

Abstract
This study presents the first messenger RNA (mRNA) therapy for metastatic ovarian cancer and cachexia-induced muscle wasting based on lipid nanoparticles that deliver follistatin mRNA predominantly to cancer clusters following intraperitoneal administration. The secreted follistatin protein, endogenously synthesized from delivered mRNA, efficiently reduces elevated activin A levels associated with aggressive ovarian cancer and associated cachexia. By altering the cancer cell phenotype, mRNA treatment prevents malignant ascites, delays cancer progression, induces the formation of solid tumors, and preserves muscle mass in cancer-bearing mice by inhibiting negative regulators of muscle mass. Finally, mRNA therapy provides synergistic effects in combination with cisplatin, increasing the survival of mice and counteracting muscle atrophy induced by chemotherapy and cancer-associated cachexia. The treated mice develop few nonadherent tumors that are easily resected from the peritoneum. Clinically, this nanomedicine-based mRNA therapy can facilitate complete cytoreduction, target resistance, improve resilience during aggressive chemotherapy, and improve survival in advanced ovarian cancer.
Keywords: lipid nanoparticles, mRNA therapy, ovarian cancer, cachexia, muscle atrophy
Graphical Abstract
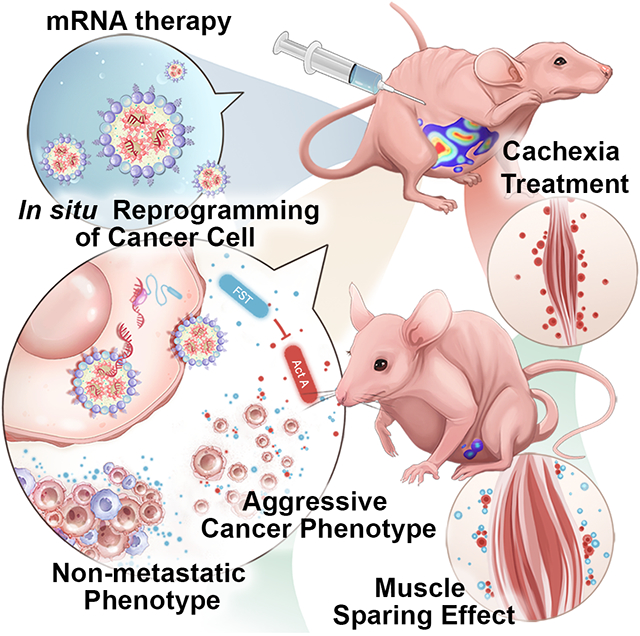
Through the suppression of negative regulators of muscle mass and modulation of ovarian cancer cluster morphology, follistatin mRNA lipid nanoparticle treatment prevents malignant ascites, enhances solid tumor form, delays cancer progression, and preserves muscle mass in cancer-bearing mice. Follistatin mRNA treatment increases mice survival while decreasing chemotherapy-induced muscular atrophy and cancer-associated cachexia.
1. Introduction
Ovarian cancer remains a disease with high mortality and five-year survival of less than 30% for cancer with distant metastases.[1,2] Typically, the disease is diagnosed at an advanced stage when metastases have already spread to the abdominal cavity. The standard of treatment for ovarian cancer is limited to a combination of primary surgical debulking with adjuvant platinum- and taxane-based chemotherapy.[3] While most patients with advanced-stage ovarian cancer respond to first-line chemotherapy, these responses are not durable, and a large proportion of patients ultimately develop and succumb to chemo-resistant disease.[4]
Disease progression, characterized by enhanced invasion and metastasis, contributes to increasing ovarian cancer burden. In addition to tumor burden, malnutrition and cancer-associated cachexia have significant implications for patients.[5] Many patients are in a state of nutritional bankruptcy and chronic wasting, which renders them poor or suboptimal candidates for primary therapeutic interventions including surgery and chemotherapy.[6] Although chemotherapy remains the frontline treatment for metastatic disease, it carries an inevitable iatrogenic burden.[7] It leads to loss of muscle mass, depletion of fat stores, fatigue, and propagation of systemic inflammation proportional to the aggressiveness of disease management.[8] Therefore, there is a clear need to identify new therapeutic targets and appropriate drug combinations that improve the efficacy and tolerability of chemotherapy.
Cytokines from the transforming growth factor β (TGF-β) family are well-known for exerting adverse modifying actions on cancer behavior. TGF-β ligand-mediated induction of metastasis, invasion, and proliferation is well documented.[9] The ligands, including activin A (ActA), myostatin, bone morphogenetic protein 7 (BMP7), and other secreted proteins, signal through downstream mediators called Smads. ActA, discovered as a signaling molecule that induces the release of follicle-stimulating hormone (FSH), has a wide arsenal of actions that range from wound healing to muscle wasting in cancer patients.[10] Its physiological role in forming primordial follicular tissue can take an abnormal developmental trajectory leading to neoplasia. Indeed, ActA levels are pathologically elevated in patients with gynecological malignancies.[11,12] Overexpressed ActA predominantly stimulates proliferation, migration, and invasion of ovarian cancer cells contributing to ovarian tumor metastasis and aggressiveness in later stages.[13,14] In addition, its elevated serum levels correlate with persistent and recurrent disease and cancer-associated comorbidities, including cancer cachexia.[15,16]
Intra-tumoral ActA and myostatin production contribute to muscle wasting which, together with the depletion of fat stores and fatigue, plays an essential role in the progression of cancer-associated cachexia.[17,18] However, unlike myostatin, ActA has a more prominent role in negatively regulating muscle mass in primates.[19] Recent studies in pre-clinical animal models and human subjects show that ActA inhibition by ligand traps results in increased muscle mass, lean body weight, and higher endurance.[20-22] For these reasons, ActA represents an attractive therapeutic target for both cancer therapy and the treatment of cancer-associated muscle wasting.
Follistatin (FST), a circulating glycoprotein, functions as an endogenous regulator for several members of the TGF-β family, including ActA.[23] FST inhibits these proteins by direct binding, sequestering them in an inactive complex and preventing their interactions with the Activin receptor type-2B (ActRIIB).[24,25] Its high target affinity and nearly irreversible binding kinetics result in complete neutralization of circulating targets.[13,24,26] In vitro work highlights the ability of recombinant FST to attenuate the stimulatory effect of ActA on the proliferation and migration of ovarian cancer cell lines.[13,27-29] Additionally, by specifically inhibiting ActA, FST leads to an increase in muscle size and strength, and the regenerative capabilities of muscles in different species.[19,24] For these reasons, FST-based treatments are promising agents for targeted inhibition of ActA.
The success of mRNA-loaded lipid nanoparticles (LNPs) in the development of SARS-CoV-2 vaccines race proved them to be the most efficient modalities for transient protein expression.[30] Although therapeutic applications of mRNA are in their infancy, they offer promising clinical potential outside the vaccine field for the management of infectious diseases. Advances in mRNA therapeutics fueled the development of novel cancer treatment strategies, including cancer vaccines and intratumoral therapies that require frequent dosage. Here, we describe the first protein-coding mRNA therapy for metastatic ovarian cancer and associated cachexia. Our studies in a murine model of metastatic ovarian cancer show that treatment with FST mRNA encapsulated in LNPs substantially increases systemic and local peritoneal levels of FST protein exerting in situ and abscopal therapeutic actions. Produced FST acting in an autocrine/paracrine and systemic fashion provides survival benefits similar to cisplatin, a platinum-based chemotherapy drug used as a standard of care in ovarian cancer treatments. Our data also reveal that systemic FST-mediated inhibition of ActA counteracts muscle loss and in combination with cisplatin provides a synergistic anticancer effect. The proposed FST mRNA LNP therapy is a highly translatable platform that outperforms recombinant protein supplementation and limits cancer progression while simultaneously enhancing resilience during an aggressive chemotherapeutic regimen.
2. Results and Discussion
2.1. Preparation, characterization, and biodistribution of mRNA-LNPs in sham and cancer-bearing mice
The purpose of this work was to develop and test the efficacy of adjuvant LNP-based mRNA therapy aiming to supplement FST protein for the treatment of metastatic ovarian cancer and cancer-associated cachexia. Because in vitro transcribed mRNA is subject to a post-transcriptional repressive control in the cytoplasm[31] and is translated to endogenous proteins with required organism-specific post-translational modifications,[32] this method allows for transient and highly-localized protein over-expression while avoiding permanent genomic changes, the immunogenicity of exogenous proteins, and dose-related toxicity.
Therefore, LNPs were constructed to realize the full potential of the developed FST mRNA therapy. The LNPs, loaded with different mRNA transcripts (i.e., FST, Enhanced Green Fluorescence Protein (EGFP), and Luciferace (Luc) mRNA), were assembled following the microfluidic mixing procedure (Figure 1A).[33] LNPs were synthesized in the ratios 50:38.5:10:1.5 of the ionizable lipid DLin-MC3-DMA, cholesterol, DSPC, and DMG-PEG-2k. A N:P ratio of 5.67 was maintained between ionizable lipids and nucleic acids to synthesize nanoparticles. The prepared FST mRNA LNPs had a spherical shape (Figure 1B), an average hydrodynamic size of 84.5 nm (Table S1) with a polydispersity index (PDI) of 0.065, ζ potential of −8 mV, and were characterized by >90% mRNA encapsulation efficiency.
Figure 1. ES-2 clusters demonstrate increased in vivo capacity for LNP uptake compared to normal tissues.

(A) Visual representation of the mRNA LNP structure. (B) A representative cryo-transmission electron microscopy (TEM) image shows the spherical shape and size distribution of LNPs. (C) Illustration of the experimental design used to create the ES-2-Luc mouse model of metastatic ovarian cancer. (D) Representative BLS images of ES-2-Luc bearing mouse (left) and sham mouse (right) 15 min after D-luciferin injection. (E) Light micrograph of a thin smear of ascites containing ES-2 clusters on a glass slide. Ascitic fluid was collected from ES-2-Luc mice. (F) Post-mortem photographs of a sham mouse and ES-2-Luc-bearing mice at the primary humane endpoint (~ day 12) showing distended abdomen due to accumulation of malignant ascites (blue arrows), decreased muscle bulk (red arrows and inset), metastases formed on omentum and mesentery (white arrows), and ovaries and gonadal fat pads (black arrows). (G) Experimental design depicting a biodistribution study with ES-2-WT-bearing mice and sham age-matched controls (not shown) administered Luc mRNA LNPs. (H) BLS images of ES-2-WT and sham mice before (top) and 4 h after (bottom) treatment with Luc mRNA LNPs. (I) Representative BLS images of tissues collected from ES-2-WT-bearing (ES-2-WT + Luc mRNA LNP) and sham (sham + Luc mRNA LNP) mice at 4 h-post injection with Luc mRNA LNP in comparison to negative (sham + PBS) and positive controls (ES-2-Luc-bearing mice + PBS). Organs are (left to right): liver, kidneys, spleen, lungs, gonadal fat pads. (J) BLS images of ascitic and physiological peritoneal fluids from ES-2-WT-bearing mice (middle) and sham mice (right), producing an active luciferase enzyme after Luc mRNA LNP treatment in comparison to a positive control (ascites of ES-2-Luc-bearing mice, left, treated with PBS).
Histopathological examination of murine livers that had received chronic mRNA LNP injections was used to assess the safety and tolerability of mRNA LNPs. Systemic toxicity was further assessed by analyzing complete blood counts with differential, blood chemistry, pancreatic and hepatic inflammation panels, and hepatic cytokines associated with inflammation (Figure S1, S2, S3). We observed no changes among treatment and control groups in gross pathology and histopathology of livers resulting from three daily injections of mRNA LNPs. Blood and hepatic gene expression studies confirmed mRNA LNP formulation to be safe for chronic administration regiments.
To evaluate mRNA delivery efficiency of the prepared LNPs and therapeutic efficacy of the expressed proteins, we used a murine model of metastatic ovarian cancer (Figure 1C-F), in which nude mice (Nu/Nu) received intraperitoneal (IP) injections of either wild type (ES-2-WT) or luciferase-producing (ES-2-Luc) ES-2 ovarian clear cell carcinoma cells. Injected cancer cells proliferated in the peritoneal cavity, resulting in malignant ascites with a suspension of cancer clusters (Figure 1E, F).
In recent decades, intraperitoneal metastases and malignant ascites were targeted by regional IP therapies exploiting enhanced endocytic characteristics of cancer cells. Multiple malignancies, including ovarian cancer, upregulate endocytic activity to enhance tumorigenesis and metastasis and to aid cancer cell metabolism.[34] We show that ES-2 cancer cells in suspension demonstrate increased in vivo capacity for LNP-mediated transfection evaluated by mRNA quantification and fluorescent imaging, presumably due to upregulated endocytosis is a consequence of increased metabolic activity in comparison to normal cells.
To determine the biodistribution profiles of mRNA-LNPs and efficiency of delivered mRNA translation to bioactive proteins, we established two experimental groups, including mice bearing ES-2-WT tumors and sham mice (Figure 1G). Mice injected with ES-2-Luc cells served as positive controls; ES-2-WT-bearing mice and sham mice injected with PBS served as negative controls. Mice received single IP injections of LNPs loaded with luciferase mRNA (Luc mRNA LNPs) with an equivalent of 10 μg mRNA per mouse. Four hours after administration of Luc mRNA LNPs, a bioluminescence signal (BLS) generated by luciferase was detected in both sham and ES-2-WT mice injected with D-luciferin (Figure 1H). The obtained results suggest that the luciferase enzyme was translated from the Luc mRNA delivered by the LNP platform to various tissues. Importantly, BLS was significantly higher in cancer-bearing mice than in sham animals. Bioluminescence imaging validated that strong BLS generated by the produced luciferase enzyme was predominantly observed in ascitic fluid and other tissues (e.g., gonadal fat pads) containing ES-2 cancer cells infiltrations (Figure 1I, J). Similar BLS distribution was confirmed by imaging the positive control mice inoculated with ES-2 cells stably transfected with Luc gene (ES-2-Luc). In contrast to cancer-bearing mice, relatively weak BLS was detected in the liver, spleen, and IP fluid of sham mice injected with Luc mRNA LNPs (Figure 1I, J). This observation supports our hypothesis that the cancer cells are the dominant cell type internalizing LNPs and translating delivered mRNA when administered intraperitoneally.
2.2. Regional mRNA LNP therapy administration targets cancer cell clusters in the peritoneal cavity
To evaluate the therapeutic efficacy of the developed FST therapy, we employed mice inoculated with ES-2 cells stably expressing luciferase enzyme (ES-2-Luc). This reporter (luciferase) was chosen in order to monitor cancer progression and therapeutic response in cancer-bearing mice using non-invasive bioluminescence imaging. Therefore, in the next step, we validated that ES-2-Luc cells, similar to the ES-2-WT cells (Figure 1J), can efficiently internalize LNP-encapsulated mRNA and translate it into bioactive proteins (Figure 2A).
Figure 2. Efficient LNP-mediated delivery of FST mRNA leads to a superior therapeutic effect, demonstrated by serum ActA reduction.

(A) Experimental design illustrating ES-2-Luc bearing mice receiving a single mRNA LNP dose (GFP mRNA, Cy5-GFP mRNA, GFP/FST mRNA, and FST mRNA, 10 μg total mRNA equivalents per dose per mouse). Four hours after IP injection, sera and ascitic fluid were collected for analysis. (B) Fluorescence images of ES-2-Luc clusters 4 h after a single injection with the LNPs loaded with the indicated mRNAs. (C) Absolute qRT-PCR quantification of GFP mRNAs in ES-2-Luc clusters 4 h after IP injection of PBS and LNPs loaded with 0 μg GFP mRNA (FST mRNA LNP), 5 μg GFP mRNA (GFP/FST mRNA LNP), and 10 μg GFP mRNA (GFP mRNA LNP). (D) Relative qRT-PCR quantification of FST mRNAs in ES-2-Luc clusters 4 h after IP injection of PBS and LNPs loaded with 10 μg FST mRNA (FSTH mRNA LNP), 5 μg FST mRNA (FSTL mRNA LNP), or 0 μg FST mRNA (GFP mRNA LNP). Expressed as mean ± SEM, n=5, *p<0.05, ***p<0.001, ****p<0.0001 analyzed by one-way ANOVA followed by Bonferroni’s post hoc test. (E) FST and (F) ActA serum protein concentrations expressed as mean ± SEM, n=5, **p<0.01, ***p<0.001, analyzed by two-way ANOVA followed by Bonferroni’s post hoc test.
ES-2-Luc-bearing mice received a single IP injection of LNPs encapsulating Cy5-tagged EGFP mRNA. The fluorescence signal generated by Cy5 allowed us to confirm mRNA LNP internalization into ES-2-Luc cancer cells, while the EGFP signal validated translation of the encapsulated mRNA into the functional encoded protein. We also used EGFP mRNA without the Cy5 tag to control for possible tag interference with cellular uptake and translation of released mRNA (Figure 2B). Four hours after injection, we collected peritoneal cavity fluid containing ES-2-Luc clusters. Fluorescence imaging and qRT-PCR analysis revealed that LNPs successfully delivered Cy5-EGFP and EGFP mRNAs to ES-2-Luc cells in the IP fluid, and both mRNAs were efficiently translated to EGFP (Figure 2B, C). The green fluorescent signal was not produced when EGFP mRNA was replaced with FST mRNA in LNPs, and the signal was observed again, when EGFP and FST mRNAs were combined in a single LNP formulation with 1:1 mRNA molar ratio (Figure 2B). qRT-PCR results displayed a concentration-dependent increase in EGFP mRNA detected in the ascites treated with FST, EGFP/FST, and EGFP mRNA LNP formulations. Therefore, we observed 3- and 3.5-fold increase in normalized EGFP mRNA transcript counts in the log scale (Figure 2C) compared to PBS and FST mRNA LNP treated mice.
Our biodistribution results are consistent with previous research demonstrating that nanoparticles efficiently accumulate in abdominal cancer tumors following intraperitoneal injection.[35,36] This route of administration increases the retention time of nanoparticles in peritoneal fluid, and nanoparticles exhibit enhanced and preferential internalization into cancer cells present in ascites and tumors due to direct contact with these cells. Preferential uptake of nanoparticles by cancer cells when compared to non-malignant cells is related to the fact that nanoparticles exploit endocytosis to enter cells, and endocytotic pathways are generally upregulated in cancer cells. We also performed in vitro and in vivo flow cytometry studies, where ES-2-WT cells were either co-cultured with RAW 264.7 murine macrophages or injected to IP cavities of nude mice and allowed to proliferate for 10 days (Figure S4). Following treatment with GFP mRNA LNPs, IP cavity fluid and cocultured ES-2-WT/RAW 264.7 cells were analyzed by flow cytometry. In both experiments, only HLA-ABC positive cells (ES-2-WT human cell line) were GFP-positive, confirming the predominant LNP uptake by ES-2 cancer cells (Figure S4).
Next, we evaluated if ES-2-Luc clusters can internalize FST mRNA LNPs and efficiently produce bioactive FST proteins (Figure 2A). Two formulations of LNPs loaded with different FST mRNA amounts (10 and 5 μg in FSTH mRNA LNP and FSTL mRNA LNP formulations, respectively) were administered into cancer-bearing mice. The IP fluid collected at 4 hours-post single injection was analyzed with qRT-PCR (Figure 2D). qRT-PCR results demonstrate that treatment with two LNP formulations containing high (10 μg) and low (5 μg) amounts of FST mRNA increased FST mRNA by ~250- and ~90-fold, respectively, in ES-2-Luc clusters compared to negative controls. The controls receiving EGFP mRNA LNPs did not display an off-target increase in endogenous FST mRNA transcription (Figure 2D). Since the FST mRNA used in the experiments translated to the secreted protein isoform (FST-315, see FST mRNA open reading frame sequence, Table S3), we quantified FST levels in serum samples with FST ELISA. Groups of ES-2-Luc-bearing mice and age-matched sham controls received a single injection of FSTH mRNA LNP, FSTL mRNA LNP, EGFP mRNA LNP formulations, or PBS in the negative control group. In sham mice, both FST mRNA formulations exhibited a concentration-dependent increase in serum FST levels (~ 3 and ~1.6-fold increase, Figure 2E). In ES-2-Luc-bearing mice, despite the high basal FST level, FSTH mRNA LNPs increased FST by ~1.6-fold.
Fortunately, in situ internalization of LNPs by cancer clusters is ideal for auto/paracrine inhibition of tumor-derived ActA protein. The effectiveness of FST mRNA LNP to inhibit ActA is evident from decreasing levels of serum ActA coincident with increased FST LNP uptake. We confirmed the synthesis of the fully functional FST protein by evaluating serum ActA levels. FSTH mRNA LNP and FSTL mRNA LNP treatments in sham mice both led to a ~2-fold decrease in serum ActA. In cancer-bearing mice, the decrease was 1.3- and 2-fold for FSTL mRNA LNP and FSTH mRNA LNP treatments, respectively (Figure 2F). The treatment with FSTL mRNA LNP in tumor-bearing mice led to a smaller decrease in serum ActA, because this formulation contained half the amount of FST mRNA loaded to LNPs. Importantly, control (injected with PBS) cancer-bearing mice had a ~4-fold increase in the basal level of ActA compared to sham mice, implying pathological roles of ActA in ovarian cancer.
2.3. Chronic regional FST mRNA LNP injections reduce tumor-derived ActA levels
The perturbation of the ActA–FST axis with over-expression of ActA is associated with a number of ovarian disorders, including carcinogenesis.[37] Similarly, previous studies reveal that FST serum levels are elevated in ovarian cancer patients to the extent that FST can be considered a tumor marker for ovarian cancer diagnosis.[38,39] Even though FST expression correlates with ActA levels, FST does not increase to the same extent as ActA. Indeed, total FST serum levels rise due to pathological increase in ActA but do not exceed the higher end of its normal physiological range in patients.[38,40] Notably, at least 90% of FST in healthy individuals is ActA-bound and biologically inactive.[41,42] Given that the maximal suppression of ActA-induced signaling was observed with an FST-to-ActA molar ratio of 4:1,[43] significant overexpression of FST is pivotal in counteracting pathological actions of ActA and its multiple adverse effects in patients with ovarian cancer.
Since the developed therapeutic as an adjuvant treatment for ovarian cancer requires multiple injections, we established a chronic daily injection schedule with FST mRNA LNPs (10 μg mRNA equivalent per dose) to study the effect of a long-term FST protein supplementation (Figure 3A). Total FST mRNA levels, including endogenous and exogenous transcripts, in the cancer clusters and various tissues, were quantified using qRT-PCR. We showed that FST mRNA was significantly increased in ES-2-Luc cell clusters and organs of mice treated with seven daily FST mRNA LNP injections (Figure 3B-G).
Figure 3. While endogenous upregulation of FST is insufficient to control serum ActA, chronic FST mRNA LNP administration substantially reduces ActA levels.

(A) Experimental design demonstrating ES-2-Luc bearing mice receiving seven daily FST mRNA LNP (10 μg mRNA) injections; the sham group is not shown on the graphic for simplicity. qRT-PCR analysis of FST transcript counts in ascites (B), liver (C), spleen (D), kidneys (E), lungs (F), and heart (G). (H) Serum ActA concentration determined by ELISA. (I) Serum FST determined by ELISA. (J) ActA protein levels in ascites. (K) ActA protein levels in ascites normalized by total protein in ascites. (L) FST protein levels in ascites. (M) FST protein levels in ascites normalized by total protein in ascites. For (B-G), results are mean ± SEM, n=10 for treatment groups (LNP) and n=5 for control groups (PBS), *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. (B), (J-M) analyzed by t-test; (C-I) analyzed by two-way ANOVA followed by Bonferroni’s post hoc test.
Similar to a single FST mRNA LNP injection (Figure 2D), a daily dosage regimen led to a significant increase of FST mRNA in ES-2-Luc clusters (Figure 3B). It is important to note that serum samples were obtained four hours after LNP injection in the single administration study but twelve hours after the final LNP injection in the chronic administration study. FST mRNA relative quantities in ES-2-Luc clusters after a single and chronic administrations were 250- and 50-fold higher than in the control mice, respectively. Importantly, we detected a pan-organ upregulation of endogenous FST mRNA and increased serum FST levels in control tumor-bearing mice compared to control sham mice (Figure 3H, I). Despite this endogenous FST upregulation, FST mRNA LNP treatment resulted in a significant FST mRNA increase in the liver, kidney, and spleen of treated sham animals (100-, 40- and a striking 13,000-fold increase, respectively, Figure 3C-E) compared to control sham mice. Treated ES-2-Luc-bearing mice exhibited a positive trend in FST mRNA counts in the liver, kidney, and spleen (~25-, ~8-, and ~12-fold increase) in contrast to control cancer-bearers. FST mRNA transcript increase in these organs of cancer-bearers was lower than in organs of sham mice, suggesting that ES-2-Luc clusters in the IP cavity are internalizing the majority of FST mRNA LNPs. FST mRNA LNP treatment substantially increased FST mRNA in the lungs and heart of both treated sham and cancer-bearing mice (Figure 3F-G). Treated sham and cancer-bearing mice displayed 8- and 4-fold increase in FST mRNA in lungs compared to sham and cancer-bearing controls. FST mRNA increase in hearts was 3-fold in both treatment groups compared to in the corresponding controls. Interestingly, the lungs and hearts had higher FST mRNA counts in treated cancer-bearing mice than treated sham mice (60 and 300 times higher than in the treated sham mice, respectively), while lower levels are observed in liver, kidney, and spleen. Because the majority of LNPs are internalized by cancer cells, relative quantification of FST mRNA is consequently decreased in the liver, kidneys, and spleen of cancer-bearing animals. The opposite trend is observed in the lungs and heart, where uptake is presumably less affected by the concentration of LNPs in the bloodstream.
Tumor-bearing mice exhibited ~10 ng mL−1 increase in serum FST after a single and chronic injections (both, ~1.3-fold increase). In sham mice, chronic injections with FST mRNA LNPs increased serum FST levels by ~18 ng mL−1 (Figure 3H), compared to a single LNP injection that increased serum FST by only ~2 ng mL−1 (Figure 2E), that is 24- and 4-fold increase, respectively. FST bioactivity in cancer-bearing and sham mice was evident from the serum ActA decrease. ActA serum levels displayed a similar 2-fold decline after a single LNP injection (Figure 3I, Figure 2F) in sham and cancer-bearing mice. FST mRNA LNP treatment led to a ~2-fold increase in FST levels in ascites resulting in a ~2-fold decrease in ascitic ActA levels (Figure 3J-M). Importantly, a 100-fold increase of FST concentration in the ascites compared to FST serum concentration strongly suggests both local FST production by cancer cells and demonstrates the applicability of the proposed treatment for efficient protein overexpression without systemic toxicity. Moreover, pooled serum samples from control and FST mRNA LNP treated mice demonstrated negative correlation between levels of FST and ActA (Figure S8). Importantly, FST mRNA LNP treatment was able to decrease ascitic concentration of myostatin, another negative muscle regulator from TGF-B family (Figure S7). Although the action of FST is multifactorial, including inactivation of ActA, myostatin, and bone morphogenic factors. However, we have focused on the impact of FST on ActA owing to its prominence in negative muscular control, especially in humans. Previous reports clearly document the association between systemic concentrations of ActA and the presence of cachexia in patients with different cancers.[44-46] It was also revealed that elevated serum levels of ActA in cachectic patients are related to cancer aggressiveness and reduced chemotherapy response.[45,46] Multiple cytokines are also capable of inducing cachexia, including but not limited to TNF-α, and IL-6.[47] However, clinical studies revealed that treatments with anti-TNF-α or anti-IL-6 antibodies provide minimal benefit on muscle mass in patients with cancer cachexia.[48,49] In contrast, treatment of cancer cachexia murine models with ActRIIB decoy receptors reduced circulating ActA levels, prevented muscle wasting and prolonged mice survival without affecting plasma levels of TNF-α and IL-6.[18]
2.4. LNP-guided FST overexpression modulates epithelial and mesenchymal characteristics of malignant ascites
Pathological actions of ActA have both local and systemic implications affecting cancer propagation and associated morbidities. In the context of local effects, we hypothesized that the observed change in the cluster morphology and size was associated with in situ reduction of available ActA, leading to a modification in ES-2-Luc cell mobility and its epithelial or mesenchymal phenotype. To evaluate the potential anticancer effect of FST mRNA LNPs, two groups of mice inoculated with ES-2-Luc cells received seven daily injections with FST mRNA LNPs (treatment group) or PBS (control group). Control mice developed abundant third-spaced fluid infiltrated with diffuse circulating ES-2-Luc cells forming microscopic clusters (Figure 4A, B). Notably, in the treatment group, we observed a minimal formation of malignant ascites (Figure 4C). Volumes of ascites in control mice were ~5-fold higher than in the FST mRNA LNP-treated mice. Instead, ES-2-Luc cells formed self-adhering tumors (aggregates) accumulating in the physiological bottlenecks of IP fluid movement (Figure 4A, B, red arrows).
Figure 4. Mice treated with FST mRNA LNPs for 7 days formed ES-2-Luc aggregates in the physiological bottlenecks of IP fluid movement with minimal apparent ascites.

(A) and (B) representative images of FST mRNA LNP treated (left) and control (right) mice. In the LNP-treated mouse, red arrows show ES-2-Luc cell aggregates; no third space/ascitic fluid is present; the control (PBS) mice formed third-spaced/ascitic fluid with a suspension of ES-2-Luc clusters (blue arrows). Black arrows in both (A) and (B) show the formation of secondary nodules on gonadal fat pads. (C) Ascites volume in control and LNP-treated mice. (D) ECM TaqMan Array heatmap for individual samples (left) and average fold change for ES-2-Luc aggregates (right); analyzed by two-way ANOVA followed by two-stage step-up method of Benjamini, Krieger and Yekutieli with FDR = 0.01. Relative expression of upregulated genes identified as true discoveries is shown on linear (E) and log (F) scales. Statistical significance between PBS and LNP groups (E, F) was determined by multiple t-tests with the Bonferroni-Dunn method, alpha = 0.05; computations assumed the samples from populations with the same scatter. Expressed as mean ± SEM; n=3 for the control group (PBS), n=6 for the treatment group (LNP). (G) Relative quantification of fibroblast (P4HB), macrophage (CD68), neutrophil (Ly6G), and immature lymphocyte (SELL) markers in control and LNP-treated cancer clusters; analyzed by two-way ANOVA followed by Bonferroni’s post hoc test on raw parametric data (dCt values); expressed as mean ± SEM, n=10 for treatment groups (LNP) and n=5 for control groups (PBS). p*<0.05, p**<0.01, p***<0.001, p****<0.0001.
In most cases, the cellular aggregates were found in suprahepatic compartments of the peritoneal cavity. Importantly, the aggregates were nonadherent to the organs or the walls of the peritoneal cavity. Both treatment and control groups had secondary cancer nodules forming on gonadal fat pads (Figure 4A, B, black arrows). We hypothesized that these differences in ES-2-Luc dissemination patterns indicated a treatment-dependent modulation of epithelial-mesenchymal plasticity of ES-2-Luc cells.
It is well documented that TGF-β family signaling is predominant mechanism in reprogramming gene expression during epithelial to mesenchymal transition (EMT).[50] EMT is associated with cancer stemness, its metastatic and invasive potentials, and resistance to treatment.[51] Mesenchymal to epithelial transition (MET), in turn, is observed in the process of colonization and formation of secondary tumors and is associated with the proliferative potential of cancer cells. For example, ActA-mediated suppression of E-cadherin expression propagates the migration of ovarian cancer cells.[52] To understand if FST mRNA LNP therapy induced epithelial or mesenchymal phenotypic changes in cancer cells, we analyzed the expression of extracellular matrix (ECM) and adhesion molecules in solid aggregates (FST mRNA LNP treatment) and diffuse circulating clusters accumulating in ascitic fluid (PBS, control group). In the cancer aggregates formed in FST mRNA LNP treated mice, we observed upregulation of nine genes involved in both epithelial-mesenchymal (EMT) and mesenchymal-epithelial (MET) transition processes (Figure 4D-F). Compared to cell suspensions from the control mice, the FST mRNA LNP treatment increased CDH1 expression (E-cadherin, pro-MET) in ES-2-Luc cell aggregates. We also observed relative over-expression of three matrix metalloproteinases (stromelysins MMP3, MMP10, and collagenase MMP1, pro-EMT), COL7A1 and ITGA2 (potentially, pro-EMT), as well as SELL, ICAM1, and VCAM1 genes. FST over-expression leading to inhibition of canonical ActA signaling and related overexpression of E-cadherin most likely changed cluster morphology to large cellular aggregates and decreased cancer cell mobility while imparting ES-2 cells with MET characteristics.
ES-2-Luc aggregates but not ES-2-Luc suspension from malignant ascites displayed upregulation of neutrophil (Ly6G), macrophage (CD68), immature lymphocyte (SELL), and fibroblast (P4BH) markers (Figure 4G), suggesting cancer clusters undergoing heterogeneous cellular infiltrations. Also, we observed a positive trend in the expression of cell cycle markers in the ES-2-Luc aggregates formed as a result of FST mRNA LNP treatment that are usually upregulated in actively proliferating cancer cells (MKI67, TOP2A, TPX2; Figure S5). To deconvolute the influence of infiltrating immune cells and fibroblasts in the ES-2-Luc aggregates on MKI67, TOP2A, TPX2 gene expression, we investigated expression of these genes in ES-2-Luc spheroids treated with FST protein (Figure S6). MKI67, TOP2A, TPX2 genes were upregulated in ES-2-Luc spheroids treated with FST protein, confirming the ability of FST inducing these genes directly.
In line with this MET-like change, the cell aggregates treated with FST mRNA LNPs exhibit a positive trend in MKI67 (Ki-67), TOP2A (DNA topoisomerase II, alpha), and TPX2 (TPX2 Microtubule Nucleation Factor) expression, genes that are upregulated in cancer cells undergoing active proliferation.[53] Other upregulated genes in FST mRNA LNP-treated clusters are recognized as pro-EMT pathway members.[54-60]. Given this differential upregulation of both pro-MET and pro-EMT genes, we hypothesized that FST treatment could trap the cells in the transitional EMT/MET phenotype, where the cells can break the connection between the true epithelial phenotype and cell cycle activation, and become more susceptible to chemotherapy.[61]
2.5. FST mRNA LNP counteracts muscle loss in ES-2-Luc bearing mice exhibiting cancer-associated cachexia
To test whether FST mRNA LNP not only exerts action on ES-2-Luc tumors but also produces bioactive FST protein inhibiting ActA with muscle-sparing effects, we assessed terminal gastrocnemius mass, muscle fiber cross-section area, and cachexia-driving gene expression in mice chronically treated with FST mRNA LNPs (Figure 5A).
Figure 5. Chronic FST mRNA LNP treatment ameliorates cancer-associated muscle bulk loss and decelerates ubiquitin-proteasome proteolysis program in cancer-bearing mice.

(A) Gastrocnemius muscles were collected from mice following daily treatment for 7 days with LNPs loaded with 10 μg FST mRNA; data were analyzed by two-way ANOVA followed by Bonferroni’s post hoc test. (B) Muscle fiber cross-section area (FCSA) and (C-E) expression of FOXO1, FBXO32, and TRIM63 in gastrocnemius. (F) Gonadal fat pad mass in control sham and treatment sham groups. (G) Relative FST mRNA counts in the gonadal fat pads of control and treatment sham groups. (H) Ucp1 expression in the gonadal fat pads of control and treatment sham groups. (C-E) were analyzed by two-way ANOVA followed by Bonferroni’s post hoc test on raw parametric data (dCt values). All data expressed as mean ± SEM; p*<0.05, p**<0.01, p***<0.001, p****<0.0001; n=10 for treatment groups (LNP) and n=5 for control groups (PBS). (F-H) analyzed by unpaired t-test (n=5) on mass or raw parametric data (dCt values); all data expressed as mean ± SEM; p***<0.001, p****<0.0001.
We observed a 37% loss of gastrocnemius muscle mass in control (PBS) cancer-bearing mice. Importantly, our data suggest that FST mRNA LNP treatment reversed bulk muscle loss in tumor-bearing animals to the level of control sham mice (~35% bulk muscle increase) associated with 2-fold decrease in serum ActA levels. We also observed a 15% increase in muscle mass in sham mice receiving FST mRNA LNP injections (Figure 5A). Muscle fiber cross-section area (CSA) analysis displays 40% decrease in CSA in non-treated cancer-bearing mice that supports 37% bulk muscle loss finding. Interestingly, FST mRNA LNP treatment led to 90% increase in CSA in cancer-bearers, that is ~50% larger CSA than in non-treated sham mice (Figure 5B). Compared to sham mice, gastrocnemius samples from tumor-bearing mice had elevated expression of E3 ubiquitin ligase-related genes Foxo1 (Foxo1), FBXO32 (Atrogin-1/Mafbx), and Trim63 (Murf1) (Figure 5 C-E). The aforementioned genes are known to induce muscle catabolism via the ubiquitin-proteasome axis in cancer-associated cachexia.[62] We observed a decreasing trend in Foxo1, FBXO32, and Trim63 expression in the treated cancer-bearing mice relative to non-treated cancer-bearing mice.
Gonadal fat pads from tumor-bearing mice were excluded from the analysis due to infiltration with ES-2-Luc cells. Interestingly, the treated sham mice displayed a significant ~2-fold increase in the adipose tissue mass of gonadal fat pads (Figure 5F). Our results also show a substantial increase in adipocyte uptake of FST mRNA LNPs (relative 3000-fold increase in FST mRNA, Figure 5G). We also observe a modest increase in the expression of Ucp1, a marker of white adipose tissue browning, known to be induced by FST signaling leading to a decrease in adiposity (Figure 5H).[63] These highly reproducible results are in dissonance with the FST function to decrease adiposity and thus require thorough investigation to determine if they reflect a true potential to slow fat stores depletion in cachexia.
2.6. Supplementation of recombinant ActA exacerbates ascites, accelerates cancer progression, and decreases survival, while recombinant FST alleviates these manifestations
Taken together, FST mRNA LNPs decreased the volume of ascites and altered dissemination patterns of ES-2 cells (Figure 4), and provided a muscle-sparing effect in cachectic mice bearing ovarian cancer xenografts (Figure 5). We hypothesized that this was due to an apparent decrease in bioactive ActA signaling (Figure 2F, 3I). We established three cancer-bearing treatment groups of mice receiving recombinant ActA injections (20 μg per kg of body weight (20 μg kg b.w.−1)) or recombinant FST injections (FSTL, 20 μg kg b.w.−1 and FSTH, 100 μg kg b.w.−1), as well as a control group receiving PBS (Figure 6A, B). As a proof of concept, daily IP injections of recombinant ActA to ES-2-Luc-bearing mice led to accelerated cancer progression and faster advancement to primary humane endpoint in the comparative survival study (Figure 6B, C). The ActA treatment group developed a more severe disease with excessive ascites and shortened survival.
Figure 6. Supplementation with recombinant ActA exacerbates ascites, accelerates cancer progression, and decreases survival, while recombinant FST alleviates these manifestations.

(A) Experimental design demonstrating ES-2-Luc bearing mice receiving daily IP recombinant protein injections. (B) Representative BLS images of ES-2-Luc mice treated with PBS, recombinant ActA (20 μg kg b.w.−1), recombinant FSTL (20 μg kg b.w.−1), and FSTH (100 μg kg b.w.−1). (C) Comparison of survival curves between control, ActA, FSTL, and FSTH recombinant protein treatments. (D) Table of significant results of post hoc Bonferroni's multiple comparison tests using median survival and standard error (n=5). (E) Representative images of mice were taken on day 12 after tumor inoculation for PBS and ActA-treatment groups and day 20 for recombinant FSTL and FSTH treatment groups.
IP injection of recombinant FSTL (20 μg kg b.w.−1) led to an insignificant change in serum FST and ActA levels (data not shown), and unaltered disease progression trajectory characterized by the formation of malignant ascites, muscle wasting (Figure 6E), and decreased mobility similar to the control group. However, FSTL and FSTH treatments had a positive impact on survival (Figure 6C, D). An increase of recombinant FST dose to 100 μg kg b.w.−1 (FSTH) led to the formation of small ES-2-Luc aggregates in the ascitic fluid and decreased volume of ascites. This group of mice had no signs of lethargy, fatigue, or change in the spinal curvature; euthanasia was performed based on the extent of abdominal distension due to the buildup of ascitic fluid. Mean survival analyzed in Figure 6C, D was 14.1±0.64, 11.1±0.73, 16±0.92, and 17.7±0.79 days for control, ActA, FSTL, and FSTH groups, respectively (represented as mean±SE). Decrease in mean survival in ActA treatment group was approaching significance (p=0.07, Figure 6D). Mice treated with FSTH displayed significant increase in mean survival compared to control and ActA treatment groups.
Although all recombinant treatments lead to the formation of ascites nearing the primary humane endpoint, FSTH postponed its formation and led to the development of macroscopic cancer cell aggregates. This promising data encouraged us to compare recombinant and mRNA-based FST supplementation treatments. Sera from the mice treated with recombinant FSTH were analyzed together with sera from mice receiving FST mRNA LNP treatment in the experiment discussed in the next section (Figure 7D, E). Recombinant FSTH treatment results indicated a significant decrease in ActA levels.
Figure 7. FST mRNA LNP treatment reprograms ES-2-Luc clusters and provides an abscopal effect alleviating cancer-associated cachexia manifestations.

(A) Design of survival experiment demonstrating ES-2-Luc-bearing mice receiving the following treatments: PBS (control group, daily), CDDP (every four days, for a maximum of 5 weeks, 2 mg kg b.w.−1 dose), recombinant FST (FSTH, 100 μg kg b.w.−1 dose, daily) or FST mRNA LNP (10 μg mRNA equivalent dose, daily), or their combination with CDDP. (B) Representative whole-body BLS images of mice in each experimental group as a function of time; imaging was performed every 5 days. (C) Representative post-mortem BLS images of mice treated with PBS, CDDP, or FST mRNA LNPs, or their combination; white arrows represent ascites, purple arrows – ES-2-Luc cell aggregates, yellow arrow – liver, red arrow – non-adherent solid ES-2-Luc tumors. (D) Representative images of mice from control, CDDP, FST mRNA LNP, and CDDP+FST mRNA LNP groups. Purple arrows show ES-2-Luc cell aggregates, white arrows – ascites, red arrows – non-adherent solid ES-2-Luc tumors. (E) Serum FST and (F) serum ActA concentrations determined by ELISA; results are mean ± SEM, n=5, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, analyzed by two-way ANOVA followed by Bonferroni’s post hoc test. (G) Muscle fiber cross-section (FCS) area; normalized to double-negative control (PBS/PBS) mice. Results are mean % ± SEM, n=500, analyzed by two-way ANOVA followed by Bonferroni’s post hoc test. aAll groups are significant with p****<0.0001, except PBS/CDDP vs. FSTH/PBS groups (**p<0.01). (H) Representative anti-laminin immunohistochemistry and (I) Picrosirius staining of muscle fibers.
2.7. FST mRNA LNPs in combination with cisplatin prolong survival, decreases tumor burden, and provides muscle sparing effect in ES-2-bearing mice
Cancer-associated inflammation and obstruction of intraperitoneal organs lead to depletion of fat stores, loss of muscle bulk, and compromised nutritional state.[5,64] These catabolic changes, together with systemic inflammation further propagating the metabolism of fat and muscles, manifest in a complex syndrome of cancer-associated cachexia. Marked by cancer-associated muscle loss, the declining muscle function in patients deteriorates even further due to the unavoidable iatrogenic burden of chemotherapy.[7] To investigate potential anti-cachexia effect of our therapy, we next performed a comparative survival study to compare the efficacy of FST mRNA LNPs to recombinant FST in combination with cisplatin chemotherapy (CDDP) (Figure 7A). We established six ES-2-Luc-bearing groups that included mice treated with CDDP (2 mg kg b.w.−1, injected every 4 days for a maximum of 5 weeks), recombinant FST (FSTH, 100 μg kg b.w.−1, daily dosage) and FST mRNA LNPs (10 μg mRNA/dose, daily dosage), as well as their combination with CDDP, and a control group receiving PBS injections.
Compared to the control group, mice treated with CDDP displayed decreased volumes of ascites, but still formed dispersed cancer clusters and exhibited wasting similar to controls. Mice treated with recombinant FSTH and FST mRNA LNP had slower cancer progression in comparison to controls, and were characterized by strikingly higher mobility and reduced apparent fatigue. Mice receiving CDDP combinations with FSTH or FST mRNA LNPs formed peritoneal aggregates of cancer cells with minimal ascites. Figure 7B shows representative images of mice from each treatment group and their cancer progression as a function of time to a primary humane endpoint (PHE).
Postmortem BLS images of mice treated with CDDP, FST mRNA LNPs, a combination of CDDP and FST mRNA LNPs, or PBS show the extent of cancer progression inside of the peritoneal cavity. Control mice displayed dispersed ES-2-Luc clusters in the ascitic fluid that gave high BLS reads (Figure 7C, white arrow). Although CDDP and FSTH treated mice exhibited slower progression of the disease, lower tumor burden, and formation of small cancer aggregates (Figure 7C, purple arrow), we still observed significant ES-2-Luc cell infiltrates in ascites (Figure 7C, white arrow). FST mRNA LNP treated mice developed ES-2-Luc aggregates accumulating in the suprahepatic regions and between the folds of the mesentery, small intestines, and other organs (Figure 7C, purple arrow). Minimal volume of ascitic fluid and binding of cells in cancer aggregates allowed us to observe organs on the image (i.e., liver, Figure 7C, yellow arrow) that would not be possible in the case of ES-2-Luc cluster suspension giving an intense and diffuse BLS signal. Combinatorial treatment with CDDP and FST mRNA LNPs led to the formation of small solid tumors nonadherent to organs (Figure 7C, red arrow) and easily removed from the IP cavity (Figure 7C, bottom image).
Figure 7D depicts ES-2-Luc cell behavior in the peritoneal cavity of the mice at primary humane endpoints showing differential response to treatments with PBS (control), CDDP, FST mRNA LNP, and CDDP+FST mRNA LNP combination. Control mice developed severe ascites with ES-2-Luc cluster suspension (white arrows). Both FST mRNA LNP- and CDDP-treated mice developed minimal ascites (white arrows) and metastatic nodules (purple arrows) forming on organs and mesenteries. However, FST mRNA LNP-treated mice also displayed nonadherent ES-2 aggregates that were easily removed from the abdominal cavity. Combinatorial CDDP+FST mRNA LNP treatment resulted in ES-2-Luc cells aggregating to solid nonadherent tumors (red arrow) with no metastatic nodules forming on organs (also confirmed on Figure 7C).
FST mRNA LNP delivered in daily doses increased serum FST levels and considerably reduced serum ActA (Figure 7E, F). An increasing concentration-dependent effect of FST was reflected by a proportionate decrease of serum ActA in the treatment groups with FST mRNA LNP and recombinant FSTH (100 μg kg b.w.−1). Serum ActA levels were decreased by 3- and 1.3-fold in FST mRNA LNP and recombinant FST treatment groups, respectively. Thus, a higher concentration of FST protein transiently expressed from the delivered FST mRNA in the immediate tumor environment not only decreased ActA levels, but also increased systemic serum FST providing therapeutic abscopal effects.
The beneficial therapeutic effect of increased systemic FST was again observed in the context of counteracting cachexia manifestations of progressing cancer and iatrogenic insults of CDDP chemotherapy. Analysis of the cross-sectional area (CSA) of individual muscle fibers indicated a 25% CSA decrease in muscle fibers of ES-2-Luc-bearing mice receiving CDDP treatments (PBS/PBS vs. PBS/CDDP, Figure 7G). FST supplementation by FSTH and to the greater extent by FST mRNA LNP treatments attenuated this detrimental effect. FSTH increased CSA by ~1.5-fold in both no chemotherapy (FSTH/PBS) and CDDP-treated (FSTH/CDDP) animals, compared to PBS/PBS and PBS/CDDP groups. FST mRNA LNP treatment led to a ~2-fold increase in CSA of muscle fibers in no chemotherapy (FST mRNA LNP/PBS) and CDDP-treated (FST mRNA LNP/CDDP) animals in comparison to PBS/PBS and PBS/CDDP groups.
We observed muscle fiber atrophy and fibrotic changes in CDDP-treated mice (PBS/CDDP group), as shown on Picrosirius muscle fiber histology with Sirius red stain highlighting regions of increased collagen deposition. Muscle fibers in CDDP-treated mice supplemented with FST protein (both recombinant FSTH and FST mRNA LNP treatments) displayed reduced fibrosis (Figure 7H, I). Although both recombinant FSTH and FST mRNA LNP treatments prevented muscle fibrosis (Figure 7G), FST mRNA LNP treatment exerted a superior muscle sparing effect compared to recombinant FSTH protein supplementation.
Although we observe similar manifestations in mice one week after ES-2-Luc cell inoculation, cancer-bearing mice treated with FST mRNA LNPs preserved their muscle bulk, as well as displayed a decreased expression of E3 ubiquitin ligase-related genes FBXO32, Trim63, and Foxo1. Thus, the effect of the FST mRNA LNP platform has a potential direct clinical translation for treating muscle catabolic manifestations of cancer-associated cachexia and muscle atrophy following chemotherapy. Since antibodies against ACTRIIB receptors were shown to ameliorate chemotherapy-dependent cachexia,[65] FST mRNA LNP treatment would be a promising adjuvant approach supplementing chemotherapeutic intervention. Similarly, in our murine model, we observed that the CDDP-treated cancer-bearing mice exhibited signs of iatrogenic muscle atrophy in addition to cancer-dependent skeletal muscle loss. However, the regional combinatorial treatment with CDDP and FST mRNA LNPs exerted an amplified therapeutic effect in terms of more effective eradication of cancer cells in addition to functional and metabolic support of the organism undergoing chemotherapeutic intervention. It is also important to note that mice treated with FST mRNA LNP/CDDP combination developed small, tightly packed nonadherent tumors easily removed from the IP cavity. This would likely be of great benefit clinically, as it would facilitate surgical resection of most ovarian tumor tissues and prevent malignant intestinal obstructions.
2.8. Synergistic effect of FST mRNA LNP treatment with CDDP chemotherapy prolongs survival of mice with ovarian cancer characterized by peritoneal metastases
The mean survival of mice treated with FST mRNA LNPs (25.8±2.8 days, mean±SE, Table S2), but not FSTH therapy (17.0±1.3 days), had differed significantly from the negative control group (12.2±1.15 days) treated with PBS (Figure 8A-D). The combination of FST mRNA LNP with CDDP (33.8±2.4 days) significantly increased the mean survival in comparison to CDDP treatment alone (21.2±1.66 days). However, there was no statistically significant difference in mean survival between negative control and CDDP groups, as well as between FST mRNA LNP alone and CDDP+FST mRNA LNP treatment groups. Importantly, the combination of CDDP with FST mRNA LNPs substantially increased the mean survival of cancer-bearing mice (33.8±2.4 days) compared to CDDP+FSTH (23.8±2.0 days) treatment (Figure 8B). These findings suggest an essential role of FST mRNA LNP treatment in increasing mean survival of cancer-bearing mice when compared to recombinant FST and further support the importance of local and systemic mRNA-mediated FST increase in addition to CDDP therapy.
Figure 8. FST mRNA LNP therapy has a potential synergistic effect with CDDP chemotherapy.

(A) Comparison of survival curves between control, FSTH, and FST mRNA LNP treatments. (B) Comparison of survival curves between control, CDDP+FSTH, and CDDP+FST mRNA LNP treatments. (C) Results of post hoc Bonferroni's multiple comparisons using median survival (D) Table of significant results of post hoc Bonferroni’s multiple comparisons using median survival (E) Bliss modeling of treatment independence for analysis of therapeutic synergy between FSTH and in combination with CDDP (F) Bliss modeling of treatment independence for analysis of therapeutic synergy between FST mRNA LNP and in combination with CDDP.
Bliss modeling of therapeutic independence demonstrated a synergistic effect of CDDP+FST mRNA LNP, but not CDDP+FSTH treatments (Figure 8E, F). Therefore, synergistic CDDP+FST mRNA LNP treatment led to a superior ~2-fold increase in mean survival compared to negative control and ~1.3-fold increase compared to single treatments used in this experiment. These findings summarize the importance of adjuvant FST mRNA LNP therapy in combination with regional administration of CDDP chemotherapy. The synergistic effect of CDDP and ActA inhibition in the treatment of ovarian cancer may derive from two distinct mechanisms. First, ActA inhibition may reduce the activity of the non-canonical ActA pathway, thereby activating ERK1/2 and p38 MAPKs through type 1 ActA receptor. This would be particularly beneficial for the treatment of chemotherapy-induced cachexia associated with ERK1/2 and p38 MAPK activation.[50, 51] Second, downregulation of the canonical ActA pathway could reduce the activation of the proteolytic program in muscle by inhibiting the SMAD 2/3 signaling that in turn mediates the expression of Atrogin-1 and Murf-1.[10] In either case, the mitigation of CDDP's adverse effects would increase the effectiveness of CDDP chemotherapy for ES-2 cancer eradication.
3. Conclusion
In conclusion, FST mRNA LNP therapy led to durable responses providing synergistic effects in combination with CDDP chemotherapy in a murine model of disseminated ovarian carcinoma. This study established a promising translatable therapeutic option for treatment of unresectable ovarian cancer with distant metastases that limits cancer progression while enhancing resilience during an aggressive chemotherapeutic intervention.
4. Experimental Section/Methods
FST, EGFP, and Luc mRNA, including fluorofore-labeled mRNA, were purchased from TriLink Biotechnologies Inc. (TriLink Biotechnologies Inc San Diego, CA, USA). DLin-MC3-DMA, cholesterol, DSPC, DMG-PEG2k were obtained from BioFine International (BioFine International, Vancouver, BC, Canada) or Avanti Polar Lipids (Avanti Polar Lipids, Alabaster, AL, USA. Cell culture reagents, including RPMI media, FBS, PBS, and penicillin and streptomycin were purchased from Gibco (Gibco BRL, Gaithersburg, MD, USA), nuclease-free water from Cytiva (Cytiva, Hyclone Laboratories, South Logan, UT, USA). D-luciferin reagent, Halt Protease Inhibitor Cocktail, Prolong Gold anti-fade mountant, Pierce BCA protein assay, Quant-iT RiboGreen RNA kit and ribosomal RNA standards were obtained from Thermo Fisher Scientific (Thermo Fisher Scientific, Waltham, MA, USA). DNA-free Kit and TRIzol reagent were obtained from Ambion (Ambion Co., Carlsbad, CA, USA). Proteinase K reagent and Qiagen RNeasy Mini kit were purchased from Qiagen (Qiagen Corporation, Hilden, Germany). TaqMan Reverse Transcription Reagents Kit, TaqMan Gene Expression Master Mix, Power SYBR Green Master Mix, and other TaqMan assay reagents were purchased from Applied Biosystems (Applied Biosystems, Foster City, CA, USA). Human Follistatin and Activin A ELISA kits were purchased from Ray Biotech (Ray Biotech, Peachtree Corners, GA, USA). All antibodies were obtained from Invitrogen (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA). PicroSirius Red Stain Kit was purchased from Abcam (Abcam, Cambridge, UK). Human recombinant follistatin protein, mouse recombinant activin A, and human recombinant activin A were purchased from Enzo Life Sciences (Enzo Life Sciences, Farmingdale, NY, USA). Cisplatin reagent was obtained from Tocris Bioscience (Tocris Bioscience, Bristol, UK).
LNP preparation and characterization:
The LNPs containing different mRNA transcripts (CleanCap FST, EGFP, and Luc mRNA) were prepared by the previously reported microfluidic procedure.[33] Briefly, LNPs were synthesized in the ratios 50:38.5:10:1.5 of the ionizable lipid DLin-MC3-DMA, cholesterol, DSPC, and DMG-PEG-2k using NanoAssemblr Spark or Benchtop (Precision Nanosystems Inc., Vancouver, Canada). Between ionizable lipids and nucleic acids, a N:P ratio of 5.67 was maintained to produce the nanoparticle that were further concentrated using Amicon Ultra centrifugal filters (Millipore, Bedford, MA, USA) after being buffer exchanged with phosphate-buffered saline (PBS; pH 7.4). Hydrodynamic size, PDI and ζ potential of the developed LNPs were evaluated by Zetasizer Nano ZS (Malvern Panalytical, Malvern, UK) according to the previously reported procedure.[66] All LNPs used in the study exhibited >90% mRNA encapsulation efficiency characterized by a modified Quant-iT RiboGreen assay as previously described.[33] Cryo-TEM micrographs were prepared by depositing 2-3 μL of LNP suspension onto a lacey carbon grid (Electron Microscopy Sciences, Cu 300 mesh), blotting for 1s using Vitrobot (FEI), and rapidly plunging the grid into liquid ethane to produce vitrified ice. The grids were then clipped and imaged on Glacios Cryo-TEM (Thermo Fisher Scientific, Cleveland, OH, USA) equipped with Gatan K3 camera (Gatan, Pleasanton, CA, USA).
In vivo studies:
All animal studies were approved by the Institutional Animal Care and Use Committees at Oregon Health and Science University and Oregon State University (approval number: IP00000690).
Animal Model:
We utilized a previously reported murine model of ovarian cancer with extensive invasive and metastatic properties.[67-69] Our experiments were performed in female nude mice (Nu/Nu, Charles River Laboratories, Wilmington, MA, USA) bearing ES-2 intraperitoneal (IP) xenograft. ES-2 human ovarian clear cell carcinoma cell line was purchased from ATCC (Manassas, VA, USA). ES-2-Luc cells were a kind gift from Glen S. Kwon from the School of Pharmacy at the University of Wisconsin–Madison. Cells were grown in RPMI (Gibco BRL, Gaithersburg, MD, USA) growth media supplemented with 10% fetal bovine serum (Gibco BRL, Gaithersburg, MD, USA) and 5% Penicillin/Streptomycin (Gibco BRL, Gaithersburg, MD, USA). All cultures were grown at 37°C with 5% CO2.
The ES-2 cell line is a human ovarian cancer that expresses an intermediate phenotype between high-grade serous cell carcinoma (HGSCC) and clear cell carcinoma (CCC), the two most aggressive types of ovarian cancer in humans.[70-72] Mixed surface epithelial carcinomas display CCC- and HGSCC-derived characteristics, including resistance to platinum-based chemotherapy, tumors with high mitotic rates, and increased mutational burden.[73,74] Compared to other mixed CCC/HGSCC cell lines, the ES-2 line is characterized by both malignant ascites and a formation of solid tumors.[75] Taken together, the rationale behind choosing the ES-2 line for this model was based on its characteristics of a short doubling time (t=19 h), invasiveness, and high metastatic potential with tropism for metastasis to organs and fat pads in the peritoneal cavity.[71,76,77] Accordingly, after IP injection of ES-2-WT or ES-2-Luc cells (3×106 cell per 100 μL in PBS), we observed the growth of malignant ascites with multi-cellular clusters in a third-spaced fluid. PBS was used for sham injection in control groups. The mice developed multiple cancer clusters, ultimately colonizing secondary sites, and forming metastatic nodules in the omentum, ovaries, and gonadal fat pads. Cancer-bearing mice also displayed a profound cachexic phenotype that was previously described in the literature.[69]
Biodistribution of Luc mRNA LNPs in sham and ES-2-WT cancer-bearing mice:
The ES-2-WT bearing mice (n=3, day ten after ES-2-WT cells injection) and the sham mice (n=3) were injected with a single Luc mRNA LNP dose (10 μg mRNA per dose). Mice injected with ES-2-Luc cells served as positive controls. ES-2-WT-bearing mice and sham mice injected with PBS served as negative controls. The positive and negative control groups (both, n=3). Four hours after injections, mice were injected with D-luciferin (150 mg kg b.w.−1 in 100 μL PBS). After fifteen minutes, BLS was captured with IVIS Lumina XRMS imaging system (Perkin Elmer, Hopkinton, MA, USA). BLS data were analyzed using Living Image software (Perkin Elmer, Hopkinton, MA, USA). Organs and ascitic fluid were collected and assessed for BLS distribution.
Biodistribution of mRNA LNPs in ES-2-Luc cancer-bearing mice:
Five groups of ES-2-Luc bearing mice (n=5, day ten after ES-2-WT cells inoculation) were injected with single doses of Cy5-EGFP, EGFP, equimolar EGFP/FST, FST mRNA LNPs (10 μg total mRNA per dose) or PBS. Four hours after injection, mice were euthanized, organs and ascitic fluid collected for further analysis. Fluorescence images of ascites were visualized using Cy5 and GFP filters on the Keyence BZ-X700 microscope (Keyence Corp., Osaka, Japan). Absolute quantification of EGFP mRNA by qRT-PCR on cells from the ascitic fluid was performed as described in the Quantitative RT-PCR section.
Quantitative RT-PCR:
Cells from ascites were centrifuged, ascitic fluid removed. Total RNA was extracted with TRIzol (Ambion Co., Carlsbad, USA) and Chloroform (Sigma-Aldrich, Merck KGaA, Darmstadt, Germany). RNA was precipitated with 200 proof ethanol (Decon Labs, Inc.; King of Prussia, PA, USA). Qiagen RNeasy Mini kit (Qiagen Corporation, Hilden, Germany) was used for further extraction following the modified manufacturer's instructions. DNA-free Kit (Ambion, Catalog #1906, Invitrogen, Carlsbad, CA, USA) was used to remove genomic DNA contaminants. Total RNA was eluted with nuclease-free water (Cytiva, Hyclone Laboratories, South Logan, UT, USA). Total RNA from liver, spleen, and lungs, and gonadal fat pads was extracted using the method above. Total RNA from fibrous tissues (muscle, kidneys) was extracted using Qiagen RNeasy Mini kit (Qiagen Corporation, Hilden, Germany) following the manufacturer's protocol. Proteinase K (Qiagen Corporation, Hilden, Germany) was employed after the tissue homogenization step. DNA-free Kit was used to remove genomic DNA contaminants.
cDNA was synthesized with TaqMan Reverse Transcription Reagents Kit (Applied Biosystems, Foster City, CA, USA). Relative quantification with qRT-PCR was performed by using TaqMan Gene Expression Master Mix (Applied Biosystems, Foster City, CA, USA) and TaqMan assay reagents (Table 1, TaqMan assay reagents) on the 7300 PCR system (Applied Biosystems). Gene expression was normalized to 18S expression.
Table 1:
TaqMan assay reagents listed as gene of interest and corresponding TaqMan assay identifier.
| Gene of interest | TaqMan assay |
| FST | Hs01086902_m1 |
| CD68 | Mm03047343_m1 |
| Ly6G | Mm04934123_m1 |
| SELL | Mm00441291_m1 |
| P4HB | Mm01243188_m1 |
| FOXO1 | Mm00490671_m1 |
| FBXO32 | Mm00499523_m1 |
| TRIM63 | Mm01185221_m1 |
| Ucp1 | Mm01244861_m1 |
| MKI67 | Hs01032443_m1 |
| TOP2A | Hs03063307_m1 |
| TPX2 | Hs00201616_m1 |
| 18S | CN: 4333760F |
Absolute quantification with qRT-PCR was performed using Power SYBR Green Master Mix (Applied Biosystems, Foster City, CA, USA). EGFP primer sequences were: forward -CACATGAAGCAGCACGACTT, reverse – GGTCTTGTAGTTGCCGTCGT; efficiency – 108%, dissociation T=82.5 °C, single peak. LoD and LoQ were calculated as previously described.[78]
For the ECM gene expression experiment, total RNA from ascitic fluid containing dispersed ES-2-Luc cells and solid ES-2-Luc aggregates was analyzed with TaqMan Array Human Extracellular Matrix & Adhesion Molecules. A 1.5-fold change threshold was used to identify over- and under-expressed genes, and a 0.01 false discovery rate was used to validate a finding of differentially expressed genes.
Evaluation of serum FST and ActA levels after single FST mRNA LNP injection:
FSTL mRNA LNP and FSTH mRNA LNP formulations were prepared by encapsulating equimolar EGFP/FST mRNAs and FST mRNAs, respectively. The mice were injected on the 10th day after ES-2-WT cells inoculation. Serum and ascites were collected for analysis. FST and ActA serum concentrations were quantified using ELISA as described below in the Enzyme-linked immunosorbent assays section. Relative quantification of FST mRNA by qRT-PCR on cells from the ascitic fluid was performed as described above in the Quantitative RT-PCR section.
Enzyme-linked immunosorbent assays:
For ELISA analysis, blood was collected by cardiac puncture in 2 mL tubes with the addition of EDTA and protease inhibitor (Halt Protease Inhibitor Cocktail; Thermo Scientific, Waltham, MA, USA) and was let to sit at room temperature for a minimum of 30 minutes. Blood samples were centrifuged for 15 minutes at 3000 g within one hour of collection (Eppendorf, 5430R, Hamburg, Germany). Serum was then transferred to clean tubes for storage or immediate analysis. Ascitic fluid was centrifuged to separate from the cellular pallet. Total protein in the ascitic supernatant was quantified with the Pierce BCA protein assay (Thermo Scientific, Waltham, MA, USA). Colorimetric quantification was performed on BioTek Synergy 2 microplate reader (BioTek, Winooski, USA) and analyzed on BioTek Gen5 Data Analysis Software. Serum and ascitic fluid were analyzed for FST or ActA concentrations using Human Follistatin and Activin A ELISA kits according to the manufacturer's protocol (Ray Biotech, Norcross, USA). Samples were diluted 1:2 and 1:10 for FST and ActA quantification, respectively. Results were read using BioTek Synergy 2 microplate reader and analyzed on BioTek Gen5 Data Analysis Software. Ascitic fluid results were normalized to total protein.
Chronic FST mRNA LNP injections:
Two control groups, ES-2-Luc-bearing and sham mice (both, n=5) and two treatment groups, ES-2-Luc-bearing and sham mice (both, n=10), received PBS or FST mRNA LNP injections (10 μg mRNA per dose), respectively. Treatment was initiated on day three after ES-2-Luc cell inoculation in cancer-bearing mice and sham PBS injection in age-matched sham mice and continued for seven days. Mice were euthanized twelve hours after the final LNP injection; serum, ascitic fluid, and organs were collected for further analysis. Relative quantification of FST mRNA in organs and ascites, and CD68, Ly6G, SELL, P4HB, MKI67, TOP2A and TPX2 mRNA transcripts in ascites was performed by qRT-PCR. Extracellular matrix and adhesion molecules assay was analyzed as described in the Quantitative RT-PCR section. FST and ActA quantification in the ascites and serum samples was performed as described above in the Enzyme-linked immunosorbent assays section. Gastrocnemius muscles and gonadal fat pads were analyses by qRT-PCR for quantification of FOXO1, FBXO32, TRIM63 and Ucp1 mRNA transcripts as described in the Quantitative RT-PCR section. Gastrocnemius muscles were flash frozen and sectioned (10 μm) on Leica CM1850 UV (Leica Biosystems Nussloch, GmbH, Germany) at −18 °C. IHC and Picrosirius staining was perfumed as described below in the Histology and immunohistochemistry section.
Flow cytometry studies:
ES-2-WT cells were co-cultured with RAW 264.7 murine macrophages in RPMI medium with 10% fetal bovine serum and 5% Penicillin/Streptomycin, and initial proportion of 3:1 of ES-2-WT cells and RAW 264.7, respectively. Nude mice were injected with ES2-WT cells (3×106 cell per 100 μL in PBS). After 10 days, the control group received PBS injection (100 uL), and the treatment group received injections with GFP mRNA LNP formulation (2ug mRNA/mouse in 100uL PBS). Ascitic fluid was collected by IP lavage. Both in vitro and in vivo samples were fixed with 1:1 acetone/methanol solution pre-cooled to −20 °C and blocked with 1:1 mixture of TruStain FcX Fc receptor blocking solutions (human and murine, Biolegend, San Diego, CA, USA) for 30 minutes at room temperature, followed by conjugated antibodies staining for HLA-ABC (HLA-ABC Monoclonal Antibody (W6/32), PE eBioscience, Thermofisher Scientific, Waltham, MA, USA) and GFP (APC Anti-GFP antibody, Abcam, Cambridge, UK). Flow cytometry was performed on BD FACSymphony S6 cell sorter (BD, Franklin lakes, NJ, USA) using laser excitation wavelength of 561 nm and 628 nm for HLA-ABC and GFP detection, respectively. Data was analyzed using FlowJo (FlowJo LLC, Ashland, OR, USA).
Histology and immunohistochemistry:
Muscle samples were flash frozen followed by 10-μm cryosectioning. For IHC studies, the slides were incubated in blocking buffer for 1 hour at room temperature, followed by primary antibody (mouse laminin, Invitrogen, rabbit, 1:200) incubation overnight at 4 °C. Secondary antibody utilized in the experiment was goat anti-rabbit AF488 (Invitrogen, 1:1000). Sections received three PBS washes between each step. Slides were mounted using Prolong Gold anti-fade media (Thermo Scientific, Waltham, MA, USA). PicroSirius staining was performed according to the manufacturer's protocol (PicroSirius Red Stain Kit, Abcam, Cambridge, UK). Histopathological examination of liver slices from the mice treated with three daily injections of Luc mRNA LNP were performed to evaluate safety and tolerability of the platform. Livers were collected into 4% PFA, stored for 24 hours before a transfer to 70% ethanol for further paraffin embedding and sectioning. Liver sections from control (PBS) and treatment (Luc mRNA LNP, 10 ug/mRNA/mouse) were then stained with hematoxylin and eosin (H&E) and evaluated for pathologically observable changes in morphology, infiltration with immune cells, fibrotic or inflammatory changes.
FST and ActA recombinant protein injections in survival experiment:
Human recombinant follistatin protein (Enzo Life Sciences, Farmingdale, NY, USA) or mouse recombinant activin A (Enzo Life Sciences, Farmingdale, NY, USA) were reconstituted according to the manufacturer's recommendations and further diluted in PBS as needed on the injection days. ActA was injected in doses of 20 μg kg b.w.−1, FST was injected in doses of 20 μg kg−1 or 100 μg kg b.w.−1 according to the experimental procedure. BLS was read as described above.
FST mRNA LNP and CDDP combinatorial treatment in survival experiment:
In the experiments requiring cisplatin (CDDP, >98% purity, Tocris Bioscience, Bristol, UK) injections, the CDDP working solution was prepared in sterile PBS. CDDP was injected in a dose of 2 mg kg b.w.−1, delivered every four days for a maximum of five weeks.
Statistical Analysis:
All experiments were performed with n = 5 unless specified otherwise. Significance was determined using paired t-test, multiple comparison t-test, two-way ANOVA using Graphpad Prism software. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
Supplementary Material
Acknowledgements
The research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Numbers R37CA234006 and R01CA237569, OHSU Knight Cancer Institute, and Friends of Doernbecher. Support was also received from the College of Pharmacy at Oregon State University, Papé Family Pediatric Research Institute at Oregon Health & Science University, and ARCS Scholarship (The Karen Irons Medicis Memorial Scholar Award, Diane and Dick Alexander). The funding sources had no involvement in study design; in the collection, analysis and interpretation of data; in the writing of the manuscript; and in the decision to submit the article for publication. The electron microscopy was performed using the Multiscale Microscopy Core (MMC) at Oregon Health & Science University with technical support from the (OHSU)-FEI Living Lab and the Center for Spatial Systems Biomedicine (OCSSB).
Footnotes
Conflict of Interest: D.L.M. is a consultant for Pfizer, Inc. and Alkermes, Inc. D.L.M. is a consultant, has received grant funding, and has equity in Tensive Controls, Inc. The other authors declare no competing interests.
Supporting Information
Supporting Information is available from the Wiley Online Library.
Contributor Information
Tetiana Korzun, Department of Pharmaceutical Sciences, College of Pharmacy, Oregon State University, 2730 S Moody Avenue, Portland, Oregon, 97201, USA; Department of Biomedical Engineering, Oregon Health & Science University, 3303 SW Bond Avenue Portland, Oregon, 97239, USA; Medical Scientist Training Program, Oregon Health & Science University, 3181 SW Sam Jackson Park Road, Portland, Oregon 97239, USA; Papé Family Pediatric Research Institute, Oregon Health & Science University, SW Sam Jackson Park Rd, Mail Code L481 Portland, Oregon, 97239, USA.
Abraham S. Moses, Department of Pharmaceutical Sciences, College of Pharmacy, Oregon State University, 2730 S Moody Avenue, Portland, Oregon, 97201, USA
Jeonghwan Kim, Department of Pharmaceutical Sciences, College of Pharmacy, Oregon State University, 2730 S Moody Avenue, Portland, Oregon, 97201, USA.
Siddharth Patel, Department of Pharmaceutical Sciences, College of Pharmacy, Oregon State University, 2730 S Moody Avenue, Portland, Oregon, 97201, USA.
Canan Schumann, Department of Pharmaceutical Sciences, College of Pharmacy, Oregon State University, 2730 S Moody Avenue, Portland, Oregon, 97201, USA.
Peter R. Levasseur, Papé Family Pediatric Research Institute, Oregon Health & Science University, SW Sam Jackson Park Rd, Mail Code L481 Portland, Oregon, 97239, USA
Parham Diba, Medical Scientist Training Program, Oregon Health & Science University, 3181 SW Sam Jackson Park Road, Portland, Oregon 97239, USA; Papé Family Pediatric Research Institute, Oregon Health & Science University, SW Sam Jackson Park Rd, Mail Code L481 Portland, Oregon, 97239, USA.
Brennan Olson, Medical Scientist Training Program, Oregon Health & Science University, 3181 SW Sam Jackson Park Road, Portland, Oregon 97239, USA; Papé Family Pediatric Research Institute, Oregon Health & Science University, SW Sam Jackson Park Rd, Mail Code L481 Portland, Oregon, 97239, USA.
Katia Graziella De Oliveira Rebola, Knight Cancer Institute, Oregon Health & Science University, 2720 S Moody Avenue, Portland, Oregon, 97201, USA.
Mason Norgard, Papé Family Pediatric Research Institute, Oregon Health & Science University, SW Sam Jackson Park Rd, Mail Code L481 Portland, Oregon, 97239, USA.
Youngrong Park, Department of Pharmaceutical Sciences, College of Pharmacy, Oregon State University, 2730 S Moody Avenue, Portland, Oregon, 97201, USA.
Ananiya A. Demessie, Department of Pharmaceutical Sciences, College of Pharmacy, Oregon State University, 2730 S Moody Avenue, Portland, Oregon, 97201, USA
Yulia Eygeris, Department of Pharmaceutical Sciences, College of Pharmacy, Oregon State University, 2730 S Moody Avenue, Portland, Oregon, 97201, USA.
Vladislav Grigoriev, Department of Pharmaceutical Sciences, College of Pharmacy, Oregon State University, 2730 S Moody Avenue, Portland, Oregon, 97201, USA; Papé Family Pediatric Research Institute, Oregon Health & Science University, SW Sam Jackson Park Rd, Mail Code L481 Portland, Oregon, 97239, USA.
Subisha Sundaram, Department of Pharmaceutical Sciences, College of Pharmacy, Oregon State University, 2730 S Moody Avenue, Portland, Oregon, 97201, USA; Papé Family Pediatric Research Institute, Oregon Health & Science University, SW Sam Jackson Park Rd, Mail Code L481 Portland, Oregon, 97239, USA.
Tanja Pejovic, Departments of Obstetrics and Gynecology and Pathology, Oregon Health & Science University, 3181 SW Sam Jackson Park Road, Portland, Oregon, 97239, USA.
Jonathan R. Brody, Knight Cancer Institute, Oregon Health & Science University, 2720 S Moody Avenue, Portland, Oregon, 97201, USA
Olena R. Taratula, Department of Pharmaceutical Sciences, College of Pharmacy, Oregon State University, 2730 S Moody Avenue, Portland, Oregon, 97201, USA
Xinxia Zhu, Papé Family Pediatric Research Institute, Oregon Health & Science University, SW Sam Jackson Park Rd, Mail Code L481 Portland, Oregon, 97239, USA.
Gaurav Sahay, Department of Pharmaceutical Sciences, College of Pharmacy, Oregon State University, 2730 S Moody Avenue, Portland, Oregon, 97201, USA.
Daniel L. Marks, Brenden-Colson Center for Pancreatic Care, Oregon Health and & Science University, 2730 S Moody Avenue, Portland, Oregon, 97201, USA; Knight Cancer Institute, Oregon Health & Science University, 2720 S Moody Avenue, Portland, Oregon, 97201, USA; Papé Family Pediatric Research Institute, Oregon Health & Science University, SW Sam Jackson Park Rd, Mail Code L481 Portland, Oregon, 97239, USA
Oleh Taratula, Department of Pharmaceutical Sciences, College of Pharmacy, Oregon State University, 2730 S Moody Avenue, Portland, Oregon, 97201, USA; Department of Biomedical Engineering, Oregon Health & Science University, 3303 SW Bond Avenue Portland, Oregon, 97239, USA.
References
- [1].Roett MA, Evans P, Am. Fam. Physician 2009, 80, 609. [PubMed] [Google Scholar]
- [2].Park HK, Ruterbusch JJ, Cote ML, Cancer Epidemiol. Biomark. Prevent 2017, 26, 1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Vermeulen CKM, Tadesse W, Timmermans M, Kruitwagen RFPM, Walsh T, Eur. J. Obstetr. Gynecol. Reproduct. Biol 2017, 219, 100. [DOI] [PubMed] [Google Scholar]
- [4].Rauh-Hain JA, Nitschmann CC, Worley MJ, Bradford LS, Berkowitz RS, Schorge JO, Campos SM, del Carmen MG, Horowitz NS, Gynecol. Oncol 2013, 129, 63. [DOI] [PubMed] [Google Scholar]
- [5].Gadducci A, Cosio S, Fanucchi A, Genazzani AR, Anticancer Res. 2001, 21, 2941. [PubMed] [Google Scholar]
- [6].Mantzorou M, Koutelidakis A, Theocharis S, Giaginis C, Nutr. Cancer 2017, 69, 1151. [DOI] [PubMed] [Google Scholar]
- [7].Mikuła-Pietrasik J, Witucka A, Pakuła M, Uruski P, Begier-Krasińska B, Niklas A, Tykarski A, Książek K, Cell. Mol. Life Sci 2019, 76, 681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Braun TP, Szumowski M, Levasseur PR, Grossberg AJ, Zhu X, Agarwal A, Marks DL, PLoS One 2014, 9, e106489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Massagué J, Cell 2008, 134, 215.18662538 [Google Scholar]
- [10].Chen JL, Walton KL, Winbanks CE, Murphy KT, Thomson RE, Makanji Y, Qian H, Lynch GS, Harrison CA, Gregorevic P, FASEB J. 2014, 28, 1711. [DOI] [PubMed] [Google Scholar]
- [11].Cobellis L, Reis FM, Luisi S, Danero S, Pirtoli L, Scambia G, Petraglia F, J. Soc. Gynecol. Investig 2004, 11, 203. [DOI] [PubMed] [Google Scholar]
- [12].Lambert-Messerlian GM, DePasquale SE, Maybruck WM, Steinhoff MM, Gajewski WH, Gynecol. Oncol 1999, 74, 93. [DOI] [PubMed] [Google Scholar]
- [13].Di Simone N, Crowley JWF, Wang QF, Sluss PM, Schneyer AL, Endocrinol. 1996, 137, 486. [DOI] [PubMed] [Google Scholar]
- [14].Steller MD, Shaw TJ, Vanderhyden BC, Ethier JF, Mol. Cancer Res 2005, 3, 50. [PubMed] [Google Scholar]
- [15].Welt CK, Lambert-Messerlian G, Zheng W, Crowley WF Jr., Schneyer AL, J. Clin. Endocrinol. Metab 1997, 82, 3720. [DOI] [PubMed] [Google Scholar]
- [16].Ries A, Schelch K, Falch D, Pany L, Hoda MA, Grusch M, Expert Opin. Ther. Targets 2020, 24, 985. [DOI] [PubMed] [Google Scholar]
- [17].Olson B, Marks DL, Grossberg AJ, J. Cachexia Sarcopenia Muscle 2020, 11, 1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zhou X, Wang JL, Lu J, Song Y, Kwak KS, Jiao Q, Rosenfeld R, Chen Q, Boone T, Simonet WS, Lacey DL, Goldberg AL, Han HQ, Cell 2010, 142, 531. [DOI] [PubMed] [Google Scholar]
- [19].Latres E, Mastaitis J, Fury W, Miloscio L, Trejos J, Pangilinan J, Okamoto H, Cavino K, Na E, Papatheodorou A, Willer T, Bai Y, Hae Kim J, Rafique A, Jaspers S, Stitt T, Murphy AJ, Yancopoulos GD, Gromada J, Nat. Commun 2017, 8, 15153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Pearsall RS, Davies MV, Cannell M, Li J, Widrick J, Mulivor AW, Wallner S, Troy ME, Spaits M, Liharska K, Sako D, Castonguay R, Keates S, Grinberg AV, Suragani RNVS, Kumar R, Sci. Rep 2019, 9, 11392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ozawa T, Morikawa M, Morishita Y, Ogikubo K, Itoh F, Koinuma D, Nygren P-Å, Miyazono K, iSci. 2021, 24, 102488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Tao JJ, Cangemi NA, Makker V, Cadoo KA, Liu JF, Rasco DW, Navarro WH, Haqq CM, Hyman DM, Clin. Cancer Res 2019, 25, 5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lee SJ, Lee YS, Zimmers TA, Soleimani A, Matzuk MM, Tsuchida K, Cohn RD, Barton ER, Mol. Endocrinol 2010, 24, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Yaden BC, Croy JE, Wang Y, Wilson JM, Datta-Mannan A, Shetler P, Milner A, Bryant HU, Andrews J, Dai G, Krishnan V, J. Pharmacol. Exp. Ther 2014, 349, 355. [DOI] [PubMed] [Google Scholar]
- [25].Lee SJ, McPherron AC, Proc. Natl. Acad. Sci. USA 2001, 98, 9306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Xia Y, Schneyer AL, J. Endocrinol 2009, 202, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Choi KC, Kang SK, Nathwani PS, Cheng KW, Auersperg N, Leung PC, Mol. Cell Endocrinol 2001, 174, 99. [DOI] [PubMed] [Google Scholar]
- [28].Mabuchi Y, Yamoto M, Minami S, Umesaki N, Oncol. Rep 2006, 16, 373. [PubMed] [Google Scholar]
- [29].Dean M, Davis DA, Burdette JE, Cancer Lett. 2017, 391, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kim J, Eygeris Y, Gupta M, Sahay G, Adv. Drug Deliv. Rev 2021, 170, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Valencia-Sanchez MA, Liu J, Hannon GJ, Parker R, Genes Dev. 2006, 20, 515. [DOI] [PubMed] [Google Scholar]
- [32].Hajj KA, Whitehead KA, Nat. Rev. Mater 2017, 2, 17056. [Google Scholar]
- [33].Patel S, Ashwanikumar N, Robinson E, Xia Y, Mihai C, Griffith JP, Hou S, Esposito AA, Ketova T, Welsher K, Joyal JL, Almarsson Ö, Sahay G, Nat. Commun 2020, 11, 983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Mellman I, Yarden Y, Cold Spring Harb. Perspect. Biol 2013, 5, a016949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Huang X, Qiu M, Wang T, Li B, Zhang S, Zhang T, Liu P, Wang Q, Qian ZR, Zhu C, Wu M, Zhao J, J. Nanobiotechnol 2022, 20, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Gao N, Bozeman EN, Qian W, Wang L, Chen H, Lipowska M, Staley CA, Wang YA, Mao H, Yang L, Theranostics 2017, 7, 1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Do T-V, Kubba LA, Antenos M, Rademaker AW, Sturgis CD, Woodruff TK, Endocrinol. 2008, 149, 3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Menon U, Riley SC, Thomas J, Bose C, Dawnay A, Evans LW, Groome NP, Jacobs IJ, BJOG 2000, 107, 1069. [DOI] [PubMed] [Google Scholar]
- [39].Ren P, Chen FF, Liu HY, Cui XL, Sun Y, Guan JL, Liu ZH, Liu JG, Wang YN, J. Int. Med. Res 2012, 40, 877. [DOI] [PubMed] [Google Scholar]
- [40].Khoury RH, Wang QF, Crowley WF Jr., Hall JE, Schneyer AL, Toth T, Midgley AR Jr., Sluss PM, J. Clin. Endocrinol. Metabol 1995, 80, 1361. [DOI] [PubMed] [Google Scholar]
- [41].Nakamura T, Sugino K, Titani K, Sugino H, J. Biol. Chem 1991, 266, 19432. [PubMed] [Google Scholar]
- [42].Mather JP, Roberts PE, Krummen LA, Endocrinol. 1993, 132, 2732. [DOI] [PubMed] [Google Scholar]
- [43].Harrison CA, Chan KL, Robertson DM, Endocrinol. 2006, 147, 2744. [DOI] [PubMed] [Google Scholar]
- [44].Loumaye A, de Barsy M, Nachit M, Lause P, Frateur L, van Maanen A, Trefois P, Gruson D, Thissen JP, J. Clin. Endocrinol. Metab 2015, 100, 2030. [DOI] [PubMed] [Google Scholar]
- [45].Loumaye A, de Barsy M, Nachit M, Lause P, van Maanen A, Trefois P, Gruson D, Thissen JP, J. Cachexia Sarcopenia Muscle 2017, 8, 768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Paajanen J, Ilonen I, Lauri H, Jarvinen T, Sutinen E, Ollila H, Rouvinen E, Lemstrom K, Rasanen J, Ritvos O, Koli K, Myllarniemi M, Clin. Lung Cancer 2020, 21, e142. [DOI] [PubMed] [Google Scholar]
- [47].Patel HJ, Patel BM, Life Sci. 2017, 170, 56. [DOI] [PubMed] [Google Scholar]
- [48].Jatoi A, Ritter HL, Dueck A, Nguyen PL, Nikcevich DA, Luyun RF, Mattar BI, Loprinzi CL, Lung Cancer 2010, 68, 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Bayliss TJ, Smith JT, Schuster M, Dragnev KH, Rigas JR, Exp. Opin. Biol. Ther 2011, 11, 1663. [DOI] [PubMed] [Google Scholar]
- [50].Lamouille S, Xu J, Derynck R, Nat. Rev. Mol. Cell Biol 2014, 15, 178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Singh A, Settleman J, Oncogene 2010, 29, 4741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Yi Y, Cheng JC, Klausen C, Leung PCK, Exp. Cell Res 2019, 382, 111471. [DOI] [PubMed] [Google Scholar]
- [53].Kalfusova A, Hilska I, Krskova L, Kalinova M, Linke Z, Kodet R, Neoplasma 2016, 63, 484. [DOI] [PubMed] [Google Scholar]
- [54].Ye T, Li J, Sun Z, Liu D, Zeng B, Zhao Q, Wang J, Xing HR, Int. J. Biol. Sci 2020, 16, 447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Taha EA, Sogawa C, Okusha Y, Kawai H, Oo MW, Elseoudi A, Lu Y, Nagatsuka H, Kubota S, Satoh A, Okamoto K, Eguchi T, Cancers 2020, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Upadhyay P, Gardi N, Desai S, Chandrani P, Joshi A, Dharavath B, Arora P, Bal M, Nair S, Dutt A, Oral Oncol. 2017, 73, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Shen CJ, Kuo YL, Chen CC, Chen MJ, Cheng YM, PLoS One 2017, 12, e0174487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Li J, Li SH, Wu J, Weisel RD, Yao A, Stanford WL, Liu SM, Li RK, Theranostics 2018, 8, 1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Wafai R, Williams ED, de Souza E, Simpson PT, McCart Reed AE, Kutasovic JR, Waltham M, Snell CE, Blick T, Thompson EW, Hugo HJ, Breast Cancer Res. 2020, 22, 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Zhang D, Bi J, Liang Q, Wang S, Zhang L, Han F, Li S, Qiu B, Fan X, Chen W, Jiao H, Ye Y, Ding Y, Front. Onco.l 2020, 10, 1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Donaldson KL, Goolsby GL, Wahl AF, Int. J. Cancer 1994, 57, 847. [DOI] [PubMed] [Google Scholar]
- [62].Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL, Cell 2004, 117, 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Singh R, Braga M, Reddy ST, Lee S-J, Parveen M, Grijalva V, Vergnes L, Pervin S, Endocrinol. 2017, 158, 1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Aust S, Knogler T, Pils D, Obermayr E, Reinthaller A, Zahn L, Radlgruber I, Mayerhoefer ME, Grimm C, Polterauer S, PLoS One 2015, 10, e0140403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Barreto R, Waning DL, Gao H, Liu Y, Zimmers TA, Bonetto A, Oncotarget 2016, 7, 43442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Duong T, Li X, Yang B, Schumann C, Albarqi HA, Taratula O, Taratula O, Nanomedicine 2017, 13, 955. [DOI] [PubMed] [Google Scholar]
- [67].Schumann C, Chan S, Millar JA, Bortnyak Y, Carey K, Fedchyk A, Wong L, Korzun T, Moses AS, Lorenz A, Shea D, Taratula O, Khalimonchuk O, Taratula O, Nanomedicine 2018, 14, 1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Guo J, Cai J, Zhang Y, Zhu Y, Yang P, Wang Z, Int. J. Oncol 2017, 51, 1199. [DOI] [PubMed] [Google Scholar]
- [69].Pin F, Barreto R, Kitase Y, Mitra S, Erne CE, Novinger LJ, Zimmers TA, Couch ME, Bonewald LF, Bonetto A, J. Cachexia Sarcopenia Muscle 2018, 9, 685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Chan JK, Teoh D, Hu JM, Shin JY, Osann K, Kapp DS, Gynecol. Oncol 2008, 109, 370. [DOI] [PubMed] [Google Scholar]
- [71].Tudrej P, Olbryt M, Zembala-Nożyńska E, Kujawa KA, Cortez AJ, Fiszer-Kierzkowska A, Pigłowski W, Nikiel B, Głowala-Kosińska M, Bartkowska-Chrobok A, Smagur A, Fidyk W, Lisowska KM, Int. J. Mol. Sci 2018, 19, 2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Kwok ALM, Wong OG-W, Wong ESY, Tsun OK-L, Chan K-K, Cheung AN-Y, J. Clin. Pathol 2014, 67, 921. [DOI] [PubMed] [Google Scholar]
- [73].Han G, Gilks CB, Leung S, Ewanowich CA, Irving JA, Longacre TA, Soslow RA, Am. J. Surg. Pathol 2008, 32. [DOI] [PubMed] [Google Scholar]
- [74].Ye S, You Y, Yang J, Cao D, Bai H, Huang H, Wu M, Chen J, Lang J, Shen K, Int J. Gynecol. Cancer 2014, 24, 1590. [DOI] [PubMed] [Google Scholar]
- [75].Shaw TJ, Senterman MK, Dawson K, Crane CA, Vanderhyden BC, Mol. Ther 2004, 10, 1032. [DOI] [PubMed] [Google Scholar]
- [76].Sugiyama T, Kamura T, Kigawa J, Terakawa N, Kikuchi Y, Kita T, Suzuki M, Sato I, Taguchi K, Cancer 2000, 88, 2584. [PubMed] [Google Scholar]
- [77].Lee W, Ko SY, Mohamed MS, Kenny HA, Lengyel E, Naora H, J. Exp. Med 2019, 216, 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Forootan A, Sjöback R, Björkman J, Sjögreen B, Linz L, Kubista M, Biomol. Detect. Quantif 2017, 12, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.