for moderate-to-severe glabellar lines. Aesthet Surg J 2020;40:93–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kestemont P, Hilton S, Andriopoulos B, Prygova I, et al. Long-term efficacy and safety of liquid AbobotulinumtoxinA formulation for moderate-to-severe glabellar lines: a Phase III, double-blind, randomized, placebo-controlled and open-label study. Aesthet Surg J 2022;42:301–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Honeck P, Weiss C, Sterry W, Rzany B, et al. Reproducibility of a four-point clinical severity score for glabellar frown lines. Br J Dermatol 2003;149:306–10. [DOI] [PubMed] [Google Scholar]
- 9.Pusic AL, Klassen AF, Scott AM, Cano SJ. Development and psychometric evaluation of the FACE-Q satisfaction with appearance scale: a new patient-reported outcome instrument for facial aesthetics patients. Clin Plast Surg 2013;40:249–60. [DOI] [PubMed] [Google Scholar]
- 10.Klassen AF, Cano SJ, Schwitzer JA, Scott AM, et al. FACE-Q scales for health-related quality of life, early life impact, satisfaction with outcomes, and decision to have treatment: development and validation. Plast Reconstr Surg 2015;135:375–86. [DOI] [PubMed] [Google Scholar]
- 11.Joseph J, Moradi A, Lorenc ZP, Coleman K, et al. AbobotulinumtoxinA for the treatment of moderate-to-severe glabellar lines: a randomized, dose-escalating, double-blind study. J Drugs Dermatol 2021;20:980–7. [DOI] [PubMed] [Google Scholar]
- 12.Brandt F, Swanson N, Baumann L, Huber B. Randomized, placebo-controlled study of a new botulinum toxin type a for treatment of glabellar lines: efficacy and safety. Dermatol Surg 2009;35:1893–901. [DOI] [PubMed] [Google Scholar]
- 13.Moy R, Maas C, Monheit G, Huber MB, et al. Long-term safety and efficacy of a new botulinum toxin type A in treating glabellar lines. Arch Facial Plast Surg 2009;11:77–83. [DOI] [PubMed] [Google Scholar]
- 14.Gubanova E, Haddad Tabet M, Bergerova Y, Moiseieva O, et al. Assessment of subject and physician satisfaction after long-term treatment of glabellar lines with AbobotulinumtoxinA (Dysport((R))/Azzalure((R))): primary results of the APPEAL noninterventional study. Aesthet Plast Surg 2018;42:1672–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.