

Abstract
A lack of practical resources in Japan has limited preclinical discovery and testing of therapies for pediatric relapsed and refractory acute lymphoblastic leukemia (ALL), which has poor outcomes. Here, we established 57 patient‐derived xenografts (PDXs) in NOD.Cg‐Prkdc scid ll2rg tm1Sug /ShiJic (NOG) mice and created a biobank by preserving PDX cells including three extramedullary relapsed ALL PDXs. We demonstrated that our PDX mice and PDX cells mimicked the biological features of relapsed ALL and that PDX models reproduced treatment‐mediated clonal selection. Our PDX biobank is a useful scientific resource for capturing drug sensitivity features of pediatric patients with ALL, providing an essential tool for the development of targeted therapies.
Keywords: biobank, leukemia, pediatric, preclinical, xenograft
We established the first Japanese biobank of ALL‐PDX. With confirming the preservation of the characteristics of primary samples and the reproduction of treatment‐induced clonal selection in PDX model, this biobank will contribute to the acceleration of the applications of PDX model for preclinical research.
Abbreviations
- ALL
acute lymphoblastic leukemia
- Ara‐C
cytarabine
- Ara‐G
nelarabine
- B‐ALL
B cell acute lymphoblastic leukemia
- BM
bone marrow
- CFB
clofarabine
- CNS
central nervous system
- CSF
cerebral spinal fluid
- NOG
NOD.Cg‐Prkdc scid ll2rg tm1Sug /ShiJic
- PDX
patient‐derived xenograft
- PE
phycoerythrin
- T‐ALL
T cell acute lymphoblastic leukemia
1. INTRODUCTION
Acute lymphoblastic leukemia (ALL) is the most frequent malignancy in childhood. Although improvements in treatment strategy have resulted in an overall survival rate >90%, patients with some ALL subtypes, such as Philadelphia chromosome‐ and KMT2A‐fusion‐positive leukemia, continue to suffer from relapse and their prognosis remains poor. 1 , 2 Therefore, there continues to be an urgent need for the development of new drugs to treat pediatric ALL; however, difficulties in accessing primary samples have limited the opportunities for drug development.
Recently, PDX models, established from primary tumor samples, have been widely used for translational drug discovery and personalized oncology studies. 3 , 4 , 5 , 6 Some large‐scale biobanks of PDX models have been established to serve as preclinical platforms; for example, PDX Finder at the National Cancer Institute, 7 EuroPDX in Europe, 8 the Public Repository of Xenografts (ProXe) at the Dana‐Faber Center Institute, 9 and the Children's Oncology Group's Childhood Solid Tumor Network. 10 Despite these large‐scale biobanks, PDX models derived from pediatric ALL are scarce. Furthermore, as ethnic differences of genes influence drug sensitivity, such as genes to drug metabolism, 11 , 12 have existed, specific national PDX libraries should be established and will be highly valuable. In Japan, the establishment of a large‐scale Japanese PDX library, consisting of a wide range of cancer subtypes, was reported recently; however, samples from leukemia patients were not successfully engrafted 13 and there is no ALL PDX library. Therefore, there is an urgent need for a Japanese biobank of pediatric ALL PDX models.
In this paper, we report a Japanese PDX biobank that provides a practical resource to accelerate research into those groups of patients with ALL who have poor outcomes.
2. MATERIALS AND METHODS
2.1. Mice
NOD.Cg‐Prkdcscidll2rgtm1Sug /ShiJic (NOG) mice, developed at the Central Institute of Experimental Animals (Kawasaki, Japan) as previously reported, 14 were used to generate PDX models. All mice were maintained under specific pathogen‐free conditions, in accordance with the guidelines of Kyoto University Graduate School and Fukushima Medical University.
2.2. Patient samples
Many of the patient samples used in this study were derived from patients enrolled in the Japanese Pediatric Leukemia/Lymphoma Study Group (JPLSG) ALL‐R14 (UMIN000019878) and IntReALL SR 2010 (jRCTs041180122) studies.
To address the deficiency of practical samples for research into pediatric ALL, the JPLSG initiated the ALL‐R14 clinical study; a prospective observational study of pediatric patients with relapsed and refractory ALL that aims to accumulate clinical information and samples from patients. Samples were also collected from the participants of IntReALL SR 2010, an international study for the treatment of standard risk childhood relapsed ALL. In these studies, primary BM or peripheral blood samples collected at registration to the study were sent to Kanagawa Children's Medical Center, where mononuclear cells were separated by Ficoll–Hypaque density gradient centrifugation and cryopreserved. Cryopreserved samples were sent and xenografted into NOG mice at Fukushima Medical University or Kyoto University. In addition, extramedullary relapsed specimens were collected and xenografted at Kyoto University. Cerebral spinal fluid and testicular samples were centrifuged (200 g, 5 min) and cells transplanted into NOG mice, without cryopreservation.
Informed consent was given for collection and use of all samples, in accordance with the Declaration of Helsinki. Approval was obtained from the local Institutional Review Board for all participating centers in the ALL‐R14 and IntReALL SR 2010 studies.
2.3. PDX mice derived from patients with pediatric ALL
All experimental studies were approved by the Animal Care and Ethics Committee of the Kyoto University Graduate School and Fukushima Medical University.
Transplantations of primary leukemic cell samples were performed using previously reported methods, 15 , 16 with some modifications. In brief, mononuclear cells isolated from primary samples (0.25–1.5 × 106 for primary BM samples) were transplanted into nonpretreated 8–12‐week‐old male NOG mice by tail vein injection. The two institutions conducting xenotransplantation (Kyoto University Graduate School and Fukushima Medical University) shared technical guidance on the method for establishing PDX mice.
2.4. Flow cytometric analysis of human leukemic cell engraftment in PDX mice
Human leukemic cell engraftment was evaluated by flow cytometry analysis of BM aspirates from transplanted NOG mice after 2 or 4 weeks. The leukemic cell surface markers, human CD19 and CD3, were used to identify B‐ALL and T‐ALL samples, respectively. Antibodies used in this study were as follows: allophycocyanin‐conjugated anti‐mouse CD45 (BD Pharmingen), PE‐conjugated anti‐human CD45 (BD Pharmingen), PE‐conjugated anti‐human CD19 (Biolegend), and fluorescein isothiocyanate‐conjugated anti‐human CD3 (BD Pharmingen).
2.5. Harvest and preservation of leukemic cells from PDX mice
Human leukemic cells were harvested from BM, CNS, and spleen from transplanted NOG mice when human leukemic cell chimerism in BM aspirates reached >90% or clinical symptoms associated with overt leukemic infiltration, such as low activity, weight loss, and limb paralysis, occurred. Cells were harvested according to previously reported methods. 17 Briefly, mice were euthanized, and then their organs were perfused with PBS before they were removed and mechanically dispersed. Engrafted leukemic cells were isolated by Ficoll–Hypaque density gradient centrifugation and cryopreserved in liquid nitrogen using CELLBANKER1 (Nippon Zenyaku Kogyo).
2.6. Characterization of PDX histology
In B‐ALL PDX mice, murine tibia, spleen, liver, brain, and testis tissue sections were analyzed morphologically, using H&E staining, and immunohistochemically. Organs were harvested, fixed in 4% paraformaldehyde, embedded in paraffin, and sectioned. Each section was deparaffinized, underwent citrate‐based antigen retrieval, and stained with rabbit anti‐human CD19 (Spring Bioscience) for B‐ALL, followed by detection using a VECTASTAIN Elite ABC Rabbit IgG Kit (Vector Laboratories) with 50% hematoxylin counterstain. A BZ‐9000 microscope (Keyence) was used for sample observation.
2.7. DNA microarray analysis
For the DNA microarray that used total RNA, a set of synthetic polynucleotides (80‐mers) (MiroDiagnostic) representing 14,400 species of human transcript sequences was printed on a glass slide using a custom array. For the RNA of the samples, SuperScript II (Invitrogen Life Technologies) and cyanine 5‐dUTP (Perkin‐Elmer Inc.) were used to synthesize labeled cDNA from 5 μg of total RNA. Using the same method for the reference RNA, cyanine 3‐dUTP (Perkin‐Elmer Inc.) was used to synthesize labeled cDNA from 5 μg of Human Universal Reference RNA Type II (MicroDiagonostic).
Hybridization was performed with a labeling and hybridization kit (MicroDiagnostic). Signals were measured using a GenePix 4000B Scanner (Axon Instruments, Inc.) and then processed into the primary expression ratios of the cyanine 5 intensity of each specimen to the cyanine 3 intensity of the human common reference RNA. Each ratio was normalized using GenePix Pro 3.0 software (Axon Instruments, Inc.). The primary expression ratios were converted into log2 values, which were designated as log ratios or converted values. The data were processed using Microsoft Excel software (Microsoft) and the MDI gene‐expression analysis software package (MicroDiagnostic).
2.8. RNA sequencing
RNA was isolated from samples using the NucleoSpin TriPrep kit (MACHEREY‐NAGEL), according to the manufacturer's instructions. RNA integrity was measured using an Agilent 2200 TapeStation and RNA Screen Tapes (Agilent Technologies). Sequencing libraries were prepared using an NEB Next Ultra II RNA Library Kit for Illumina (New England Biolabs), according to the manufacturer's protocol, and prepared libraries were run on an Illumina HiSeq × high‐throughput sequencing system. Paired‐end reads were aligned to the hg19 human genome assembly using STAR. 18 Fusion transcripts were detected using Genomon version 2.6.2 (https://github.com/Genomon‐Project/) and filtered by excluding fusions: (a) mapping to repetitive regions, (b) with fewer than four spanning reads, (c) that occurred out of frame, or (d) that had junctions not located at known exon–intron boundaries. The expression level of each RefSeq gene was calculated from mapped read counts using HTSeq and normalized using the Bioconductor package, DESeq2 version 1.28.1.23. Supercomputing resources were provided by the Human Genome Center, Institute of Medical Science, The University of Tokyo. Hierarchical clustering was applied and performed using Ward's method to calculate Euclidian distances. Cluster stability was ascertained by consensus clustering using the R package, ConsensusClusterPlus, with 500 iterations.
2.9. In vitro drug sensitivity assays
Cryopreserved leukemic cells harvested from the spleens of PDX mice were recovered in RPMI1640 medium supplemented with 20% FBS, and disseminated at 1.0 × 104 cells per 10 μl in each well of a 384‐well plate. Then, cells were treated with drugs at predetermined concentrations. After 96 h incubation at 37°C in a fully humidified 5% CO2 atmosphere, live cell counts were determined using the Cell Titer Glo Assay (Promega), a chemiluminescent assay that detects live cells based on metabolic activity and ATP content.
2.10. In vivo dasatinib treatment assay
After substantial engraftment of leukemic cells was confirmed by BM aspiration, mice (n = 2 per group) were treated with dasatinib (LC laboratories, Woburn, MA) or vehicle. Dasatinib was formulated in 0.5% (w/v) methylcellulose solution (Wako, Japan) and delivered orally at 35 mg/kg for 5 days (Monday to Friday) on/2 days off. Leukemic cell chimerism was monitored by flow cytometry analysis of BM aspirates, as described above. Once leukemic cell chimerism in BM reached <5%, the dasatinib dose was reduced to 20 mg/kg. After chimerism had recovered to >90%, mice were sacrificed by cervical dislocation, the femur, tibia, ilium, and brain were removed and mechanically dispersed, and leukemic cells were then isolated by Ficoll–Hypaque density gradient centrifugation and used for FISH analysis.
2.11. FISH analysis
Cells isolated from BM and CNS samples from PDX mice treated with dasatinib were sent to the cytogenetics laboratory and processed following standard procedures. FISH was performed using the LSI BCR‐ABL dual color, dual fusion translocation probe (Abbot), according to the manufacturer's instructions. Cells with a t(9;22) translocation exhibited a yellow fusion signal, whereas normal cells showed a pattern of two red and two green signals. Analysis of 1000 cells in metaphase was conducted for each case, and the percentages of cells with double, triple, and quadruple fusion signals were calculated.
2.12. Statistical analysis
Survival analysis was conducted by log‐rank test with Bonferroni correction. Results are expressed as mean values with the standard deviation. p‐values for comparisons of two groups were determined using a two‐sided t‐test. Two‐sided p‐value <0.05 was considered statistically significant.
3. RESULTS
3.1. Establishment of a biobank of pediatric ALL PDX models
In total, 60 primary samples were transplanted into NOG mice; 34 in Fukushima Medical University, 21 in Kyoto University, and five in both institutes, to assess the concordance of PDX sample quality between the two institutions. The engraftment success rate of first passage samples was 93.3% (57/60), including 54 B‐ALL and 3 T‐ALL samples. Of established models, 54 were derived from primary BM samples, two from CSF, and one from testis. Relevant clinical details are provided in Table 1.
TABLE 1.
Clinical characterstics of samples
| B or T | Cytogenic abnormality | Chromosomal number | Age (y)/Gender | Source | Blast (%) | Disease stage | Treatment response after relapse | |
|---|---|---|---|---|---|---|---|---|
| ALL #1 | B‐ALL | Other abnormality a | 44 | 11/F | BM | 85.8 | 1st relapse, late relapse | CR |
| ALL #2 | B‐ALL | Other abnormality a | 54 | 5/M | BM | 59.2 | 2nd relapse | CRp |
| ALL #3 | B‐ALL | ETV6‐RUNX1 | 46 | 6/M | BM | 97.0 | 1st relapse, early relapse | CR |
| ALL #4 | B‐ALL | MEF2D‐BCL9 | 52 | 12/F | BM | 48.4 | 1st relapse, extremely early relapse | CR |
| ALL #5 | B‐ALL | ETV6‐RUNX1 | 46 | 6/M | BM | 98.0 | 1st relapse | NR |
| ALL #6 | B‐ALL | Other abnormality a | 46 | 15/M | BM | 52.0 | 1st relapse, extremely early relapse | PD |
| ALL #7 | B‐ALL | Normal | 46 | 3/M | BM | 79.7 | 1st relapse, extremely early relapse | PR |
| ALL #8 | B‐ALL | Normal | 46 | 10/M | BM | 97.0 | 1st relapse, extremely early relapse | CR |
| ALL #9 | B‐ALL | Other abnormality a | 47 | 6/F | BM | 89.8 | 1st relapse, early relapse | CR |
| ALL #10 | B‐ALL | BCR‐ABL | 46 | 7 /M | BM | 70.2 | 1st relapse, late relapse | CR |
| ALL #11 | B‐ALL | Other abnormality a | 55 | 11/M | BM | 62.8 | 1st relapse, late relapse | NR |
| ALL #12 | B‐ALL | Other abnormality a | 55 | 7/M | BM | NR | Second relapse | Relapse |
| ALL #13 | B‐ALL | Other abnormality a | 58 | 11/M | BM | 98.4 | 1st relapse, late relapse | SD |
| ALL #14 | B‐ALL | Other abnormality a | 45 | 10/F | BM | NR | 1st relapse, early relapse | CR |
| ALL #15 | B‐ALL | NR | 56 | 16/M | BM | 93.4 | 1st relapse, extremely early relapse | PD |
| ALL #16 | B‐ALL | BCR‐ABL | NR | 7/F | CSF | 80.0 | 1st relapse, extremely early relapse | PD |
| ALL #17 | B‐ALL | KMT2A‐AFF1 | NR | 0.4/F | BM | 72.3 | Diagnostic | CR |
| ALL #18 | B‐ALL | KMT2A‐MLLT1 | NR | 2/M | BM | 75.4 | Diagnostic | CR |
| ALL #19 | B‐ALL | KMT2A‐AFF1 | NR | 10/M | BM | NR | 1st relapse, extremely early relapse | CR |
| ALL #20 | B‐ALL | KMT2A‐AFF1 | 45 | 10/M | BM | 98.6 | Diagnostic | CR |
| ALL #21 | B‐ALL | BCR‐ABL | 46 | 6/F | BM | NR | Diagnostic | Relapse |
| ALL #22 | B‐ALL | Other abnormality a | NR | NR | BM | NR | Diagnostic | NR |
| ALL #23 | B‐ALL | ETV6‐RUNX1 | NR | NR | BM | NR | Diagnostic | NR |
| ALL #24 | B‐ALL | MEF2D‐BCL9 | NR | NR/ F | BM | NR | 1st relapse | NR |
| ALL #25 | B‐ALL | ETV6‐ABL1 | 46 | 10/M | Testis | NR | 1st relapse, extremely early relapse | NR |
| ALL #26 | T‐ALL | Normal | 46 | 3/M | CSF | NR | 1st relapse | CR |
| ALL #27 | T‐ALL | NR | NR | 13/M | BM | 58.8 | 1st relapse, late relapse | PR |
| ALL #28 | T‐ALL | Other abnormality a | 47 | 9/M | BM | 91.8 | 1st relapse, early relapse | CR |
| ALL #29 | B‐ALL | Other abnormality a | 58 | 9/F | BM | NR | 1st relapse, late relapse | CR |
| ALL #30 | B‐ALL | Other abnormality a | 47 | 17/F | BM | NR | 1st relapse, late relapse | CR |
| ALL #31 | B‐ALL | Other abnormality a | 45 | 4/M | BM | 98.0 | 1st relapse, extremely early relapse | NR |
| ALL #32 | B‐ALL | Other abnormality a | 53 | 5 /M | BM | 79.0 | 1st relapse, early relapse | CR |
| ALL #33 | B‐ALL | ETV6‐RUNX1 | 46 | 15/F | BM | 90.0 | 1st relapse, late relapse | PR |
| ALL #34 | B‐ALL | Other abnormality a | 46 | 9/F | BM | 70.0 | 1st relapse, extremely early relapse | PR |
| ALL #35 | B‐ALL | Normal | 46 | 11/F | BM | 40.4 | 1st relapse, extremely early relapse | PR |
| ALL #36 | B‐ALL | ETV6‐RUNX1 | 46 | 8/F | BM | 70.0 | 2nd relapse | NR |
| ALL #37 | B‐ALL | Other abnormality a | 51 | 7/F | BM | 94.1 | 1st relapse, late relapse | CR |
| ALL #38 | B‐ALL | Other abnormality a | 46 | 14/F | BM | 89.0 | 1st relapse, early relapse | NR |
| ALL #39 | B‐ALL | ETV6‐RUNX1 | 46 | 6/F | BM | 80.0 | 1st relapse, early relapse | CR |
| ALL #40 | B‐ALL | Other abnormality a | 46 | 9/F | BM | 97.8 | 1st relapse, early relapse | SD |
| ALL #41 | B‐ALL | Other abnormality a | 48 | 25/M | BM | 90.0 | >3rd relapse | SD |
| ALL #42 | B‐ALL | Other abnormality a | 55 | 4/F | BM | 95.0 | 1st relapse, early relapse | CR |
| ALL #43 | B‐ALL | BCR‐ABL | 46 | 14/M | BM | 36.8 | 1st relapse, extremely early relapse | CR |
| ALL #44 | B‐ALL | Other abnormality a | 46 | 20/F | BM | 90.0 | 1st relapse, late relapse | NR |
| ALL #45 | B‐ALL | Other abnormality a | 48 | 17/F | BM | 90.0 | >3rd relapse | PR |
| ALL #46 | B‐ALL | Other abnormality a | 49 | 17/F | BM | 75.8 | second relapse | SD |
| ALL #47 | B‐ALL | Other abnormality a | 46 | 18/F | BM | 96.6 | 1st relapse, late relapse | CR |
| ALL #48 | B‐ALL | Other abnormality a | 46 | 13/M | BM | 65.6 | 1st relapse, late relapse | CR |
| ALL #49 | B‐ALL | Other abnormality a | 46 | 3/M | BM | 96.4 | 1st relapse, extremely early relapse | SD |
| ALL #50 | B‐ALL | Other abnormality a | 47 | 10/M | BM | 64.3 | 1st relapse, late relapse | NR |
| ALL #51 | B‐ALL | Other abnormality a | 46 | 14/M | BM | 61.6 | 1st relapse, late relapse | CR |
| ALL #52 | B‐ALL | Normal | 46 | 9/F | BM | 97.8 | 1st relapse, early relapse | PD |
| ALL #53 | B‐ALL | Other abnormality a | 46 | 13/M | BM | NR | 1st relapse, late relapse | CR |
| ALL #54 | B‐ALL | Normal | 46 | 8/M | BM | NR | 1st relapse, late relapse | CR |
| ALL #55 | B‐ALL | BCR‐ABL | 46 | 12/F | BM | 93.0 | 1st relapse, late relapse | CR |
| ALL #56 | B‐ALL | Other abnormality a | 45 | 10/F | BM | NR | 1st relapse, early relapse | CR |
| ALL #57 | B‐ALL | Normal | 46 | 15/F | BM | 66.0 | 1st relapse, late relapse | CR |
| ALL #58 | B‐ALL | BCR‐ABL | 45 | 8/M | BM | 18.6 | 2nd relapse | CRp |
| ALL #59 | B‐ALL | Normal | 46 | 7/M | BM | NR | 1st relapse, late relapse | CR |
| ALL #60 | B‐ALL | ETV6‐RUNX1 | NR | 8/M | Testis | NR | 1st relapse, late relapse | NR |
Abbreviations: CR, complete remission; CRp, complete remission with incomplete platelet recovery; NR, not reported; PD, progressive disease; PR, partial response; SD, stable disease.
Other abnormality denotes another abnormality except for BCR‐ABL, ETV6‐RUNX1, KMT2A rearrangement, RUNX1‐RUNX1T1, E2A‐PBX1, IGH‐CMYC, IL3‐IGH, JAK mutation, and CRLF2 mutation.
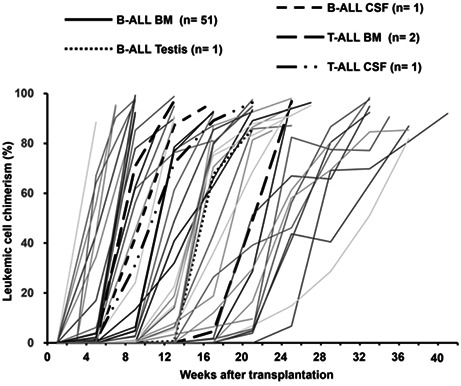
The timing of engraftment, defined as the detection of >0.1% human CD19 or CD3 cells in tibial BM aspirates by flow cytometry, ranged from 14 to 168 days (Figure 1A). To assess the usefulness of PDX models as incubators of rare primary samples, we calculated the fold expansion of primary leukemic cells in each PDX mouse. In primary B‐ALL samples derived from BM, mean leukemic cell expansion was 28.9 (±6.9)‐fold, whereas T‐ALL BM primary samples expanded 19.5‐fold per mouse. In particular, leukemic cells from extramedullary relapsed samples were effectively expanded in PDX mice, despite the fact that very few primary cells could be collected from patients (Figure 1B).
FIGURE 1.
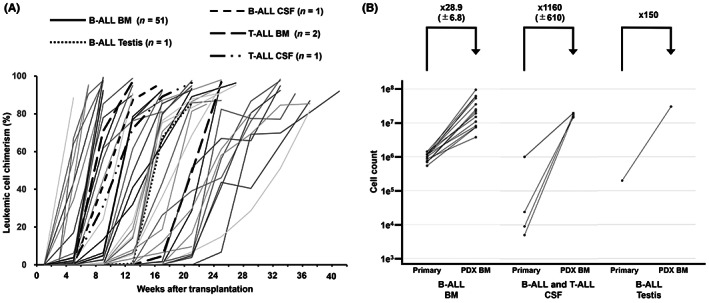
Establishment of a biobank of pediatric ALL PDX models. (A) Engraftment of leukemia cells in PDX mice over time. Primary leukemic cells were transplanted into recipient mice, and the proportions of human CD19‐ or CD3‐positive ALL cells were detected in BM by flow cytometry. Each line represents data from a single mouse. (B) In vivo expansion rate of leukemic cells. Numbers above are mean expansion rates with standard deviation. BM (B‐ALL, n = 14), CSF (B‐ALL, n = 3; T‐ALL, n = 2), and testis (B‐ALL, n = 1).
3.2. PDX mice preserve the characteristics of primary samples
To determine whether ALL PDX models could reproduce the pattern of human leukemic infiltration, we conducted histopathological and immunohistochemical examinations of ALL PDX mouse specimens. Microscopic examination confirmed that PDX models preserved the histopathological characteristics of leukemic infiltration in both hematopoietic and extramedullary sites. Human leukemic cells infiltrated spleen diffusely; in liver, large clusters of leukemic cells were observed in the portal area, whereas in the brain, leukemic infiltration was observed along the subarachnoid space, and leukemic cells were present in the interstitium of testicle specimens. Engraftment patterns in each organ were consistent with previously reported pathology of human patients with ALL 19 , 20 (Figure 2A–D).
FIGURE 2.
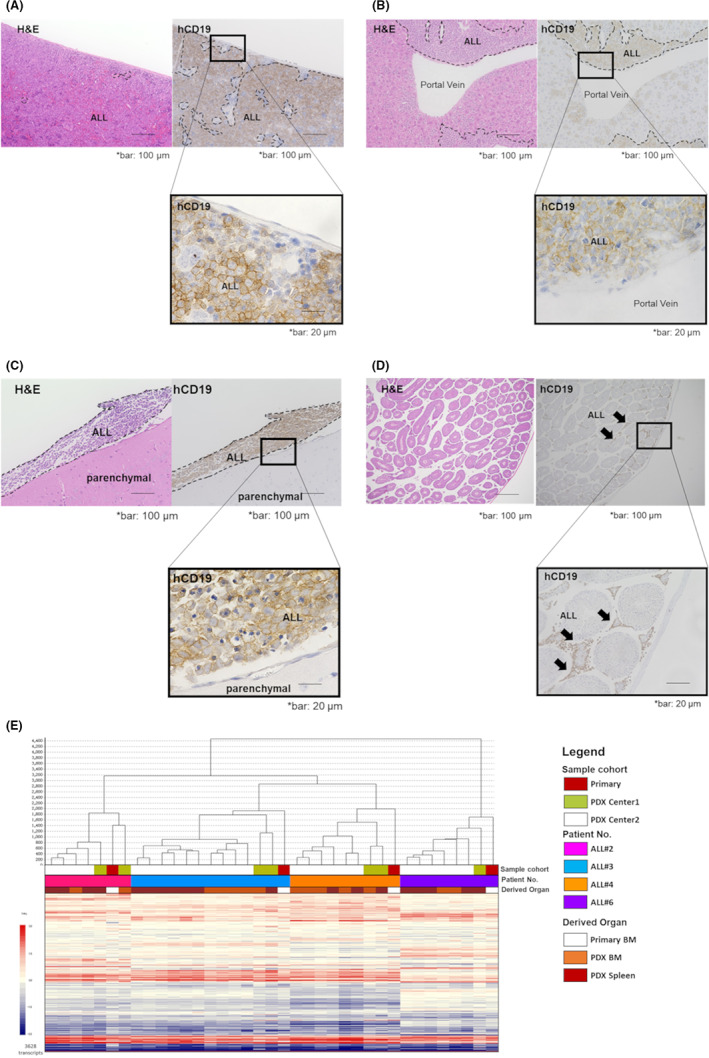
PDX mice preserve the characteristics of primary samples. (A–D) Immunohistochemical analysis of leukemic cell proliferation in (A) spleen, (B) liver, (C) brain, and (D) testis from a xenograft mouse generated from a sample from patient ALL#25 relapsed in testis. (E) Unsupervised hierarchical clustering of expression of patterns using DNA microarray data among four primary ALL samples and the corresponding PDX cells derived from BM and spleen.
To evaluate the stability of the genetic features of PDX cells generated from the same ALL samples at two different institutes, we compared gene‐expression data from four PDX mouse strains using DNA microarray analysis. As expected, unsupervised clustering of samples showed that each individual primary sample and the corresponding PDX cells were clustered together, regardless of both the organ and the institute from which PDX cells were derived. These data demonstrated that there was less variation between PDX cells produced from the same primary sample than there was between different primary sample strains. (Figure 2E).
3.3. PDX cells retain gene‐expression features corresponding to their cytogenetic abnormalities
It is established that cytogenetic abnormalities, such as specific fusion genes and chromosomal aneuploidies, are related to sensitivity to molecular targeted therapy in pediatric ALL. 21 To apply PDX cells for evaluation of the efficacy of novel therapeutic agents, it is necessary to determine whether PDX cells retain molecular profiles corresponding to their cytogenetic abnormalities. Therefore, we analyzed gene‐expression patterns in 16 PDX samples with specific cytogenetic abnormalities by RNA‐seq. As expected, the expression patterns of PDX cells clustered according to the cytogenetic abnormalities present in primary samples (Figure 3).
FIGURE 3.
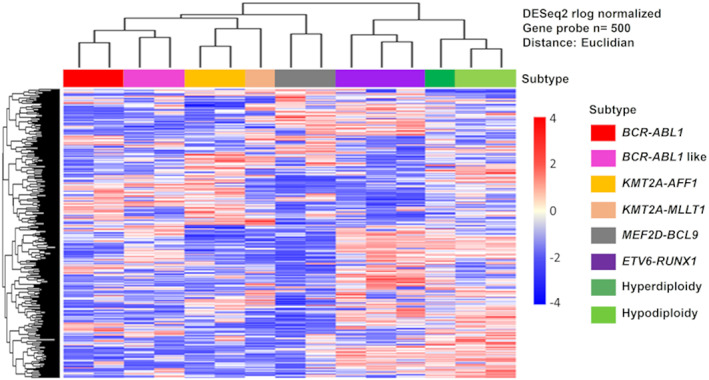
PDX cells retain gene‐expression features corresponding to their cytogenetic abnormalities. Unsupervised hierarchical clustering of expression of patterns of 500 genes among 16 PDX cells with cytogenetic abnormalities.
Furthermore, using these 16 PDX samples, we acquired in vitro drug sensitivity data for 80 anti‐neoplastic compounds, 22 that showed that KMT2A fusion‐positive samples tended to be highly sensitive to the MEK inhibitors, trametinib and selumetinib, relative to cells with other cytogenetic characteristics (Figure S1). These results are consistent with some previous reports showing high sensitivity of KMT2A fusion‐positive leukemia to treatment with MEK inhibitor. 23 , 24 Furthermore, our data imply that the established PDX samples reflect both gene‐expression and drug‐sensitivity features consistent with their cytogenetic abnormalities.
3.4. PDX cells are useful resources for in vitro drug testing
To assess the level of concordance between the in vitro and in vivo anticancer drug responses of PDX samples, we compared drug sensitivities between B‐ALL and T‐ALL PDX samples by treating PDX mice with Ara‐C or Ara‐G. As expected, during in vivo experiments, B‐ALL PDX mice showed significantly improved survival duration in only the Ara‐C‐treated group, whereas T‐ALL PDX mice showed improvements in both Ara‐C‐ and Ara‐G‐treated groups as was previously described 20 (Figure S2). Next, we examined whether in vitro drug sensitivity of PDX cells was consistent with the observed in vivo drug responses; the same drug sensitivity tendencies were observed (Figure 4A,B). In addition, we compared in vitro drug sensitivity data between three pairs of primary samples and PDX cells of KMT2A‐rearranged B‐ALL. The PDX cells are resembling primary samples in the pattern of in vitro drug sensitivity. (Figure S3) These suggest that in vitro drug screening assays could be useful substitutes for costly and time consuming in vivo experiments (Figure 4C).
FIGURE 4.
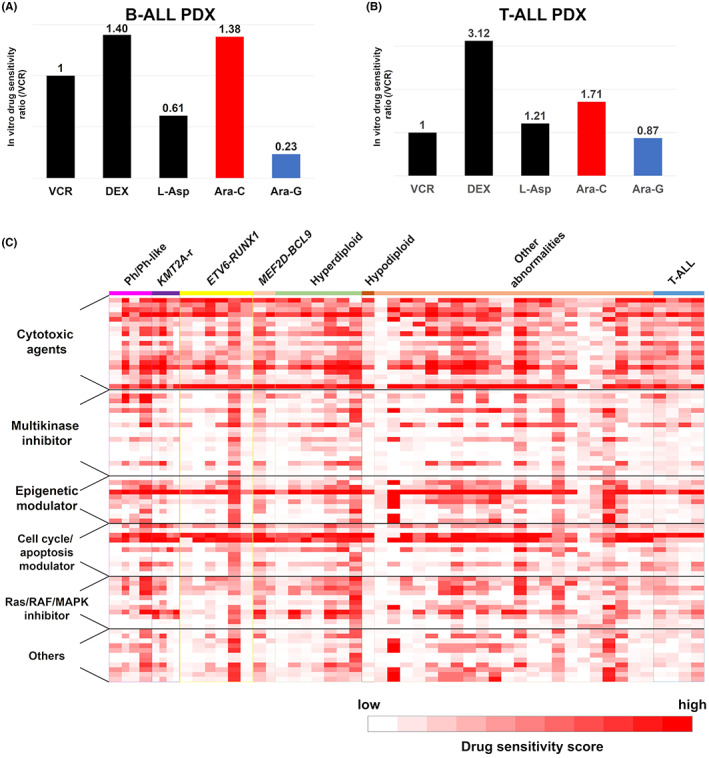
PDX cells are useful resources for in vitro drug testing. (A, B) In vitro drug treatment of leukemic cells from the same B‐ALL (A) and T‐ALL (B) xenograft mice. Single agent VCR, DEX, l‐Asp, Ara‐C, or Ara‐G therapies were assessed and sensitivity to each drug is shown as the ratio of that to VCR. (C) Heatmap of in vitro responses of 80 drugs tested against ALL PDX cells. Responses are shown for each drug against each PDX tested as scored according to the following formula: drug effective score = [log125 × {1 − (survival rate at less than ×1/125 concentration)} + log25 × {1 − (survival rate at less than ×1/25 concentration)} + log5 × {1 − (survival rate at less than ×1/5 concentration)} + {1 − (survival rate at less than ×1 concentration)}] × 100/(log125 + log25+ log5 + 1).
3.5. Treatment‐induced clonal selection reproduced in a patient‐derived mouse model
Due to the difference in microenvironment, a major drawback of applying PDX models for analysis of responses to anti‐tumor compounds is uncertainty over whether mechanisms of drug resistance will be reproducible in mice. 25 , 26 , 27 , 28 Therefore, we compared the clonal architecture in a PDX model and its dynamics with that in the patient from which the cells were derived. A 6‐year‐old girl, diagnosed with Philadelphia chromosome‐positive ALL, suffered a secondary disease relapse during maintenance therapy with dasatinib after hematopoietic transplantation. 29 Leukemic cells at diagnosis and at secondary relapse were analyzed by FISH to detect the BCR‐ABL fusion. At diagnosis, a major subpopulation (740/1000 cells) showed triple fusion signals, whereas at secondary relapse 900/1000 cells showed quadruple fusion signals (Figure 5A). We established PDX mice using a diagnostic sample from this patient. In serial transplantation, untreated PDX mice showed prompt engraftment and PDX mice treated with dasatinib relapsed once they reached <5% of leukemic cells in BM. FISH analysis revealed that, in untreated mice, the proportion of leukemic cells with quadruple BCR‐ABL fusion signals was consistent with that of the primary sample. By contrast, in relapsed PDX mice treated with dasatinib, cells with quadruple signals became dominant (740/1000 cells), consistent with findings in relapsed samples in the clinical setting (Figure 5C). This result supports the idea that PDX models have the potential to reproduce treatment‐induced clonal selection and to be a valuable tool for preclinical therapy evaluation.
FIGURE 5.
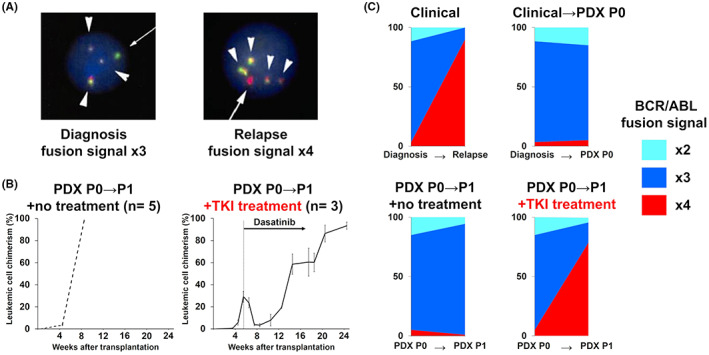
Treatment‐induced clonal selection reproduced in a patient‐derived mouse model. (A) Multiplexed‐FISH analysis showing the BCR‐ABL1 fusion signal in patient samples collected at diagnosis (left) and at relapse (right). Original magnification ×100. Arrowheads, yellow fusion signal; arrows, probe signals. Probe combinations were as follows: BCR, green; ABL1, red. (B) Human leukemic chimerism levels in the BM of PDX mice over time in untreated (left, n = 5) and dasatinib treated (right, n = 3) mice. Data are presented as means with standard deviations. (C) Patterns of cytogenetic subclonal populations. Clonal stability, with no change in the dominant clone between transplantation without treatment, is observed in upper right and lower left charts. Clonal selection with a minor population becoming dominant at relapse in the clinical setting was reproduced in PDX mice (lower right chart).
4. DISCUSSION
Faithful preclinical models are critical tools for cancer research to facilitate the study of tumor biology and anticancer therapy. Recently, use of PDX models has been widespread in preclinical cancer drug development. 3 , 4 , 5 , 6 , 30 Some large‐scale repositories and PDX model resources have been established in the USA and Europe; however, PDX models of pediatric tumor are not yet adequate, due to the rarity of the disease. Here, we established a domestic open‐resource biobank of pediatric ALL PDX models in Japan.
One of the advantages of our PDX biobank is that most of the primary samples were derived from patients with relapsed and refractory ALL. PDX models of patients with relapsed and refractory pediatric ALL are in high demand because prognosis remains poor for such patients. In our PDX biobank, the success rate of engraftment was higher than previously reported, 31 , 32 , 33 , 34 possibly attributable to the fact that our PDX biobank mainly comprises relapsed and refractory ALL samples because leukemic cell growth in immunodeficient mice is reported to be associated with clinical relapse. 35 Another advantage is that our biobank contains some PDX models derived from extramedullary relapsed ALL. We previously demonstrated that leukemic cells that have infiltrated the CNS show differential biological features compared with those in the BM. 15 Although a considerable proportion of ALL relapse occurs in extramedullary organs, such as the CNS and testis, 1 PDX models derived from extramedullary relapse ALL samples are very difficult to establish, mainly due to the small amounts of available primary samples. PDX models serve as efficient in vivo incubators for such scarce samples.
Although previous studies have reported that leukemic cells engrafted in PDX models retain many of the phenotypic and genotypic features of the original specimens, 36 , 37 there remain several possible issues with the reproducibility of PDX models for drug development. 27 Transplanted leukemic cells in the murine microenvironment may be exposed to some pressure for selection of subclones through engraftment. 25 , 38 Therefore, to validate whether PDX samples are reliable for drug development and biological analysis, we confirmed that leukemic infiltrations maintained their histopathological features in both hematopoietic and extramedullary organs. In addition, we confirmed the concordance of the genetic profiles between primary and PDX cells by DNA microarray analysis. Our data demonstrate that differences in organs from which PDX cells are derived do not influence the genetic features of primary samples. In addition, we have shown that serial transplantation had preserved clonal composition analyzed by FISH after serial transplantation. As previously reported, 39 this means that molecular signatures during serial transplantation of PDX models demonstrated high stability. These data enhance the utility of PDX mice as in vivo incubators because large amounts of PDX cells can be collected from the spleens of PDX mice. Moreover, our data show that PDX cells generated at two different institutes retained identical qualities. As the quality of primary cells influences the engraftment rate, it is preferable that multiple centers can establish identical PDX mice to prevent the deterioration of primary cells during transportation.
A significant limitation of PDX models as a preclinical platform is the fact that in vivo studies are not well suited for high‐throughput drug screening due to their cost and time‐consuming nature. Furthermore, it was established that treatment response in pediatric ALL is related to the presence of the cytogenetic abnormalities, which are each associated with distinct genetic expression patterns. We used RNA‐seq analysis to demonstrate that PDX cells retained the molecular features corresponding to the cytogenetic abnormalities observed in primary samples. These data indicate that our PDX cells can be used for in vitro screening of candidate compounds by pathway analysis. Furthermore, we show that the results of in vitro drug sensitivity screening using PDX cells were consistent with those found in vivo, and added to the profile of in vitro drug responses, including molecular targeted agents. Recently, several clinical studies have reported that drug efficacy tests showed high concordance between the PDX model and patients' responses. 40 , 41 Additionally, we have demonstrated the similarities in in vitro drug sensitivity patterns between primary and PDX cells. Our data suggest that in vitro drug screening, followed by in vivo response validation, will be a cost‐effective and time‐saving method for drug development.
In addition, we demonstrated the reproducibility of changes in clonal composition observed during the clinical course of a patient with Philadelphia chromosome‐positive ALL. As enhanced BCR‐ABL signaling is a reported mechanism of resistance to tyrosine kinase inhibitors in leukemia, 42 the clonal selection of quadruple fusion signals was likely to be the main cause of relapse in the patient. Although it has already been reported that treatment‐induced clonal selection in chronic lymphoblastic leukemia PDX models was reproduced in chronic lymphoblastic leukemia PDX models, our result is the first data showing that ALL PDX models have the potential to reproduce the treatment‐induced selection of clones resistant to anti‐tumor agents, providing a valuable tool for preclinical testing of novel therapeutic strategies against refractory subclones.
5. CONCLUSIONS
Here, we report our new national biobank of pediatric ALL and its value as a preclinical tool for further drug development. Now, to make our PDX models available to academic and scientific communities in Japan, we are planning to produce a catalog of PDX samples, presenting the characteristics of each line, which can be easily browsed in a web portal site.
DISCLOSURE
The authors declare no conflict of interest.
Seishi Ogawa and Junko Takita are Editorial Board Members for Cancer Science.
ETHICS STATEMENT
Many of the patient samples used in this study were derived from patients enrolled in the Japanese Pediatric Leukemia/Lymphoma Study Group (JPLSG) ALL‐R14 (UMIN000019878) and IntReALL SR 2010 (jRCTs041180122) studies.
Informed consent was given for collection and use of all samples, in accordance with the Declaration of Helsinki. Approval was obtained from the local Institutional Review Board for all participating centers in the ALL‐R14 and IntReALL SR 2010 studies. All animal experimental studies were approved by the Animal Care and Ethics Committee of the Kyoto University Graduate School and Fukushima Medical University.
Supporting information
Figure S1.
Figure S2.
Figure S3.
ACKNOWLEDGMENTS
The authors would like to thank the patients and families for their cooperation in this study. This work was supported by a grant for cancer research from Kyoto preventive medical center and the Takeda Science Foundation.
Tanaka K, Kato I, Dobashi Y, et al. The first Japanese biobank of patient‐derived pediatric acute lymphoblastic leukemia xenograft models. Cancer Sci. 2022;113:3814‐3825. doi: 10.1111/cas.15506
REFERENCES
- 1. Nguyen K, Devidas M, Cheng SC, et al. Factors influencing survival after relapse from acute lymphoblastic leukemia: a Children's oncology group study. Leukemia. 2008;22(12):2142‐2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pui CH, Sandlund JT, Pei D, et al. Improved outcome for children with acute lymphoblastic leukemia: results of Total therapy study XIIIB at St Jude Children's research hospital. Blood. 2004;104(9):2690‐2696. [DOI] [PubMed] [Google Scholar]
- 3. Gao H, Korn JM, Ferretti S, et al. High‐throughput screening using patient‐derived tumor xenografts to predict clinical trial drug response. Nat Med. 2015;21(11):1318‐1325. [DOI] [PubMed] [Google Scholar]
- 4. Izumchenko E, Meir J, Bedi A, Wysocki PT, Hoque MO, Sidransky D. Patient‐derived xenografts as tools in pharmaceutical development. Clin Pharmacol Ther. 2016;99(6):612‐621. [DOI] [PubMed] [Google Scholar]
- 5. Jones L, Carol H, Evans K, et al. A review of new agents evaluated against pediatric acute lymphoblastic leukemia by the pediatric preclinical testing program. Leukemia. 2016;30(11):2133‐2141. [DOI] [PubMed] [Google Scholar]
- 6. Pauli C, Hopkins BD, Prandi D, et al. Personalized in vitro and in vivo cancer models to guide precision medicine. Cancer Discov. 2017;7(5):462‐477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Conte N, Mason JC, Halmagyi C, et al. PDX Finder: a portal for patient‐derived tumor xenograft model discovery. Nucleic Acids Res. 2019;47(D1):D1073‐D1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hidalgo M, Amant F, Biankin AV, et al. Patient‐derived xenograft models: an emerging platform for translational cancer research. Cancer Discov. 2014;4(9):998‐1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Townsend EC, Murakami MA, Christodoulou A, et al. The public repository of xenografts enables discovery and randomized phase II‐like trials in mice. Cancer Cell. 2016;29(4):574‐586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stewart E, Federico S, Karlstrom A, et al. The childhood solid tumor network: a new resource for the developmental biology and oncology research communities. Dev Biol. 2016;411(2):287‐293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Moriyama T, Nishii R, Perez‐Andreu V, et al. NUDT15 polymorphisms alter thiopurine metabolism and hematopoietic toxicity. Nat Genet. 2016;48(4):367‐373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tanaka Y, Kato M, Hasegawa D, et al. Susceptibility to 6‐MP toxicity conferred by a NUDT15 variant in Japanese children with acute lymphoblastic leukaemia. Br J Haematol. 2015;171(1):109‐115. [DOI] [PubMed] [Google Scholar]
- 13. Yagishita S, Kato K, Takahashi M, et al. Characterization of the large‐scale Japanese patient‐derived xenograft (J‐PDX) library. Cancer Sci. 2021;112(6):2454‐2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ito M, Hiramatsu H, Kobayashi K, et al. NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood. 2002;100(9):3175‐3182. [DOI] [PubMed] [Google Scholar]
- 15. Kato I, Nishinaka Y, Nakamura M, et al. Hypoxic adaptation of leukemic cells infiltrating the CNS affords a therapeutic strategy targeting VEGFA. Blood. 2017;129(23):3126‐3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Takahashi N, Higa A, Hiyama G, et al. Construction of in vitro patient‐derived tumor models to evaluate anticancer agents and cancer immunotherapy. Oncol Lett. 2021;21(5):406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kato I, Niwa A, Heike T, et al. Identification of hepatic niche harboring human acute lymphoblastic leukemic cells via the SDF‐1/CXCR4 axis. PLoS One. 2011;6(11):e27042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA‐seq aligner. Bioinformatics. 2013;29(1):15‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kay HE. Testicular infiltration in acute lymphoblastic leukaemia. Br J Haematol. 1983;53(4):537‐542. [DOI] [PubMed] [Google Scholar]
- 20. Sasaki H. Practical guide for cancer research using patient‐derived experimental model (Japanese). YODOSHA; 2019:293. [Google Scholar]
- 21. Irving JA, Enshaei A, Parker CA, et al. Integration of genetic and clinical risk factors improves prognostication in relapsed childhood B‐cell precursor acute lymphoblastic leukemia. Blood. 2016;128(7):911‐922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Goto H, Yoshino Y, Ito M, et al. Aurora B kinase as a therapeutic target in acute lymphoblastic leukemia. Cancer Chemother Pharmacol. 2020;85(4):773‐783. [DOI] [PubMed] [Google Scholar]
- 23. Chu SH, Song EJ, Chabon JR, et al. Inhibition of MEK and ATR is effective in a B‐cell acute lymphoblastic leukemia model driven by Mll‐Af4 and activated Ras. Blood Adv. 2018;2(19):2478‐2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kerstjens M, Driessen EM, Willekes M, et al. MEK inhibition is a promising therapeutic strategy for MLL‐rearranged infant acute lymphoblastic leukemia patients carrying RAS mutations. Oncotarget. 2017;8(9):14835‐14846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Belderbos ME, Koster T, Ausema B, et al. Clonal selection and asymmetric distribution of human leukemia in murine xenografts revealed by cellular barcoding. Blood. 2017;129(24):3210‐3220. [DOI] [PubMed] [Google Scholar]
- 26. Davies NJ, Kwok M, Gould C, et al. Dynamic changes in clonal cytogenetic architecture during progression of chronic lymphocytic leukemia in patients and patient‐derived murine xenografts. Oncotarget. 2017;8(27):44749‐44760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Klco JM, Spencer DH, Miller CA, et al. Functional heterogeneity of genetically defined subclones in acute myeloid leukemia. Cancer Cell. 2014;25(3):379‐392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nowak D, Liem NL, Mossner M, et al. Variegated clonality and rapid emergence of new molecular lesions in xenografts of acute lymphoblastic leukemia are associated with drug resistance. Exp Hematol. 2015;43(1):32‐43. e31‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Masuda T, Maeda S, Shimada S, et al. RUNX1 transactivates BCR‐ABL1 expression in Philadelphia chromosome positive acute lymphoblastic leukemia. Cancer Sci. 2021;113:529‐539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Peterson JK, Houghton PJ. Integrating pharmacology and in vivo cancer models in preclinical and clinical drug development. Eur J Cancer. 2004;40(6):837‐844. [DOI] [PubMed] [Google Scholar]
- 31. Brabetz S, Leary SES, Grobner SN, et al. A biobank of patient‐derived pediatric brain tumor models. Nat Med. 2018;24(11):1752‐1761. [DOI] [PubMed] [Google Scholar]
- 32. Bruna A, Rueda OM, Greenwood W, et al. A biobank of breast cancer explants with preserved intra‐tumor heterogeneity to screen anticancer compounds. Cell. 2016;167(1):260‐274. e222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liem NL, Papa RA, Milross CG, et al. Characterization of childhood acute lymphoblastic leukemia xenograft models for the preclinical evaluation of new therapies. Blood. 2004;103(10):3905‐3914. [DOI] [PubMed] [Google Scholar]
- 34. Meyer LH, Debatin KM. Diversity of human leukemia xenograft mouse models: implications for disease biology. Cancer Res. 2011;71(23):7141‐7144. [DOI] [PubMed] [Google Scholar]
- 35. Meyer LH, Eckhoff SM, Queudeville M, et al. Early relapse in ALL is identified by time to leukemia in NOD/SCID mice and is characterized by a gene signature involving survival pathways. Cancer Cell. 2011;19(2):206‐217. [DOI] [PubMed] [Google Scholar]
- 36. Wang K, Sanchez‐Martin M, Wang X, et al. Patient‐derived xenotransplants can recapitulate the genetic driver landscape of acute leukemias. Leukemia. 2017;31(1):151‐158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wong NC, Bhadri VA, Maksimovic J, et al. Stability of gene expression and epigenetic profiles highlights the utility of patient‐derived paediatric acute lymphoblastic leukaemia xenografts for investigating molecular mechanisms of drug resistance. BMC Genomics. 2014;15:416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ben‐David U, Ha G, Tseng YY, et al. Patient‐derived xenografts undergo mouse‐specific tumor evolution. Nat Genet. 2017;49(11):1567‐1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Richter A, Roolf C, Sekora A, et al. The molecular subtype of adult acute lymphoblastic leukemia samples determines the engraftment site and proliferation kinetics in patient‐derived xenograft models. Cell. 2022;11(1):150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xu H, Zheng H, Zhang Q, et al. A multicentre clinical study of sarcoma personalised treatment using patient‐derived tumour xenografts. Clin Oncol (R Coll Radiol). 2022. doi: 10.1016/j.clon.2022.06.002 [DOI] [PubMed] [Google Scholar]
- 41. Cheng Y, Qin SK, Li J, et al. A multicenter clinical study: personalized medication for advanced gastrointestinal carcinomas with the guidance of patient‐derived tumor xenograft (PDTX). J Cancer Res Clin Oncol. 2022;148(3):673‐684. [DOI] [PubMed] [Google Scholar]
- 42. Wassmann B, Pfeifer H, Scheuring UJ, et al. Early prediction of response in patients with relapsed or refractory Philadelphia chromosome‐positive acute lymphoblastic leukemia (Ph+ALL) treated with imatinib. Blood. 2004;103(4):1495‐1498. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1.
Figure S2.
Figure S3.