

Abstract
The potential for tumor occurrence triggered by cancer stem cells (CSCs) has emerged as a significant challenge for human colorectal cancer therapy. However, the underlying mechanism of CSC development remains controversial. Our study provided evidence that the bulk of tumor cells could dedifferentiate to CSCs and reacquire CSC‐like phenotypes if cultured in the presence of extracellular matrix reagents, such as Matrigel and fibrin gels. In these 3D gels, CD133− colorectal cancer cells can regain tumorigenic potential and stem‐like phenotypes. Mechanistically, the 3D extracellular matrix could mediate cytoskeletal F‐actin bundling through biomechanical force associated receptors integrin β1 (ITGB1), contributing to the release of E3 ligase tripartite motif protein 11 (TRIM11) from cytoskeleton and degradation of the glycolytic rate‐limiting enzyme phosphofructokinase (PFK). Consequently, PFK inhibition resulted in enhanced glycolysis and upregulation of hypoxia‐inducible factor 1 (HIF1α), thereby promoting the reprogramming of stem cell transcription factors and facilitating tumor progression in patients. This study provided novel insights into the role of the extracellular matrix in the regulation of CSC dedifferentiation in a cytoskeleton/glycolysis‐dependent manner.
Keywords: colorectal cancer, dedifferentiation, extracellular matrix, glycolysis, integrin β1
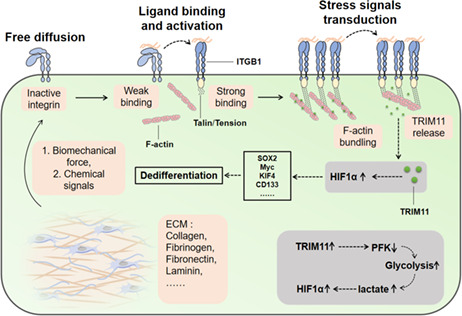
F‐actin bundling was modulated by the extracellular matrix through biomechanical forces via the receptor integrin β1, contributing to the release of E3 ligase Tripartite motif protein 11 (TRIM11) from the cytoskeleton and to the degradation of the glycolytic rate‐limiting enzyme phosphofructokinase (PFK). Inhibiting PFK contributed to enhanced glycolysis and upregulation of hypoxia‐inducible factor 1 (HIF1α), thereby reprogramming stem cell transcription factors and facilitating the dedifferentiation of colorectal cancer cells.
Abbreviations
- CSCs
cancer stem cells
- ITGB1
integrin β1
- TRIM11
tripartite motif protein 11
- PFK
phosphofructokinase
- HIF1α
hypoxia‐inducible factor 1
- NAs
neutralizing antibodies
- LatA
latrunculin A
1. INTRODUCTION
Colorectal cancer is one of the most common archenteric malignant diseases and the fourth most deadly cancer worldwide. 1 Currently, many patients with colorectal cancer suffer recurrence after the initial surgery or chemotherapy treatment, and in many of these patients, the disease progresses to metastatic colorectal cancer. 2 , 3 Around 70% of all colorectal cancer–associated deaths is caused by metastasis in the liver. 4 Some studies have linked cancer stem cells (CSCs) to a higher potential of metastasis or tumor recurrence in colorectal cancer. 5 , 6 In fact, CSCs are believed to have the ability to self‐renew and differentiate and to be responsible for tumorigenicity and distant metastasis. 7 However, the concept of CSCs remains controversial.
Cancer stem cells, also called tumor‐initiating cells, are cells within tumor tissues that have self‐renewal capacity and contribute to tumorigenicity and heterogeneity. 8 Unlike differentiated cancer cells, CSCs persist indefinitely, and they frequently are insensitive to chemotherapy. 9 The concept of CSCs supports the idea that a unique subpopulation of CSCs could sustain tumor growth, due to their capability of self‐renewal, and differentiate to the bulk of tumor cells. Hence, CSCs could contribute to the generation of palingenetic CSCs or differentiated cancer cells and cause tumor heterogeneity in vivo. Though CSCs have self‐renewal potential, palingenetic CSCs do not necessarily come from primary CSCs. A series of cancers disobey the unidirectional hierarchical CSCs concept, which elicits the possibility of “plasticity of cancer cells,” in which dedifferentiated tumor cells could reacquire the potential of renewal and exhibit stem‐like phenotypes 10 , 11 in a process named “dedifferentiation.” Recently, specific findings have supported this “dedifferentiation” hypothesis. Over 25% of the patients presented CD133− or differentiated melanoma cells. These cells have shown self‐renewal and tumorigenicity in immunodeficient mice. 12 , 13 Bidirectional conversion between differentiated tumor cells and CSCs has also been observed in intestinal tumors, in which surrounding inflammation signals could promote Wnt signaling activation and result in the reprogramming of tumor cells. 14 Additionally, it is poorly understood whether extrinsic factors in the tumor microenvironment, such as the extracellular matrix, could play a role in tumor remodeling. Overall, there is still a lack of direct evidence to support the concept of CSCs or tumor cell differentiation and explain how differentiated cells are converted to CSCs to affect tumor progression.
In this study, we showed that tumor cells are plastic. Differentiated CD133− colorectal cancer cells can dedifferentiate and reacquire CSC phenotypes in 3D Matrigel and fibrin gels. Simultaneously, 2D culture using flasks eliminated the tumorigenic potential of colorectal CSCs, demonstrating the bidirectional conversion between CSCs and differentiated cells. Mechanistically, we proved that the 3D matrix could mediate F‐actin bundling and release TRIM11 to degrade PFK, resulting in enhanced glycolysis and reprogramming of HIF1α‐induced stem cell transcription factors. Meanwhile, aberrant expression of ITGB1/TRIM11/HIF1α tightly correlated with tumor recurrence and cancer progression in the clinic. Our study demonstrated the dedifferentiation process and explored the role of cytoskeleton/glycometabolism in regulating the dedifferentiation process in colorectal cancer.
2. MATERIALS AND METHODS
2.1. 3D cell culture
HCT116 and SW480 were cultured on a flask (rigid dish) or 3D fibrin gels. For conventional flask cell culture, HCT116 and SW480 cells were cultured in a rigid dish with RPIM 1640 medium with 10% FBS (AusGene X, FBS500‐S) at 37°C with 5% CO2. 3D fibrin gels culture was formed according to the previous method: 1:1 fibrin and tumor cell solution were conducted, resulting in 1 mg/ml fibrin and 10,000 cells/ml in the mixture. A total of 250 μl cell/fibrin mixture was seeded in each well of a precooled 24‐well plate and mixed well with pre‐added 5 μl thrombin (0.1 U/μl, Searun Holdings Company). After 2 hours of incubation at 37 °C, 1 ml of RPMI1640 medium containing 10% FBS was added.
To form 3D matrix gels, 20 μl of Matrigel (CORNING, 356234) was diluted with 20 μl of RPMI 1640 medium containing 1000 HCT116 or SW480 cells and seeded in a 24‐well plate. The gels were placed in a CO2 incubator at 37°C for 1 hour for solidification and then overlaid with 1 ml of RPMI 1640 complete medium.
2.2. Glucose consumption and lactic acid measurement
The flask‐ or 3D‐cultured cells were seeded in six‐well plates for 24 hours. Then the RPMI 1640 medium was replaced with 2 ml of fresh medium. After 24 hours, the supernatant was collected, and glucose consumption was measured using a glucose and sucrose assay kit (Sigma‐Aldrich, MAK013).
For lactic acid measurement, the flask‐ or 3D‐cultured cells were collected after culturing for the same length of the indicated time, and then lactic acid production of cells was measured by colorimetry according to the instructions of a Lactate Colorimetric Assay Kit II (Biovision, K627‐100). The glucose and glucose consumption were normalized to μmol/106 cells.
2.3. Animal experiment
Six‐week‐old NSG mice were obtained from HFK Bio. All animal experiments were conducted per standard procedures and approved by the Animal Ethics Committee of the Second Xiangya Hospital of Central South University. To establish a tumorigenicity assay mouse model, 1 × 104 HCT116 and SW480 cells were subcutaneously injected into the mice. The sample size was n = 20. The tumor number was calculated per group.
2.4. Statistical analyses
All statistical analyses were performed using GraphPad Prism 6 or SPSS version 20. Experimental data are presented as mean ± SEM. Student's t test was performed when only two groups were compared. For the correlation analysis, Pearson's correlation analysis was used. Survival analysis was performed using the Kaplan‐Meier method. The TCGA data sets were used to evaluate the expression of ITGB1, HIF1α, and TRIM11 transcripts in patients with low expression and high expression. Differences between groups were denoted as ns (not significant), *p < 0.05, and **p < 0.01.
3. RESULTS
3.1. 3D gels induced dedifferentiation of colorectal cancer cells
Cancer stem cells can self‐renew and differentiate into non‐CSCs, which are responsible for tumor initiation and sustained growth. However, studies in various organ systems implicated that differentiated cancer cells can dedifferentiate into stem‐like cancer cells, eventually resulting in tumor occurrence. 15 To investigate the transition between CSCs and differentiated tumor cells, CD133 was used as a stem marker to sort colorectal CSCs from HCT116 and SW480 cells (Figure 1A). Consistent with previous studies, our colony formation and in vivo tumorigenic analysis revealed that CD133+ HCT116/SW480 cells exhibited a stronger ability to form colonies (Figure S1A) and higher tumorigenesis (Figure S1B) compared with CD133− cells. This result was consistent with the traditional hypothesis of CSCs. However, a series of studies indicated that CD133− cancer cells, also defined as differentiated tumor cells, could also initiate tumors and dedifferentiate into stem‐like status. 16 In fact, spheroid colonies formed by CD133− HCT116/SW480 cells were observed (Figure S1A). Therefore, we supported the hypothesis that cancer cells are plastic, harboring the potential of transition between CSCs/differentiated cancer cells and that extracellular cues might influence the stem‐like phenotypes of cancer cells.
FIGURE 1.
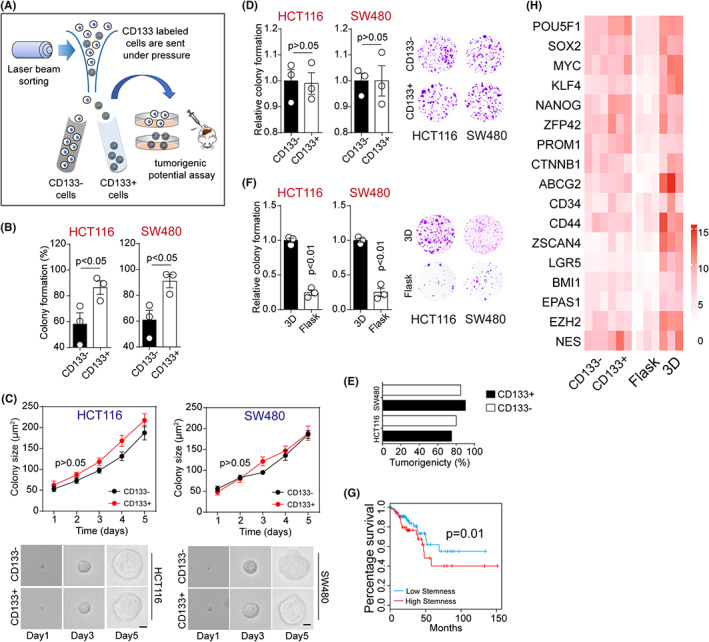
3D gels induced dedifferentiation of colorectal cancer cells. A, CD133− and CD133+ cells were sorted by flow cytometry, and tumorigenic potential was determined by colony formation and tumor bearing mice assay. B, Colony formation rates of CD133− and CD133+ HCT116/SW480 cells in 3D fibrin gel at 5 days. HCT116, p = 0.49; S4480, p = 0.29. C, Colony sizes of CD133− and CD133+ HCT116/SW480 cells in 3D fibrin gel. The scale bar is 25 μm. D, Colony formation of 3D fibrin–cultured CD133− and CD133+ HCT116/SW480 cells was examined in flask. E, The tumorigenic rates of mice subcutaneously injected with 104 3D fibrin–cultured CD133+/− HCT116/SW480 cells. F, 3D fibrin gels–cultured HCT116/SW480 cells were collected and cultured in flask or 3D fibrin gels for 5 d again. After that, the colony formation capability of tumor cells was examined in flask. HCT116, p = 0.001; SW480, p = 0.004. G, The survival analysis of colorectal patients with high/low stem‐associated gene signature in TCGA database (n = 455). H, The heatmap of the stem‐associated genes in CD133+/− HCT116 and flask/3D fibrin–cultured HCT116 cells is shown. ΔCT = CT (target gene) – CT (GADPH). The data are presented as means ± SEM of three independent experiments
When cultured on 3D substrates, including matrix or fibrin gels, tumor cells usually formed spheroid colonies and acquired stem‐like characteristics. 17 To elucidate the dedifferentiation potential of differentiated tumor cells, we purified CD133− and CD133+ HCT116/SW480 cells and seeded 1000 cells on 3D fibrin gels for 5 days. Although a more substantial colony formation capacity was observed on 3D gels in CD133+ HCT116/SW480 cells, the vast majority of CD133+/− tumor cells (CD133− cells, ~60% and CD133+ cells, ~90%) succeeded in forming spheroid colonies (Figure 1B). Intriguingly, 3D‐cultured CD133− and CD133+ HCT116/SW480 cells displayed similar colony growth curves (Figure 1C), prompting us to speculate that those CD133− cells might dedifferentiate into the stem‐like status and possessed similar characteristics to those of CD133+ cells. To further confirm our hypothesis, we collected these 3D‐cultured CD133+/− cells and assessed their spheroid formation and tumorigenic capability. As shown in Figure 1D,E, CD133− and CD133+ HCT116/SW480 cells exhibited a similar ability to form colonies in 2D (flask) (Figure 1D) and neoplasm (Figure 1E), suggesting that CD133− differentiated cancer cells reacquired stem‐like characteristics after 3D culture. Consistently, enhanced colony formation potential and dedifferentiation of CD133− HCT116/SW480 cells were found in the 3D Matrigel culture system (Figure S1C). We then seeded those dedifferentiated cells in a flask. After 5 days, cells were collected and the spheroid colony formation capability was examined again. As expected, the flask culture weakened the colony formation capacity of dedifferentiated HCT116/SW480 cells, when compared with permanent 3D‐cultured cells (Figure 1F), suggesting that tumor cells were plastic and could transition between CSCs and differentiated tumor cells. Subsequently, we sought to elucidate the biological difference between CD133+ sorted or dedifferentiated tumor cells at the genetic levels. As previously reported by Miranda et al., a stemness score based on the stem cell transcriptome presented a higher correlation with CSCs in solid cancers. 18 Consistently, survival analysis based on the TCGA database implicated that colorectal cancer patients with high stem scores exhibited poor prognosis (Figure 1G). Thus, we next examined the stem cell transcriptome by real‐time PCR in HCT116/SW480 cells cultured in flask/3D or CD133 sorted cells. We found a moderate difference between the CD133− and CD133+ groups. However, a smaller internal similarity was found between flask‐ and 3D fibrin–cultured dedifferentiated cancer cells (Figure 1H), indicating that tumor cells are plastic and those dedifferentiated 3D cells represented more inherent features of CSCs. These results suggested that the extracellular matrix is capable of reverting CSCs and differentiated cells into each other in a dynamic status.
3.2. The activation of ITGB1 signaling contributed to the dedifferentiation of tumor cells
Integrin‐mediated cell adhesions have been widely demonstrated to mediate ECM signals transduction to cancer cells. 19 This prompted us to explore the roles of integrins in the dedifferentiation process of cancer cells. Hence, we examined the expression of integrin β1 ~ 8 in HCT116/SW480 cells cultured in flask or 3D fibrin gels. It is noteworthy that 3D‐cultured cells showed an increased expression of ITGB1 as determined by real‐time PCR (Figure 2A). Elevated expression of ITGB1 was also found in 3D fibrin gels– and Matrigel‐cultured HCT116/SW480 cells at the protein level (Figures 2B and S2A). This observation suggested that ITGB1 might play a central role in the dedifferentiation process of colorectal cancer cells. Subsequently, to confirm the role of ITGB1 in cell dedifferentiation, ITGB1‐neutralizing antibodies (NAs) were used to treat HCT116/SW480 cells cultured in 3D gels. As expected, ITGB1 NAs remarkably suppressed colony formation (Figure 2C) and growth (Figure 2D) of HCT116/SW480 cells in 3D fibrin gels. In addition, HCT116/SW480 cells treated with ITGBs NAs revealed a weakened colony formation in flask (Figure 2E) and in vivo tumorigenic potential (Figure 2F). Similar suppressive effects were observed in a 3D Matrigel (Figure S2B,C). Intriguingly, ITGB1 NAs did not affect the colony formation rates of HCT116/SW480 cells cultured in flasks (Figure S2D). These results suggested that cancer cells in 3D gels reacquired CSC phenotypes in an ITGB1‐dependent manner.
FIGURE 2.
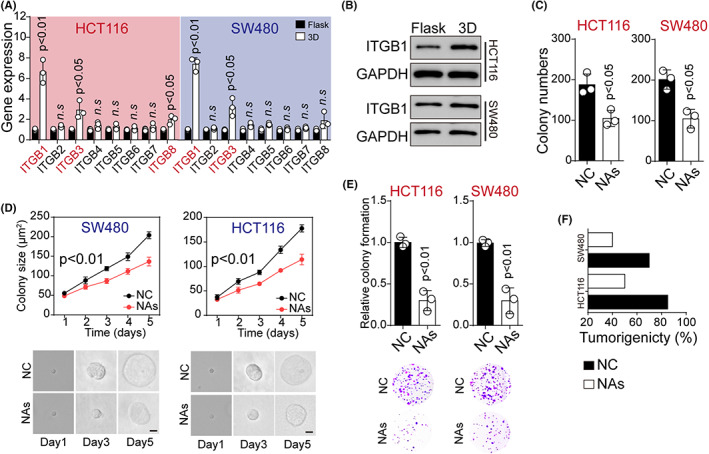
The activation of ITGB1 signaling contributed to dedifferentiation of tumor cells. A, The relative expression of ITGB1 ~ 8 at mRNA level in HCT116/SW480 cells cultured in flask or 3D fibrin gel (HCT116: ITGB1, p = 0.001; ITGB3, p = 0.018; ITGB8, p = 0.042; SW480. ITGB1: p = 0.01; ITGB3, p = 0.012). B, Western blotting assay of ITGB1 in HCT116 or SW480 cells in flask or 3D fibrin gel. C, Colony formation of HCT116/SW480 cells treated with NC (negative control) or ITGB1 neutralizing antibodies (NAs) (10 μg/ml) was calculated in 3D fibrin gels (HCT116, p = 0.02; SW480, p = 0.03). D, Colony sizes of HCT116/SW480 cells treated with NC or ITGB1 NAs (10 μg/ml) in 3D fibrin gel at different times (HCT116, p = 0.003; SW480, p = 0.004). The scale bar is 25 μm. E, Colony formation analysis of 3D fibrin–cultured HCT116/SW480 cells treated with NC or ITGB1 NAs (10 μg/ml) in flask (HCT116, p = 0.005; SW480, p = 0.004). The scale bar is 2 mm. F, Tumorigenic rates of mice subcutaneously injected with 104 HCT116/SW480 cells cultured in 3D fibrin gels (treated with NC or 10 μg/ml ITGB1 NAs). The data are presented as means ± SEM of three independent experiments
3.3. 3D gels culture promoted glycolysis to mediate dedifferentiation of cancer cells
Next, we sought to understand the mechanism of the ITGB1‐associated dedifferentiation process in cancer cells. As previously reported, tumor cells with elevated integrin expression displayed stem‐like phenotypes and invasive properties, accompanied by changes in metabolic signatures. 20 , 21 , 22 Importantly, CSCs could shift from mitochondrial aerobic glucose metabolism to temporary glycolysis, contributing to the activation of HIF‐associated pro‐stem signaling pathways. 23 Hence, we decided to elucidate the role of glucose metabolism in the dedifferentiation of cancer cells. To do this, we used cancer cells cultured in flask or 3D fibrin gels and examined the glucose metabolism and lactate production in these cells. ELISA analysis revealed a systemic glycolysis upregulation in HCT116 cells cultured in 3D fibrin gels, as shown by the increased glucose uptake (Figure 3A) and lactate production (Figure 3B). The glycolysis rate is regulated by the expression of glycolytic enzymes, such as HK1, HK2, PFK, ALDA, LDHA, and PDH. Here, we found significant PFK downregulation in the dedifferentiated HCT116 cells cultured in 3D fibrin gels, as determined by real‐time PCR (Figure 3C). Consistently, PFK downregulation was also found in differentiated HCT116/SW480 cells at the protein level (Figure 3D). As previously reported, PFK catalyzed a rate‐limiting step glycolysis via phosphorylating fructose 6‐phosphate, thereby suppressing the glycolysis process. 24 , 25 Hence, we presented the hypothesis that 3D culture could promote cancer dedifferentiation by remodeling glucose metabolism in an ITGB1/PFK‐dependent manner.
FIGURE 3.
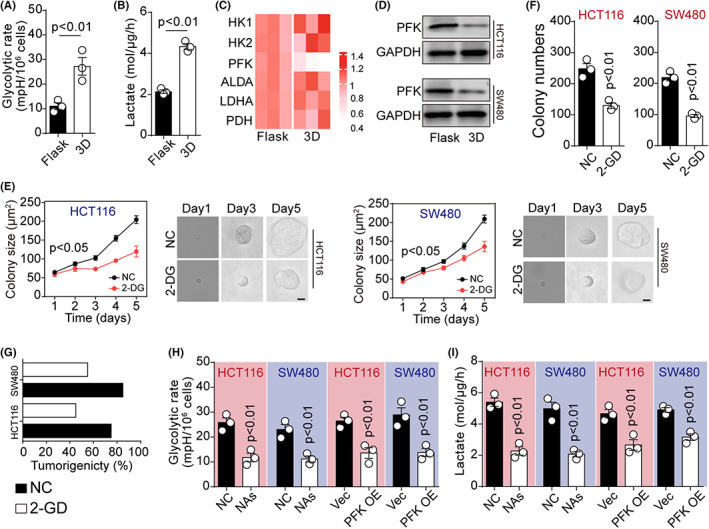
3D gels culture promoted glycolysis to mediate dedifferentiation of cancer cells. A, Glycolytic rates of HCT116 cells in flask or 3D gels (HCT116, p = 0.003; SW480, p = 0.006). B, A total of 1.5 × 104 of flask‐ or 3D fibrin gels–cultured HCT116 cells were seeded in a 96‐well plate in 100 μl medium. Twenty‐four hours later, the culture medium was collected and the level of lactate was detected (p = 0.005). C, The heatmap of HK1, HK2, PFK, ALDA, LDHA, and PDH expression at mRNA level was detected by qPCR in HCT116 cells cultured in flask or 3D fibrin gels. ΔCT = CT (target gene) – CT (GADPH). D, Western blotting assay of PFK in HCT116 or SW480 cells cultured in flask or 3D fibrin gels. E, Colony sizes of HCT116/SW480 cells in 3D fibrin gels treated with NC or 2‐DG (2 μM) at different times (HCT116, p = 0.03; SW480, p = 0.03). The scale bar is 25 μm. F, HCT116 or SW480 cells were seeded in 3D fibrin gels and treated with NC or 2‐DG (2 μM) for 5 days. Then, the colony formation capability was examined in flask (HCT116, p = 0.002; SW480, p = 0.003). G, The tumorigenic rates of mice subcutaneously injected with 104 HCT116 or SW480 cells in (E). H, Glycolytic rates of 3D fibrin–cultured HCT116/SW480 cells treated with NC or ITGB1 neutralizing antibodies (NAs) (10 μg/ml), or 3D‐cultured vector/PFK overexpression HCT116/SW480 cells (p = 0.002, 0.003, 0.003, 0.002). I, The level of lactate secretion was determined in 3D fibrin–cultured HCT116/SW480 cells treated with NC or ITGB1 NAs (10 μg/ml), or 3D‐cultured vector/PFK overexpression HCT116/SW480 cells (p = 0.001, 0.002, 0.002, 0.003). The data are presented as means ± SEM of three independent experiments
To corroborate this hypothesis, we treated 3D fibrin gels–cultured cells with 2‐deoxyglucose, a well‐known glycolysis inhibitor, 26 and assessed the spheroid formation and tumorigenic capacity of cancer cells. In fact, 2‐deoxyglucose treatment significantly suppressed colony growth in 3D fibrin gels (Figure 3E). However, the cells cultured in 3D gels showed a weak colony formation (Figure 3F) and in vivo tumorigenic potential (Figure 3G) after 2‐deoxyglucose treatment, suggesting that 3D gels promoted the dedifferentiation process by regulating glycolysis of cancer cells. Subsequently, we sought to determine the roles of ITGB1 and PFK in glycolysis. To assess the influence of PFK in this context, PFK was overexpressed in HCT116/SW480 cells seeded in 3D fibrin gels (Figure S3A,B). In addition, ITGB1 NAs were added to the HCT116/SW480 cells cultured in 3D gels. Both PFK overexpression and ITGB1 NA treatment resulted in reduced glucose utilization and lactate production in 3D cultured cancer cells (Figure 3H,I), suggesting suppression of glycolysis. These findings showed that 3D culture could promote the dedifferentiation process via regulating glycolysis in cancer cells. Moreover, the results suggested that the alteration of glucose metabolism was mediated by the expression of ITGB1 and PFK.
3.4. F‐actin bundling released TRIM11 to degrade PFK
As our previous results have described the roles of ITGB1 and PFK in glycolysis, we next aimed to understand how ITGB1 acted downstream of PFK in colorectal cancer cells. Compelling studies have shown that cells could sense extracellular biomechanical force through actomyosin and cytoskeleton contraction. 27 , 28 Intriguingly, cytoskeletal F‐actin bundling has been previously reported to regulate glycolysis through E3 ligase TRIM proteins, which targeted PFKP for proteasomal degradation. 29 Importantly, reduced expression and distribution of F‐actin were observed in HCT116/SW480 cells cultured with 3D fibrin gels compared with the flask culture group. In addition, treatment with ITGB1 NA disrupted F‐actin binding (Figure 4A), indicating a potential role for F‐actin in the regulation of cell dedifferentiation. Subsequently, we targeted F‐actin using the actin‐monomer‐sequestering compound latrunculin A (LatA) to treat flask‐cultured HCT116/SW480 cells. LatA treatment decreased F‐actin lengthening and distribution (Figure 4B) and downstream PFK expression in cancer cells (Figure 4C). Those findings suggested that ITGB1 might interact with F‐actin and transduce extracellular biomechanical signals to regulate PFK expression. Increasing evidence suggests that PFK degradation is mediated by the disassembly of stress sensing cytoskeleton (F‐actin), which colocalized with TRIM proteins to suppress the PFK ubiquitination process induced by TRIM. 29 Using anti‐F‐actin to pull down F‐actin binding proteins in flask‐cultured HCT116 cells, three candidates, TRIM22, TRIM11, and TRIM58, were identified by mass spectrometry (Figure 4D). Next, we analyzed the correlation between TRIM proteins and overall survival of colorectal cancer patients. Poor overall survival was observed in patients with high TRIM11 expression (Figure 4E–G), suggesting that TRIM11 might participate in the development of colorectal cancer. Intriguingly, we also found a strong colocalization of F‐actin and TIRM11 in HCT116/SW480 cells cultured in a flask, while cells cultured in 3D gels showed TIRM11 in the cytoplasm instead (Figure 4H), indicating the potential role of TRIM11 involved in ITGB1/F‐actin–associated cell behavior. To confirm our hypothesis that F‐actin bundles sequestered TRIM11, which degraded PFKP to reduce PFK expression in cancer cells, we overexpressed TRIM11 in HCT116/SW480 cells (Figure S4A,B) and seeded them in 3D fibrin gels. In these cells, TRIM11 was widely distributed in the cytoplasm (Figure S4C,D). Notably, we found that PFKP was efficiently precipitated by a Flag‐antibody against TRIM11 (Figure 4I), and reduced expression of PFKP was found in TRIM11‐overexpressing HCT116/SW480 cells cultured in 3D gels (Figure 4J), indicating that TRIM11 interacted with PFKP mediating its downregulation. Next, we further confirmed the TRIM11‐mediated PFKP ubiquitination process. The proteasome inhibitor MG132‐treated 3D cells displayed PFKP upregulation compared with PBS‐treated 3D cells (Figure S4E). Treatment with MG132 stabilized the PFKP proteins suggesting degradation by the proteasome. Indeed, we also found that 3D culture increased PFKP ubiquitination, while flask culture decreased ubiquitination (Figure S4F). Those results suggested that 3D gels culture and ITGB1 mediated F‐actin bundling to release TRIM11 to degrade PFKP, resulting in PFK downregulation.
FIGURE 4.
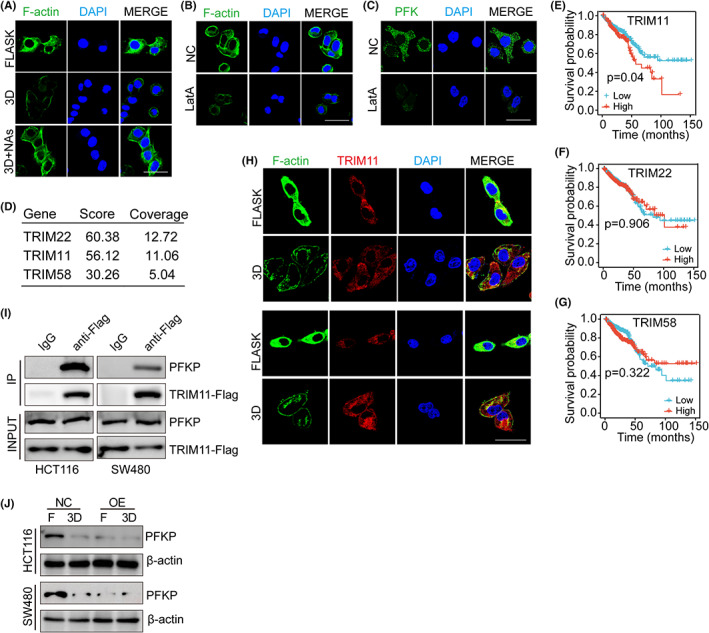
F‐actin binding released TRIM11 to degrade PFK. A, Immunofluorescence staining of F‐actin in HCT116 cells cultured in flask or 3D fibrin environment (containing 10 μg/ml ITGB1 neutralizing antibodies [NAs] or not). The scale bar is 20 μm. B, Immunofluorescence staining of F‐actin in HCT116 cells treated with NC or LatA (1 μM). The scale bar is 20 μm. C, Immunofluorescence staining of PFK in HCT116 cells treated with NC or LatA (1 μM). The scale bar is 20 μm. D, The HCT116 cells were lysed and immunoprecipitated with anti‐F‐actin or IgG antibody. The proteins immunoprecipitated were run on SDS‐PAGE gel and processed for mass spectrometry. The candidate genes are listed. E, Kaplan‐Meier analysis of TRIM11 mRNA expression in patients from TCGA database (N = 455). F, Kaplan‐Meier analysis of TRIM22 mRNA expression in patients from TCGA database (N = 455). G, Kaplan‐Meier analysis of TRIM58 mRNA expression in patients from TCGA (N = 455). H, Immunofluorescence staining of F‐actin and TRIM11 in HCT116 cells (up) or SW480 cells (down) cultured in flask or 3D fibrin environment. The scale bar is 20 μm. I, The Western blotting analysis of PFKP and TRIM11from total HCT116/SW480 cell lysates (input) and proteins immunoprecipitated (IP) with anti‐FLAG or control immunoglobulin G (IgG) cells. J, Expression of PFKP in HCT116 or SW480 cells transfected with NC plasmid or pCMV‐TRIM11 in flask or 3D fibrin environment
3.5. Upregulation of HIF1α by glycolysis promoted stem‐like phenotypes in colorectal cancer cells
As previously reported, the lactate produced by tumor cells during the glycolysis process could enhance CSCs properties via HIF1α signaling. 30 Therefore, we determined whether 3D culture could upregulate HIF1α expression and promote glycolysis. As expected, 3D fibrin culture or lactate treatment significantly increased HIF1α levels in both HCT116 and SW480 cells. Moreover, suppression of HIF1α expression occurred in the PFK overexpression group, despite 3D gel culture (Figure 5A,B), suggesting that elevated glycolysis resulted in HIF1α activation and dedifferentiation of colorectal cancer cells. To further assess the role of HIF1α in the dedifferentiation process, HCT116/SW480 cells were cultured in 3D fibrin gels and treated with the HIF1α inhibitor GN44028, and colony formation capability of tumor cells was examined. HIF1α inhibition contributed to the reduction of spheroid colony formation (Figure 5C) and tumorigenicity (Figure 5D) compared with the dedifferentiated cells in 3D gels. Overexpression of HIF1α (Figure S5A,B) remarkably upregulated the stem‐associated signaling molecules (Figure 5E), as shown in Figure 1E, indicating that 3D gels promoted dedifferentiation and CSCs properties through HIF1α signals. As HIF1α upregulated the stem‐associated signaling molecules that highly correlated with the overall survival in patients, we further evaluated the influence of HIF1α on colorectal cancer patients using the TCGA database. Indeed, patients with high HIF1α expression exhibited a poor overall survival (Figure 5F), suggesting a correlation between HIF1α and colorectal cancer prognosis. Together, these findings suggested that HIF1α upregulation induced by glycolysis promoted stem‐like phenotypes in colorectal cancer cells.
FIGURE 5.
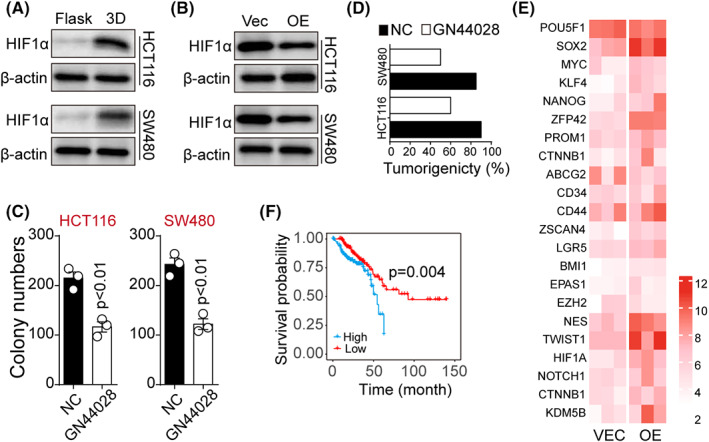
Upregulation of HIF1α by glycolysis promoted stem‐like phenotypes in colorectal cancer cells. A, Western blotting assay of HIF1α in flask or 3D fibrin gel–cultured HCT116/SW480 cells. B, Western blotting assay of HIF1α in 3D gels–cultured PFK overexpression (or not) HCT116/SW480 cells. C, Colony formation of 3D fibrin–cultured HCT116/SW480 cells treated with NC or GN44028 (5 μM). HCT116, p = 0.0032; SW480, p = 0.002; D, Tumorigenic rates of mice subcutaneously injected with 104 3D fibrin gels–cultured HCT116/SW480 treated with NC or GN44028 (5 μM). E, The heatmap of the stem‐associated signaling molecules in HIF1α overexpression (or not) HCT116/SW480 cells. F, Kaplan‐Meier cumulative survival analysis of colorectal cancer patients from TCGA database; patients were divided into a high HIF1α expression group (n = 169) and a low HIF1α expression group (n = 166). The data are presented as means ± SEM of three independent experiments
3.6. Aberrant expression of ITGB1/TRIM11/HIF1α correlated with tumor recurrence and development in the clinic
Our previous results confirmed the effects of the ITGB1/TRIM11/HIF1α axis on regulating cell dedifferentiation and stemness. We thus considered whether these experimental findings could be clinically relevant for the prognosis of colorectal cancer patients in the clinic. Hence, we retrieved information and tumor specimens from 24 colorectal cancer patients (Figure 6A), which were divided into nonrecurrent tumor (Non‐R) and recurrent tumor groups (Rec) according to the follow‐up visit. Due to the crucial role of the extracellular matrix in regulating cell dedifferentiation, immunohistochemistry was performed to assess major extracellular matrix–associated proteins (collagen I, fibronectin, fibrinogen, and laminin) expression in these tumor tissues. Intriguingly, increased collagen I and fibrinogen were observed in Rec tumor tissues compared with the Non‐R group (Figure 6B). Our previous results have suggested that the extracellular matrix could mediate biomechanical signaling receptor ITGB1 activation and cytoskeleton bundling to promote cell dedifferentiation and stemness. Furthermore, collagen I and fibrinogen served as significant solid elements in the extracellular matrix, as they can produce biomechanical signals and mediate cell dedifferentiation. 31 , 32 , 33 In addition, we performed immunofluorescence and confocal analysis to estimate ITGB1/TRIM11/HIF1α protein level in those colorectal tumor samples in order to assess the relevance of our research data in clinical tumor recurrence. Remarkably, we found high ITGB1/TRIM11/HIF1α protein expression in recurrent tumor tissues compared with the nonrecurrent tumor group (Figure 6C,D). More importantly, we detected a significant positive correlation between progression‐free survival time and ITGB1/HIF1α expression (Figure 6E,F). However, no significant differences were observed in the progression‐free survival analysis in TRIM11high or TRIM11low patients (Figure 6G). This suggested that activation of ITGB1/TRIM11/HIF1α signaling might tightly correlate with tumor recurrence and influence colorectal cancer development in patients. These findings generally agreed with our experimental data and strengthened the concept that extracellular matrix/ITGB1/TRIM11/HIF1α/dedifferentiation signaling pathways could be significant mechanisms for promoting cell stemness and tumor progression in colorectal cancer.
FIGURE 6.
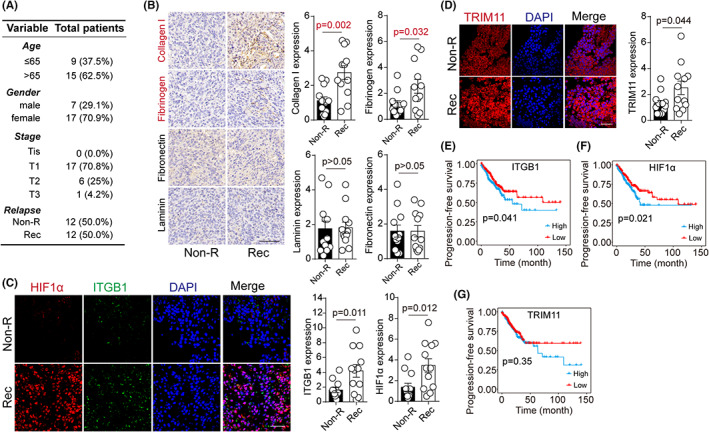
Aberrant expression of ITGB1/TRIM11/HIF1α correlated with tumor recurrence and development in clinic. A, Information of 24 colorectal cancer patients, divided into Non‐R (nonrecurrent) and R (recurrent) groups. B, Immunohistochemistry of collagen, fibrinogen, fibronectin, and laminin in tumor tissues from Non‐R and R patients. Protein expression was quantified in 24 patients. C, Immunofluorescence of HIF1α and ITGB1 in tumor tissues from Non‐R and R patients. Protein expression was quantified in 24 patients. D, Immunofluorescence of TRIM11 in tumor tissues from Non‐R and R patients. Protein expression was quantified in 24 patients. E‐G, Progression‐free survival analysis in 549 colorectal cancer patients, divided into high/low ITGB1, HIF1α, and TRIM11 groups. The data are presented as means ± SEM of three independent experiments
4. DISCUSSION
Increasing evidence exists that CSCs within tumors may cause chemoresistance and tumor recurrence in patients after interventions. This has attracted increasing attention to improving clinic outcomes by eliminating CSCs. 34 , 35 However, few signaling pathways have been reported to be involved in controlling CSCs generation/development. Our study clarified the novel concept that the bulk of tumor cells could switch to CSCs status through a matrix‐dependent dedifferentiation process and explored the underlying mechanism of dedifferentiation in colorectal cancer cells.
It has been documented that the lower stage of tumor differentiation confers to poor prognosis in the clinic. 36 , 37 Dedifferentiated tumor cells usually exhibit a strong tumorigenic potential and express increased stem‐associated genes, such as SOX2 and CD133. 38 , 39 However, little is known about the dedifferentiation process in non‐CSCs. Furthermore, the mechanism of microenvironment‐induced tumor heterogeneity remains elusive. Intriguingly, recent studies stated that extracellular matrix–based 3D gels could promote the tumorigenic potential and mediate the CSCs selection in the bulk of tumor cells. 40 , 41 In line with those observations, we found that colorectal cancer cells seeded in 3D Matrigel/fibrin gels reacquired stem‐like characteristics when compared with flask culture. Notably, a similar CSCs status transition was observed in differentiated colorectal cancer cells (CD133−). This indicates the presence of a dedifferentiation process in tumor cells rather than CSCs selection induced by 3D gels. Accordingly, dedifferentiated cells could revert to differentiated status in the flask culture, supporting our concept of bidirectional conversion between differentiated cells and CSCs.
Various 3D gels culture systems, including collagen, Matrigel, and hydrogel, have been applied to tumor cell culture and CSC research. 42 Using 3D Matrigel and fibrin gels, we further demonstrated the process of dedifferentiation in colorectal cancer cells. Notably, both 3D fibrin and Matrigel succeeded in inducing the generation of stem‐like phenotypes in cancer cells. This allowed us to elucidate the mechanism of the dedifferentiation process in 3D gels through biomechanical signaling pathways instead of chemical signals induced by compounds. In line with our hypothesis, immunohistochemical analysis of clinical tumor tissues revealed a potential correlation between tumor recurrence and solid extracellular matrix components (collagen I and fibrinogen) but not soluble components such as laminin or fibronectin. Compelling studies have suggested that the transduction of extracellular signals is mainly mediated by integrin receptors on the cell membrane. 20 Furthermore, integrin expression is closely associated with the activation of pro‐survival signaling pathways in tumor cells. 43 For instance, Grzesiak and his colleagues demonstrated that extracellular collagen could mediate the malignant phenotype of pancreatic cancer through integrin β1. 44 Ritzenthaler reported that the fibronectin‐integrin axis promotes the growth of lung cancer cells in a PI3K/AKT‐dependent manner. 45 Consistently, 3D Matrigel– or fibrin gel–cultured colorectal CSCs, which exhibited stronger stem‐like characteristics, revealed significant upregulation of ITGB1 compared with the cells in flask culture. More importantly, our study also observed an alteration of the cytoskeleton, as dedifferentiated colorectal cells revealed enhanced binding of F‐actin. This finding further confirmed our hypothesis that extracellular matrix could transduce biomechanical signals to mediate the dedifferentiation process of cancer cells, which was dependent on the biomechanical receptor integrins and deformation of the cytoskeleton induced by biomechanical force. Our study demonstrated the role of the cytoskeleton in regulating cell stemness.
Aberrant metabolic characteristics of CSCs have been widely documented. CSCs are preferentially transformed from aerobic glucose metabolism to glycolysis or primarily glycolytic. 46 For instance, radiotherapy‐resistant clone spherical nasopharyngeal cancer cells displayed a clear glycolytic metabolism. 47 In liver cancer, glycolysis was also preferred in CD133+/CD49f+ CSCs. 48 In line with those observations, our study demonstrated increased glycolysis induced by 3D Matrigel/fibrin gels culture to promote the differentiation and facilitate the stem‐like phenotypes of colorectal cells. We confirmed that PFK, a rate‐limiting enzyme related to the glycolytic pathway, was significantly downregulated in 3D Matrigel/fibrin‐cultured cancer cells, resulting in stemness upregulation and the process of dedifferentiation. Mechanistically, we found that F‐actin bundling could regulate glycolysis through E3 ligase TRIM proteins, which targeted PFKP for proteasomal degradation causing PFK downregulation. This finding was consistent with a previous report by Jin Suk Park showing mechanical regulation of glycolysis induced by cytoskeleton and TRIM21. 48 Our study also determined the role of TRIM11 in PFK degradation and highlighted the correlation between ITGB1 and glucose metabolism. Collectively, these findings indicated that the dedifferentiation process is regulated by ITGB1 and cytoskeleton and is tightly related to glycolysis. We provided the first evidence to disclose the correlation between extracellular matrix, cytoskeleton, glucose metabolism, and tumor stemness.
In conclusion, our study provided evidence that tumor cells are plastic, and dedifferentiated tumor cells could reacquire CSC phenotypes in the presence of extracellular cues. Blockade of the ITGB1/F‐actin/TRIM11/PFK/HIF1α axis efficiently suppressed the process of dedifferentiation, providing a potential therapeutic tool for colorectal cancer.
FUNDING INFORMATION
This work received no fundings.
DISCLOSURE
The authors declare no competing interests.
ETHICAL STATEMENT
Approval of the research protocol by an Institutional Reviewer Board: N/A.
Informed consent: N/A.
Registry of the study/trial: N/A.
Animal Studies: The animal experiments were approved by the Ethics Committee of the Second Xiangya Hospital of Central South University and conducted in accordance with the ARRIVE guidelines as well as the National Institutes of Health guide for the care and use of Laboratory animals.
Supporting information
Figure S1
Han T, Jiang Y, Wang X, et al. 3D matrix promotes cell dedifferentiation into colorectal cancer stem cells via integrin/cytoskeleton/glycolysis signaling. Cancer Sci. 2022;113:3826‐3837. doi: 10.1111/cas.15548
DATA AVAILABILITY STATEMENT
All data that support the findings of this study are available from the corresponding authors upon reasonable request.
REFERENCES
- 1. Janney A, Powrie F, Mann EH. Host‐microbiota maladaptation in colorectal cancer. Nature. 2020;585:509‐517. [DOI] [PubMed] [Google Scholar]
- 2. Cheung P, Xiol J, Dill MT, et al. Regenerative reprogramming of the intestinal stem cell state via hippo signaling suppresses metastatic colorectal cancer. Cell Stem Cell. 2020;27:590‐604.e599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Li C, Sun YD, Yu GY, et al. Integrated omics of metastatic colorectal cancer. Cancer Cell. 2020;38:734‐747.e739. [DOI] [PubMed] [Google Scholar]
- 4. Zhang Y, Song J, Zhao Z, et al. Single‐cell transcriptome analysis reveals tumor immune microenvironment heterogenicity and granulocytes enrichment in colorectal cancer liver metastases. Cancer Lett. 2020;470:84‐94. [DOI] [PubMed] [Google Scholar]
- 5. Fumagalli A, Oost KC, Kester L, et al. Plasticity of Lgr5‐negative cancer cells drives metastasis in colorectal cancer. Cell Stem Cell. 2020;26:569‐578.e567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zeuner A, Todaro M, Stassi G, De Maria R. Colorectal cancer stem cells: from the crypt to the clinic. Cell Stem Cell. 2014;15:692‐705. [DOI] [PubMed] [Google Scholar]
- 7. Ayob AZ, Ramasamy TS. Cancer stem cells as key drivers of tumour progression. J Biomed Sci. 2018;25:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nassar D, Blanpain C. Cancer stem cells: basic concepts and therapeutic implications. Annu Rev Pathol. 2016;11:47‐76. [DOI] [PubMed] [Google Scholar]
- 9. Vlashi E, Pajonk F. Cancer stem cells, cancer cell plasticity and radiation therapy. Semin Cancer Biol. 2015;31:28‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang L, Shi H, Chen H, et al. Dedifferentiation process driven by radiotherapy‐induced HMGB1/TLR2/YAP/HIF‐1α signaling enhances pancreatic cancer stemness. Cell Death Dis. 2019;10:724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li J, Stanger BZ. How tumor cell dedifferentiation drives immune evasion and resistance to immunotherapy. Cancer Res. 2020;80:4037‐4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kloskowski T, Jarząbkowska J, Jundziłł A, et al. CD133 antigen as a potential marker of melanoma stem cells: in vitro and in vivo studies. Stem Cells Int. 2020;2020:8810476‐8810410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sharma BK, Manglik V, O'Connell M, et al. Clonal dominance of CD133+ subset population as risk factor in tumor progression and disease recurrence of human cutaneous melanoma. Int J Oncol. 2012;41:1570‐1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Krausova M, Korinek V. Wnt signaling in adult intestinal stem cells and cancer. Cell Signal. 2014;26:570‐579. [DOI] [PubMed] [Google Scholar]
- 15. Li W, Zhang L, Guo B, et al. Exosomal FMR1‐AS1 facilitates maintaining cancer stem‐like cell dynamic equilibrium via TLR7/NFκB/c‐Myc signaling in female esophageal carcinoma. Mol Cancer. 2019;18:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Aghajani M, Mansoori B, Mohammadi A, Asadzadeh Z, Baradaran B. New emerging roles of CD133 in cancer stem cell: signaling pathway and miRNA regulation. J Cell Physiol. 2019;234:21642‐21661. [DOI] [PubMed] [Google Scholar]
- 17. Liu J, Tan Y, Zhang H, et al. Soft fibrin gels promote selection and growth of tumorigenic cells. Nat Mater. 2012;11:734‐741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Miranda A, Hamilton PT, Zhang AW, et al. Cancer stemness, intratumoral heterogeneity, and immune response across cancers. Proc Natl Acad Sci U S A. 2019;116:9020‐9029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kechagia JZ, Ivaska J, Roca‐Cusachs P. Integrins as biomechanical sensors of the microenvironment. Nat Rev Mol Cell Biol. 2019;20:457‐473. [DOI] [PubMed] [Google Scholar]
- 20. Cooper J, Giancotti FG. Integrin signaling in cancer: Mechanotransduction, stemness, epithelial plasticity, and therapeutic resistance. Cancer Cell. 2019;35:347‐367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hamidi H, Ivaska J. Every step of the way: integrins in cancer progression and metastasis. Nat Rev Cancer. 2018;18:533‐548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sung JS, Kang CW, Kang S, et al. ITGB4‐mediated metabolic reprogramming of cancer‐associated fibroblasts. Oncogene. 2020;39:664‐676. [DOI] [PubMed] [Google Scholar]
- 23. Semenza GL. Hypoxia‐inducible factors: coupling glucose metabolism and redox regulation with induction of the breast cancer stem cell phenotype. EMBO J. 2017;36:252‐259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fernandes PM, Kinkead J, McNae I, Michels PAM, Walkinshaw MD. Biochemical and transcript level differences between the three human phosphofructokinases show optimisation of each isoform for specific metabolic niches. Biochem J. 2020;477:4425‐4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Murillo‐López J, Zinovjev K, Pereira H, et al. Studying the phosphoryl transfer mechanism of the E. coli phosphofructokinase‐2: from X‐ray structure to quantum mechanics/molecular mechanics simulations. Chem Sci. 2019;10:2882‐2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pajak B, Siwiak E, Sołtyka M, et al. 2‐deoxy‐d‐glucose and its analogs: from diagnostic to therapeutic agents. Int J Mol Sci. 2019;21:234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Calvo F, Ege N, Grande‐Garcia A, et al. Mechanotransduction and YAP‐dependent matrix remodelling is required for the generation and maintenance of cancer‐associated fibroblasts. Nat Cell Biol. 2013;15:637‐646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cho S, Vashisth M, Abbas A, et al. Mechanosensing by the lamina protects against nuclear rupture, DNA damage, and cell‐cycle arrest. Dev Cell. 2019;49:920‐935.e925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Park JS, Burckhardt CJ, Lazcano R, et al. Mechanical regulation of glycolysis via cytoskeleton architecture. Nature. 2020;578:621‐626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Masoud GN, Li W. HIF‐1α pathway: role, regulation and intervention for cancer therapy. Acta Pharm Sin B. 2015;5:378‐389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kwaan HC, Lindholm PF. Fibrin and fibrinolysis in cancer. Semin Thromb Hemost. 2019;45:413‐422. [DOI] [PubMed] [Google Scholar]
- 32. Martins Cavaco AC, Dâmaso S, Casimiro S, Costa L. Collagen biology making inroads into prognosis and treatment of cancer progression and metastasis. Cancer Metastasis Rev. 2020;39:603‐623. [DOI] [PubMed] [Google Scholar]
- 33. Xu S, Xu H, Wang W, et al. The role of collagen in cancer: from bench to bedside. J Transl Med. 2019;17:309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cazet AS, Hui MN, Elsworth BL, et al. Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nat Commun. 2018;9:2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yoon C, Park DJ, Schmidt B, et al. CD44 expression denotes a subpopulation of gastric cancer cells in which hedgehog signaling promotes chemotherapy resistance. Clin Cancer Res. 2014;20:3974‐3988. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 36. Rayess HM, Dezube A, Bawab I, et al. Tumor differentiation as a prognostic factor for major salivary gland malignancies. Otolaryngol Head Neck Surg. 2017;157:454‐461. [DOI] [PubMed] [Google Scholar]
- 37. Clarke MF. Clinical and therapeutic implications of cancer stem cells. N Engl J Med. 2019;380:2237‐2245. [DOI] [PubMed] [Google Scholar]
- 38. Costa CD, Justo AA, Kobayashi PE, et al. Characterization of OCT3/4, nestin, NANOG, CD44 and CD24 as stem cell markers in canine prostate cancer. Int J Biochem Cell Biol. 2019;108:21‐28. [DOI] [PubMed] [Google Scholar]
- 39. Vora P, Venugopal C, Salim SK, et al. The rational development of CD133‐targeting immunotherapies for glioblastoma. Cell Stem Cell. 2020;26:832‐844.e836. [DOI] [PubMed] [Google Scholar]
- 40. Malanchi I, Santamaria‐Martínez A, Susanto E, et al. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature. 2011;481:85‐89. [DOI] [PubMed] [Google Scholar]
- 41. Xiao H, Jiang N, Zhou B, Liu Q, Du C. TAZ regulates cell proliferation and epithelial‐mesenchymal transition of human hepatocellular carcinoma. Cancer Sci. 2015;106:151‐159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ozturk S, Gorgun C, Gokalp S, Vatansever S, Sendemir A. Development and characterization of cancer stem cell‐based tumoroids as an osteosarcoma model. Biotechnol Bioeng. 2020;117:2527‐2539. [DOI] [PubMed] [Google Scholar]
- 43. Yin L, Fang F, Song X, et al. The pro‐adhesive and pro‐survival effects of glucocorticoid in human ovarian cancer cells. J Mol Endocrinol. 2016;57:61‐72. [DOI] [PubMed] [Google Scholar]
- 44. Grzesiak JJ, Tran Cao HS, Burton DW, et al. Knockdown of the β(1) integrin subunit reduces primary tumor growth and inhibits pancreatic cancer metastasis. Int J Cancer. 2011;129:2905‐2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ritzenthaler JD, Han S, Roman J. Stimulation of lung carcinoma cell growth by fibronectin‐integrin signalling. Mol Biosyst. 2008;4:1160‐1169. [DOI] [PubMed] [Google Scholar]
- 46. Ma Z, Cui X, Lu L, et al. Exosomes from glioma cells induce a tumor‐like phenotype in mesenchymal stem cells by activating glycolysis. Stem Cell Res Ther. 2019;10:60. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 47. Zhang J, Jia L, Tsang CM, Tsao SW. EBV infection and glucose metabolism in nasopharyngeal carcinoma. Adv Exp Med Biol. 2017;1018:75‐90. [DOI] [PubMed] [Google Scholar]
- 48. Zhang HL, Wang MD, Zhou X, et al. Blocking preferential glucose uptake sensitizes liver tumor‐initiating cells to glucose restriction and sorafenib treatment. Cancer Lett. 2017;388:1‐11. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Data Availability Statement
All data that support the findings of this study are available from the corresponding authors upon reasonable request.