

Abstract
Pembrolizumab plus chemotherapy with or without bevacizumab demonstrated prolonged progression‐free survival (PFS) and overall survival (OS) versus chemotherapy in patients with persistent, recurrent, or metastatic cervical cancer in the phase 3, randomized, double‐blind, placebo‐controlled KEYNOTE‐826 study. We report outcomes in patients enrolled in Japan. Patients received pembrolizumab 200 mg or placebo Q3W for up to 35 cycles plus chemotherapy (paclitaxel 175 mg/m2 + cisplatin 50 mg/m2 or carboplatin AUC 5) with or without bevacizumab 15 mg/kg. Dual primary endpoints were PFS per RECIST v1.1 by investigator assessment and OS in the global population; these were evaluated in patients with tumors with PD‐L1 combined positive score (CPS) ≥1, all‐comers, and PD‐L1 CPS ≥10. Fifty‐seven patients from Japan were randomized (pembrolizumab plus chemotherapy, n = 35; placebo plus chemotherapy, n = 22). Pembrolizumab plus chemotherapy improved PFS versus placebo plus chemotherapy in patients with PD‐L1 CPS ≥1 (n = 51; hazard ratio [HR; 95% CI], 0.36 [0.16–0.77]), all‐comers (n = 57; 0.45 [0.22–0.90]), and patients with PD‐L1 CPS ≥10 (n = 25; 0.36 [0.12–1.07]). HRs (95% CI) for OS were 0.38 (0.14–1.01), 0.41 (0.17–1.00), and 0.37 (0.10–1.30), respectively. Incidence of grade 3–5 AEs was 94% in the pembrolizumab group and 100% in the placebo group. Consistent with findings in the global KEYNOTE‐826 study, pembrolizumab plus chemotherapy with or without bevacizumab may prolong survival versus placebo plus chemotherapy with or without bevacizumab and had a manageable safety profile in Japanese patients with persistent, recurrent, or metastatic cervical cancer.
Keywords: bevacizumab, cervical cancer, chemotherapy, Japan, pembrolizumab
Pembrolizumab plus chemotherapy with or without bevacizumab demonstrated prolonged progression‐free survival and overall survival compared with chemotherapy in patients with persistent, recurrent, or metastatic cervical cancer in the phase 3 KEYNOTE‐826 study. In this subset analysis of patients enrolled in Japan in the KEYNOTE‐826 study, pembrolizumab plus chemotherapy with or without bevacizumab was associated with prolonged progression‐free survival and overall survival in this setting.

1. INTRODUCTION
The incidence of cervical cancer in Japan is high, 1 with approximately 12,785 new cases diagnosed and 4213 deaths in 2020. 2 The relatively higher prevalence of cervical cancer in Japan compared with many developed countries 3 may be attributable in part to low rates of cervical cancer screening and suspension of proactive recommendations for the human papilloma virus (HPV) vaccine. 4 Treatment options for patients with cervical cancer include surgery, radiation, and chemotherapy depending on disease stage. 5 , 6 , 7 The standard of care first‐line therapy for patients with persistent, recurrent, or metastatic disease has been a regimen consisting of paclitaxel combined with cisplatin or carboplatin with or without bevacizumab. 5 , 8 , 9 , 10 , 11 However, contraindications to bevacizumab are common among patients with metastatic cancer, 12 and all patients ultimately experience disease progression, underscoring an unmet need for treatment options for persistent, recurrent, or metastatic cervical cancer.
Pembrolizumab is a monoclonal antibody that binds to the programmed death 1 (PD‐1) receptor, blocking its interaction with programmed death ligand 1 (PD‐L1) and programmed death ligand 2 (PD‐L2) and thereby inhibiting tumor cells from evading immune surveillance. 13 Pembrolizumab monotherapy has demonstrated activity in patients with PD‐L1–positive cervical cancer. 14 , 15 , 16 The activity of pembrolizumab monotherapy in patients with previously treated advanced cervical cancer was evaluated in the phase 1b KEYNOTE‐028 study, which enrolled patients with PD‐L1–positive disease, 14 and the phase 2 KEYNOTE‐158 study, which enrolled patients irrespective of tumor PD‐L1 expression. 15 The objective response rate (ORR) was 17% in the KEYNOTE‐028 study and 12% in the KEYNOTE‐158 study, with all responses occurring in patients with PD‐L1–positive disease. Albeit, all of the patients in KEYNOTE‐028 and a majority (84%) of the patients with cervical cancer enrolled in the KEYNOTE‐158 study had PD‐L1–positive disease. In both studies, pembrolizumab monotherapy had a manageable safety profile.
KEYNOTE‐826 is a global, phase 3, randomized, double‐blinded, placebo‐controlled study that assessed pembrolizumab plus chemotherapy with or without bevacizumab versus placebo plus chemotherapy with or without bevacizumab as first‐line treatment for persistent, recurrent, or metastatic cervical cancer. 17 At the protocol‐specified first interim analysis, both progression‐free survival (PFS) based on Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 as assessed by the investigator and overall survival (OS) were significantly prolonged in the pembrolizumab group versus the placebo group when assessed sequentially in patients with tumors with a PD‐L1 combined positive score (CPS) ≥1, all‐comers (i.e., the intent‐to‐treat population), and patients with tumors with PD‐L1 CPS ≥10. The hazard ratios (HRs) for PFS were 0.62 (95% CI, 0.50–0.77; P < 0.001) in patients with PD‐L1 CPS ≥1, 0.65 (95% CI, 0.53–0.79; P < 0.001) among all‐comers, and 0.58 (95% CI, 0.44–0.77; P < 0.001) in patients with PD‐L1 CPS ≥10. The HRs for OS were 0.64 (95% CI, 0.50–0.81; P < 0.001) in patients with PD‐L1 CPS ≥1, 0.67 (95% CI, 0.54–0.84; P < 0.001) among all‐comers, and 0.61 (95% CI, 0.44–0.84; P = 0.001) in patients with PD‐L1 CPS ≥10. The safety profile for the pembrolizumab group was as expected for pembrolizumab and platinum‐based chemotherapy with or without bevacizumab in this setting, and no new safety signals for pembrolizumab were identified.
We present efficacy and safety results from patients enrolled in Japan in the KEYNOTE‐826 study to assess whether clinical outcomes in Japanese patients were consistent with those in the overall study population.
2. MATERIALS AND METHODS
2.1. Patients
Eligibility criteria for enrollment in the KEYNOTE‐826 study (and the study protocol) have been previously published. 17 Briefly, eligible patients were ≥18 years of age and had persistent, recurrent, or metastatic adenocarcinoma, adenosquamous carcinoma, or squamous cell carcinoma of the cervix that had not been treated with systemic chemotherapy and was not amenable to curative treatment. Prior radiotherapy, including chemoradiotherapy, was permitted if completed at least 2 weeks before randomization and treatment‐related toxicity had resolved. Patients had an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, measurable disease per RECIST version 1.1 as assessed by the investigator, and provided an adequate archival or newly obtained (preferred) tumor tissue sample for evaluation of tumor PD‐L1 expression status.
2.2. Trial design and regimens
KEYNOTE‐826 is a global, phase 3, randomized, double‐blinded, placebo‐controlled study (ClinicalTrials.gov, NCT03635567). Patients were randomly assigned (1:1) to receive pembrolizumab 200 mg or placebo once every 3 weeks (Q3W) for up to 35 cycles. All patients received chemotherapy (paclitaxel 175 mg/m2 plus either cisplatin 50 mg/m2 or carboplatin AUC 5) Q3W for six cycles; however, patients with ongoing clinical benefit who were tolerating combination chemotherapy could continue chemotherapy beyond six cycles with sponsor consultation. Patients could receive bevacizumab 15 mg/kg Q3W according to local practice at the investigator's discretion. All study medication was administered intravenously.
Treatment was continued until the maximum number of cycles for each component, radiographic progression, unacceptable toxicity, use of prohibited therapy (e.g., new antineoplastic therapy or nonpalliative radiotherapy), a decision by the investigator to discontinue the regimen, or withdrawal of consent by the patient. Randomization was stratified by metastasis at initial diagnosis (yes vs. no), investigator decision to use bevacizumab (yes vs. no), and tumor PD‐L1 status (CPS <1 vs. CPS 1 to <10 vs. CPS ≥10).
2.3. Assessments
Tumor imaging was conducted at baseline, every 9 weeks through week 54, and every 12 weeks thereafter. Adverse events (AEs) were monitored from the time of randomization through 30 days after the discontinuation of study treatment (90 days for serious AEs). AEs were graded by the investigator according to the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.0. Tumor PD‐L1 expression was assessed using PD‐L1 IHC 22C3 pharmDx (Agilent Technologies, Carpinteria, CA, USA) at a central laboratory and evaluated according to CPS, defined as the number of PD‐L1‐staining cells (tumor cells, lymphocytes, and macrophages) divided by the total number of viable tumor cells, multiplied by 100. 18
2.4. Endpoints
The dual primary endpoints of the study were PFS (time from randomization to first documented disease progression or death due to any cause, whichever occurred first) per RECIST version 1.1 by investigator assessment and OS (time from randomization to death due to any cause). Secondary endpoints included ORR (proportion of patients who had a best overall response of either confirmed complete response or partial response per RECIST version 1.1 by investigator), duration of response (DOR; for patients with a complete or partial response, time from first documented complete or partial response until first documented disease progression per RECIST version 1.1 by investigator or death due to any cause), PFS per RECIST version 1.1 by blinded independent central review (BICR), and the 12‐month PFS rate (the proportion of patients that are PFS event‐free at 12 months per RECIST version 1.1 as assessed by investigator).
2.5. Statistical analysis
For the analysis of patients enrolled in Japan, efficacy was assessed in patients with PD‐L1 CPS ≥1 tumors, all‐comers (i.e., the intent‐to‐treat population), and patients with PD‐L1 CPS ≥10 tumors. Safety was assessed in the as‐treated population (i.e., all randomly assigned patients who received at least one dose of pembrolizumab or placebo). This study was not designed to test hypotheses in the Japan subgroup and analyses in the Japan population were not alpha‐controlled. PFS per RECIST version 1.1 by investigator, OS, and DOR were estimated using the Kaplan–Meier method. For median OS and PFS, 95% CIs are provided. The magnitude of the difference between the pembrolizumab group and placebo group was estimated using an unstratified Cox proportional hazards model with Efron's method of tie handling. The between‐group difference in ORR and corresponding 95% CI were estimated using the unstratified Miettinen and Nurminen method.
3. RESULTS
3.1. Patients and treatment
The global study was carried out across 151 study sites in 19 countries, including 13 sites in Japan. Between January 8, 2019, and December 23, 2019, 57 patients from Japan were randomized to treatment (pembrolizumab group, n = 35; placebo group, n = 22). As of the data cutoff date of May 3, 2021, 18 patients in the pembrolizumab group discontinued all therapy (due to progressive disease [n = 13], AEs [n = 3], and complete response [n = 2]); at the data cutoff date, 17 patients continued to receive at least one study treatment (Figure 1). In the placebo group, 18 patients discontinued therapy (due to progressive disease and clinical progression [n = 17] and AEs [n = 1]); four patients continued to receive at least one study treatment.
FIGURE 1.

Patient disposition in the Japan subset
The median time from randomization to data cutoff in all‐comers in the Japan subset was 23.2 months (range, 16.4–27.8 months). Baseline characteristics were generally well balanced between the pembrolizumab and placebo groups (Table 1). Most patients had PD‐L1–positive disease: 30 patients (86%) in the pembrolizumab group and 21 (95%) in the placebo group had PD‐L1 CPS ≥1. The majority of patients had squamous cell carcinoma (pembrolizumab group, n = 27 [77%]; placebo group, n = 16 [73%]). A summary of study drug exposure is outlined in Table S1. Bevacizumab was administered to 22 patients (63%) in the pembrolizumab group and 17 patients (77%) in the placebo group during the study.
TABLE 1.
Demographics and baseline disease characteristics (Japan intent‐to‐treat population) a
| Characteristic |
Pembrolizumab + Chemo N = 35 |
Placebo + Chemo N = 22 |
|---|---|---|
| Age | ||
| Median (range), years | 54 (26–82) | 50 (33–78) |
| ≥65 years | 9 (26) | 5 (23) |
| ECOG performance status | ||
| 0 | 29 (83) | 16 (73) |
| 1 | 6 (17) | 6 (27) |
| Disease stage at initial diagnosis b | ||
| I | 8 (23) | 4 (18) |
| II | 13 (37) | 1 (5) |
| IIIA | 1 (3) | 2 (9) |
| IIIB | 2 (6) | 1 (5) |
| IVB | 11 (31) | 14 (64) |
| Disease status at trial entry | ||
| Metastatic c | 4 (11) | 10 (46) |
| Persistent or recurrent with distant metastases | 24 (69) | 9 (41) |
| Persistent or recurrent without distant metastases | 7 (20) | 3 (14) |
| Histologic type | ||
| Adenocarcinoma | 6 (17) | 5 (23) |
| Adenosquamous carcinoma | 2 (6) | 1 (5) |
| Squamous cell carcinoma | 27 (77) | 16 (73) |
| PD‐L1 combined positive score | ||
| <1 | 5 (14) | 1 (5) |
| 1 to <10 | 15 (43) | 11 (50) |
| ≥10 | 15 (43) | 10 (46) |
| Previous therapy | ||
| Chemoradiotherapy and surgery | 6 (17) | 1 (5) |
| Radiotherapy and surgery | 2 (6) | 1 (5) |
| Chemoradiotherapy only | 17 (49) | 9 (41) |
| Radiotherapy only | 2 (6) | 1 (5) |
| Surgery only | 4 (11) | 0 |
| None | 4 (11) | 10 (45) |
| Bevacizumab use during the trial | ||
| Yes | 22 (63) | 17 (77) |
| No | 13 (37) | 5 (23) |
Abbreviations: ECOG, Eastern Cooperative Oncology Group; PD‐L1, programmed death ligand 1.
Data are presented as n (%) unless specified otherwise.
Stage at initial diagnosis determined using FIGO 2009/NCCN 2017 criteria.
Metastatic includes patients with paraaortic lymph node involvement.
3.2. Efficacy
Hazard ratios for PFS per RECIST version 1.1 by investigator favored the pembrolizumab group versus the placebo group for patients in the PD‐L1 CPS ≥1 population, all‐comers, and patients in the PD‐L1 CPS ≥10 population. Among patients in the PD‐L1 CPS ≥1 population, median PFS was not reached (NR; 95% CI, 12.2 months–NR) in the pembrolizumab group versus 11.9 months (95% CI, 6.4–14.5 months) in the placebo group (HR, 0.36; 95% CI, 0.16–0.77). Among all‐comers, median PFS was NR (95% CI, 10.2 months–NR) in the pembrolizumab group versus 11.5 months (95% CI, 8.1–13.6 months) in the placebo group (HR, 0.45; 95% CI, 0.22–0.90). In the PD‐L1 CPS ≥10 population, median PFS was NR (95% CI, 6.3 months–NR) in the pembrolizumab group versus 12.3 months (95% CI, 4.0–14.5 months) in the placebo group (HR, 0.36; 95% CI, 0.12–1.07; Figure 2). HRs for PFS were similar when assessed by BICR: 0.52 (0.23–1.17) in the PD‐L1 CPS ≥1 population, 0.62 (0.29–1.30) among all‐comers, and 0.41 (0.13–1.26) in the PD‐L1 CPS ≥10 population.
FIGURE 2.
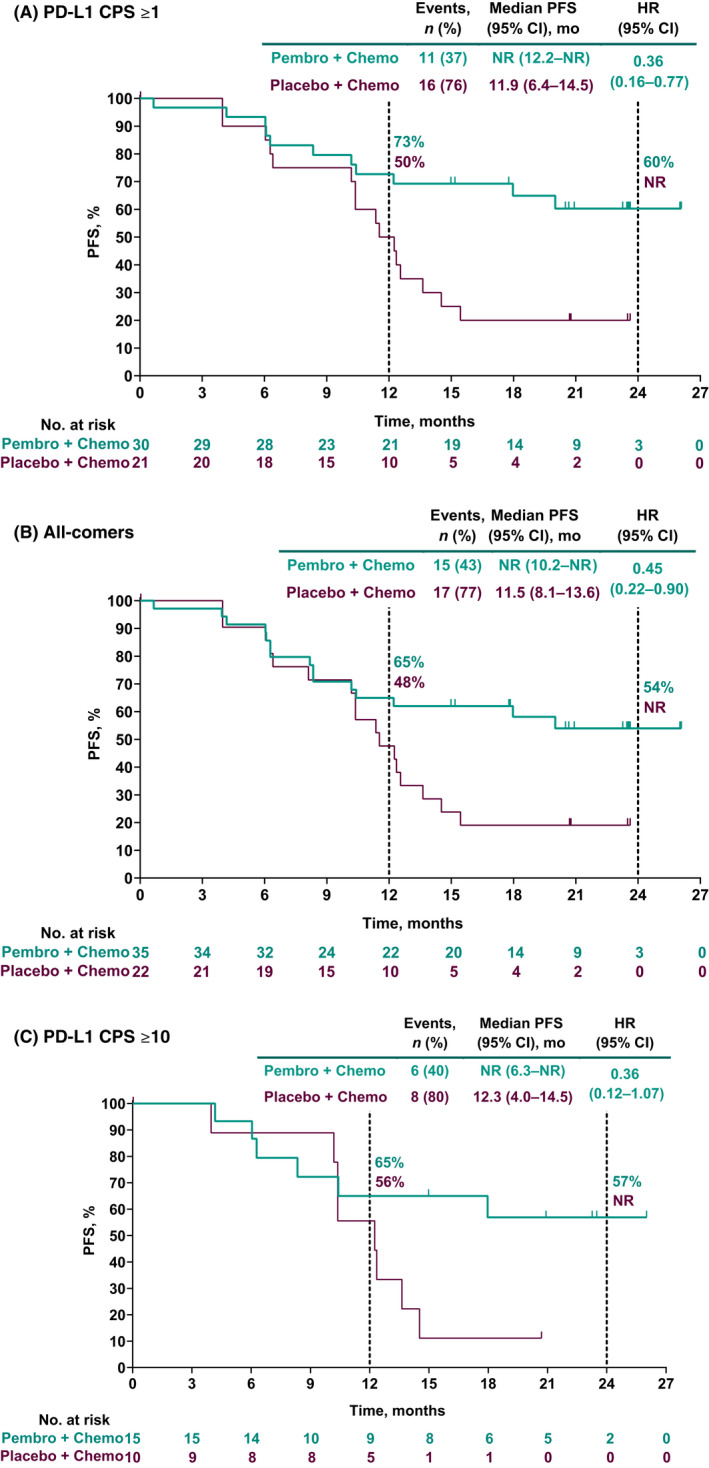
Progression‐free survival (PFS) per RECIST version 1.1 by investigator in (A) patients with programmed death ligand 1 (PD‐L1) combined positive score (CPS) ≥1 tumors, (B) all‐comer patients, and (C) patients with PD‐L1 CPS ≥10 tumors. HR, hazard ratio; NR, not reached
Similar to PFS per RECIST version 1.1 by investigator, HRs for OS favored the pembrolizumab group versus the placebo group across all three study populations. As of data cutoff, 9 of 35 patients (26%) in the pembrolizumab group and 11 of 22 patients (50%) in the placebo group had died (all‐comers population). Among patients in the PD‐L1 CPS ≥1 population, all‐comers, and the PD‐L1 CPS ≥10 population, HRs (95% CI) for OS were 0.38 (0.14–1.01), 0.41 (0.17–1.00), and 0.37 (0.10–1.30), respectively. For the pembrolizumab group versus the placebo group, 24‐month OS estimates were 75% versus 56% for patients in the PD‐L1 CPS ≥1 population, 72% versus 51% for all‐comers, and 72% versus 50% for patients in the PD‐L1 CPS ≥10 population (Figure 3).
FIGURE 3.
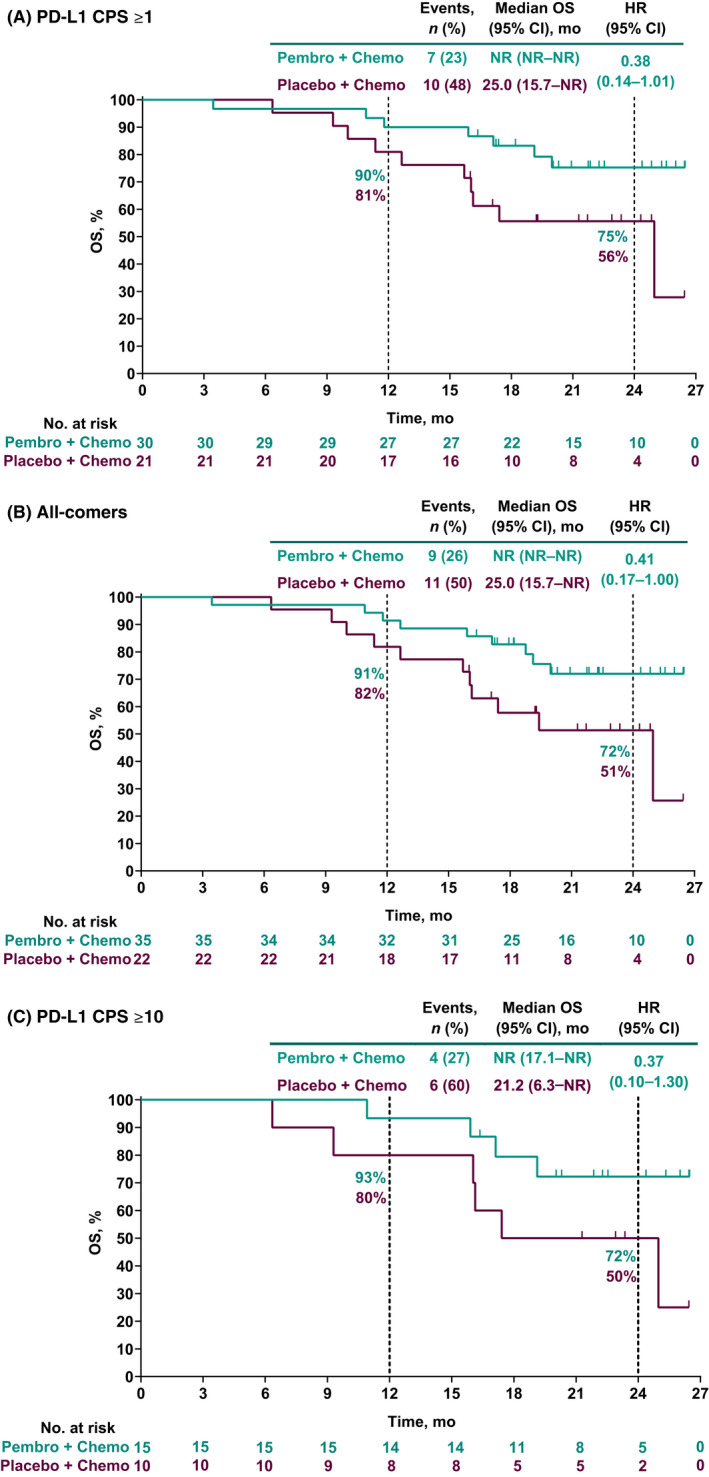
Overall survival (OS) in (A) patients with programmed death ligand 1 (PD‐L1) combined positive score (CPS) ≥1 tumors, (B) all‐comer patients, and (C) patients with PD‐L1 CPS ≥10 tumors. HR, hazard ratio; NR, not reached; PFS, progression‐free survival
The confirmed ORR was higher and DOR was longer in the pembrolizumab group compared with the placebo group across the three study populations (Table 2). Among patients in the PD‐L1 CPS ≥1 population, the ORR for patients in the pembrolizumab group was 80% (95% CI, 61%–92%) versus 71% (95% CI, 48%–89%) in the placebo group. Median DOR was NR (range, 4.0+ to 24.1+ months) in the pembrolizumab group and 10.5 months (range, 4.2 to 21.7+ months) in the placebo group. Among all‐comers, ORR was 77% (95% CI, 60%–90%) in the pembrolizumab group versus 68% (95% CI, 45%–86%) in the placebo group; median DOR was NR (range, 4.0+ to 24.1+ months) in the pembrolizumab group and 10.5 months (range, 4.2 to 21.7+ months) in the placebo group. Among the PD‐L1 CPS ≥10 population, ORR was 87% (95% CI, 60%–98%) in the pembrolizumab group versus 60% (95% CI, 26%–88%) in the placebo group; median DOR was NR (range, 4.0+ to 24.1+ months) and 9.4 months (range, 8.2 to 18.7+ months), respectively.
TABLE 2.
Summary of confirmed objective response per RECIST version 1.1 by investigator assessment
| PD‐L1 CPS ≥1 | All‐Comer | PD‐L1 CPS ≥10 | ||||
|---|---|---|---|---|---|---|
| Pembrolizumab | Placebo | Pembrolizumab | Placebo | Pembrolizumab | Placebo | |
| N = 30 | N = 21 | N = 35 | N = 22 | N = 15 | N = 10 | |
| Objective response rate, % (95% CI) a | 80 (61–92) | 71 (48–89) | 77 (60–90) | 68 (45–86) | 87 (60–98) | 60 (26–88) |
| Difference vs. placebo, % (95% CI) b | 9 (−15 to 34) | 9 (−14 to 33) | 27 (−8 to 59) | |||
| Best overall response, n (%) | ||||||
| Complete response | 12 (40) | 5 (24) | 12 (34) | 5 (23) | 6 (40) | 1 (10) |
| Partial response | 12 (40) | 10 (48) | 15 (43) | 10 (46) | 7 (47) | 5 (50) |
| Stable disease | 5 (17) | 5 (24) | 7 (20) | 6 (27) | 2 (13) | 3 (30) |
| Progressive disease | 1 (3) | 0 | 1 (3) | 0 | 0 | 0 |
| Not evaluable | 0 | 0 | 0 | 0 | 0 | 0 |
| No assessment | 0 | 1 (5) | 0 | 1 (5) | 0 | 1 (10) |
| Median time to response (range), months | 2.1 (1.9–10.2) | 2.1 (1.9–4.2) | 2.1 (1.9–10.2) | 2.1 (1.9–4.2) | 2.2 (1.9–4.4) | 2.0 (1.9–2.2) |
| Median duration of response (range), months c |
NR (4.0+ to 24.1+) |
10.5 (4.2 to 21.7+) |
NR (4.0+ to 24.1+) |
10.5 (4.2 to 21.7+) |
NR (4.0+ to 4.1+) |
9.4 (8.2 to 18.7+) |
Note: “+” indicates there was no progressive disease by the time of last disease assessment.
Abbreviations: CPS, combined positive score; PD‐L1, programmed death ligand 1; RECIST, Response Evaluation Criteria in Solid Tumors.
Includes patients with best objective response with confirmation as complete response or partial response.
Based on Miettinen and Nurminen method.
From product‐limit (Kaplan–Meier) method for censored data.
3.3. Safety
The median duration of treatment was 15.6 months (range, 0.0–26.0 months) in the pembrolizumab group and 12.5 months (0.0–24.1 months) in the placebo group. All patients in both the pembrolizumab group and the placebo group experienced at least one AE (Table 3). Grade 3–5 AEs occurred in 33 patients (94%) in the pembrolizumab group and 22 patients (100%) in the placebo group. There were no grade 5 AEs in either treatment group. The most frequently occurring AEs included alopecia (pembrolizumab group, 89% vs. placebo group, 77%), decreased neutrophil count (71% vs. 73%), peripheral sensory neuropathy (57% vs. 77%), and anemia (63% vs. 55%). The most frequently occurring grade 3–5 AEs were decreased neutrophil count (pembrolizumab, 60% vs. placebo, 59%), anemia (40% vs. 27%), and decreased white blood cell count (34% vs. 32%). Serious AEs occurred in 15 patients (43%) in the pembrolizumab group and 10 patients (46%) in the placebo group. Toxicity led to discontinuation for 11 patients (31%) in the pembrolizumab group and five patients (23%) in the placebo group.
TABLE 3.
Summary of AEs (Japan all patients as‐treated population) a
| Adverse event |
Pembrolizumab + Chemo N = 35 |
Placebo + Chemo N = 22 |
||
|---|---|---|---|---|
| Any grade | 35 (100) | 22 (100) | ||
| Grade 3–5 b | 33 (94) | 22 (100) | ||
| Serious | 15 (43) | 10 (46) | ||
| Led to discontinuation | 11 (31) | 5 (23) | ||
| AEs occurring in ≥25% of patients | Any grade | Grade 3–5 | Any grade | Grade 3–5 |
| Alopecia | 31 (89) | 0 | 17 (77) | 0 |
| Neutrophil count decreased | 25 (71) | 21 (60) | 16 (73) | 13 (59) |
| Anemia | 22 (63) | 14 (40) | 12 (55) | 6 (27) |
| Peripheral sensory neuropathy | 20 (57) | 0 | 17 (77) | 1 (5) |
| White blood cell count decreased | 19 (54) | 12 (34) | 10 (46) | 7 (32) |
| Constipation | 17 (49) | 0 | 8 (36) | 0 |
| Arthralgia | 15 (43) | 0 | 11 (50) | 0 |
| Nausea | 14 (40) | 0 | 13 (59) | 1 (5) |
| Platelet count decreased | 13 (37) | 4 (11) | 8 (36) | 1 (5) |
| Stomatitis | 12 (34) | 0 | 4 (18) | 0 |
| Diarrhea | 10 (29) | 0 | 6 (27) | 0 |
| Peripheral neuropathy | 9 (26) | 0 | 3 (14) | 0 |
| Alanine aminotransferase increased | 6 (17) | 3 (9) | 7 (32) | 3 (14) |
| Myalgia | 6 (17) | 0 | 6 (27) | 0 |
| Hypertension | 6 (17) | 1 (3) | 12 (55) | 6 (27) |
| Aspartate aminotransferase increased | 5 (14) | 1 (3) | 7 (31) | 0 |
| Immune‐mediated AEs and infusion reactions | Any grade | Grade 3 | Any grade | Grade 3 |
| Any | 15 (43) | 5 (14) | 5 (23) | 2 (9) |
| Infusion reactions | 5 (14) | 0 | 4 (18) | 2 (9) |
| Hyperthyroidism | 4 (11) | 0 | 0 | 0 |
| Hypothyroidism | 3 (9) | 0 | 1 (5) | 0 |
| Thyroiditis | 3 (9) | 1 (3) | 0 | 0 |
| Severe skin reactions | 3 (9) | 3 (9) | 0 | 0 |
| Vasculitis | 2 (6) | 0 | 0 | 0 |
| Colitis | 2 (6) | 0 | 0 | 0 |
| Hepatitis | 2 (6) | 2 (6) | 0 | 0 |
| Myocarditis | 1 (3) | 1 (3) | 0 | 0 |
| Myositis | 1 (3) | 0 | 0 | 0 |
| Adrenal insufficiency | 1 (3) | 1 (3) | 0 | 0 |
Abbreviation: AE, adverse event.
Data are presented as n (%).
There were no grade 5 AEs in either treatment group.
Immune‐mediated AEs and infusion reactions occurred in 43% of patients in the pembrolizumab group and 23% of patients in the placebo group. Grade 3 immune‐mediated AEs and infusion reactions occurred in 14% of patients in the pembrolizumab group and 9% of patients in the placebo group; there were no grade 4 or 5 events. The most common immune‐mediated AEs were hyperthyroidism (pembrolizumab group, 11% vs. placebo group, 0%), hypothyroidism (9% vs. 5%), severe skin reactions (9% vs. 0%), and thyroiditis (9% vs. 0%). Infusion reactions occurred in 14% of patients in the pembrolizumab group and 18% of patients in the placebo group.
4. DISCUSSION
In this subset analysis of the phase 3 KEYNOTE‐826 study evaluating outcomes among patients enrolled in Japan, pembrolizumab plus chemotherapy with or without bevacizumab was associated with prolonged PFS per RECIST version 1.1 by investigator and OS compared with placebo plus chemotherapy with or without bevacizumab as first‐line therapy for persistent, recurrent, or metastatic cervical cancer. PFS outcomes determined by the investigator were consistent with those determined by BICR. These favorable efficacy outcomes for the pembrolizumab group versus the placebo group are consistent with those from the KEYNOTE‐826 global study showing that the addition of pembrolizumab to chemotherapy with or without bevacizumab significantly improved both PFS and OS (the primary study endpoints), independent of PD‐L1 CPS status. 17 Based on results from the global study, pembrolizumab plus chemotherapy with or without bevacizumab was recently approved by the US Food and Drug Administration for the treatment of patients with persistent, recurrent, or metastatic cervical cancer whose tumors express PD‐L1 (CPS ≥1). 19
The magnitude of the treatment effect favoring the pembrolizumab group over the placebo group was more pronounced for the Japan subset than for the global study population. This apparent greater magnitude of benefit among Japanese patients was observed across all three study populations (i.e., patients with PD‐L1 CPS ≥1 disease, all‐comers, and patients with PD‐L1 CPS ≥10 disease); albeit, the 95% CIs for PFS and OS were wider in the Japan subset compared with the global study because of a smaller number of patients. These data provide support for improved survival with the addition of pembrolizumab to chemotherapy with or without bevacizumab in Japanese patients. Furthermore, the finding from the current analysis suggesting a trend for treatment benefit irrespective of tumor PD‐L1 expression is consistent with the global study. However, it is important to note that there were very few patients with PD‐L1‐negative tumors in the Japan subset (five in the pembrolizumab group and one in the placebo group) and that all the HRs for PFS and OS were similar in each population, with wide 95% CIs, precluding a definitive conclusion being drawn from these data.
Data from the Japan subset showed an ORR and DOR treatment effect that favored the pembrolizumab group over the placebo group across all three populations. This is consistent with the global study, which demonstrated a higher ORR and longer DOR in the pembrolizumab group compared with placebo in patients with PD‐L1 CPS ≥1 (ORR, 68.1% vs. 50.2%; DOR, 18.0 vs. 10.4 months), all‐comers (65.9% vs. 50.8%; 18.0 vs. 10.4 months), and PD‐L1 CPS ≥10 (69.6% vs. 49.1%; 21.1 vs. 9.4 months). 17
As in the global population, the safety profile of pembrolizumab plus chemotherapy with or without bevacizumab was as anticipated given the previously reported toxicity with the individual treatment components, and no new safety signals were identified. As anticipated (given the mechanism of action of pembrolizumab), the incidence of immune‐mediated AEs was higher in the pembrolizumab group than in the placebo group. The overall safety profile of this treatment regimen among Japanese patients was consistent with that in the KEYNOTE‐826 global study. 17 Among patients in the Japan subset, grade 3–5 AEs occurred in 94% of patients in the pembrolizumab group and 100% of patients in the placebo group, compared with 82% and 75%, respectively, in the global study. Notwithstanding these higher rates of grade 3–5 toxicity, there was no evidence of treatment discontinuation due to toxicity in Japanese patients. Rates of treatment discontinuation due to AEs in the Japan subset were 31% in the pembrolizumab group and 23% in the placebo group versus 37% and 27%, respectively, in the global study. Additionally, it is important to note that no fatal AEs occurred in either treatment group among patients enrolled in Japan. The higher incidence of grade 3–5 AEs in the Japan subset may reflect differences in the demographic and clinical characteristics of the Japanese population compared with the global population. Notably, the proportion of patients aged ≥65 years was higher in the Japan subset (25%) than in the global study population (16%). Additionally, the Japan subset also had a higher proportion of patients with an ECOG performance status of 0 (79%) and rate of bevacizumab use (68%) than in the global population (56% and 63% respectively). 17
Other studies have reported results of outcomes with paclitaxel and carboplatin with or without bevacizumab in Japanese patients with metastatic or recurrent cervical cancer. In the phase 3 JCOG0505 study of paclitaxel plus carboplatin versus paclitaxel plus cisplatin in 253 patients in Japan with metastatic or recurrent cervical cancer, median OS was 18.3 versus 17.5 months and median PFS was 6.9 versus 6.2 months, respectively. 9 Similar findings were also observed in the subsequent JCOG1311 study, that evaluated conventional paclitaxel and carboplatin with or without bevacizumab versus dose‐dense paclitaxel and carboplatin with or without bevacizumab in 122 patients in Japan with stage IVB, recurrent, or persistent cervical carcinoma (median OS, 19.6 vs. 16.4 months; median PFS, 7.7 vs. 7.2 months). 20 The median OS and PFS reported in the aforementioned studies are shorter than those observed in the placebo group in the current study, probably due to the limited number of patients in this group. 9 , 20
Limitations of this subset analysis include the small size of the Japan population, consisting of 57 patients in total. The small size of the patient population limits our ability to draw definitive conclusions from this subset analysis and to perform subset analyses. It is important to note that all analyses are descriptive; no alpha was assigned. In addition, there was an imbalance in the number of patients between the pembrolizumab and placebo groups (n = 35 vs. 22) in the Japan subset.
In conclusion, pembrolizumab plus chemotherapy with or without bevacizumab may prolong PFS and OS with manageable toxicity in Japanese patients with persistent, recurrent, or metastatic cervical cancer, consistent with the findings in the global KEYNOTE‐826 study.
FUNDING INFORMATION
Funding for this research was provided by Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA (MSD).
DISCLOSURE
Kan Yonemori has received lecture fees, honoraria, or other fees from Pfizer, Eisai, Chugai Pharmaceutical Co., Ltd, AstraZeneca, Takeda, Fuji Pharma. Takashi Iwata has received research funds from Merck & Co., Inc., Rahway, NJ, USA. Aikou Okamoto has received lecture fees, honoraria, or other fees from AstraZeneca K.K., Takeda Pharmaceutical Company Ltd; research funds from MSD K.K., Eisai Co., Ltd, Takeda Pharmaceutical Company Ltd, Daiichi Sankyo Co., Ltd; scholarship or research grant from Taiho Pharmaceutical Co., Ltd, Chugai Pharmaceutical Co., Ltd, Mochida Pharmaceutical Co., Ltd, Kaken Pharmaceutical Co., Ltd, Gyne Mom Co., Ltd, Terumo Corporation. Kenichi Harano has received research funds from Merck and Daiichi Sankyo. Keiko Yamamoto, Takeshi Maruko, and Hiroyuki Ugai are employees of MSD K.K., Tokyo, Japan. Cumhur Tekin is an employee of MSD. Nicoletta Colombo has received remuneration from Roche, PharmaMar, AstraZeneca, Clovis, MSD, GSK, Pfizer, Immunogen, Mersana, Eisai, Oncherna, Novartis, Nuvation. Keiichi Fujiwara has received lecture fees, honoraria, or other fees from Daiichi Sankyo, Genmab, Nanocarrier; research grants (to institution) from AstraZeneca, Eisai, Genmab, MSD, Takeda. Kosei Hasegawa has received lecture fees, honoraria, or other fees from MSD K.K., AstraZeneca, Chugai Pharmaceutical Co., Ltd; research funding from MSD K.K., Ono Pharmaceutical. Kimio Ushijima has received research funding from Merck. All other authors have no conflicts of interest to disclose. The study was funded by Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA. All authors had access to the data from the study and had final responsibility for the decision to submit for publication.
ETHICS STATEMENT
Approval of the research protocol by an Institutional Reviewer Board: The trial protocol was approved by the appropriate ethics body at each site, and the trial was conducted in accordance with Good Clinical Practice guidelines and the Declaration of Helsinki.
CONSENT
Informed consent was obtained from all patients.
Supporting information
Table S1
ACKNOWLEDGMENTS
Medical writing assistance was provided by Lisa Denny, PhD, of ICON plc (Blue Bell, PA, USA). This assistance was funded by Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA (MSD). Additionally, the authors thank the following MSD personnel: Gursel Aktan, Steve Keefe, and Sarper Toker for trial design; Amy Blum, Susan Galligan, and Karyn O'Flaherty for trial support; Kan Li, Ying Zhang, and Jing Zhao for statistical support; and Yusuke Kajimoto for publication support.
Nishio S, Yonemori K, Usami T, et al. Pembrolizumab plus chemotherapy in Japanese patients with persistent, recurrent or metastatic cervical cancer: Results from KEYNOTE‐826. Cancer Sci. 2022;113:3877‐3887. doi: 10.1111/cas.15479
The trial is registered with Clinicaltrials.gov: NCT03635567.
DATA AVAILABILITY STATEMENT
Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA (MSD) is committed to providing qualified scientific researchers access to anonymized data and clinical study reports from the company's clinical trials for the purpose of conducting legitimate scientific research. MSD is also obligated to protect the rights and privacy of trial participants and, as such, has a procedure in place for evaluating and fulfilling requests for sharing company clinical trial data with qualified external scientific researchers. The MSD data‐sharing website (available at: http://engagezone.msd.com/ds_documentation.php) outlines the process and requirements for submitting a data request. Applications will be promptly assessed for completeness and policy compliance. Feasible requests will be reviewed by a committee of MSD subject matter experts to assess the scientific validity of the request and the qualifications of the requestors. In line with data privacy legislation, submitters of approved requests must enter into a standard data‐sharing agreement with MSD before data access is granted. Data will be made available for request after product approval in the USA and the European Union (EU) or after product development is discontinued. There are circumstances that may prevent MSD from sharing requested data, including country‐ or region‐specific regulations. If the request is declined, it will be communicated to the investigator. Access to genetic or exploratory biomarker data requires a detailed, hypothesis‐driven statistical analysis plan that is collaboratively developed by the requestor and MSD subject matter experts; after approval of the statistical analysis plan and execution of a data‐sharing agreement, MSD will either perform the proposed analyses and share the results with the requestor or will construct biomarker covariates and add them to a file with clinical data that are uploaded to an analysis portal so that the requestor can perform the proposed analyses.
REFERENCES
- 1. Arbyn M, Weiderpass E, Bruni L, et al. Estimates of incidence and mortality of cervical cancer in 2018: a worldwide analysis. Lancet Glob Health. 2020;8(2):e191‐e203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bruni L, Albero G, Serrano B, et al. Human papillomavirus and related diseases in Japan. Summary report 22 October 2021. ICO/IARC Information Centre on HPV and Cancer (HPV Information Centre); 22 October, 2021.
- 3. World Health Organization International Agency for Research on Cancer (IARC) . Cancer Today. https://gco.iarc.fr/today/home. Accessed January 5, 2022.
- 4. Hanley SJ, Yoshioka E, Ito Y, Kishi R. HPV vaccination crisis in Japan. Lancet. 2015;385(9987):2571. [DOI] [PubMed] [Google Scholar]
- 5. Ebina Y, Mikami M, Nagase S, et al. Japan Society of Gynecologic Oncology guidelines 2017 for the treatment of uterine cervical cancer. Int J Clin Oncol. 2019;24(1):1‐19. [DOI] [PubMed] [Google Scholar]
- 6. Marth C, Landoni F, Mahner S, et al. Cervical cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow‐up. Ann Oncol. 2017;28(Suppl 4):iv72‐iv83. [DOI] [PubMed] [Google Scholar]
- 7. National Comprehensive Cancer Network (NCCN) . NCCN clinical practice guidelines in oncology (NCCN guidelines). Cervical cancer. Version 1.2022. https://www.nccn.org/professionals/physician_gls/pdf/cervical.pdf. Accessed October 29, 2021.
- 8. Moore DH, Blessing JA, McQuellon RP, et al. Phase III study of cisplatin with or without paclitaxel in stage IVB, recurrent, or persistent squamous cell carcinoma of the cervix: a gynecologic oncology group study. J Clin Oncol. 2004;22(15):3113‐3119. [DOI] [PubMed] [Google Scholar]
- 9. Kitagawa R, Katsumata N, Shibata T, et al. Paclitaxel plus carboplatin versus paclitaxel plus cisplatin in metastatic or recurrent cervical cancer: the open‐label randomized phase III trial JCOG0505. J Clin Oncol. 2015;33(19):2129‐2135. [DOI] [PubMed] [Google Scholar]
- 10. Tewari KS, Sill MW, Penson RT, et al. Bevacizumab for advanced cervical cancer: final overall survival and adverse event analysis of a randomised, controlled, open‐label, phase 3 trial (Gynecologic Oncology Group 240). Lancet. 2017;390(10103):1654‐1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tewari KS, Sill MW, Long HJ III, et al. Improved survival with bevacizumab in advanced cervical cancer. N Engl J Med. 2014;370(8):734‐743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Skelton WP IV, Castagno J, Cardenas‐Goicoechea J, et al. Bevacizumab eligibility in patients with metastatic and recurrent cervical cancer: a retrospective review. Clin Med Insights Oncol. 2018;12:1179554918779587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Khoja L, Butler MO, Kang SP, Ebbinghaus S, Joshua AM. Pembrolizumab. J Immunother Cancer. 2015;3:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Frenel JS, Le Tourneau C, O'Neil B, et al. Safety and efficacy of pembrolizumab in advanced, programmed death ligand 1‐positive cervical cancer: results from the phase Ib KEYNOTE‐028 trial. J Clin Oncol. 2017;35(36):4035‐4041. [DOI] [PubMed] [Google Scholar]
- 15. Chung HC, Ros W, Delord JP, et al. Efficacy and safety of pembrolizumab in previously treated advanced cervical cancer: results from the phase II KEYNOTE‐158 study. J Clin Oncol. 2019;37(17):1470‐1478. [DOI] [PubMed] [Google Scholar]
- 16. Kranawetter M, Rohrich S, Mullauer L, et al. Activity of pembrolizumab in recurrent cervical cancer: case series and review of published data. Int J Gynecol Cancer. 2018;28(6):1196‐1202. [DOI] [PubMed] [Google Scholar]
- 17. Colombo N, Dubot C, Lorusso D, et al. Pembrolizumab for persistent, recurrent, or metastatic cervical cancer. N Engl J Med. 2021;385(20):1856‐1867. [DOI] [PubMed] [Google Scholar]
- 18. Kulangara K, Zhang N, Corigliano E, et al. Clinical utility of the combined positive score for programmed death ligand‐1 expression and the approval of pembrolizumab for treatment of gastric cancer. Arch Pathol Lab Med. 2019;143(3):330‐337. [DOI] [PubMed] [Google Scholar]
- 19. US Food and Drug Administration . FDA approves pembrolizumab combination for the first‐line treatment of cervical cancer. https://www.fda.gov/drugs/resources‐information‐approved‐drugs/fda‐approves‐pembrolizumab‐combination‐first‐line‐treatment‐cervical‐cancer. Accessed October 29, 2021.
- 20. Ishikawa M, Shibata T, Iwata T, et al. A randomized phase II/III trial of conventional paclitaxel and carboplatin with or without bevacizumab versus dose‐dense paclitaxel and carboplatin with or without bevacizumab, in stage IVB, recurrent, or persistent cervical carcinoma (JCOG1311): primary analysis. Gynecol Oncol. 2021;162(2):292‐298. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Data Availability Statement
Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA (MSD) is committed to providing qualified scientific researchers access to anonymized data and clinical study reports from the company's clinical trials for the purpose of conducting legitimate scientific research. MSD is also obligated to protect the rights and privacy of trial participants and, as such, has a procedure in place for evaluating and fulfilling requests for sharing company clinical trial data with qualified external scientific researchers. The MSD data‐sharing website (available at: http://engagezone.msd.com/ds_documentation.php) outlines the process and requirements for submitting a data request. Applications will be promptly assessed for completeness and policy compliance. Feasible requests will be reviewed by a committee of MSD subject matter experts to assess the scientific validity of the request and the qualifications of the requestors. In line with data privacy legislation, submitters of approved requests must enter into a standard data‐sharing agreement with MSD before data access is granted. Data will be made available for request after product approval in the USA and the European Union (EU) or after product development is discontinued. There are circumstances that may prevent MSD from sharing requested data, including country‐ or region‐specific regulations. If the request is declined, it will be communicated to the investigator. Access to genetic or exploratory biomarker data requires a detailed, hypothesis‐driven statistical analysis plan that is collaboratively developed by the requestor and MSD subject matter experts; after approval of the statistical analysis plan and execution of a data‐sharing agreement, MSD will either perform the proposed analyses and share the results with the requestor or will construct biomarker covariates and add them to a file with clinical data that are uploaded to an analysis portal so that the requestor can perform the proposed analyses.