

Abstract
The rearrangement of anaplastic lymphoma kinase (ALK) occurs in 3%‐5% of patients with non–small cell lung cancer (NSCLC) and confers sensitivity to ALK–tyrosine kinase inhibitors (TKIs). For the treatment of patients with ALK‐rearranged NSCLC, various additional ALK‐TKIs have been developed. Ceritinib is a second‐generation ALK‐TKI and has shown great efficacy in the treatment of patients with both newly diagnosed and crizotinib (a first‐generation ALK‐TKI)‐refractory ALK‐rearranged NSCLC. However, tumors can also develop ceritinib resistance. This may result from secondary ALK mutations, but other mechanisms responsible for this have not been fully elucidated. In this study, we explored the mechanisms of ceritinib resistance by establishing ceritinib‐resistant, echinoderm microtubule‐associated protein‐like 4 (EML4)‐ALK–positive H3122 cells and ceritinib‐resistant patient‐derived cells. We identified a mechanism of ceritinib resistance induced by bypass signals that is mediated by the overexpression and activation of fibroblast growth factor receptor 3 (FGFR3). FGFR3 knockdown by small hairpin RNA or treatment with FGFR inhibitors was found to resensitize the resistant cells to ceritinib in vitro and in vivo. FGFR ligands from either human serum or fetal bovine serum were able to activate FGFR3 and induce ceritinib resistance. A detailed analysis of ceritinib‐resistant patient‐derived specimens confirmed that tyrosine‐protein kinase Met (cMET) amplification induces ceritinib resistance. Amplified cMET counteractivated EGFR and/or Her3 and induced ceritinib resistance. These results reveal multiple ceritinib resistance mechanisms and suggest that ceritinib resistance might be overcome by identifying precise resistance mechanisms.
Keywords: ALK kinase, ceritinib, drug resistance, FGFR3, lung cancer
For the treatment of ALK rearranged non‐small cell lung cancer, 5 ALK inhibitors approved and prognosis has been markedly improved, but still the emergence of drug resistance is a major obstacle. In this study it was newly found that FGFR3 overexpression and activation mediated ALK‐TKI resistance as a bypass pathway activation mediated resistance.
Abbreviations
- ALK
anaplastic lymphoma kinase
- EGFR
epidermal growth factor receptors
- ERK
extracellular signal–regulated kinases
- FGFR
fibroblast growth factor receptor
- FISH
fluorescence in situ hybridization
- RTK
receptor tyrosine kinase
- TKI
tyrosine kinase inhibitors
1. INTRODUCTION
Worldwide, approximately 1.8 million people die of lung cancer annually. This makes it the leading cause of cancer deaths. 1 As sequencing technologies have developed, multiple oncogenic driver mutations and genetic alterations in patients with lung cancer have been identified. Among these is anaplastic lymphoma kinase (ALK) rearrangement, which occurs in 3%‐5% of those with lung adenocarcinoma. 2 ALK rearrangement is most frequently seen in the echinoderm microtubule‐associated protein‐like 4 (EML4)‐ALK fusion gene of individuals with non–small cell lung cancer (NSCLC). As this was first identified in 2007, numerous other ALK fusion genes with various fusion partners have been identified in patients with lung cancer. 3 , 4 In cases of ALK‐rearranged lung cancer, the patients tend to be relatively young and nonsmokers or light smokers. Their tumors are usually adenocarcinomas. 2 , 5 Crizotinib is a first‐generation ALK inhibitor that was approved for the treatment of patients with ALK‐rearranged NSCLC in 2011 by the Food and Drug Administration (FDA) in the United States and in 2012 by the Pharmaceuticals and Medical Devices Agency (PMDA) in Japan. 6 , 7 , 8
Crizotinib often produced drastic regression of ALK‐rearranged NSCLC. 7 However, more than 50% of patients relapsed within a year due to the development of crizotinib‐resistant tumors. 7 , 8 , 9 The main causes of crizotinib resistance are mutations in the ALK kinase domain, activation of bypass pathways such as epidermal growth factor receptors (EGFRs), and amplification of c‐Kit tyrosine kinase receptors with upregulation of its stem cell factor ligand. 9 , 10 , 11 , 12 , 13 , 14 , 15 To overcome crizotinib‐resistant mutations, multiple next‐generation ALK inhibitors have been developed, including alectinib, brigatinib, lorlatinib, and ceritinib. 16 , 17 , 18 , 19 Ceritinib was approved by the FDA in April 2014 and by the PMDA in March 2016. In clinical trials, patients with ALK‐rearranged NSCLC, including post‐crizotinib treatment patients, responded well to ceritinib. 17 The potency of ceritinib is >10‐fold higher than that of crizotinib and has been found efficacious against multiple crizotinib‐resistant mutations both in vitro and in vivo. 20 However, tumors also eventually develop ceritinib resistance due to ceritinib‐resistant mutations in the ALK kinase domain such as G1202R or F1174C/V. 20 Unfortunately, little is known about the ceritinib resistance mechanisms especially bypass pathway–mediated resistance.
We established ceritinib‐resistant H3122 (EML4‐ALK–harboring cell line) cells in vitro by treating them with increasing concentrations of ceritinib over 6 months. In addition to assessing the ceritinib resistance of these cell lines, we examined the clinical specimens taken from ceritinib‐resistant patients. After comprehensive analyses, we identified a novel ceritinib resistance mechanism mediated by FGFR3 activation. This finding highlights the importance of determining the resistance mechanisms in the ALK‐TKI resistant patients to ensure an appropriate treatment strategy.
2. MATERIAL AND METHODS
Detailed information is shown in Appendix S2.
2.1. Patients
Clinical specimens were collected from patients with ALK‐rearranged NSCLC who acquired ceritinib resistance. The patients submitted written informed consent for all genetic and cell biological analyses, which were performed in accordance with the protocols approved by the institutional review board (IRB) of the Japanese Foundation for Cancer Research (#2013–1093).
2.2. Cell lines
H3122 human NSCLC cell line (harboring EML4‐ALK variant 1) was obtained in 2010, which was originally established from the lung cancer patient as previously described. 21 Ba/F3, immortalized murine bone marrow–derived pro‐B cells were obtained from the RIKEN BRC Cell Bank (RIKEN BioResource Center) in 2012.
2.3. Reagents
Ceritinib, alectinib, lorlatinib, cabozantinib, and zoligratinib were purchased from ActiveBiochem. Crizotinib, brigatinib, and infigratinib were purchased from Biochempartner. AZD4547 was purchased from Selleck, and PHA665752 was purchased from Tocris Bioscience.
2.4. Cell growth assay
Cell viability was measured using the CellTiter‐Glo assay reagent (Promega) and Centro LB 960 microplate luminometer (Berthold Technologies).
2.5. Immunoblotting
Cell lysis and immunoblotting were performed as previously described. 9 , 22
2.6. Sequencing and qRT‐PCR
Sequencing was bidirectionally conducted using Sanger sequencing, and qRT‐PCR was performed using FastStart Essential DNA Green Master (Roche) according to the manufacture's protocol.
2.7. Lentivirus transduction
Viruses were produced in 293FT cells as previously described. 13
2.8. Phospho–receptor tyrosine kinase (RTK) array
The RTK array was performed using a human phospho‐RTK array kit (R&D Systems) according to the manufacturer's protocol.
2.9. Mouse experiments
Female BALB/c‐nu/nu (nude) mice were purchased from Charles River Laboratories, Yokohama, Japan. All animal procedures were performed in accordance with protocols approved by the JFCR Animal Care and Use Committee.
2.10. FISH analysis
FISH analyses to detect ALK, CEP2, and MET were conducted using formalin‐fixed, paraffin‐embedded tissues with in‐house probes made from BAC clones (the exact clone names are available upon request).
2.11. Microarray analysis
RNA was purified using the RNeasy mini kit (Qiagen). A total of 100 ng of extracted RNA was labeled and hybridized onto the GeneChip PrimeView human gene expression array (Affymetrix Inc.). Microarray data have been deposited in the Gene Expression Omnibus database (www.ncbi.nlm.nih.gov/geo) under accession numbers GSE77764.
2.12. Statistical analysis
All data are presented as mean ± standard deviation. Statistical analysis was performed using the two‐tailed Student's t test or Mann‐Whitney U test (for mice experiments). Significant p values are defined as *p < 0.05.
3. RESULTS
3.1. Establishment of ceritinib‐resistant H3122 cells
H3122 cells from the patient with NSCLC harbored EML4‐ALK fusion genes and were highly sensitive to ALK inhibitors (Figure 1A). Ceritinib has shown remarkable clinical effects as a first, second, and later line of treatment. 17 , 23 , 24 However, the tumors eventually develop ceritinib resistance mediated by mutations, such as ALK‐G1202R, ALK‐F1174C/V, or P‐glycoprotein upregulation. 20 , 25 To identify the ceritinib resistance mechanisms, we first treated H3122 cells with increasing concentrations of ceritinib for 6‐12 months and established four ceritinib‐resistant H3122 LR cell lines (Figure 1B). The IC50 of ceritinib for the H3122 cells after 3 days of treatment was approximately 8 nM. However, the H3122 LR1 and LR5 cells survived after continuous exposure to 100 nM ceritinib, and the H3122 LR2 and LR3 cells even grew after exposure to ceritinib concentrations of 1 μM. The H3122 LR1 and LR5 cells showed moderate resistance to ceritinib, crizotinib, and alectinib (Figure 1C,D). Neither exhibited ALK tyrosine kinase mutations. We performed an immunoblotting analysis and found EML4‐ALK overexpression and phospho‐ALK upregulation in the H3122 LR1 and LR5 cells (Figure 1E). We quantified the number of ALK copies using qRT‐PCR with genomic DNA and identified ALK gene amplification in both the H3122 LR1 and LR5 cells (Figure 1F). In contrast, ALK amplification was not observed in the H3122 LR2 and LR3 cells. FISH analysis clearly showed that ALK gene amplification was caused by gene amplification of the EML4‐ALK locus within chromosome 2 in H3122 LR1 and H3122 LR5 cells because we detected more ALK 3' split signals to CEP2 signals in H3122 LR1 and H3122 LR5 cells than in H3122 and H3122 LR3 cells (Figure S1). These results suggest that ALK gene amplification induced moderate resistance to ceritinib.
FIGURE 1.
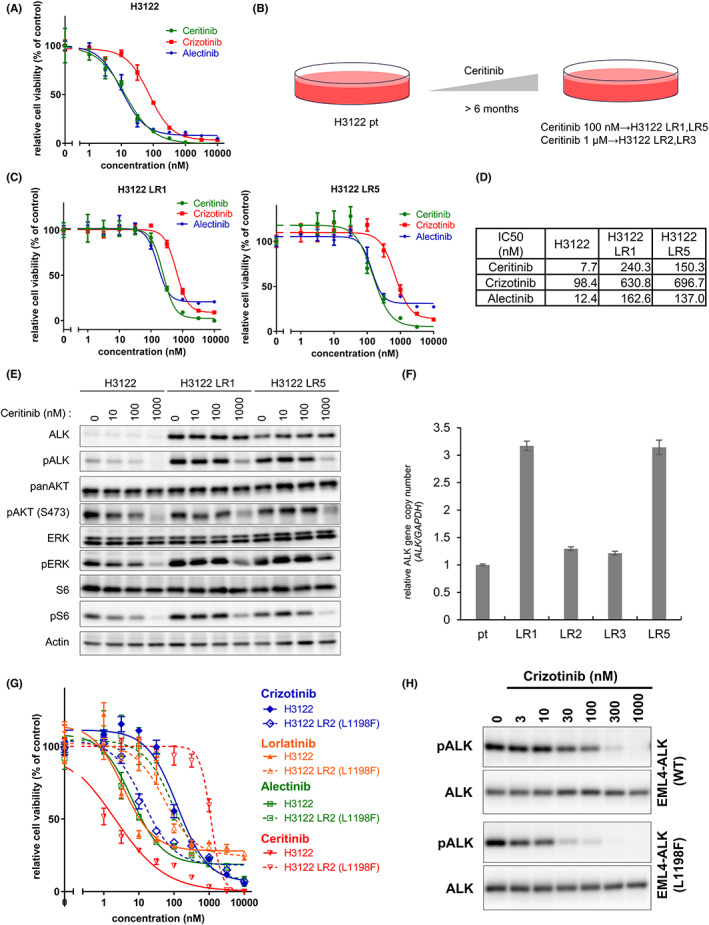
Anaplastic lymphoma kinase (ALK) amplification in the H3122 LR1 and LR5 and ALK L1198F mutation in the H3122 LR2 conferred intermediate and high resistance to ceritinib, respectively. A, C, G, The H3122 and H3122 LR cells were treated with the indicated concentrations of ALK inhibitors for 72 h. Calculated IC50 values are shown in (D). B, The ceritinib (LDK378)‐resistant H3122 cells were established by treating the parental H3122 (H3122 pt) cells with increasing concentrations of ceritinib over 6 months. E, The H3122 and H3122 LR1 and LR5 cells were treated with the indicated concentrations of ceritinib or crizotinib for 6 h. After incubation, the cells were lysed and analyzed by immunoblotting. F, Genomic DNA was extracted from five cell lines, and each relative ALK gene copy number was analyzed by qRT‐PCR. H, The Ba/F3 expressing EML4‐ALK‐WT or ‐L1198F cells were treated with the indicated concentrations of ceritinib for 4 h. After incubation, the cells were lysed and analyzed by immunoblotting.
3.2. Identification of L1198F in the H3122 LR2 cells
The H3122 LR2 cells demonstrated much higher ceritinib resistance than the H3122 cells (Figure S2A), and the phospho‐ALK levels of the H3122 LR2 cells were maintained even at a ceritinib concentration of 1 μM (Figure S2B). This suggested that the H3122 LR2 cells harbored a ceritinib resistance mutation. Thus, we sequenced the ALK gene from the H3122 LR2 cells and 10 clones isolated from ceritinib‐resistant H3122 LR2 cells and found a c3592t (L1198F) mutation in the ALK (Figure S2C). Several studies have reported that the L1198F mutation confers resistance to various other ALK inhibitors, but not crizotinib. 26 , 27 , 28 The H3122 LR2 cells were found to be resistant to lorlatinib and alectinib as well as ceritinib but sensitive to crizotinib (Figure S2D). Similarly, Ba/F3 cells expressing EML4–ALK‐L1198F showed high ceritinib resistance (Figure S3A,B). The IC50 of all next‐generation ALK inhibitors (ceritinib, alectinib, lorlatinib, and brigatinib) were higher in Ba/F3‐EML4–ALK‐L1198F than in Ba/F3‐EML4–ALK‐WT, but the IC50 of crizotinib was lower in Ba/F3‐EML4–ALK‐L1198F than in Ba/F3‐EML4–ALK‐WT (Figure S3C–E).
3.3. Induction of H3122 LR3 ceritinib resistance by FGFR3 activation
We next evaluated the ceritinib‐resistant H3122 LR3 cells and found that they were also highly resistant to other ALK inhibitors (Figure 2A). The H3122 LR3 cells did not show ALK amplification or an EML4‐ALK mutation. Therefore, we inferred that a different resistance mechanism such as activation of another RTK had induced the ceritinib resistance in these cells. To identify the activation of other RTKs, we performed a phospho‐RTK array to compare the H3122 cells and the H3122 LR3 cells with and without ceritinib exposure. In the H3122 LR3 cells, we detected more phospho‐EGFR, vascular endothelial growth factor receptor (VEGFR)2, and FGFR3 than in the H3122 cells (Figure 2B). On the basis of this result, we treated the H3122 LR3 cells with ceritinib and several RTK inhibitors corresponding to specific RTK (erlotinib, EGFR inhibitor; cabozantinib, VEGFR inhibitor; and infigratinib, FGFR inhibitor). Interestingly, only combined treatment with ceritinib and infigratinib resensitized the cells to ceritinib (Figure 2C). Immunoblotting analysis showed that the downstream signaling of ALK was maintained during ceritinib treatment alone but decreased when ceritinib was combined with infigratinib (Figure 2D). AZD4547, another FGFR inhibitor, also resensitized the H3122 LR3 cells to ceritinib (Figure S4A). Additionally, the combination of other ALK inhibitors with infigratinib was effective against the H3122 LR3 cells but did not affect the parental H3122 cells (Figure S4B–D). Infigratinib alone did not suppress the growth of either the H3122 or H3122 LR3 cells even at concentrations as high as 3 μM (Figure S3E). All 10 single clones of the H3122 LR3 cells were also resensitized to ceritinib when it was combined with infgratinib (Figure S4F). Additionally, the H3122 LR3 cells grew faster with higher fetal bovine serum (FBS) concentrations than the H3122 cells, and the induction of ceritinib resistance was found to be dependent on FBS (Figure S5A,B). The dependence on serum was similarly observed when we used human serum instead of FBS (Figure S6). These results suggested that ligand‐dependent overactivation of FGFR was a significant factor in the ALK inhibitor resistance of the H3122 LR3 cells.
FIGURE 2.
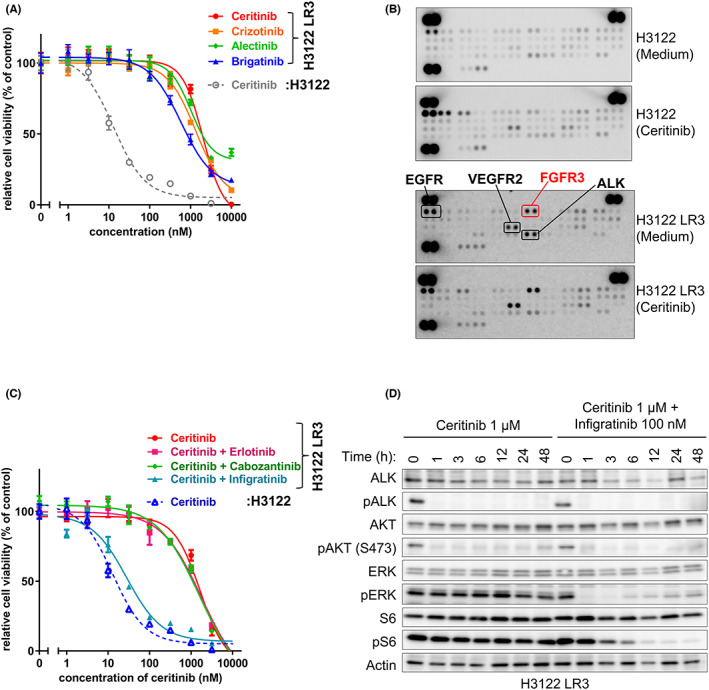
Ceritinib resistance was induced by ligand‐dependent activation of FGFR3 in the H3122 LR3 cells. A, C, The H3122 and H3122 LR3 cells were treated with various concentrations of ceritinib with or without erlotinib (1 μM), cabozantinib (1 μM), or infigratinib (100 nM) for 72 h. B, The phosphorylation levels of 49 RTKs in the H3122 and H3122 LR3 cells were measured using an RTK array with cell lysates treated with or without 1 μM of ceritinib for 8 h. D, The H3122 LR3 cells were treated with ceritinib (1 μM) with or without infigratinib (100 nM) for 1–48 h. After incubation, the cells were lysed and analyzed by immunoblotting.
3.4. Resensitization of the H3122 LR3 cells to ceritinib by FGFR3 small hairpin RNA (shRNA) knockdown
The FGFR family comprises five variants, and all of these but FGFR5 have tyrosine kinase domains. 29 , 30 We quantified the mRNA expression of FGFR1‐4 in the parental H3122 and H3122 LR3 cells using quantitative PCR and found overexpression of FGFR3 in the H3122 LR3 cells (Figure 3A), whereas FGFR3 gene amplification was not observed (Figure S7). The mRNA expression of FGFR3 ligands (FGF1, FGF2, FGF4, FGF9, and FGF18) was also similar between the parental H3122 and H3122 LR3 cells (Figure S8). Additionally, we performed gene expression analysis using cDNA microarray to confirm whether other molecules or pathways were related to the ceritinib resistance of the H3122 LR3 cells (Figure S9A–C). In the microarray analysis, FGFR3 was the gene with the greatest increase in expression. Next, to examine whether the overexpressed FGFR3 was responsible for the ceritinib resistance in the H3122 LR3 cells, we performed an shRNA knockdown of FGFR3 in the H3122 LR3 cells (Figure 3B). The FGFR3 expression level was much lower in the H3122 LR3‐sh374 and H3122 LR3‐sh199837 and slightly lower in the H3122 LR3‐sh413000 than in the H3122 LR3 and the H3122 LR3‐sh‐control cells. We found that the FGFR3 knockdown resensitized the cells to ceritinib according to the decrease in FGFR3 expression levels of the shFGFR3 (Figure 3C). Immunoblotting analysis showed the downstream signaling in the FGFR3 knockdown cells treated with 1 μM of ceritinib to be reduced more than those of the H3122 LR3 and the sh‐control cells (Figure 3D). Also, ceritinib resistance was not induced in the H3122 LR3‐sh374 even in the medium containing 10% human serum (Figure 3E). Therefore, we concluded that FGFR3 overexpression induces serum‐derived FGFR ligand–dependent ceritinib resistance.
FIGURE 3.
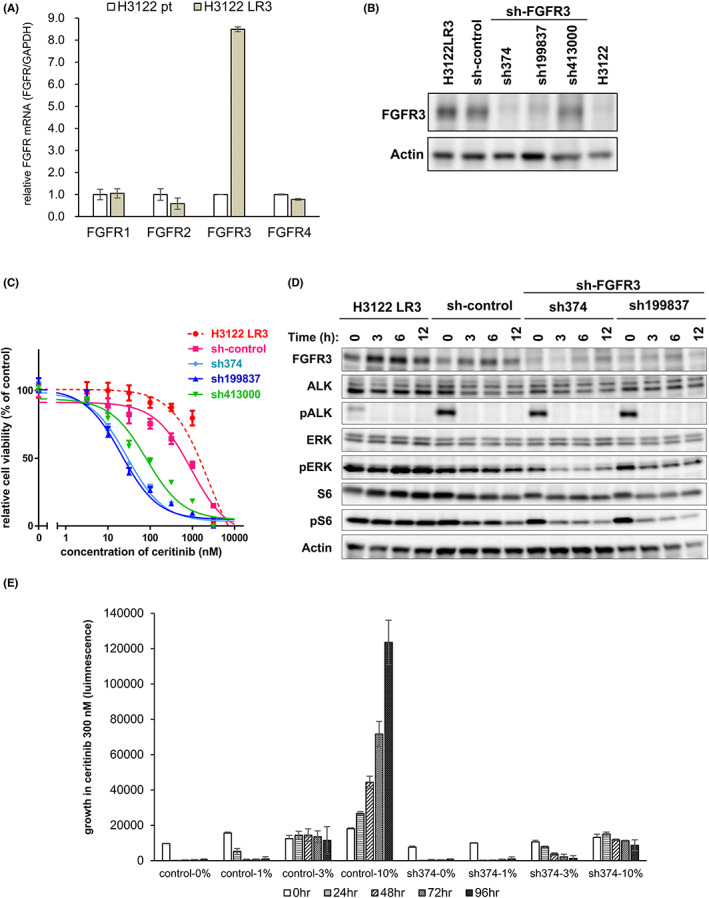
FGFR3 knockdown in H3122 LR3 cells restored sensitivity to ceritinib. A, Quantitative real‐time PCR was used to measure FGFR (FGFR1‐4) mRNA expression levels in the H3122 and H3122 LR3 cells. The H3122 LR3 mRNA expression levels (FGFRs/GAPDH) relative to those in the H3122 parental cells are shown in the bar graph. B, FGFR3 protein expression in the indicated shRNA‐infected H3122 LR3 cells was analyzed by immunoblotting. C, H3122 LR3 and shFGFR3 H3122 LR3 cells were treated with the indicated concentrations of ceritinib for 72 h. D, The H3122 LR3 and shFGFR3 H3122 LR3 cells were treated with 1 μM ceritinib for the indicated amount of time (0–2 h). After incubation, the cells were lysed and analyzed by immunoblotting. E, H3122 LR3 control cells and H3122 LR3‐sh374 cells were plated in RPMI‐1640 containing the indicated concentrations of human AB serum (Mediatech). Beginning the next day, they were cultured with 300 nM ceritinib for 0–96 h.
3.5. Resistance of H3122 LR3 cells suppressed by ALK inhibitor combined with FGFR inhibitor
We next performed an experiment to confirm whether combined treatment with ALK inhibitor and FGFR inhibitor was also effective in vivo. However, we found that treatment with ceritinib and infigratinib markedly decreased the body weight of mice (data not shown), so we adopted another combination strategy: alectinib and zoligratinib (an FGFR inhibitor). 31 The combined treatment with alectinib and zoligratinib also effectively inhibited the growth of the H3122 LR3 cells in vitro (Figure 4A), whereas zoligratinib treatment alone did not affected cell growth (Figure S10). We inoculated the H3122 LR3 cells into nude mice and administered the combined drug treatment daily over 6 days (Figure 4B). An RTK array on the tumor cells found greater phosphorylation of FGFR3 in the H3122 LR3 tumors than in the H3122 tumors (Figure S11A). As with the in vitro results, the combined treatment with alectinib and zoligratinib was also effective in vivo (Figure 4C). Similarly, an H3122 LR3‐sh‐control xenograft tumor was resistant to ceritinib, but an H3122 LR3‐sh374 xenograft was sensitive to ceritinib in vivo (Figure S11B). In conclusion, combined treatment with ALK inhibitors and FGFR inhibitors can overcome FGFR3‐mediated resistance mechanisms.
FIGURE 4.
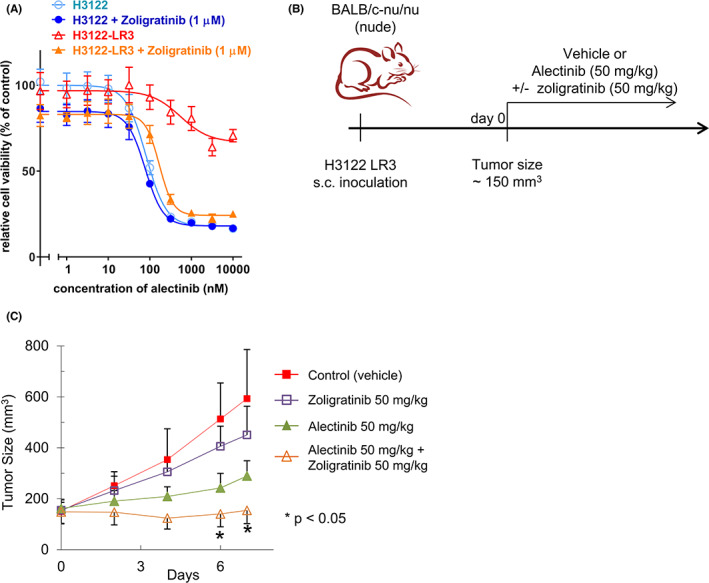
Combined treatment with alectinib and the FGFR inhibitor, zoligratinib, effectively resensitized H3122 LR3 cells to ceritinib in vivo. A, The H3122 LR3 cells were treated with various concentrations of alectinib with or without 1 μM of zoligratinib for 72 h. B, Schematic representation of the treatment schedule. After the tumors reached sizes of approximately 150 mm3, the mice were randomized by tumor size, and daily treatment with 50 mg/kg of alectinib with or without 50 mg/kg of zoligratinib was initiated. The tumor volumes were measured as 0.5 × length × width × height. The mean tumor volumes are shown in (C). *p < 0.05.
3.6. Ceritinib resistance mechanisms other than ALK mutations in the clinical specimens
In patients treated with crizotinib, only one‐third developed resistance due to ALK secondary mutations or ALK fusion gene amplification. 32 We have analyzed the clinical specimens treated with ceritinib, and reported several ceritinib resistance mechanisms (Table S1). Of the tumor samples, 42% (5/12) carried ALK resistance mutations such as F1174C, F1174V, and G1202R. This is consistent with a previous report that analyzed biopsy specimens from ceritinib‐resistant patients. 33 Of the 58% (7/12) of our samples without ALK mutations, we found P‐glycoprotein overexpression in three samples and tyrosine‐protein kinase Met (cMET) gene amplification in one sample (JFCR‐059‐2). 34 In two of the remaining three samples, we were unable to establish cell lines, but we successfully established a cell line in the third sample, JFCR‐093. In our recent study, we found that treatment with GSK3 inhibitors or Src inhibitors sensitizes the resistant cells established from JFCR‐093 to ALK inhibitors. 35 JFCR‐059‐2 patient had been treated with first‐line crizotinib and had shown a marked partial response, as measured by the Response Evaluation Criteria in Solid Tumors, lasting 14 months (Figure S12A). After relapsing on crizotinib, the patient received chemotherapy (cisplatin, pemetrexed plus bevacizumab) for 3 months, followed by bevacizumab for 4 months, and then with docetaxel for 10 weeks. The patient was then enrolled in a phase II trial for ceritinib and showed a significant response (Figure S12B, left and middle). However, 8 months later, the disease progressed because of ceritinib resistance (Figure S12B, right). To identify the ceritinib resistance mechanism, we established the JFCR‐059‐2 using the malignant fluid cell line. Through FISH analysis, we confirmed the presence of ALK rearrangement (Figure S12C, left). However, we did not observe any secondary mutations in ALK in the ceritinib‐resistant tumor sample. FISH analysis of EGFR and cMET revealed cMET amplification in the JFCR‐059‐2 cells (Figure S12C, right). Additionally, strong phospho‐cMET and phospho‐EGFR, and intermediate phospho‐human HER3 signals were observed in the JFCR‐059‐2 cells in a phospho‐RTK array (Figure S12D). We found that treatment with PHA665752 (cMET inhibitor) but not with erlotinib (EGFR inhibitor) sensitized the JFCR‐059‐2 cells to ceritinib (Figure S12E). Additionally, PHA665752 treatment inhibited phospho‐cMET and phospho‐EGFR, suggesting that the phosphorylation of EGFR was mediated by cMET (Figure S12F). Because crizotinib can inhibit both ALK and cMET, 36 we treated the cells with crizotinib, other ALK‐tyrosine kinase inhibitors (TKIs; ceritinib, alectinib, or lorlatinib), or alectinib with PHA665752 and examined the downstream signaling by immunoblotting. This revealed that the downstream phospho‐AKT, ‐ERK, and ‐S6 were inhibited according to the extent of the cMET inhibition by crizotinib or PHA‐665752 and ALK inhibition. However, a high concentration of crizotinib was required to inhibit both the ALK‐ and cMET‐mediated downstream signaling (Figure S12F). Next, we subcutaneously inoculated JFCR‐059‐2 cells into nude mice and treated them with ALK inhibitors in combination with or without crizotinib. As expected, alectinib or lorlatinib single treatment did not induce tumor shrinkage, but crizotinib single treatment did induce tumor regression by inhibiting both ALK and cMET; however, the tumors were not completely diminished by the single treatment. On the other hand, half dose of alectinib or lorlatinib combined with half dose of crizotinib almost completely diminished the tumor (Figure S13). These results suggested that crizotinib with other ALK‐inhibitor combination might be effective for the treatment of cMET amplification–mediated ALK‐TKI resistance. The analysis of clinically developed ceritinib‐resistant specimens suggested that cell‐line establishment or tumor xenograft establishment were deemed effective means of identifying the resistance mechanisms resulting from bypass pathway activation.
4. DISCUSSION
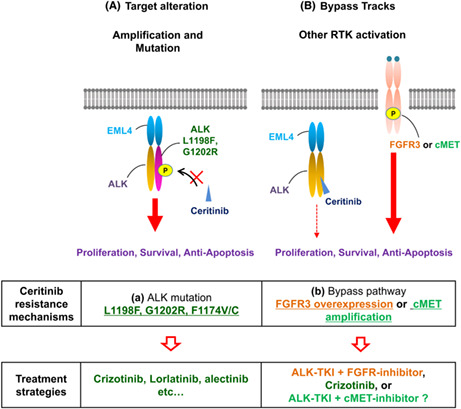
Anaplastic lymphoma kinase inhibitors have been successfully developed following the identification of ALK‐rearranged NSCLC in 2007. 3 , 37 Today, five ALK inhibitors (crizotinib, ceritinib, alectinib, brigatinib, and lorlatinib) are available for clinical use. Ceritinib has shown great clinical efficacy against ALK–rearranged NSCLC, regardless of treatment history of crizotinib. 17 However, as with other ALK inhibitors, ceritinib resistance has been observed in patients treated with ceritinib, so it is important to identify all such mechanisms and to determine how they can be overcome. In this study, we conducted comprehensive analyses of ceritinib resistance using cell line models and clinical specimens from ceritinib‐refractory patients with advanced ALK‐positive NSCLC and identified FGFR3 activation–mediated ceritinib resistance mechanism (Figure 5).
FIGURE 5.
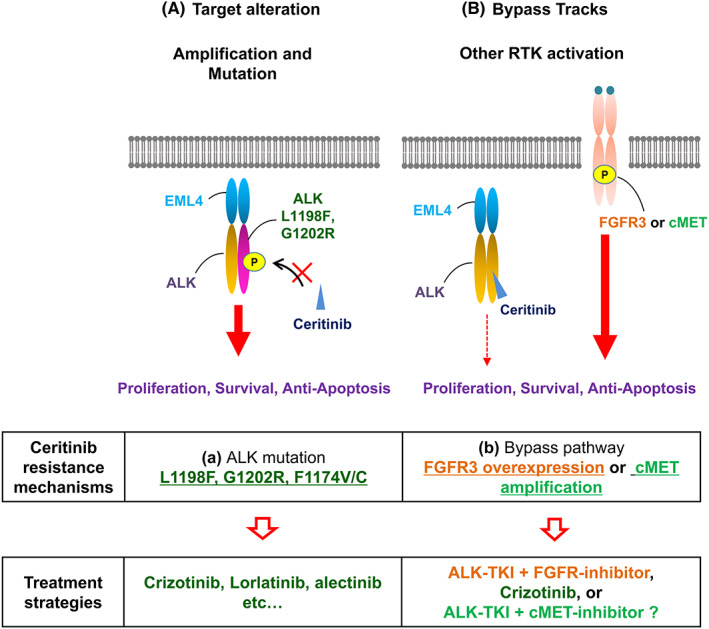
New ceritinib resistance mechanisms and therapeutic strategies to overcome them were identified. A diagrammatic representation of the identified ceritinib resistance mechanisms mediated by anaplastic lymphoma kinase (ALK) mutation (left) and bypass pathway activation resulting from FGFR3 or cMET overexpression.
First, we found that ALK amplification and L1198F mutation conferred intermediate and high ceritinib resistance, respectively (Figure 1). An L1198F mutation, with an additional C1156Y mutation, has previously been identified in a lorlatinib‐refractory patient. In our previous research, we have used MP‐CAFEE to create computational simulations that revealed the free binding of ceritinib to L1198F mutations is higher than that of crizotinib or gilteritinib (a multikinase inhibitor approved for FLT3‐mutated acute myeloid leukemia). 28 , 38 Our results indicate that crizotinib can be a treatment option for patients with a treatment history of second‐ or third‐generation ALK inhibitors with L1198F mutation in ALK.
Second, we found that FGFR3 bypass pathway activation caused ceritinib resistance in the H3122 LR3 cells (Figure 2). Bypass pathway activation is one of the most common resistance mechanisms against crizotinib, in which it is mediated by EGFR or cKIT activation. 9 , 10 , 14 However, there have been few reports on ceritinib resistance mediated by bypass pathway activation. FGFR1‐4 have tyrosine kinase domains, and many reports have noted that their abnormality is related to various types of cancer development, including breast, gastric, and colorectal cancers. 39 , 40 , 41 Additionally, other groups have used cell line models to demonstrate that FGFR1 contributes to resistance against EGFR‐TKIs in EGFR‐mutated lung cancer. 42 , 43 Because FGFR abnormalities have been observed in many types of cancer, multiple FGFR inhibitors have been developed, and several of these are currently available for clinical use. 44 , 45 In the present study, FGFR3 overexpression conferred ligand‐dependent high resistance to ALK inhibitors, and FGFR3 shRNA knockdown resensitized the H3122 LR3 cells to ceritinib (Figure 3). As ceritinib and infigratinib treatment caused drastic weight loss, we chose alectinib with zoligratinib. This combination treatment was tolerable for the mice and induced marked shrinkage of H3122 LR3 tumor (Figure 4C). In addition to FGFR abnormalities described above, recent studies revealed that FGFR fusion proteins can induce resistance to EGFR TKIs. 46 , 47 Therefore, we analyzed the existence of FGFR3 fusion protein in H3122 LR3 cells, but no FGFR3 fusion proteins were detected. However, this result does not preclude the possibility that FGFR3 fusion can induce resistance to ALK TKIs. Further studies and development of appropriate combination therapy from the viewpoints of both efficacy and safety will be required.
Third, we summarized 12 clinically developed ceritinib‐resistant specimens and realized that over half had ceritinib resistance mechanisms other than ALK mutations or amplification (Table S1). One tumor sample carried MET gene amplification (Figure S12). cMET has been reported to induce alectinib resistance. Also, autocrine activation of cMET by HGF upregulation causes resistance to alectinib. 48 , 49 , 50 Additionally, MET amplification has been observed in a few alectinib‐resistant clinical specimens. 51 , 52 , 53 Interestingly, these reports suggest that crizotinib cannot completely overcome this resistance despite its ability to inhibit cMET activation. Indeed, a result of phase II trial showed limited efficacy of crizotinib to patients with ALK‐positive NSCLC treated with alectinib immediately before crizotinib monotherapy. 54 In our study, cMET activation was observed in JFCR‐059‐2 and was found to induce EGFR and HER3 tyrosine phosphorylation, as previously reported in MET amplification–mediated EGFR‐TKI gefitinib resistance. 55 However, in JFCR‐059‐2 cells, combination of alectinib or lorlatinib with crizotinib (as a cMET inhibitor) could induce almost complete eradication of tumor in an in vivo model. Thus, at least in JFCR‐059‐2 cells, EGFR activation is one of the downstream of cMET‐mediated growth signaling activation, and cMET and ALK inhibition can overcome the resistance. To overcome these bypass pathway–mediated resistances, combination therapies will be required, and further studies are needed to test the efficacy and toxicity of combination therapy in vivo and in clinical trials.
In the present study, we analyzed ceritinib resistance mechanisms using ceritinib‐resistant H3122 cells and ceritinib‐resistant patient‐derived cells and showed that over half of patients harbor non‐ALK alteration resistance mechanisms. We also found FGFR3 activation to be one of the bypass pathway resistance mechanisms. Because ceritinib has been approved for all lines of therapy, we expect increasing research attention will be paid to ceritinib resistance in the near future. As FGFR3 overexpression induced the resistance to all the current ALK inhibitors, this resistance mechanism might be found not only in ceritinib‐refractory patients, but also other ALK‐TKI–resistant patients. Further studies are needed to comprehensively illustrate the diverse resistance mechanisms and to identify therapeutic strategies for overcoming this resistance.
FUNDING INFORMATION
This study was supported in part by MEXT/JSPS KAKENHI (grant number JP17H06327 [to N.F.], JP19H03524, JP20K21554, and 22 K18383 [to R. Katayama]); a grant from the AMED (grant number JP21ck0106695h0001 and JP22ama221201h0001 [to R. Katayama]); a grant from the Princess Takamatsu Cancer Research Fund (to R. Katayama); and a grant from the Nippon Foundation (to N. Fujita).
DISCLOSURE
KT, NF, RK are editorial board members of Cancer Science. MN reports receiving lecture fees from Ono Pharmaceutical, Bristol‐Myers Squibb, AstraZeneca, Takeda Pharm, Chugai, Pharm, Eli Lilly, and Pfizer, and research grants from Pfizer, Chugai, Novartis, and Astellas. KT received personal fees from Chugai, Kyowa Kirin, MSD, TAKEDA, Janssen, Eizai, Cellgene, Yakult, Taiho, Nichirei, Nippon Shinyaku, and Meiji, and research fundings from Kyowa Kirin, Fujirebio, and Daiichi Sankyo. NY reports receiving lecture fees from Chugai and Ono Pharmaceutical. NF reports receiving a research funding from Api Co Ltd, and Toppan Printing. RK reports receiving a research funding from Chugai Pharma, TAKEDA, and Toppan Printing. The other authors (TS, SK, SKL, ST, and SB) have no conflict of interest.
ETHICS STATEMENT
Approval of the research protocol by an Institutional Review Board: Japanese Foundation for Cancer Research (#2013–1093). Informed consent: Written informed consent for all genetic and cell biological analyses was obtained from the patients. Registry name and the registration no. of the study/trial: N/A. Animal studies: All animal procedures were performed in accordance with protocols approved by the JFCR Animal Care and Use Committee.
Supporting information
Appendix S1
Appendix S2
ACKNOWLEDGMENTS
We would like to thank S. Mori, T. Mashima, and S. Sato from the JFCR for their help with our microarray analysis (S.M. and T.M.), mice experiments (S.S.). We would also like to thank the patient whose cells were used in this research, who participated in this study.
Sakashita T, Yanagitani N, Koike S, et al. Fibroblast growth factor receptor 3 overexpression mediates ALK inhibitor resistance in ALK‐rearranged non–small cell lung cancer. Cancer Sci. 2022;113:3888‐3900. doi: 10.1111/cas.15529
REFERENCES
- 1. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209‐249. doi: 10.3322/caac.21660 [DOI] [PubMed] [Google Scholar]
- 2. Cheng L, Li Y, Zhang SB, Teng XD. Molecular pathology of lung cancer: key to personalized medicine. Zhonghua Bing Li Xue Za Zhi. 2012;41(10):715‐720. doi:10.3760/cma.j.issn.0529‐5807.2012.10.019 [DOI] [PubMed] [Google Scholar]
- 3. Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4‐ALK fusion gene in non‐small‐cell lung cancer. Nature. 2007;448(7153):561‐566. [DOI] [PubMed] [Google Scholar]
- 4. Katayama R, Lovly CM, Shaw AT. Therapeutic targeting of anaplastic lymphoma kinase in lung cancer: a paradigm for precision cancer medicine. Clin Cancer Res. 2015;21(10):2227‐2235. doi:10.1158/1078‐0432.CCR‐14‐2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shaw AT, Yeap BY, Mino‐Kenudson M, et al. Clinical features and outcome of patients with non‐small‐cell lung cancer who harbor EML4‐ALK. J Clin Oncol. 2009;27(26):4247‐4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shaw AT, Camidge DR, Engelman JA. Clinical activity of crizotinib in advanced non‐small cell lung cancer (NSCLC) harboring ROS1 gene rearrangement. J Clin Oncol. 2012;30(supple):abstr 7508. [Google Scholar]
- 7. Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK‐positive lung cancer. N Engl J Med. 2013;368(25):2385‐2394. [DOI] [PubMed] [Google Scholar]
- 8. Solomon BJ, Mok T, Kim DW, et al. First‐line crizotinib versus chemotherapy in ALK‐positive lung cancer. N Engl J Med. 2014;371(23):2167‐2177. doi:10.1056/NEJMoa1408440 [DOI] [PubMed] [Google Scholar]
- 9. Katayama R, Shaw AT, Khan TM, et al. Mechanisms of acquired crizotinib resistance in ALK‐rearranged lung cancers. Sci Transl Med. 2012;4(120):120ra17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Camidge DR, Pao W, Sequist LV. Acquired resistance to TKIs in solid tumours: learning from lung cancer. Nat Rev Clin Oncol. 2014;11(8):473‐481. doi:10.1038/nrclinonc.2014.104 [DOI] [PubMed] [Google Scholar]
- 11. Choi YL, Soda M, Yamashita Y, et al. EML4‐ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med. 2010;363(18):1734‐1739. [DOI] [PubMed] [Google Scholar]
- 12. Doebele RC, Pilling AB, Aisner DL, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non‐small cell lung cancer. Clin Cancer Res. 2012;18(5):1472‐1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Katayama R, Khan TM, Benes C, et al. Therapeutic strategies to overcome crizotinib resistance in non‐small cell lung cancers harboring the fusion oncogene EML4‐ALK. Proc Natl Acad Sci U S A. 2011;108(18):7535‐7540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sasaki T, Koivunen J, Ogino A, et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res. 2011;71(18):6051‐6060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sasaki T, Okuda K, Zheng W, et al. The neuroblastoma‐associated F1174L ALK mutation causes resistance to an ALK kinase inhibitor in ALK‐translocated cancers. Cancer Res. 2010;70(24):10038‐10043. doi:10.1158/0008‐5472.CAN‐10‐2956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Seto T, Kiura K, Nishio M, et al. CH5424802 (RO5424802) for patients with ALK‐rearranged advanced non‐small‐cell lung cancer (AF‐001JP study): a single‐arm, open‐label, phase 1‐2 study. Lancet Oncol. 2013;14(7):590‐598. [DOI] [PubMed] [Google Scholar]
- 17. Shaw AT, Kim DW, Mehra R, et al. Ceritinib in ALK‐rearranged non‐small‐cell lung cancer. N Engl J Med. 2014;370(13):1189‐1197. doi:10.1056/NEJMoa1311107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim DW, Tiseo M, Ahn MJ, et al. Brigatinib in patients with Crizotinib‐refractory anaplastic lymphoma kinase‐positive non‐small‐cell lung cancer: a randomized, multicenter phase II trial. J Clin Oncol. 2017;35(22):2490‐2498. doi:10.1200/JCO.2016.71.5904 [DOI] [PubMed] [Google Scholar]
- 19. Solomon BJ, Besse B, Bauer TM, et al. Lorlatinib in patients with ALK‐positive non‐small‐cell lung cancer: results from a global phase 2 study. Lancet Oncol. 2018;19(12):1654‐1667. doi:10.1016/S1470‐2045(18)30649‐1 [DOI] [PubMed] [Google Scholar]
- 20. Friboulet L, Li N, Katayama R, et al. The ALK inhibitor ceritinib overcomes crizotinib resistance in non‐small cell lung cancer. Cancer Discov. 2014;4(6):662‐673. doi:10.1158/2159‐8290.CD‐13‐0846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Koivunen JP, Mermel C, Zejnullahu K, et al. EML4‐ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clin Cancer Res. 2008;14(13):4275‐4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Katayama R, Koike S, Sato S, Sugimoto Y, Tsuruo T, Fujita N. Dofequidar fumarate sensitizes cancer stem‐like side population cells to chemotherapeutic drugs by inhibiting ABCG2/BCRP‐mediated drug export. Cancer Sci. 2009;100(11):2060‐2068. doi:10.1111/j.1349‐7006.2009.01288.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gainor JF, Tan DS, De Pas T, et al. Progression‐free and overall survival in ALK‐positive NSCLC patients treated with sequential Crizotinib and Ceritinib. Clin Cancer Res. 2015;21(12):2745‐2752. doi:10.1158/1078‐0432.CCR‐14‐3009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Soria JC, Tan DSW, Chiari R, et al. First‐line ceritinib versus platinum‐based chemotherapy in advanced ALK‐rearranged non‐small‐cell lung cancer (ASCEND‐4): a randomised, open‐label, phase 3 study. Lancet. 2017;389(10072):917‐929. doi:10.1016/S0140‐6736(17)30123‐X [DOI] [PubMed] [Google Scholar]
- 25. Katayama R, Sakashita T, Yanagitani N, et al. P‐glycoprotein mediates ceritinib resistance in anaplastic lymphoma kinase‐rearranged non‐small cell lung cancer. EBioMedicine. 2016;3:54‐66. doi:10.1016/j.ebiom.2015.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shaw AT, Friboulet L, Leshchiner I, et al. Resensitization to Crizotinib by the Lorlatinib ALK resistance mutation L1198F. N Engl J Med. 2016;374(1):54‐61. doi:10.1056/NEJMoa1508887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ceccon M, Mologni L, Giudici G, et al. Treatment efficacy and resistance mechanisms using the second‐generation ALK inhibitor AP26113 in human NPM‐ALK‐positive anaplastic large cell lymphoma. Mol Cancer Res. 2015;13(4):775‐783. doi:10.1158/1541‐7786.MCR‐14‐0157 [DOI] [PubMed] [Google Scholar]
- 28. Mizuta H, Okada K, Araki M, et al. Gilteritinib overcomes lorlatinib resistance in ALK‐rearranged cancer. Nat Commun. 2021;12(1):1261. doi:10.1038/s41467‐021‐21396‐w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ahmad I, Iwata T, Leung HY. Mechanisms of FGFR‐mediated carcinogenesis. Biochim Biophys Acta. 2012;1823(4):850‐860. doi:10.1016/j.bbamcr.2012.01.004 [DOI] [PubMed] [Google Scholar]
- 30. Beenken A, Mohammadi M. The FGF family: biology, pathophysiology and therapy. Nat Rev Drug Discov. 2009;8(3):235‐253. doi:10.1038/nrd2792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nakanishi Y, Akiyama N, Tsukaguchi T, et al. The fibroblast growth factor receptor genetic status as a potential predictor of the sensitivity to CH5183284/Debio 1347, a novel selective FGFR inhibitor. Mol Cancer Ther. 2014;13(11):2547‐2558. doi:10.1158/1535‐7163.MCT‐14‐0248 [DOI] [PubMed] [Google Scholar]
- 32. Katayama R. Drug resistance in anaplastic lymphoma kinase‐rearranged lung cancer. Cancer Sci. 2018;109(3):572‐580. doi:10.1111/cas.13504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gainor JF, Dardaei L, Yoda S, et al. Molecular mechanisms of resistance to first‐ and second‐generation ALK inhibitors in ALK‐rearranged lung cancer. Cancer Discov. 2016;6(10):1118‐1133. doi:10.1158/2159‐8290.CD‐16‐0596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yanagitani N, Uchibori K, Koike S, et al. Drug resistance mechanisms in Japanese anaplastic lymphoma kinase‐positive non‐small cell lung cancer and the clinical responses based on the resistant mechanisms. Cancer Sci. 2020;111(3):932‐939. doi:10.1111/cas.14314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shimizu Y, Okada K, Adachi J, et al. GSK3 inhibition circumvents and overcomes acquired lorlatinib resistance in ALK‐rearranged non‐small‐cell lung cancer. NPJ Precis Oncol. 2022;6(1):16. doi:10.1038/s41698‐022‐00260‐0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ou SH, Kwak EL, Siwak‐Tapp C, et al. Activity of crizotinib (PF02341066), a dual mesenchymal‐epithelial transition (MET) and anaplastic lymphoma kinase (ALK) inhibitor, in a non‐small cell lung cancer patient with de novo MET amplification. J Thorac Oncol. 2011;6(5):942‐946. doi:10.1097/JTO.0b013e31821528d3 [DOI] [PubMed] [Google Scholar]
- 37. Cameron L, Solomon B. Treatment of ALK‐rearranged non‐small cell lung cancer: recent Progress and future directions. Drugs. 2015;75(10):1059‐1070. doi:10.1007/s40265‐015‐0415‐9 [DOI] [PubMed] [Google Scholar]
- 38. Okada K, Araki M, Sakashita T, et al. Prediction of ALK mutations mediating ALK‐TKIs resistance and drug re‐purposing to overcome the resistance. EBioMedicine. 2019;41:105‐119. doi:10.1016/j.ebiom.2019.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jang JH, Shin KH, Park JG. Mutations in fibroblast growth factor receptor 2 and fibroblast growth factor receptor 3 genes associated with human gastric and colorectal cancers. Cancer Res. 2001;61(9):3541‐3543. [PubMed] [Google Scholar]
- 40. Murase H, Inokuchi M, Takagi Y, Kato K, Kojima K, Sugihara K. Prognostic significance of the co‐overexpression of fibroblast growth factor receptors 1, 2 and 4 in gastric cancer. Mol Clin Oncol. 2014;2(4):509‐517. doi:10.3892/mco.2014.293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yoshimura N, Sano H, Hashiramoto A, et al. The expression and localization of fibroblast growth factor‐1 (FGF‐1) and FGF receptor‐1 (FGFR‐1) in human breast cancer. Clin Immunol Immunopathol. 1998;89(1):28‐34. [DOI] [PubMed] [Google Scholar]
- 42. Azuma K, Kawahara A, Sonoda K, et al. FGFR1 activation is an escape mechanism in human lung cancer cells resistant to afatinib, a pan‐EGFR family kinase inhibitor. Oncotarget. 2014;5(15):5908‐5919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ware KE, Hinz TK, Kleczko E, et al. A mechanism of resistance to gefitinib mediated by cellular reprogramming and the acquisition of an FGF2‐FGFR1 autocrine growth loop. Oncogenesis. 2013;2:e39. doi:10.1038/oncsis.2013.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dieci MV, Arnedos M, Andre F, Soria JC. Fibroblast growth factor receptor inhibitors as a cancer treatment: from a biologic rationale to medical perspectives. Cancer Discov. 2013;3(3):264‐279. doi:10.1158/2159‐8290.CD‐12‐0362 [DOI] [PubMed] [Google Scholar]
- 45. Szymczyk J, Sluzalska KD, Materla I, Opalinski L, Otlewski J, Zakrzewska M. FGF/FGFR‐dependent molecular mechanisms underlying anti‐cancer drug resistance. Cancers (Basel). 2021;13(22):5796. doi:10.3390/cancers13225796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Haura EB, Hicks JK, Boyle TA. Erdafitinib overcomes FGFR3‐TACC3‐mediated resistance to Osimertinib. J Thorac Oncol. 2020;15(9):e154‐e156. doi:10.1016/j.jtho.2019.12.132 [DOI] [PubMed] [Google Scholar]
- 47. Raphael A, Dudnik E, Hershkovitz D, et al. FGFR Fusions as an acquired resistance mechanism following treatment with epidermal growth factor receptor tyrosine kinase inhibitors (EGFR TKIs) and a suggested novel target in advanced non‐small cell lung cancer (aNSCLC). J Clin Med. 2022;11(9):2475. doi:10.3390/jcm11092475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kogita A, Togashi Y, Hayashi H, et al. Activated MET acts as a salvage signal after treatment with alectinib, a selective ALK inhibitor, in ALK‐positive non‐small cell lung cancer. Int J Oncol. 2015;46(3):1025‐1030. doi:10.3892/ijo.2014.2797 [DOI] [PubMed] [Google Scholar]
- 49. Isozaki H, Ichihara E, Takigawa N, et al. Non‐small cell lung cancer cells acquire resistance to the ALK inhibitor alectinib by activating alternative receptor tyrosine kinases. Cancer Res. 2016;76(6):1506‐1516. doi:10.1158/0008‐5472.CAN‐15‐1010 [DOI] [PubMed] [Google Scholar]
- 50. Tanimoto A, Yamada T, Nanjo S, et al. Receptor ligand‐triggered resistance to alectinib and its circumvention by Hsp90 inhibition in EML4‐ALK lung cancer cells. Oncotarget. 2014;5(13):4920‐4928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Berger LA, Janning M, Velthaus JL, et al. Identification of a high‐level MET amplification in CTCs and cfTNA of an ALK‐positive NSCLC patient developing evasive resistance to Crizotinib. J Thorac Oncol. 2018;13(12):e243‐e246. doi:10.1016/j.jtho.2018.08.2025 [DOI] [PubMed] [Google Scholar]
- 52. Tsuji T, Ozasa H, Aoki W, et al. Alectinib resistance in ALK‐rearranged lung cancer by dual salvage signaling in a clinically paired resistance model. Mol Cancer Res. 2019;17(1):212‐224. doi:10.1158/1541‐7786.MCR‐18‐0325 [DOI] [PubMed] [Google Scholar]
- 53. Makimoto G, Ohashi K, Tomida S, et al. Rapid Acquisition of Alectinib Resistance in ALK‐positive lung cancer with high tumor mutation burden. J Thorac Oncol. 2019;14(11):2009‐2018. doi:10.1016/j.jtho.2019.07.017 [DOI] [PubMed] [Google Scholar]
- 54. Harada D, Isozaki H, Kozuki T, et al. Crizotinib for recurring non‐small‐cell lung cancer with EML4‐ALK fusion genes previously treated with alectinib: a phase II trial. Thorac Cancer. 2021;12(5):643‐649. doi:10.1111/1759‐7714.13825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316(5827):1039‐1043. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Appendix S2