

Abstract
The application of advanced molecular technology has significantly expanded lymphoma classification, allowing risk stratification and treatment optimization. Limited evidence suggests the presence of a genetic predisposition in lymphoma, indicating the potential for better individualized clinical management based on a novel lymphoma classification. Herein, we examined the impact of germline pathogenic variants in 27 cancer‐predisposing genes with lymphoma risk and explored the clinical characteristics of pathogenic variant carriers. This study included 2,066 lymphoma patients and 38,153 cancer‐free controls from the Japanese population. Following quality control of sequencing data, samples from 1,982 lymphoma patients and 37,592 controls were further analyzed. We identified 309 pathogenic variants among 4,850 variants in the 27 cancer‐predisposing genes. Pathogenic variants in the following four cancer‐predisposing genes were associated with a high risk of lymphoma: ATM (odds ratio [OR], 2.63; 95% confidence interval [CI], 1.25–5.51; p = 1.06 × 10−2), BRCA1 (OR, 5.88; 95% CI, 2.65–13.02; p = 1.27 × 10−5), BRCA2 (OR, 2.94; 95% CI, 1.60–5.42; p = 5.25 × 10−4), and TP53 (OR, 5.22; 95% CI, 1.43–19.02; p = 1.23 × 10−2). The proportion of carriers of these genes was 1.6% of lymphoma patients. Furthermore, pathogenic variants in these genes were especially associated with a higher risk of mantle cell lymphoma (OR, 21.57; 95% CI, 7.59–61.26; p = 8.07 × 10−9). These results provide novel insights concerning monogenic form into lymphoma classification. Some lymphoma patients may benefit from surveillance and targeted treatment, such as other neoplasms.
Keywords: cancer‐predisposing gene, case‐control study, germline pathogenic variant, lymphoma, mantle cell lymphoma
We found that pathogenic variants in the four cancer‐predisposing genes (ATM, BRCA1, BRCA2, and TP53) were associated with lymphoma risk. Pathogenic variants in these genes were especially associated with a higher risk of mantle cell lymphoma. These results would provide novel insights concerning monogenic form into lymphoma classification.
Abbreviations
- CI
confidence interval
- GATK
Genome Analysis Toolkit
- MCL
mantle cell lymphoma
- OR
odds ratio
1. INTRODUCTION
Lymphoma is one of the most common hematopoietic neoplasms, with an estimated 627,000 new cases worldwide in 2020; 1 however, it is associated with heterogeneous subtypes. Lymphoma is classified into approximately 70 subtypes using the information on morphology, immune‐phenotyping, and molecular genetics of tumors based on the World Health Organization classification. 2 The detailed classification has important implications for diagnosis, prognosis, and treatment. 2 Several studies suggested the presence of a genetic predisposition in lymphoma. For example, a previous report found that germline pathogenic variants in BRCA2 increased the risk of non‐Hodgkin lymphoma five‐fold. 3 Our previous study across 14 cancer types also found that germline pathogenic variants in BRCA1 tended to be associated with lymphoma risk, although the results were not statistically significant. 4 A population‐based study reported that lymphoma patients share the familial risks of the same lymphoma subtypes, 5 indicating the presence of a genetic predisposition to the lymphoma subtype. However, because there has been insufficient evaluation of other cancer‐predisposing genes and insufficient assessment of how these genetic predispositions differ across the subtype of lymphoma, the classification with germline pathogenic variants has not yet been divided in the World Health Organization classification of lymphoid neoplasms, 2 unlike myeloid neoplasms. 6
Herein, we evaluate the association between germline pathogenic variants in 27 cancer‐predisposing genes and the lymphoma risk and then explore the clinical characteristics of pathogenic variant carriers to provide evidence for appropriate diagnosis and treatment based on novel insights concerning monogenic form in lymphoma classification.
2. MATERIALS AND METHODS
2.1. Study subjects
All patients were enrolled from BioBank Japan, 7 , 8 which is a multi‐institutional hospital registry that collected peripheral blood samples and clinical information from participants with 51 common diseases from all over Japan between 2003 and 2018. Clinical information of individuals was collected through interviews or medical record surveys using a standard questionnaire at the point of entry to Biobank Japan. In the present study, we analyzed 2,066 individuals with lymphoma diagnoses registered to BioBank Japan or a past personal history of lymphoma. We also analyzed 38,153 individuals registered to BBJ with non‐malignant diseases; controls were selected to have no past personal or family history of cancer to improve statistical power. All participants provided written informed consent. The study was approved by the ethics committees of the Institute of Medical Sciences, The University of Tokyo, and the RIKEN Center for Integrative Medical Sciences.
2.2. Sequencing and bioinformatics analysis
We analyzed 27 cancer‐predisposing genes (APC, ATM, BARD1, BMPR1A, BRCA1, BRCA2, BRIP1, CDK4, CDKN2A, CDH1, CHEK2, EPCAM, HOXB13, NBN, NF1, MLH1, MSH2, MSH6, MUTYH, PALB2, PMS2, PTEN, RAD51C, RAD51D, SMAD4, STK11, and TP53), which were evaluated in our previous study. 9 This is our original cancer‐predisposing genes panel, which includes 25 genes from the Myriad myRisk Hereditary Cancer Test, 10 and an additional two genes (not included in the panel), namely NF1 and HOXB13, which are associated with breast/ovarian 11 and prostate 12 cancer risk, respectively. We analyzed all coding regions and two base pair flanking intronic sequences of all 27 genes (84,822 base pairs), except exons 10–15 of PMS2 due to the homology with the PMS2 pseudogene. 13 A multiplex polymerase chain reaction‐based targeted sequencing method was used to sequence the target region. 14 Sequence reads allocated to each individual were aligned to the human reference sequence (hg19) using the Burrows–Wheeler Aligner (ver. 0.7.17) and processed using the Genome Analysis Toolkit (GATK, ver. 3.7–0). For quality control, we selected individuals in whom more than 98% of the targeted region was covered with 20 or more sequencing reads. A part of the sequencing data came from our other studies (Figure S1).
We called germline variants in each individual separately using the UnifiedGenotyper and HaplotypeCaller of GATK. Genotypes for all individuals were jointly determined for each variant based on the sequencing read ratios of the reference and alternative alleles. When the alternative allele fraction was between 0 and 0.15, 0.25 and 0.75, and 0.85 and 1.00, we considered the individual a homozygote of the reference allele, heterozygote, and homozygote of the alternative allele, respectively. We excluded variants with call rates <98%, variants that did not follow the Hardy–Weinberg equilibrium in controls (p < 1.0 × 10−6), and variants with a minor allele frequency ≥0.1% in controls. The details are described in our previous studies. 4 , 9
We defined loss of function as “HIGH” impact using SnpEff impact prediction. 15 HIGH impact includes the variants that are assumed to have high impact in the protein, probably causing protein truncation, loss of function, or triggering nonsense‐mediated decay. 15 We defined “pathogenic variants” in this study as either loss‐of‐function variants determined by the SnpEff ver 4.3 t., 15 or pathogenic variants identified by ClinVar (ver. 2022‐03‐06). 16
2.3. Statistical analysis
First, we identified genes associated with lymphoma risk. We estimated ORs and the corresponding 95% confidence intervals (CIs) using a logistic regression model adjusted for age at entry and sex to evaluate the impact of each cancer‐predisposing gene on the lymphoma risk. As for MUTYH, germline pathogenic variant status was defined as an individual with biallelic variants. 10 Carriers with a germline pathogenic variant were defined in the dominant models for the other genes. In these gene‐based analyses, Bonferroni correction was applied considering multiple comparisons (p < 1.83 × 10−3 [=0.05/27]).
Second, to explore the clinical characteristics of the germline pathogenic variant carriers, we focused on genes that showed p < 0.05 in the gene‐based analyses. We compared the characteristics between pathogenic variant carriers and non‐carriers among lymphoma patients using the Mann–Whitney U‐test for continuous variables and Fisher's exact test or the χ2‐test for discrete variables. We evaluated the heterogeneous impact of pathogenic variants across the lymphoma subtypes with specific diagnoses using the above logistic regression model, Cochran's Q statistic, and the I 2 statistic.
All statistical tests were two‐sided, and p < 0.05 was considered statistically significant. Bonferroni correction was applied if necessary. All statistical analyses were performed using Stata version 16.0 (Stata Corp.) and R version 3.5.2 (R Foundation for Statistical Computing).
3. RESULTS
3.1. Characteristics of the study subjects
The characteristics of the study population are shown in Table 1. The median (interquartile range) age at entry was 66 (56–73) years in lymphoma patients and 64 (54–72) years in controls. The proportion of men was 56.5% in lymphoma patients and 53.1% in controls. The proportions of subtypes of patients were as follows: diffuse large B‐cell lymphoma, 39.9%; follicular lymphoma, 17.0%; Hodgkin lymphoma, 7.6%; extranodal marginal zone lymphoma of mucosa‐associated lymphoid tissue, 6.9%; mantle cell lymphoma (MCL), 2.1%; peripheral T‐cell lymphoma, 1.6%; and angioimmunoblastic T‐cell lymphoma, 1.2%. The proportions were comparable to the population‐based cancer registry data in Japan (Figure S2). 17
TABLE 1.
Characteristics of the study subjects
| Lymphoma patients (N = 2,066) | Control (N = 38,153) | |
|---|---|---|
| Age at entry, median (interquartile range) | 66 (56; 73) | 64 (54; 72) |
| Sex (%) | ||
| Male | 1,167 (56.5) | 20,242 (53.1) |
| Female | 899 (43.5) | 17,911 (46.9) |
| Subtype of lymphoma (%) | ||
| Diffuse large B‐cell lymphoma | 825 (39.9) | ‐ |
| Follicular lymphoma | 352 (17.0) | ‐ |
| Hodgkin lymphoma | 157 (7.6) | ‐ |
| MALT lymphoma | 143 (6.9) | ‐ |
| Mantle cell lymphoma | 44 (2.1) | ‐ |
| Peripheral T‐cell lymphoma | 33 (1.6) | ‐ |
| Angioimmunoblastic T‐cell lymphoma | 25 (1.2) | ‐ |
| Burkitt lymphoma | 14 (0.7) | ‐ |
| Extra nodal T/NK‐cell lymphoma | 13 (0.6) | ‐ |
| Anaplastic large cell lymphoma | 13 (0.6) | ‐ |
| Adult T‐cell lymphoma | 12 (0.6) | ‐ |
| B‐cell lymphoma NOS | 110 (5.3) | ‐ |
| T/NK‐cell lymphoma NOS | 19 (0.9) | ‐ |
| Other lymphoma | 77 (4.2) | ‐ |
| Unknown | 229 (11.1) | ‐ |
Abbreviations: MALT lymphoma, extranodal marginal zone lymphoma of mucosa‐associated lymphoid tissue; MCL, mantle cell lymphoma; NOS, not otherwise specified.
3.2. Annotation of germline variants
The average number of sequence reads among all study subjects was 994x. Sequencing coverages in 27 cancer‐predisposing genes among all subjects are summarized in Table S1. Following quality control of sequencing data, we included 1,982 lymphoma patients and 37,592 controls. All subjects had more than 99.8% of the targeted region covered by 20 or more sequencing reads. We annotated 309 of the 4,850 germline variants as pathogenic (Table S2). All germline pathogenic variants are shown in the Table S3.
3.3. Gene‐based association analyses between cancer‐predisposing genes and lymphoma risk
The results of the gene‐based association analyses are shown in Table 2 (all results are shown in Table S4). We observed a significant association in BRCA1 (OR, 5.88; 95% CI, 2.65–13.02; p = 1.27 × 10−5) and BRCA2 (OR, 2.94; 95% CI, 1.60–5.42; p = 5.25 × 10−4) and marginal associations (p < 0.05) in ATM (OR, 2.63; 95% CI, 1.25–5.51; p = 1.06 × 10−2) and TP53 (OR, 5.22; 95% CI, 1.43–19.02; p = 1.23 × 10−2). The proportion of carriers of these genes accounts for 1.6% of lymphoma patients (ATM: 0.4%, BRCA1: 0.4%, BRCA2: 0.6%, and TP53: 0.2%).
TABLE 2.
Gene‐based association analyses between cancer‐predisposing genes and lymphoma risk
| Number of pathogenic variant carrier (%) | OR (95% CI) a | p value a | ||
|---|---|---|---|---|
| Lymphoma patients (N = 1,982) | Control (N = 37,592) | |||
| BRCA1 | 8 (0.40) | 26 (0.07) | 5.88 (2.65–13.02) | 1.27 × 10−5 |
| BRCA2 | 12 (0.61) | 79 (0.21) | 2.94 (1.60–5.42) | 5.25 × 10−4 |
| ATM | 8 (0.40) | 59 (0.16) | 2.63 (1.25–5.51) | 1.06 × 10−2 |
| TP53 | 3 (0.15) | 10 (0.03) | 5.22 (1.43–19.02) | 1.23 × 10−2 |
| RAD51D | 11 (0.56) | 122 (0.33) | 1.72 (0.92–3.19) | 0.088 |
| MSH2 | 1 (0.05) | 3 (0.01) | 7.00 (0.72–67.54) | 0.093 |
| NBN | 1 (0.05) | 50 (0.13) | 0.39 (0.05–2.80) | 0.346 |
| EPCAM | 1 (0.05) | 8 (0.02) | 2.66 (0.33–21.36) | 0.359 |
| PALB2 | 2 (0.10) | 20 (0.05) | 1.98 (0.46–8.47) | 0.359 |
| CHEK2 | 1 (0.05) | 35 (0.09) | 0.55 (0.08–4.04) | 0.559 |
| BARD1 | 1 (0.05) | 19 (0.05) | 1.03 (0.14–7.74) | 0.974 |
3.4. Clinical characteristics of the lymphoma patients with germline pathogenic variants
To explore the clinical characteristics of the germline pathogenic variant carriers, we focused on the four genes that have significant or marginal associations with lymphoma risk (ATM, BRCA1, BRCA2, and TP53). All carriers with pathogenic variants among lymphoma patients are summarized in Table 3. Regarding family history of cancer, the proportion of lymphoma patients with a family history of breast or ovarian cancer was higher among carriers (breast cancer: carriers, 22.6%; non‐carriers, 4.9% [p = 7.00 × 10−4], ovarian cancer: carriers, 6.5%; non‐carriers, 0.5% [p = 1.42 × 10−2]), while we did not observe a statistically difference between carriers and non‐carriers in a family history of lymphoma (carriers, 0.0%; non‐carriers, 2.8% [p = 1.000]). An individual with ataxia telangiectasia, an autosomal recessive inherited disorder of ATM, has a higher risk of B‐ and T‐cell lymphoid neoplasms. 18 In the present study, eight pathogenic variant carriers of ATM were observed among the lymphoma patients. However, all were heterozygous carriers. In addition, we did not find any information supporting a family history of ataxia telangiectasia, leukemia, or lymphoma from the available data. Regarding age at diagnosis or sex, we did not observe any difference between pathogenic variant carriers and non‐carriers.
TABLE 3.
Clinical characteristics of lymphoma patients with germline pathogenic variants
| Lymphoma patients (n = 1,982) | p value | ||
|---|---|---|---|
| Pathogenic variant carriers (n = 31) | Pathogenic variant non‐carriers (n = 1,951) | ||
| Age at diagnosis, median (interquartile range) a | 61 (54; 73) | 62 (53; 70) | 0.721 |
| Sex (%) b | |||
| Male | 22 (71.0) | 1,105 (56.6) | 0.143 |
| Female | 9 (29.0) | 846 (43.4) | |
| Family history of cancer (%) | |||
| Lymphoma | 0 (0.0) | 54 (2.8) | 1.000 |
| Leukemia | 0 (0.0) | 36 (1.9) | 1.000 |
| Breast cancer | 7 (22.6) | 95 (4.9) | 7.00 × 10−4 |
| Ovarian cancer | 2 (6.5) | 10 (0.5) | 1.42 × 10−2 |
| Pancreatic cancer | 0 (0.0) | 67 (3.4) | 0.624 |
| Prostate cancer | 1 (3.2) | 57 (2.9) | 0.605 |
| Colon cancer | 1 (3.2) | 178 (9.1) | 0.356 |
| Lung cancer | 3 (9.7) | 168 (8.6) | 0.746 |
| Gastric cancer | 4 (12.9) | 413 (21.2) | 0.374 |
| Subtype of lymphoma (%) b | |||
| Diffuse large B‐cell lymphoma | 14 (45.2) | 777 (39.8) | 7.10 × 10−2 |
| Follicular lymphoma | 4 (12.9) | 329 (16.9) | |
| Hodgkin lymphoma | 1 (3.2) | 153 (7.8) | |
| MALT lymphoma | 2 (6.5) | 136 (7.0) | |
| Mantle cell lymphoma | 4 (12.9) | 40 (2.1) | |
| Peripheral T‐cell lymphoma | 0 (0.0) | 30 (1.5) | |
| Angioimmunoblastic T‐cell lymphoma | 0 (0.0) | 24 (1.2) | |
| Burkitt lymphoma | 0 (0.0) | 14 (0.7) | |
| Extra nodal T/NK‐cell lymphoma | 0 (0.0) | 12 (0.6) | |
| Anaplastic large cell lymphoma | 0 (0.0) | 13 (0.7) | |
| Adult T‐cell lymphoma | 0 (0.0) | 12 (0.6) | |
| B‐cell lymphoma NOS | 1 (3.2) | 104 (5.3) | |
| T/NK‐cell lymphoma NOS | 1 (3.2) | 18 (0.9) | |
| Other lymphoma | 0 (0.0) | 71 (3.64) | |
| Unknown | 4 (12.9) | 218 (11.2) | |
Note: Other evaluations were performed using Fisher's exact test.
Pathogenic variant carriers were defined in ATM, BRCA1, BRCA2, or TP53.
MALT lymphoma, extranodal marginal zone lymphoma of mucosa‐associated lymphoid tissue; NOS, not otherwise specified.
Evaluated by Mann–Whitney U‐test.
Evaluated using the χ2‐test.
3.5. Heterogeneous impacts of germline pathogenic variants across the subtypes of lymphoma
We observed the heterogeneous impact of germline pathogenic variants in four genes on the lymphoma risk across subtypes (Figure 1; p heterogeneity = 0.020, I 2 = 69.5%). The impact of pathogenic variants on the risk of MCL was particularly large (MCL [OR, 21.57; 95% CI, 7.59–61.26; p = 8.07 × 10−9], diffuse large B‐cell lymphoma [OR, 3.96; 95% CI, 2.28–6.86; p = 9.55 × 10−7], extranodal marginal zone lymphoma of mucosa‐associated lymphoid tissue [OR, 3.26; 95% CI, 0.80–13.30; p = 0.099], follicular lymphoma [OR, 2.66; 95% CI, 0.98–7.21; p = 0.055], and Hodgkin lymphoma [OR, 1.29; 95% CI, 0.18–9.26; p = 0.802]). In the present study, 9.1% (95% CI, 3.4%–22.4%) of MCL patients had pathogenic variants (in ATM, BRCA1, or BRCA2, which are involved in the DNA damage repair pathway). 19 Although we evaluated the combined impact of the four genes on the risk of MCL due to the limited sample size, the point estimate on the risk of MCL was high even when it was evaluated according to gene (ATM: OR, 32.34, BRCA1: OR, 32.15, and BRCA2: OR, 11.33; Figure S3).
FIGURE 1.
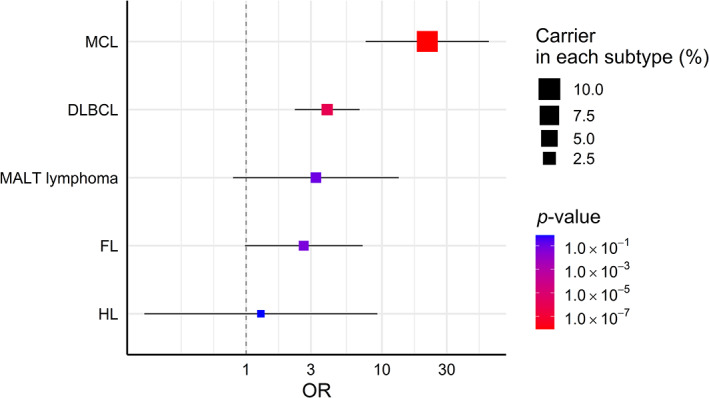
Heterogeneous impacts of germline pathogenic variants across the subtypes of lymphoma. The association of germline pathogenic variants with the lymphoma risk in each subtype was evaluated using a logistic regression model adjusted for age and sex. Error bars represent 95% CIs. Pathogenic variant carriers were defined in ATM, BRCA1, BRCA2, and TP53. CI, confidence interval; DLBCL, diffuse large B‐cell lymphoma; FL, follicular lymphoma; HL, Hodgkin lymphoma; MALT lymphoma, extranodal marginal zone lymphoma of mucosa‐associated lymphoid tissue; MCL, mantle cell lymphoma; OR, odds ratio
4. DISCUSSION
This is the largest study to evaluate the association between germline pathogenic variants in cancer‐predisposing genes and lymphoma risk in the Japanese population. We observed that the lymphoma risk was significantly associated with BRCA1 and BRCA2 and marginally associated with ATM and TP53. Furthermore, these genes were associated with a particularly high risk of MCL.
We observed a significant association of germline pathogenic variants in BRCA1 and BRCA2 with lymphoma risk. Our previous study found that pathogenic variants in BRCA1 tended to be associated with lymphoma risk, although it was not statistically significant. 4 To overcome this challenge, we increased the number of lymphoma patient subjects to detect the association in the present study. Cancer risk profiles for BRCA1 and BRCA2 are expanded to esophageal cancer, biliary tract cancer, and gastric cancer, 4 in addition to four cancers (breast cancer, ovarian cancer, prostate cancer, and pancreatic cancer). 11 In the present study, we revealed that pathogenic variants in BRCA1 and BRCA2 are also associated with lymphoma risk, indicating the expansion of the cancer risk profile of BRCA1 and BRCA2. We compared the germline pathogenic variants of BRCA1/2 found in this study with those observed in biliary duct (BRCA1), female breast (BRCA1/2), male breast (BRCA2), esophageal (BRCA2), gastric (BRCA1/2), ovarian (BRCA1/2), pancreatic (BRCA1/2), and prostate cancer (BRCA2) carriers from our previous study. 4 Sixteen pathogenic variants of BRCA1/2 were observed in lymphoma patients. Four of the 16 (25%) variants were found only in lymphoma patients (Table S5). A previous report suggested that the impacts of some pathogenic variant locations in BRCA1/2 were different for different cancer types. 20 However, these four variants found in only lymphoma patients were not concentrated in a specific location. Some lymphoma patients may be identified through surveillance of carriers with germline pathogenic variants in high‐risk genes. Although further evaluation is required, they may benefit from poly ADP‐ribose polymerase inhibitors, as in other cancers. 11
The most interesting finding in our present study was the heterogeneous impact of germline pathogenic variants, which were especially associated with a high risk of MCL. In the present study, 9.1% of MCL patients had germline pathogenic variants, which are involved in the DNA damage response pathway. 19 These findings are relevant to previous tumor genomic evaluations. A previous study identified several somatic driver mutations for MCL, which were most frequently involved in the DNA damage response pathway. 21 This suggests that alterations in the mechanisms involved in genome stability would be important for MCL pathogenesis. Furthermore, other reports mentioned that alternations in the DNA damage response pathway were observed independently of proliferation activity, implying that they might be involved in the early phase of MCL lymphomagenesis. 22 These findings indicate that some genetic variants observed in MCL may be derived from germline variants, and germline pathogenic variants in the DNA damage response pathway may predispose to MCL. 22 However, the associations between the germline variants and the risk of MCL were not evaluated. In our present study, we found that germline pathogenic variants, in particular, predispose to the risk of MCL. This provides evidence for further development of lymphoma classification, indicating the presence of a monogenic form.
Previous studies suggested the presence of a genetic predisposition to lymphoma. In genome‐wide association studies, over 250 common genetic variants were significantly associated with lymphoma risk, but the risk of most associated variants was low (median OR, 1.26; interquartile range, 1.15–1.42), 23 and clinical applications of those variants, such as risk stratification, are insufficient. 5 , 24 In general, the frequency and impact of germline variants are inversely correlated, while rare variants are known to cause monogenic form. 25 Regarding rare variants with high risk, a few studies have evaluated the association of lymphoma risk in whole‐genome sequencing or whole‐exome sequencing from the aspect of the germline in a limited sample size. 26 To increase the statistical power in the present study, we focused on the cancer‐predisposing genes in a larger number of samples to identify germline pathogenic variants associated with a high lymphoma risk and heterogeneous impact of the pathogenic variant across the subtypes.
A limitation of this study should be noted: in the exploration of the characteristics of germline pathogenic variant carriers, the potential of alpha errors cannot be ruled out. Although our present study is one of the largest studies, further extensive evaluations to confirm our results are warranted.
In conclusion, we identified significant associations between BRCA1 and BRCA2 and marginal associations of ATM and TP53 with lymphoma risk. Moreover, these genes were especially associated with an increased risk of MCL. Our results would provide novel insights concerning monogenic form into lymphoma classification. Some lymphoma patients may benefit from surveillance and targeted treatment like other neoplasms.
FUNDING INFORMATION
This research was supported by the Japan Agency for Medical Research and Development (AMED) under Grant No. JP19kk0305010.
DISCLOSURE
Keitaro Matsuo, Teruhiko Yoshida, Koichi Matsuda, Yoshinori Murakami, and Hidewaki Nakagawa are Editorial Board Members of Cancer Science. The remaining authors have no conflicts of interest concerning this study.
ETHICS STATEMENT
Approval of the research protocol by an Institutional Reviewer Board: The study was approved by the ethics committees of the Institute of Medical Sciences, The University of Tokyo, and the RIKEN Center for Integrative Medical Sciences.
INFORMED CONSENT
All participants provided written informed consent.
Supporting information
Figure S1
Figure S2
Figure S3
Table S1
Table S2
Table S3
Table S4
Table S5
ACKNOWLEDGMENTS
We thank the individuals who participated in this study. We acknowledge T. Aoi, N. Hakozaki, M. Yamaguchi, the staff of the Laboratory for Genotyping Development in RIKEN, the RIKEN‐IMS Genome Platform, and the BioBank Japan Project.
Usui Y, Iwasaki Y, Matsuo K, et al. Association between germline pathogenic variants in cancer‐predisposing genes and lymphoma risk. Cancer Sci. 2022;113:3972‐3979. doi: 10.1111/cas.15522
DATA AVAILABILITY STATEMENT
The sequence data used in this study is submitted to the NBDC human database (https://humandbs.biosciencedbc.jp/en) and will be available as JGAS000347 under the NBDC Data Sharing Policy (controlled‐access data Type‐1).
REFERENCES
- 1. International Agency for Research on Cancer . Cancer Today. Accessed November 4, 2021. https://gco.iarc.fr/today/explore
- 2. Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375‐2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang Z, Wilson CL, Armstrong GT, et al. Association of germline BRCA2 mutations with the risk of pediatric or adolescent Non‐Hodgkin lymphoma. JAMA Oncol. 2019;5:1362‐1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Momozawa Y, Sasai R, Usui Y, et al. Expansion of cancer risk profile for BRCA1 and BRCA2 pathogenic variants. JAMA Oncol. 2022;8:871‐878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sud A, Chattopadhyay S, Thomsen H, et al. Analysis of 153 115 patients with hematological malignancies refines the spectrum of familial risk. Blood. 2019;134:960‐969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391‐2405. [DOI] [PubMed] [Google Scholar]
- 7. Hirata M, Kamatani Y, Nagai A, et al. Cross‐sectional analysis of BioBank Japan clinical data: a large cohort of 200,000 patients with 47 common diseases. J Epidemiol. 2017;27:S9‐S21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nagai A, Hirata M, Kamatani Y, et al. Overview of the BioBank Japan Project: study design and profile. J Epidemiol. 2017;27:S2‐S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fujita M, Liu X, Iwasaki Y, et al. Population‐based screening for hereditary colorectal cancer variants in Japan. Clin Gastroenterol Hepatol. 2020. doi: 10.1016/j.cgh.2020.12.007. Online ahead of print. [DOI] [PubMed] [Google Scholar]
- 10. Buys SS, Sandbach JF, Gammon A, et al. A study of over 35,000 women with breast cancer tested with a 25‐gene panel of hereditary cancer genes. Cancer. 2017;123:1721‐1730. [DOI] [PubMed] [Google Scholar]
- 11. National Comprehensive Cancer Network . Genetic/Familial High‐Risk Assessment: Breast, Ovarian, and Pancreatic (Version 1); 2022. Accessed November 3, 2021. https://www.nccn.org/professionals/physician_gls/pdf/genetics_bop.pdf
- 12. Ewing CM, Ray AM, Lange EM, et al. Germline mutations in HOXB13 and prostate‐cancer risk. N Engl J Med. 2012;366:141‐149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nakagawa H, Lockman JC, Frankel WL, et al. Mismatch repair gene PMS2: disease‐causing germline mutations are frequent in patients whose tumors stain negative for PMS2 protein, but paralogous genes obscure mutation detection and interpretation. Cancer Res. 2004;64:4721‐4727. [DOI] [PubMed] [Google Scholar]
- 14. Momozawa Y, Akiyama M, Kamatani Y, et al. Low‐frequency coding variants in CETP and CFB are associated with susceptibility of exudative age‐related macular degeneration in the Japanese population. Hum Mol Genet. 2016;25:5027‐5034. [DOI] [PubMed] [Google Scholar]
- 15. Cingolani P, Platts A, Wang IL, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso‐2; iso‐3. Fly (Austin). 2012;6:80‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Landrum MJ, Lee JM, Benson M, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44:D862‐D868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chihara D, Ito H, Matsuda T, et al. Differences in incidence and trends of haematological malignancies in Japan and the United States. Br J Haematol. 2014;164:536‐545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Riaz IB, Faridi W, Patnaik MM, Abraham RS. A systematic review on predisposition to lymphoid (B and T cell) neoplasias in patients with primary immunodeficiencies and immune dysregulatory disorders (inborn errors of immunity). Front Immunol. 2019;10:777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pilié PG, Tang C, Mills GB, Yap TA. State‐of‐the‐art strategies for targeting the DNA damage response in cancer. Nat Rev Clin Oncol. 2019;16:81‐104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kuchenbaecker KB, Hopper JL, Barnes DR, et al. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA. 2017;317:2402‐2416. [DOI] [PubMed] [Google Scholar]
- 21. Nadeu F, Martin‐Garcia D, Clot G, et al. Genomic and epigenomic insights into the origin, pathogenesis, and clinical behavior of mantle cell lymphoma subtypes. Blood. 2020;136:1419‐1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jares P, Colomer D, Campo E. Genetic and molecular pathogenesis of mantle cell lymphoma: perspectives for new targeted therapeutics. Nat Rev Cancer. 2007;7:750‐762. [DOI] [PubMed] [Google Scholar]
- 23. GWAS Catalog. Accessed November 3, 2021. https://www.ebi.ac.uk/gwas/
- 24. Cerhan JR, Slager SL. Familial predisposition and genetic risk factors for lymphoma. Blood. 2015;126:2265‐2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747‐753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rotunno M, Barajas R, Clyne M, et al. A systematic literature review of whole exome and genome sequencing population studies of genetic susceptibility to cancer. Cancer Epidemiol Biomarkers Prev. 2020;29:1519‐1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Figure S3
Table S1
Table S2
Table S3
Table S4
Table S5
Data Availability Statement
The sequence data used in this study is submitted to the NBDC human database (https://humandbs.biosciencedbc.jp/en) and will be available as JGAS000347 under the NBDC Data Sharing Policy (controlled‐access data Type‐1).