

Abstract
Tumor-infiltrating B and plasma cells (TIBs) are prevalent in lung adenocarcinoma (LUAD), however they are poorly characterized. We performed paired single-cell RNA and B cell receptor (BCR) sequencing of 16 early-stage LUADs and 47 matching multi-region normal tissues. By integrative analysis of ~50,000 TIBs, we define 12 TIB subsets in the LUAD and adjacent normal ecosystems and demonstrate extensive remodeling of TIBs in LUADs. Memory B cells and plasma cells (PCs) were highly enriched in tumor tissues with more differentiated states and increased frequencies of somatic hypermutation. Smokers exhibited markedly elevated PCs and with distinct differentiation trajectories. BCR clonotype diversity increased but clonality decreased in LUADs, smokers, and with increasing pathologic stage. TIBs were mostly localized within CXCL13+ lymphoid aggregates and immune cell sources of CXCL13 production evolved with LUAD progression and included elevated fractions of CD4 regulatory T-cells. This study provides a spatial landscape of TIBs in early-stage LUAD.
Keywords: Lung adenocarcinoma (LUAD), Tumor-infiltrating B and plasma cells (TIBs), Plasma cells (PCs), Single-cell RNA sequencing (scRNA-seq), Single-cell B cell receptor sequencing (scBCR-seq), Spatial transcriptomics (ST), Tumor microenvironment (TME), CXCL13, Tertiary lymphoid structure (TLS), Somatic hypermutation (SHM)
INTRODUCTION
Lung adenocarcinoma (LUAD) remains the most frequently diagnosed histological subtype of lung cancer and accounts for most cancer deaths related to smoking (1). Improved clinical screening strategies have led to increased diagnosis of LUADs at earlier pathological stages (1). Surgery is the standard treatment for localized LUADs, yet, the overall 5-year survival remains below 50%. According to the statistics from American Cancer Society, the 5-year survival rate drops to about 7% for patients who developed distant metastases. Therefore, strategies to treat LUAD in its earliest stages are urgently needed. Such advances are severely limited by our lagging knowledge of the earliest changes in the tumor microenvironment (TME) during LUAD pathogenesis and that could thus lead to ideal targets for interception.
T cells have been, for the most part, the center of attention in efforts to understand the immunobiology of lung cancer (2,3). Other cell types, in particular, tumor-infiltrating B and plasma cells (TIBs), have been mostly overlooked and their roles in pathogenesis of solid tumors such as LUAD remain poorly understood. TIBs have been detected in different solid tumor types (4). Recent studies from our group and others demonstrated that TIBs can strongly impact patient responses to anti-cancer chemo- and immunotherapies as well as clinical outcomes in various cancers including melanoma and lung cancer (5,6). TIBs were also detected in recent single-cell studies, but their phenotypes and states were not extensively characterized (6,7). It is possible that these important studies lacked the power to perform in-depth profiling of TIBs due to an inadequate number of sequenced B cells. We recently profiled epithelial and TME cells from five patients with early-stage LUADs and found that the fractions of TIBs, including both B cells and plasma cells (PCs), were immensely increased in LUADs relative to their matched normal lung tissues (6), thereby suggesting potential roles for TIBs in LUAD pathogenesis.
To better understand the landscape of TIBs in early LUAD pathogenesis, we performed paired single-cell RNA- and BCR-sequencing (scRNA/BCR-seq) of 16 LUADs and 47 matched multi-region normal lung tissues from patients with early-stage disease. We studied BCRs from ~73K cells and transcriptomes of ~50K cells, the largest single-cell dataset on TIBs to date. We interrogated the spatial atlas of TIBs at unprecedented resolution exploring their transcriptional states, differentiation and maturation status, geospatial characteristics, clonotypic properties, as well as cellular interactions and co-localization patterns with other TME cell populations. We also correlated TIB characteristics with major clinicopathological features including smoking, driver gene mutation and tumor stage, and assessed their clinical significance such as patient survival and response to immunotherapy. This study provides a much needed and detailed understanding of the prevailing nature and functional phenotypes of TIBs in early LUAD development.
RESULTS
Single-cell profiling of TIBs in LUADs and matched multi-region normal lung tissues
We performed integrative scRNA-seq and scBCR-seq on cells obtained from 16 early-stage LUADs and 47 matched multi-region normal lung tissues with varying spatial proximities from the tumors (i.e., adjacent, 0.5cm from tumor edge; intermediate, 3–5cm from tumor edge; distant, periphery of the lobe) (Fig.1A; Supplementary Table S1). Fractions of B lineage subsets among EPCAM-negative (TME) cells were markedly enriched in LUADs compared to their matched normal lung tissues, especially in smokers (Fig.1B; p = 1.5×10−8). To validate this finding and further examine spatial distribution of TIBs, we performed multiplexed fluorescence (mIF) with a panel of 8 markers on 20 available tissues from 5 of the 16 patients (see Methods). Lymphoid aggregates (LAs) were detected in tumor samples from all patients including the tumor compartment and stroma (Fig.1C), as well as the non-tumoral stromal compartment from 2 patients. Consistently, the total intensities of B cells were significantly higher in tumors compared to their matched normal tissues (Fig.1D; p = 6.0×10−4).
Figure 1. Single-cell profiling of B lineage cells in tumor and matched multi-regional normal lung tissues in 63 samples from 16 patients with early-stage LUADs.
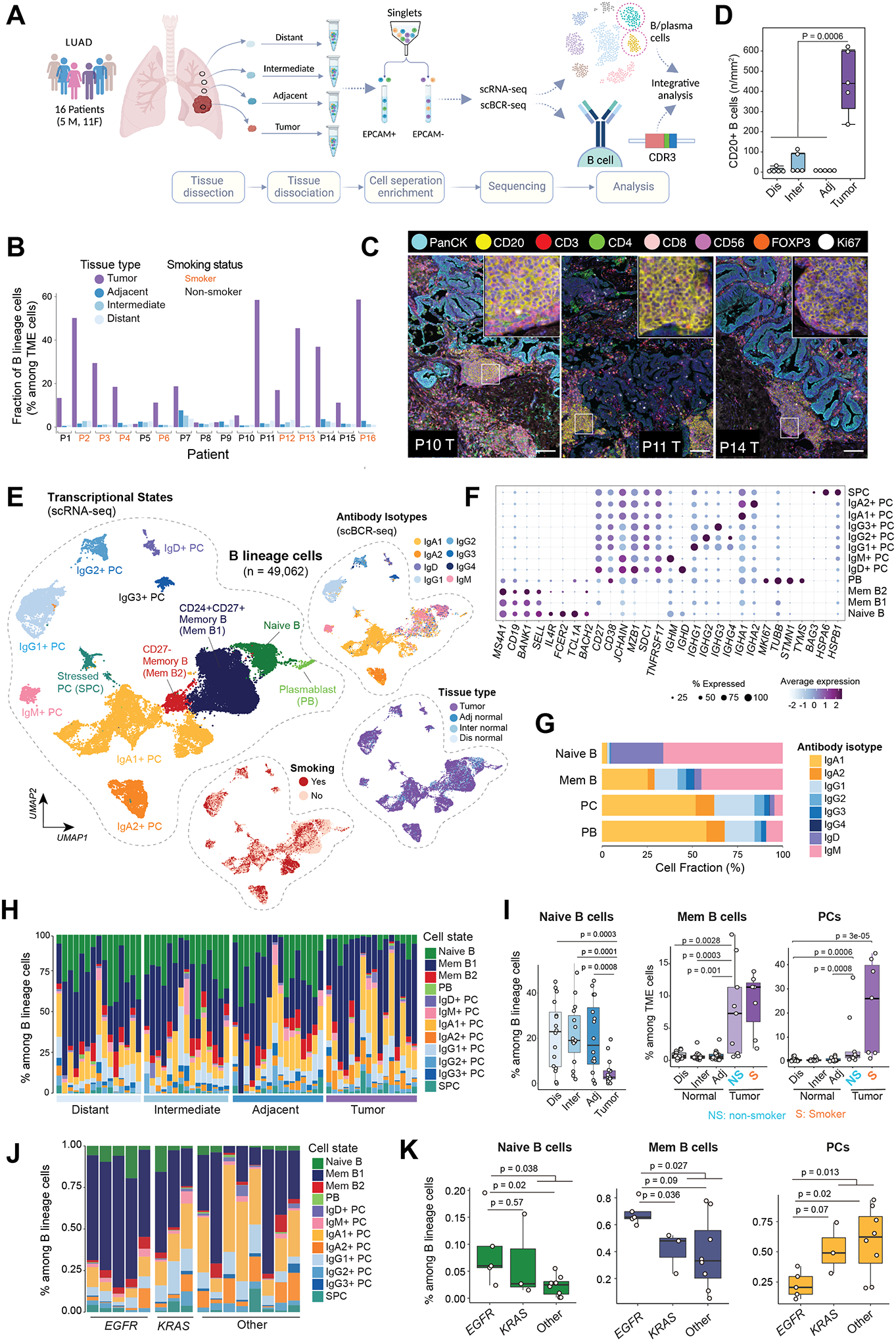
(A) A schematic view of the experimental design, created with BioRender.com. Single-cell RNA sequencing (scRNA-seq) and paired B cell receptor sequencing (scBCR-seq) were performed on EPCAM-negative immune and stromal cell compartments in the tumor microenvironment (TME). Single-cell data generated on B lineage cells were extracted and included in this study. (B) Bar graph showing increased fractions of B lineage cells (among TME cells, i.e., EPCAM-negative cells) in tumor tissues when compared to matched multi-regional normal lung tissues (adjacent, intermediate, distant normal) collected from the same patient (p=1.5e-08, the Mann-Whitney test). (C) Representative images of multiplex immunofluorescence (mIF) showing CD20+ lymphoid aggregates adjacent to areas of PanCK+ tumor cells. mIF was done with a panel of 8 markers on available tissues (n = 20) from 5 of the 16 patients. Scale bar: 100 μm. The inserts are zoomed-in view of CD20+ lymphoid aggregates. (D) Quantification of CD20+ B cells in tumor and multi-region normal lung tissue using mIF. (E) Uniform manifold approximation and projection (UMAP) embeddings of the 49,062 B lineage cells that passed quality control. Cells are color coded by their inferred cell types/states based on transcriptional profiles (left), antibody isotypes using scBCR-seq data (top right), their corresponding spatial location (middle right), and patient smoking status (bottom right). PC, plasma cell; Mem, memory; PB, plasmablast; SPC, stressed plasma cell; adj, adjacent normal; inter, intermediate normal; dis normal, distant normal (same as in panel A). (F) Bubble plot showing proportions and average expression levels of select marker genes for 12 B cell and PC clusters as defined in panel E. More information on cluster-specific marker genes are provided in the Supplementary Table S2. Bubble size indicates the percentage of cells expressing a specific gene in a given cluster, and the color depicts the average expression level of the gene in a cluster of interest and relative to all other cell clusters. (G) Bar graph showing the cellular composition of antibody isotypes in 4 major cell subsets. (H) Bar graph showing the landscape of B lineage cell compositions across all tumor and normal samples grouped by their spatially defined locations (with increasing proximity to tumor from left to right). (I) Boxplot displaying decreased relative fractions of naïve B cell (left, among B lineage cells) and increased fractions of memory B cells (middle) and plasma cells (right) among TME cells in tumor tissues when compared to multi-region normal lung tissues. In the two plots on the right, tumor samples were stratified by patient’s smoking status. NS, non-smoker; S, smoker. P values were determined by Mann-Whitney tests. (J) Bar graph showing the landscape of B lineage cell compositions across all tumor (LUAD) samples grouped by mutation status of LUAD driver gene (e.g., KRAS, EGFR). (K) Boxplots comparing the relative fractions of major B cell subtypes within tumor samples grouped by driver gene mutations. KRAS and EGFR drive mutations were identified using whole-exome sequencing. Mem B, memory B cells; PCs, plasma cells.
We then sought to define the various transcriptional states and BCR isotypes of TIBs. After rigorous filtering (see Methods), a total of 49,062 B lineage cells with scRNA-seq data were retained for subsequent analyses. Unsupervised clustering revealed 12 different cell clusters (Fig.1E–F; Supplementary Table S2) including one each naïve B, plasmablast (PB), stressed PC (SPC) cluster, two switched memory B and seven PC clusters. Naïve B cells highly expressed MS4A1 (CD20), IGHD, IL4R and FCER2 (CD23), without detectable expression of CD27 and CD38 (Fig.1F). The two switched memory B cell clusters differed in their expression of CD27 and CD24. Both naïve and memory B cells showed high expression of MHC class II genes, as expected. PCs exhibited distinct profiles such as high expression of CD38, SDC1 (CD138), TNFRSF17 (BCMA), MZB1, JCHAIN, immunoglobin (Ig) isotype genes with no/low expression of MHC class II genes (Fig.1F and S1A–B). Seven PC clusters were characterized by differential expression of main classes and subclasses of immunoglobin isotype genes (Fig.1E; Supplementary Table S2). Cells of the PB cluster were CD38+CD138+IgD−CD27+ and they showed relatively higher expression of MHC class II genes when compared to PCs, and the highest expression of MKI67 as well as other cell cycle-related genes (Fig.1F). We also identified a rare IgD+ PC cluster which was previously reported to be associated with exposure to common bacteria and airborne antigens (8). For cells with scBCR-seq data (n = 72,949), we defined Ig heavy-chain (IgH) isotypes based on the sequences of Ig constant regions. Overall, the antibody isotypes detected by scBCR-seq agreed well with that inferred from scRNA-seq (Fig.1E, top right). Specifically, the naïve B cell cluster was dominated by IgM and IgD isotypes, memory B cell clusters showed various different isotypes, and PC clusters showed a high enrichment of their corresponding IgH isotypes (Fig.1E, G). Like PCs, the PB cluster was dominated by IgA1/2 and IgG1/2 isotypes (Fig.1G).
Markedly increased TIBs in LUADs and highly abundant PCs particularly in smokers
We then examined the abundance and cellular compositions of B lineage cells in LUADs and matched normal lung tissues (Fig.1H and S1C). The relative fractions of naïve B cells among total B lineage cells were significantly decreased with increasing proximity to the tumors. In contrast, we noted markedly elevated fractions of memory B cells and PCs in LUADs (Fig. 1I), with the latter especially prominent in smokers relative to non-smokers (p = 0.07). There was a consistent trend towards increased fractions of IgA+ and IgG+ PCs in LUADs of smokers, albeit not reaching statistical significance (Fig. S1D). We next integrated TIB characteristics with genomic data including tumor mutation burden (TMB) and driver mutations identified using whole-exome sequencing (9). No significant differences in TIB compositions were observed between LUADs with high and low TMB. Of note, EGFR-mutant LUADs had significantly lower fractions of PCs and higher fractions of naïve and memory B cells (among total TIBs) compared to KRAS-mutant or KRAS/EGFR double wild-type LUADs (Fig.1J–K).
Next, we sought to validate our observations in independent patient cohorts, first by analyzing a large public scRNA-seq dataset from Kim N et al. which comprised primary LUADs, lymph node and brain metastases, together with normal lung and lymph node tissues obtained from 44 LUAD patients (10). In line with our cohort, fractions of memory B cells and PCs were significantly increased in primary LUADs compared to normal lung tissues (Fig.2A and S2A). We next analyzed another scRNA-seq cohort composed of 30 early-stage LUADs from Leader et al. (11). Very much in line with our cohort, fractions of PCs were significantly increased in smokers versus non-smokers (Fig.2B). We also performed immune deconvolution analysis of bulk RNA-seq data from The Cancer Genome Atlas (TCGA) LUAD cohort (12) using CIBERSORTx (13). Corroboratively, LUADs displayed lower fractions of naïve B cells (Fig.S2B), whereas the fractions of PCs were markedly increased in ever-smoker versus non-smoker LUADs (Fig.2C, left). Of note, PC fractions were markedly increased in smoker but not non-smoker LUADs and the magnitude of change was greater in current or reformed smokers who quit smoking ≤15 years than those reformed >15 years, when compared to their matched normal lung tissues (Fig.2C, right). These observations indicate the potential roles of PCs in the immunopathology of LUAD, particularly in smokers.
Figure 2. PC signature in LUADs predicts better survival and response to immunotherapy.
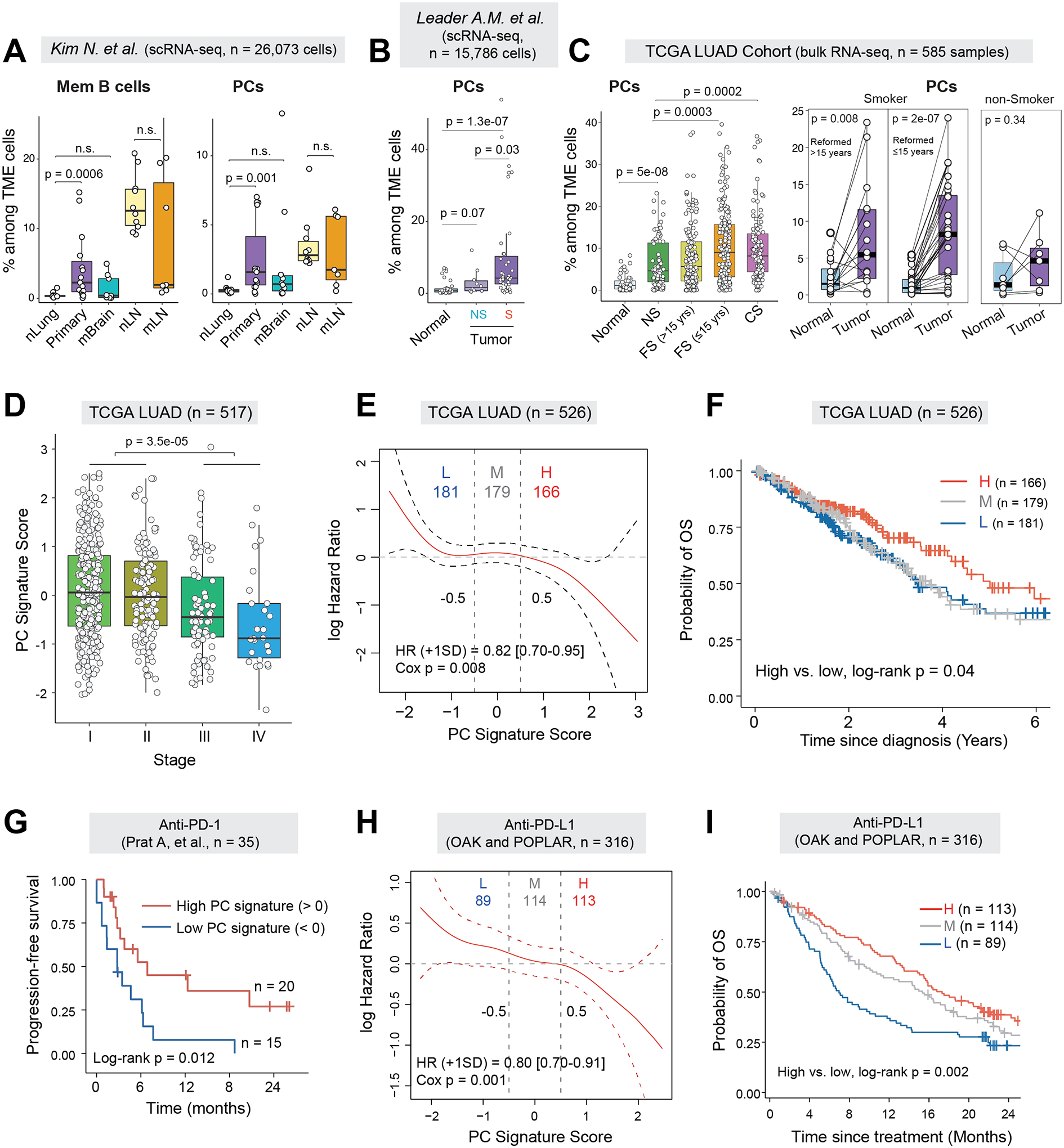
(A) Boxplots comparing cellular fractions of memory B cells (left) and plasma cells (right) among total TME cells in a public scRNA-seq dataset from Kim N. et al (10). P values were determined by Mann-Whitney tests. nLung, normal lung; Primary, primary LUAD; mBrain, brain metastases; nLN, normal lymph node (LN); mLN, lymph node metastases; n.s., not statistically significant. (B) Box plots showing the relative fractions of PCs among the TME cells in the NSCLC scRNA-seq dataset from Leader AM et.al. Only LUAD patients were included in analysis. (C) Boxplots comparing the estimated cell fractions of PCs between tumor and normal lung tissues from the TCGA LUAD cohort (left) and in a subset of patients with tumor-normal pairs (n = 52) (right). Samples were stratified by patient smoking status. NS, non-smoker; FS, former smoker; CS, current smoker. (D) Boxplot showing the expression of PC signature scores across different pathological stages of LUAD in the TCGA LUAD cohort. (E) Estimates for the dependence of all-time risk of death on plasma cell abundance in tumor (expression of plasma cell signature) in TCGA LUAD cohort. The solid curve (red) was generated using the Cox proportional hazards model and the dotted curves indicate the 95% CI of log hazard ratio. (F) Kaplan-Meier curves displaying differences in overall survival (OS) probability between TCGA LUAD patients whose tumors had high (H), medium (M) or low (L) levels of PC gene signature. (G) Kaplan-Meier curves displaying differences in progression-free survival (PFS) between patients whose pre-treatment tumors had high or low levels of PC gene signature in the Prat A et al. cohort receiving anti-PD-1 treatment (14). (H) Estimates for the dependence of all-time risk of death on plasma cell abundance in tumor of combined cohorts of OAK and POPLAR receiving anti-PD-L1 treatment. The solid curve (red) was generated using the Cox proportional hazards model and the dotted curves indicate the 95% CI of log hazard ratio. (I) Kaplan-Meier curves displaying differences in OS probability between anti-PD-L1 treated patients whose pre-treatment tumors had high, medium and low levels of PC gene signature in the combined cohorts.
High PC signature in LUADs predicts better survival and response to immunotherapy
To assess the clinical significance of TIBs, we first constructed cell type-specific gene signatures for major TIB subsets using our scRNA-seq data in the manner we previously described (6) and confirmed its reliability in deconvolution analysis (Fig.S2C–D; Supplementary Table S3; Methods). We next analyzed the bulk expression data from the TCGA LUAD cohort and found that PC signature scores were gradually decreased with advanced pathological stages (Fig.2D). Both the Cox proportional hazards model and the Kaplan-Meier model revealed that relatively high PC signature scores were significantly associated with better overall survival (OS) (Fig.2E–F). Among the 22 immune cell subsets evaluated, we found that PC signature was distinctively and significantly associated with better OS in the TCGA LUAD cohort (Fig. S3A).
We then assessed the correlation of PC signature with patient response to immunotherapy by analyzing public datasets from 4 reported clinical trials (14–16). Analysis of the Prat cohort (14) with 35 patients and the combined cohort from Prat et al. (14) and Hwang et al. (15) with 56 NSCLC patients treated with anti-PD-1 therapy showed that relatively higher PC signature in pre-treatment tumors was associated with better response to anti-PD-1 therapy, a durable clinical benefit (DCB) and significantly better progression-free survival (PFS) (Fig.2G and Fig.S3B–D). Likewise, analysis of the large LUAD cohorts (n = 316 patients) from POPLAR (17) and OAK (18), two trials using atezolizumab for PD-L1 blockade, showed very consistent results. Relatively higher expression of the PC signature at baseline was significantly associated with better response to anti-PD-L1 therapy (Fig.S3E) and improved OS (Fig.2H–I) as reported by the original study (16). We also observed significantly higher expression of PC signature in patients with DCB when compared to those with no durable benefit (NDB) (Fig.S3F). Moreover, we found that the relative abundance of IgA+ cells but not IgG+ cells, as measured by the expression of IGHA/IGHG genes relative to PC signature genes, were significantly lower in responders versus non-responders (Fig.S3G).
Enrichment of highly differentiated PCs in LUADs and long-lived PCs in smokers
We next aimed to perform in-depth analysis of the differentiation states of PCs. First, we reconstructed the developmental trajectory of PCs using Monocle 3 (19) and pseudotime analysis revealed various different states of PC differentiation including long-lived PCs, short-lived PCs, and PBs (Fig.3A). Long-lived PCs were characterized by high expression of STAT3, IKZF3 (Aiolos) and CD37 (Fig.3B), consistent with their functions in supporting various aspects of PC longevity (20–22). The expression levels of IgA/IgG transcripts were also higher (Fig.3B and S4A), consistent with the established antibody production in long-lived PCs. In addition, and in support of their antibody secreting functions (23), most long-lived PCs lost expression of membrane Ig variable (IgV) genes and BCR (Fig.3C, left). The cellular differentiation states inferred by CytoTRACE (24) was in line with the Monocle 3 results (Fig.3A) and we observed gradually decreased CytoTRACE scores, i.e., increased differentiation, as the pseudotime progressed (Fig.3C, middle). Intriguingly, fully differentiated, long-lived PCs were prominently enriched in LUADs of smokers versus non-smokers (Fig.3C, right, p < 0.0001).
Figure 3. Inference of cell differentiation states and antibody isotypes of PCs and memory B cells in LUADs and matched normal lung tissues.
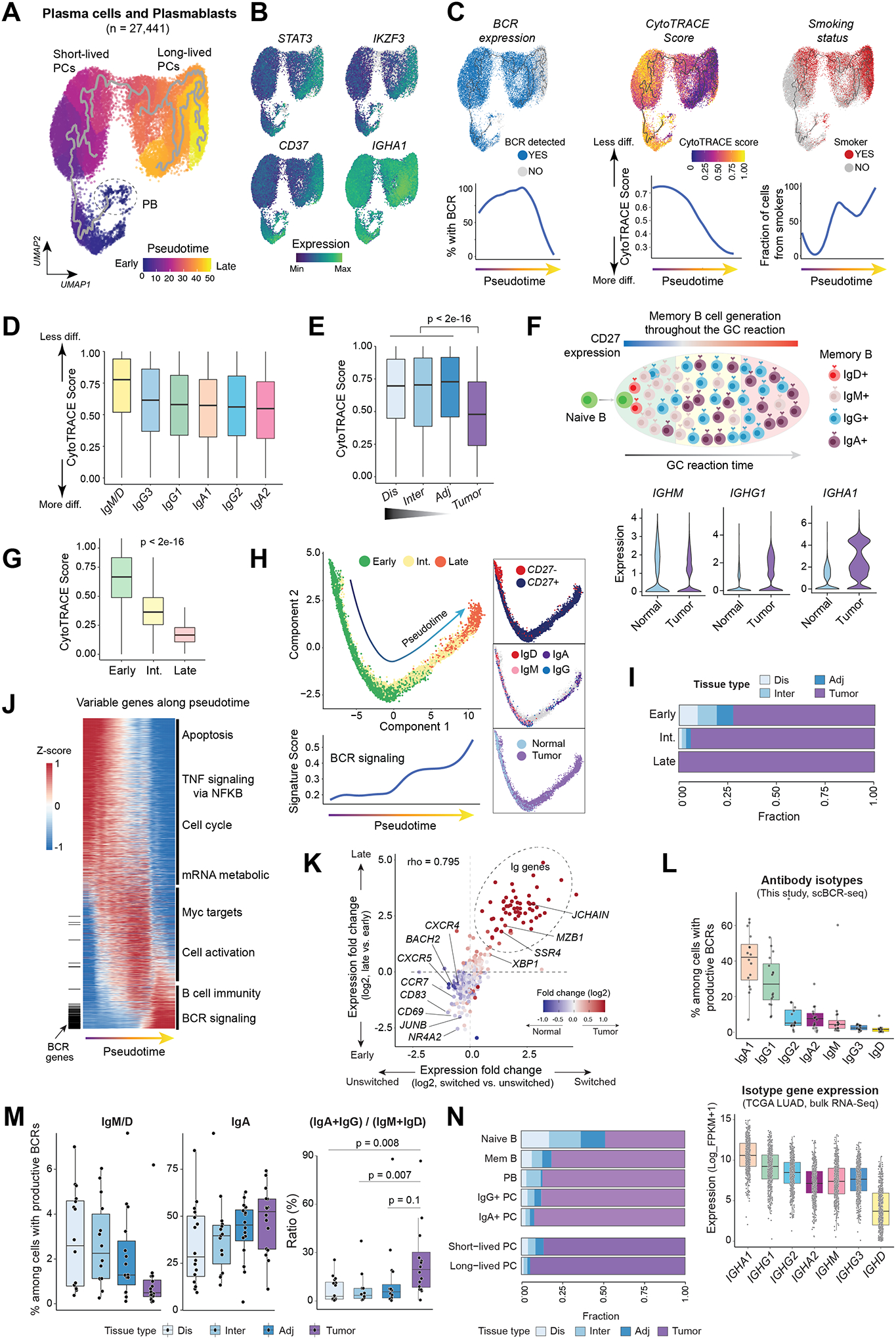
(A) Monocle 3 pseudotime trajectory analysis of PCs revealed different states of PC differentiation and maturation. PB, plasmablasts. UMAP is colored by inferred pseudotime. (B) The same UMAP as in (A) but with cells color-coded according to gene expression levels of four markers associated with long-lived PCs. (C) (top) The same UMAP as in (A) but with cells color-coded (from left to right) by their BCR expression, corresponding CytoTRACE score, and patient smoking status, respectively. CytoTRACE scores were computed using scRNA-seq data and BCR expression was dichotomized based on the presence of productive BCR clonotype by analyzing scBCR-seq data. Loess smooth curves (bottom) showing (from left to right) fractions of cells with productive BCR, the distribution of CytoTRACE scores, and fractions of smokers, respectively, by pseudotime. Less diff., less differentiated; More diff., more differentiated. (D) Box plots displaying the distribution of CytoTRACE scores across PCs with different antibody isotypes defined using paired scBCR-seq and scRNA-seq data. (E) Comparison of CytoTRACE scores across LUADs and multi-region normal tissues with differing spatial proximities from the tumors (as in Fig. 1A). P value was determined by Mann-Whitney test. (F) A schematic illustration (top) of memory B cell generation in a germinal center (GC) and violin plots (bottom) showing differences in the expression levels of IgH isotypes between LUADs and normal lung tissues. (G) The distribution of CytoTRACE scores in memory B cells at 3 different developmental stages. P value was calculated using the Mann-Whitney test. Int., intermediate. (H) Monocle 3 trajectory reconstruction analysis of B cell differentiation (left). Cells are color-coded by developmental stage, CD27 expression, IgH isotypes, or tissue type (right, top to bottom). A regression line was fitted along pseudotime by a generalized additive model for signature scores of BCR signaling (bottom left). (I) Bar graph showing the difference in tissue compositions across memory B cells at 3 different developmental stages. (J) Pseudotime heatmap ordering of the top 3,000 highly variable genes along the trajectory of memory B cell differentiation. Locations of BCR genes are indicated as a segment (in black) on the left and enriched pathways are labelled on the right. (K) Scatter plot showing gene expression fold change (log2) between late- and early-stage memory B cells (y axis) against the corresponding values of switched and unswitched memory B cells (x axis). Each dot indicates a gene and is color-coded by its corresponding expression fold change between LUADs and normal lung tissues. (L) (top) Box plot showing cellular fractions of antibody isotypes among all B lineage cells with productive V(D)J rearrangements based on paired scBCR-seq data. Each dot indicates a LUAD sample. (bottom) Relative abundances of immunoglobin heavy chain (IgH) isotypes in the TCGA LUAD cohort inferred from bulk RNA-seq data based on the normalized expression levels of each IgH gene. (M) Cellular fractions of IgM+/D+, IgA+ cells (left and middle, respectively) and ratios of IgA+IgG to IgM+IgD abundance (right) across tumor and normal samples. P values were calculated using paired t-tests. (N) Bar graph showing differences in tissue compositions across 7 defined B cell and PC subsets.
When investigating PCs by antibody subclass, IgM/IgD+ PCs were the least differentiated, followed by IgG3+, IgG1+, IgA1+, IgG2+, and finally IgA2+ PCs which had the lowest scores (i.e., most differentiated) (Fig.3D), consistent with their exact order during isotype switching. We also noted increased fractions of IgA1/IgA2+ PCs in LUADs (Fig.S4B, p < 2×10−16) and PCs from LUADs were significantly more differentiated relative to those from normal lung tissues (Fig.3E). Consistent with CytoTRACE-based differentiation inference, we noted that the number of genes expressed in LUAD PCs (but not other TME cells) was significantly lower relative to those of normal lung tissues (Fig.S4C; p < 2×10−16). Notably, we corroborated our findings in two independent scRNA-seq datasets from Lambrechts et al. (25) and Kim et al. (10) showing enrichment of more differentiated PCs in LUADs (Fig.S4D).
Memory B cells in LUADs are more frequently class-switched and at late-GC stage
Memory B cells are generated during the germinal center (GC) reaction, in parallel to PC differentiation (Fig.3F, top), and their differentiation is tightly controlled at the transcriptional level. Relative to matched normal lung tissues, we observed dramatically increased expression of IgA/IgG genes in memory B cells from tumor tissues (Fig.3F, bottom and S5A), indicating that memory B cells in LUADs are mainly class-switched and at late-GC stage. To further investigate the maturation status and transcriptional heterogeneity of memory B cells in LUADs, we defined three developmental stages−early-, intermediate-, and late-GC stages, based on their expression of IgA/IgG genes (Fig.S5B), followed by CytoTRACE cell differentiation analysis. Consistently, compared to memory B cells at early-GC stage, cells at late-GC stage were likely more differentiated, indicated by significantly lower CytoTRACE scores (Fig.3G). Additionally, memory B cells early along the trajectory were mostly CD27−, unswitched, had the lowest BCR signature scores, and were mostly derived from normal lung tissues, whereas late-trajectory cells were CD27+, switched, had the highest BCR signature scores, and were nearly exclusive to tumor tissues (Fig.3H–I).
Analysis of expression dynamics of the top 3,000 variable genes along the trajectory revealed distinct biological processes associated with memory B cell differentiation (Fig.3J; Supplementary Table S4). For example, early (relative to inferred pseudotime) upregulated genes were enriched for apoptosis, TNFα signaling via NFκB, and cell cycle pathways, whereas late upregulated genes included many IgV genes and were enriched for BCR signaling. We also found upregulated genes (e.g., XBP1, MZB1, and JCHAIN) and downregulated genes (e.g., CXCR5, CCR7, CD69 and BACH2) in late- relative to early-stage memory B cells, consistent with previously reported roles of these genes in B cell differentiation (26–29) (Fig.3K and S5C; Supplementary Table S5). In addition, the upregulated differentially expressed genes (DEGs) in tumor tissues (vs. matched normal lung), late-stage (vs. early-stage) and switched (vs. unswitched) memory B cells were largely overlapping (Fig.3K; Supplementary Table S5), implying that memory B cells in LUADs were likely resulting at a late stage along the differentiation trajectory. In agreement with this notion, IgA1+ and IgG1+ cells were the most abundant class-switched subsets indicated by scBCR-seq (Fig.3L, top), and this observation was confirmed in the large TCGA LUAD cohort based on isotype-specific gene expression (Fig.3L, bottom). In addition, we observed gradually decreased fractions of IgM+/IgD+ cells and increased fractions of IgA+ cells with increasing proximity to LUADs and consequently, significantly increased ratios of switched to unswitched isotypes in tumor tissues (Fig.3M). Our observations suggest that B cells and PCs with various differentiation states exhibit distinct transcriptional and phenotypic profiles as well as geospatial heterogeneity in the lung (Fig.3I and 3N).
Higher BCR clonotype diversity and lower clonality in LUADs, smokers, and with increasing pathological stage
To better understand the immunoglobin repertoires in LUADs and neighboring tissues, we analyzed the paired scBCR-seq data generated on the same cDNA libraries and identified 72,949 cells with productive VDJ rearrangements. Relative to normal lung tissues, we observed markedly increased numbers of unique BCR clonotypes, i.e., higher diversity in their BCR repertoires, in tumor tissues (Fig.4A and S6A). We detected increased numbers of expanded BCR clonotypes in tumor tissues but at relatively lower clonal frequencies, i.e., lower unevenness in clonal size distributions of the BCR repertoires than in normal tissues where a small number of expanded clones dominated the repertoires (Fig.4B, left). Likewise, we also observed lower BCR clonal unevenness in smokers versus non-smokers (Fig.4B, middle) and with increasing pathologic stage (Fig.4B, right).
Figure 4. Characterization of BCR clonotype diversity, clonality, and somatic hypermutations in lung tissues based on proximity from primary LUADs.
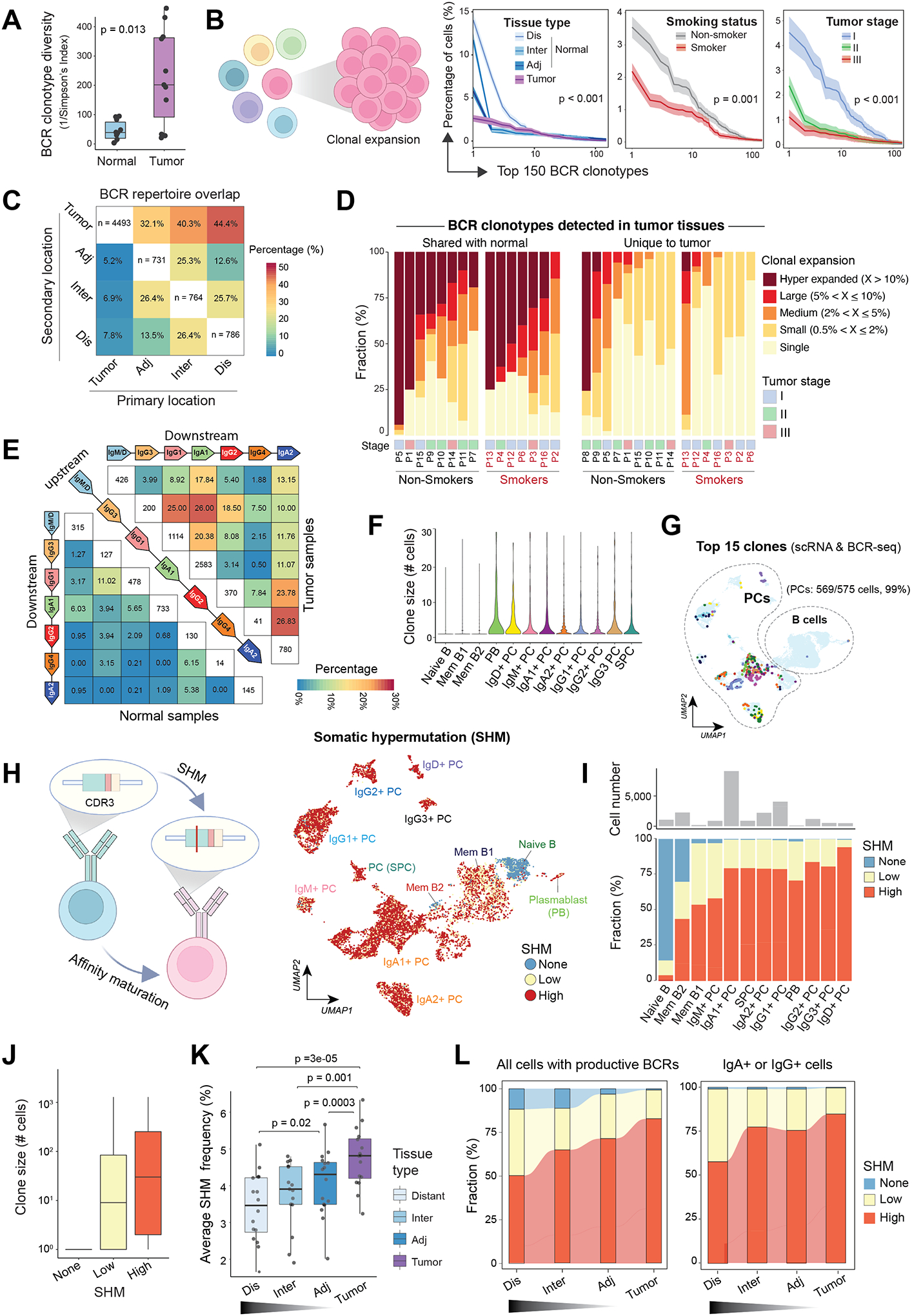
(A) Boxplots showing differences in B cell receptor (BCR) clonotype diversity scores between LUADs and matched normal lung tissues. Each dot indicates a LUAD patient. P value was determined by the Mann-Whitney test. (B) (left) A schematic illustration of B cell clonal expansion. (right) The clone size distribution of BCR repertoire across groups of different (from left to right) tissue types (location), smoking status, or tumor stages. The top 150 BCR clonotypes are shown. Kolmogorov–Smirnov tests were used for pairwise comparisons. (C) BCR repertoire overlap across spatially defined locations. A BCR clonotype was defined as shared if it was detected in B cells from two or more different samples collected from the same patient. Heatmap showing how the BCR repertoire in samples from a given geospatial location (i.e., the primary location on x axis) overlap with that from another location (i.e., the secondary location on y axis). (D) The landscape of BCR clonal expansion in all patients, stratified by their tissue specificity, i.e., whether a BCR clonotype detected in LUADs shared with their matched normal lung (left) or unique to tumor tissues (right). BCR clonotypes were classified into 5 categories, namely, hyper-expanded, large, medium, small, and single based on their corresponding clonotype size, and their sample-level compositions are shown in bar graphs. Patient number, smoking status, and tumor stage are annotated at the bottom. (E) BCR repertoire overlap across groups of cells with different antibody isotypes for LUAD (top right) and normal lung tissues (bottom left). Groups are ordered according to the genomic coordinates (5’ to 3’) of Ig subclass (i.e., IgG3, IgG1, IgA1, IgG2, IgG4 and IgA2) coding genes. Heatmap showing the fractions of BCR clonotypes belonging to an upstream isotype (each row) that are shared with a downstream isotype (columns). (F) Violin plot showing the distribution of BCR clonotype size across 12 B cell and PC states as defined in Figure 1E. (G) The same UMAP as in Figure 1E showing B cells and PCs with the 15 most expanded BCR clonotypes (same colors as in panel F). Cells with both scRNA-seq and scBCR-seq data were analyzed. (H) (left) A schematic illustration of BCR somatic hypermutation (SHM). (right) The same UMAP as in Figure 1E but cells are color-coded by levels of SHM in their corresponding BCRs. None, germline sequence with no detected SHM. Cells with both scRNA-seq and scBCR-seq data are shown. (I) Distribution of SHM frequencies across 12 B cell and PC subsets as defined in Figure 1E. (J) Box plot showing the distribution of BCR clonotypes size across 3 groups of cells with different SHM frequencies in their corresponding BCRs. (K) Box plot showing average SHM frequencies in tumor and normal tissues with spatially defined locations. P values were determined by paired t tests. (L) Alluvial plots showing the distribution of cells with different SHM frequencies across spatially defined locations, among all cells with productive BCRs based on scBCR-seq data (left) or among IgA+ or IgG+ cells only (right). Panels B and H were created with BioRender.com.
We next aimed to interrogate whether the geospatial distribution of BCR clonotypes in tumor and normal lung tissues could inform of the trafficking of B cells. We measured how the BCR repertoire in a sample from a given geospatial location (e.g., LUAD) overlaps with that from another location (e.g., distant normal lung). On average, 38.9% of BCR clonotypes from normal lung tissues were shared with that from LUADs, contrary to BCR clonotypes from LUADs which were largely unique with a much lower proportion of shared clones (6.6%) with their neighboring tissues (Fig.4C). Notably, when splitting BCR clonotypes detected in LUADs based on whether or not they were unique to tumors, we observed that BCR clonotypes that were shared with normal tissues were highly expanded, in contrast to tumor-unique clonotypes which were less expanded and largely composed of small clones and singletons, particularly in smokers (Fig.4D).
Distinct profiles of Ig class switching, VDJ gene usage and expanded PCs in tumors
We next explored the overlap in BCR repertoires between clonotypes of an upstream isotype with its downstream isotype following the genomic coordinates (5’ to 3’) of Ig subclass (i.e., IgG3, IgG1, IgA1, IgG2, IgG4 and IgA2) coding genes (Fig.4E). Relative to normal lung tissues, an upstream antibody isotype had increased percentages of shared clonotypes with its direct downstream isotype in LUADs, implying more frequent Ig class switching. We also examined V(D)J gene usage and noted LUAD enrichment of IGHV genes and IGHV-J pairs that are infrequently used in cells from normal lung tissues (Fig. S6B–C), suggesting that B cells in LUADs may have undergone unique V(D)J rearrangements. Lastly, by integrating the scRNA-seq and scBCR-seq data, we found that cells of most expanded clones were PCs (Fig.4F). The top 15 most expanded clones were nearly exclusively mapped to clusters of PCs (Fig. 4G and S6D). Lastly, compared to randomly grouped B cells and size-controlled clones, B cells of the same clone were more likely to share a common transcriptional state, antibody isotype, or spatial location (Fig. S6E).
Geospatial characteristics of BCR somatic hypermutation (SHM) in LUADs
The affinity maturation of B cells in GCs is a result of iterative rounds of clonal expansion coupled with somatic hypermutation (SHM), followed by affinity-based selection of antigen-specific B cells (30) (Fig. 4H, left). Typically, high SHM frequency indicates strong antigen-specific affinity (31). We quantified SHM levels using scBCR-seq data (see Methods) and based on which, we classified cells into 4 groups: none- (no SHM), low- (mutation frequency ≤3%) and high-SHM (>3%). As expected, we observed no SHM in BCRs of naïve B cells, low or medium level of SHM in BCRs of memory B cells, and high level of SHM in BCRs of PCs (Fig. 4H, right; 4I and S7A). Memory B cells at the late-GC stage displayed higher SHM frequencies relative to those at early- or intermediate-GC stages (Fig. S7B–E). The clonotype size was relatively larger in cells with high SHM frequency (Fig. 4J). The rare subset of IgD+ PC exhibited the highest SHM frequency (Fig. 4I and S7A), consistent with the hypothesis that IgD+ B cells develop in super antigen-driven GC reactions (32). Notably, we observed gradually increased SHM frequencies with increasing proximity to tumors and in both switched and unswitched cells (Fig. 4K–L and S7F). Further analysis of expanded clones (n>10 cells) revealed higher SHM frequencies in BCRs of tumor-resident clones relative to normal-resident or shared clones (Fig. S7G). In addition, we noted lower BCR SHM frequencies in PCs of EGFR-mutant LUADs (Fig. S7H–I). Consistently, quantification of tissue enrichment of all cells with scBCR-seq data based on the ratio of observed to expected cell number (Ro/e) showed that the none- and low-SHM cells were enriched in EGFR-mutant LUADs (Fig. S7J).
The B cell chemotactic CXCL13-CXCR5 axis in early LUAD development
To understand the recruitment of TIBs in LUADs, we examined the expression of known attractants of B cells including CXCL13, which is essential for B cell recruitment and formation of tertiary lymphoid structures (TLSs) (33,34) (Fig. 5A) and other chemokines and their receptors including CCR7-CCL19/CCL21, CXCR4-CXCL12, CXCR3-CXCL9/10/11, and CCR6-CCL20. Among them, the CXCL13-CXCR5 chemokine axis appeared to be predominant in early-stage LUADs. Both CXCL13+ T cells and CXCR5+ B cells among TME cells were highly enriched in LUADs (Fig. 5B) and we noted significantly increased relative proportions of CXCL13+ cells among total T cells in tumors compared to normal lung tissues in this cohort (Fig. S8A) and the finding was corroborated in 3 public scRNA-seq cohorts (Fig. S8B–D) (10,11,35). The expression of CXCL13 was also higher in tumors than in normal lung tissues of TCGA LUAD cohort (Fig. S8E).
Figure 5. Interplay between TIBs and other TME cells in LUADs.
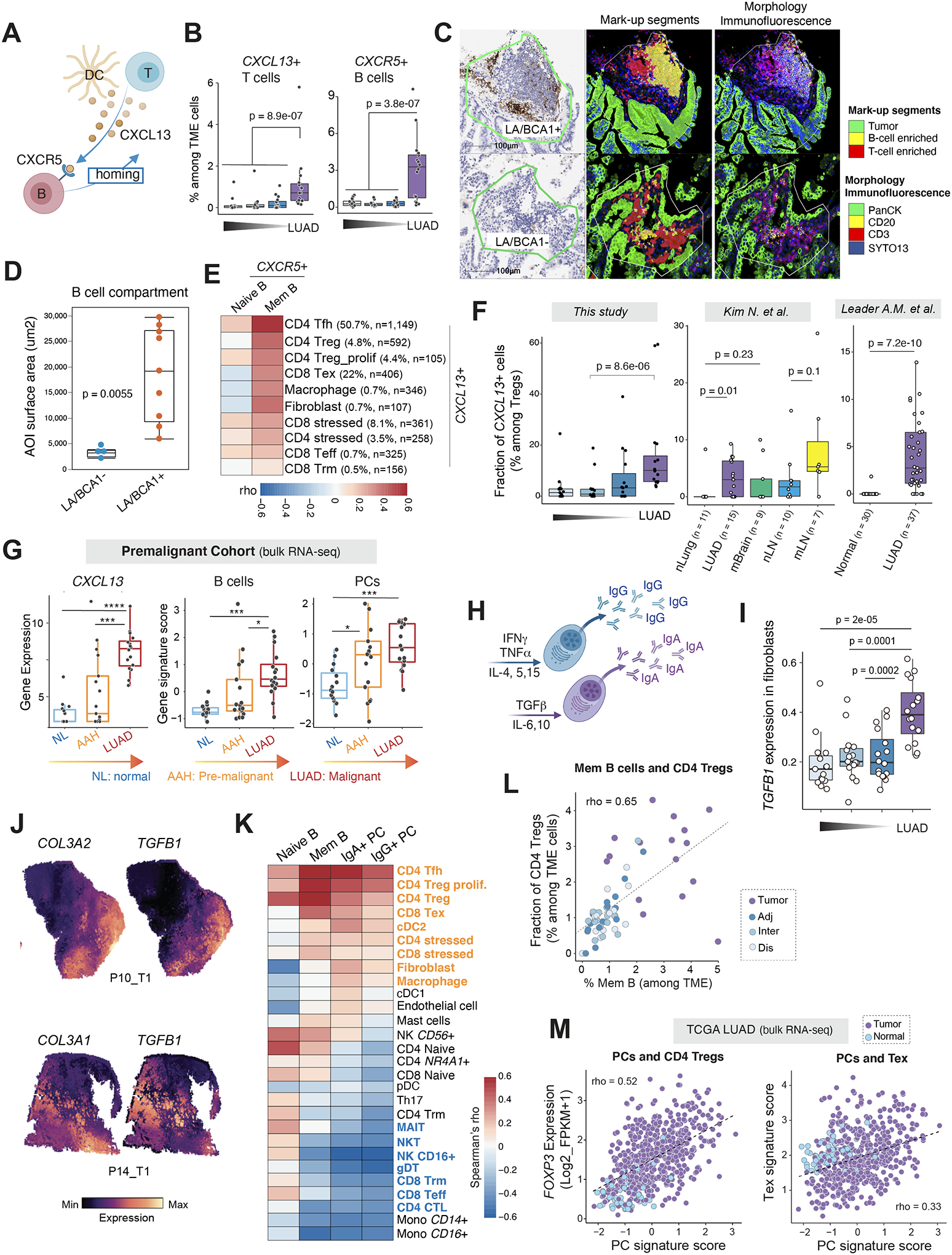
(A) A schematic illustration of B cell recruitment via the CXCL13-CXCR5 axis. (B) Fractions of CXCL13+ T cells and CXCR5+ B lineage cells, respectively, among all TME cells in LUADs and the multi-region normal lung tissues with spatially defined locations. (C) Representative DSP images showing AOI segmentation strategy. Each ROI was segmented into 3 AOIs: B cell, T cell or tumor cell-enriched based on expression of the morphology markers. ROI, region of interest; AOI, area of illumination. (D) Comparative analysis of B cell abundance between CXCL13 (BCA1)-positive and -negative lymphoid aggregates (LAs). B cell abundance in each ROI was quantified by measuring the AOI surface area of the B cell compartment. P value was calculated by the Wilcoxon test. (E) Co-occurrence relationships between CXCR5+ naïve and memory B cells and other CXCL13+ TME subsets identified using scRNA-seq. Spearman’s correlation analysis was used to statistically evaluate co-occurrence relationships of different TME cell subsets based on their corresponding cell fractions. A heatmap was plotted based on the Spearman’s correlation coefficient (rho). Ten other TME cell subsets with >100 cells or with a fraction of CXCL13-expressing cells > 0.5% were included in the heatmap. Mem, memory. Tfh, T follicular helper cells; Treg, regulatory T cells; Tex, exhausted T cells; Teff, effector T cells; Trm, resident memory T cells. (F) Fractions of CXCL13+ cells among Tregs in this study (left) and two public scRNA-seq datasets (middle and right). (G) Gene expression levels of CXCL13 and gene signature scores of B cells and PCs in a premalignant cohort with bulk RNA-seq data. P values were determined by the Mann-Whitney test. ***, P < 0.001; ****, P < 0.0001. (H) A schematic illustration of cytokines regulating PC isotype switching and antibody secretion. (I) Increased TGFB1 expression in tumor-associated fibroblasts. Wilcoxon test was used to calculate the p values. (J) Spatial transcriptomics (ST)-based mapping of TGFB1-expressing to fibroblast-enriched regions from 2 LUAD patients using the Visium platform (10X Genomics). (K) Co-occurrence relationships between 4 TIB subsets and 27 other TME cell populations identified using scRNA-seq. Spearman’s correlation analysis was used to statistically evaluate co-occurrence relationships of different TME cell subsets based on their corresponding cell fractions. Heatmap was plotted based on the Spearman’s correlation coefficient (rho). (L) Scatter plot displaying the correlation between the fractions of CD4 Tregs and that of memory B cells in the LUADs and normal lung tissues from all 16 patients in this scRNA-seq cohort. Spearman’s correlation independent of logarithmic transformation is shown. Samples are labelled by their spatial locations. (M) Scatter plots displaying the correlation between the gene signature scores of PCs and expression levels of the Treg marker gene, FOXP3 (top), or expression levels of the T cell exhaustion related gene signature (bottom), in samples from the TCGA LUAD cohort. Samples are labelled by their tissue types (tumor or normal). Tregs, regulatory T cells; Tex, exhausted T cells. Panels A and H were created with BioRender.com.
Our mIF data suggested that lymphoid aggregates (LAs) accounted for the majority of TIBs detected in LUADs (Fig. 1C). However, due to the lack of CXCL13 in the mIF panel, we were not able to determine the spatial relationship between CXCL13+ T cells and TIBs in LUADs. We then characterized CXCL13 protein (BCA1 antibody) expression along with lineage markers that define T-cell (CD3), B-cell (CD20) and tumor compartments (pan cytokeratin) using the nanoString GeoMx digital spatial profiler (DSP) platform in 4 LUADs and paired normal-appearing lung tissues (see Methods). For each case, a total of 13 regions of interest (ROIs) were selected including 4 ROIs for CXCL13-positive LAs (LA/BCA+), 3 ROIs for CXCL13-negative LAs (LA/BCA-), 3 ROIs that covered tumor and tumor stroma without LAs (T/LA-/BCA-), and 3 ROIs that covered normal lung tissues (Fig. S9A–F). Each of these selected ROIs were then segmented into 3 areas of illumination (AOI): B-cell, T-cell or tumor-cell enriched based on expression of the morphology markers (Fig. 5C). No or very few B cells were observed in normal lung or T/LA-/BCA- ROIs. Relative to the LA/BCA- ROIs, we observed prominently increased B cell abundance in the LA/BCA1+ ROIs (Fig. 5D). To obtain a more comprehensive picture of the spatial distribution of LAs, we further performed spatial transcriptomics (ST) on 4 samples from two patients in our scRNA-seq cohort using the Visium platform (10X Genomics). Consistent with the mIF and DSP data, the majority of LAs identified were within the tumor compartment and B cells in LUADs were largely co-localized with CXCR5+ T follicular helper (Tfh) cells and CXCL13+ T cells within LAs (Fig. S9G).
While T cells were overall the main source of CXCL13 production, our single-cell analysis suggested that the majority of CXCL13+ cells were CD4 Tfh and CD8 exhausted T cells (Tex), followed by CD4 regulatory T cells (Treg) and proliferative Tregs, whereas only a very small subset of CD8 effector T (Teff) cells and CD8 resident memory T (Trm) cells expressed CXCL13 (Fig. 5E). We also detected CXCL13 expression in stressed CD4/CD8 T cells and in a small subset of LAMP3+ DCs, macrophages and fibroblasts. Analysis of these ten CXCL13-expressing subsets (with >100 CXCL13+ cells and the percentage of CXCL13 expression >0.5%) revealed a strong correlation between CXCL13+ CD4 Tfh, Treg, CD8 Tex, fibroblasts and macrophages on one hand, and CXCR5+ memory B cells (Fig. 5E).
We then examined whether the cellular sources of CXCL13 production in LUADs evolve as disease develops and progresses. Consistently across this cohort and 3 additional public scRNA-seq datasets, we observed significantly increased relative proportions of CXCL13+ cells among Tregs in LUADs versus normal lung tissues (Fig. 5F and S8D). Increased expression of CXCL13 was also found in TCGA LUAD cohort whereby it was significantly higher in tumors, regardless of their pathological stage, relative to normal lung tissues (Fig. S8E). CXCL13 expression was highest in early-stage LUADs and its expression gradually decreased with advancing pathological stage. We also interrogated our precancer bulk RNA-seq cohort (36) and noted that CXCL13 levels were progressively increased from normal lung to atypical adenomatous hyperplasia (AAH), the earliest precursor lesion of LUAD (37), and up to invasive LUAD (Fig.5G, left). Consistently, B cell and PC signature scores were also gradually increased along this pathological spectrum (Fig.5G, middle and right).
Expression of immunomodulatory genes and TIB phenotypic heterogeneity
We next analyzed expression of immune biomarkers using the GeoMx DSP data we generated on 43 protein markers (Supplementary Table S6). We determined the differences in immune biomarker expression in the B cell compartment between tumor-associated LAs with and without CXCL13 expression (LA/BCA+ and LA/BCA-, respectively). Relative to LA/BCA- ROIs, we found that the B cell compartment of LA/BCA+ ROIs showed significantly increased expression of CD27, CD40, and decreased expression of CTLA-4, LAG3, as well as PD-L1 (Fig. S10A). In addition, increased IgA+ PCs in LUADs prompted us to interrogate how cytokines produced by various cell populations within TME regulate B cell class-switch recombination (CSR) and generation of PCs. We then quantified expression levels of cytokines that induce IgG- (IFN-γ, TNFα) and IgA (TGF-β, IL-6 and IL-10) CSR (Fig. 5H and S10B) in immune and stromal cell subsets (30,38–41). We noted the limitation of scRNA-seq in detecting the transcripts of interleukin (likely due to the “dropout” events) and therefore focused our analysis on TGFB1, which is known to be critical in promoting IgA isotype switching in human B cells (30,38–41). Although TGFB1 was broadly expressed by various cell types (Fig. S10B), it is noteworthy that, relative to matched normal lung tissues, TGFB1 was significantly higher in cancer-associated fibroblasts (CAFs) in LUADs in our cohort (Fig. 5I) and the dataset from Kim et al (Fig. S10C) (10). In addition, we performed spatial mapping of TGFB1-expressing cells using the Visium data and observed strong TGFB1 expression, particularly, in spatial regions enriched with fibroblasts, as indicated by high expression of canonical markers of fibroblasts (Fig. 5J). These results suggest that TGFB1 upregulation in CAFs is likely associated with increased fractions of IgA+ PCs in LUADs.
Memory B cells and IgA+ PCs strongly co-occur with CD4 Tregs and CD8 Tex, and they negatively correlate with cytotoxic T cells
Lastly, to better understand how the various TIB subsets may influence the functional phenotypes of the TME, we first measured the expression of immunomodulatory genes (Supplementary Table S7) in TIB subsets. TGFB1, GPR183 (EBI2), CD24, and LTB were highly enriched in memory B cells, whereas CCR10, VSIR (VISTA), TNFRSF18 (GITR), and LGALS3 (Galectin 3) were expressed by IgA+ PCs (Fig. S10D), thereby pointing towards their immunosuppressive potential. We further examined cell-state co-association relationships with 28 other immune and stromal cell subsets defined using our scRNA-seq data and found that memory B cells and IgA+ PCs strongly co-occured with CD4+ Tfh, Treg, and CD8+ Tex cells (Fig. 5K–L). IgA+ PCs also strongly co-occured with cDC2, fibroblasts and macrophages. In contrast, IgA+ PCs showed significant negative correlations with cytotoxic lymphocytes (CTLs) including CD4+ CTLs, CD8 Teff, and CD8 Trm cells (Fig. 5K). With immune deconvolution analysis of the bulk RNA-seq data, we also observed positive correlations between the signature scores of PCs and Treg/Tex in TCGA LUAD cohort (Fig. 5M). These results suggest distinct co-association relationships and crosstalk between TIBs and various TME cells in LUADs.
DISCUSSION
Single-cell sequencing has been applied to interrogate the highly complex immune microenvironment of LUADs (10,11,25,35), and we and others have demonstrated that the local TME consists of immune cells with diverse phenotypes and complex crosstalk (6,42). However, analysis of the immune biology of LUADs and other solid tumors has been largely T cell-focused and the characteristics of TIBs are relatively understudied. Most of previous single-cell studies classified TIBs into two main cell types, B cells and PCs, with each one treated as a homogenous subset (10,25,42,43). This study leverages our unique scRNA-seq and scBCR-seq data generated on spatially defined tumor and matched normal lung tissues collected from surgically resected LUADs and presents a spatially resolved single-cell atlas of TIBs in early-stage LUAD (Fig. 6). Our study by deep profiling of the various transcriptional and differentiation states, BCR clonotypes, antibody isotypes, SHM frequencies, spatial features of B cells and PCs, and co-occurrence patterns with other TME cells using multiple independent approaches, highlights the heterogeneous nature of TIBs in LUADs and their crosstalk with the TME, and it presents a unique attempt to comprehensively characterize TIBs in solid tumors.
Figure 6. Schematic highlighting key discoveries of this study.

A schematic cartoon depicts the geospatial changes (from left to right) in cellular composition of TIBs, BCR clonal diversity and clonality, frequency of BCR somatic hypermutation (SHM), and cell differentiation states of B cells and PCs, respectively, in LUADs when compared to their matched normal lung tissues. Part of this figure was created with BioRender.com.
We demonstrate extensive remodeling of TIBs in the TME of LUADs, characterized by markedly increased fractions of memory B cells and PCs with more differentiated states. Our analyses suggest that various microenvironmental factors may have contributed to the altered TIB landscapes in LUADs. First, we showed significantly increased TGFB1 expression in CAFs which is in line with the highly enriched IgA+ PCs in LUADs given the critical role of TGFB1 in promoting IgA isotype switching in human B cells (30,38–41). In addition, we noted massive changes in TIBs in smoker LUADs such as enrichment of the fully differentiated, long-lived, IgA-producing PCs, and decreased B cell clonality, pointing towards a link to cigarette smoke exposure (44). We also showed that the majority of TIBs detected in LUADs were within LAs as well as underscored prominent differences in B cell abundance between CXCL13+ and CXCL13- LAs and in immune biomarker expression such as upregulated CD27, CD40 and downregulated CTLA-4, LAG3 in the B cell compartment of CXCL13+ LAs. Our findings suggest that the presence of CXCL13+ T cells in the TME may have not only contributed to B cell recruitment, but also to phenotypic heterogeneity of TIBs in the LUAD TME. The progressively increased CXCL13 expression along the evolution continuum from AAH to invasive LUAD supports a multifaceted role of CXCL13 in early-stage LUAD. In addition to Tfh cells, we found multiple other cellular sources of CXCL13 production in the TME such as CD8 Tex and CD4 Tregs cells. Our observation of significantly increased relative proportions of CXCL13+ cells among Tregs in LUADs versus normal lung tissues across multiple scRNA-seq cohorts intriguingly points to evolutionary dynamics of CXCL13 production by various TME cell subsets in LUAD.
The generation of a cancerization “field” has been considered to present an initiating phase in tumor development from a particular niche and field carcinogenesis has been described in various malignancies including those of the lung (45). Previous studies from our and other groups have identified cellular and molecular alterations in histological normal-appearing regions that are adjacent to solid tumors and that are less prevalent/absent in relatively more distant normal regions (6,45,46), but previous analyses of the field of cancerization were, for the most part, focused on the epithelial compartment. The multiregional sampling strategy of this study is unique and allowed us to interrogate geospatial distribution of TIBs in LUAD tissues as well as in adjacent (to tumor) and relatively more distant normal lung tissues. By characterizing the landscape of TIBs within the LUAD field, we found significant geospatial changes such as gradually decreased fractions of naïve B cells and elevated fractions of class-switched memory B cells and PCs with increasing tumor proximity (Fig. 5). Notably, we found increased BCR clonal diversity and frequencies of SHM across normal tissues with closer proximity to the LUADs, together with significant changes in cell differentiation states. These findings allow us to surmise that TIBs and, thus, the mounting of B-cell-mediated immune responses, may be actively associated with development of the LUAD from a particular niche in the lung, as opposed to somewhere else in the mutagenized (by smoking) lung. Of note, over one third of the BCR clonotypes detected in normal tissues were shared with those in the LUAD tissue from the same patient. Although their antigen specificity is to be determined, our results underscore a phenomenon of B cell trafficking from distant to more proximal (to the tumor) regions within the field, a supposition that warrants further investigation, for instance in higher spatial resolution and in a longitudinal setting.
Notably, our findings of the prominent increase in the fractions of PCs in smokers with more long-lived PCs are in agreement with the observation of the decreased fractions of PCs and lower SHM rates in LUADs harboring EGFR mutations (which are seen common in non-smokers) and our results indicate a superior role of PCs in smoking-related LUADs. In addition, our analyses pointed towards highly abundant IgA+ PCs in LUADs whereby IgA+ PCs strongly co-occurred with activated and proliferative Tregs, CD8+ Tex, CAFs and microphages and negatively correlated with CTLs, which are consistent with multiple lines of evidence supporting the immunosuppressive and pro-tumorigenic role of IgA+ PCs (30), but further functional studies are needed to determine their exact roles in LUAD pathogenesis.
In terms of their clinical significance, our analysis clearly points to the PC (not B cell) subset, as the PC signature derived from this study notably showed the strongest correlation with patient survival across multiple LUAD cohorts and significantly associated with patient response to anti-PD-1/PD-L1-based immunotherapy. This observation is in accordance with a recent study demonstrating that intratumoral PCs predict outcomes in NSCLC patients treated with atezolizumab (16). Our findings also lender support for the PC signature as a potential biomarker for prediction of OS benefit and the efficacy of PD-1/PD-L1 blockade in LUAD patients.
While single-cell sequencing provides a robust approach that enables whole transcriptome profiling of TIBs and integration with BCR repertoire, our study has limitations. First, the interleukin transcripts are poorly captured in scRNA-seq likely due to a high drop-out rate and consequently, IL10 which is important to characterize the reported regulatory B cells (Bregs) is not informative and the fraction of IL-10+ Bregs was very low (0.87%). Another reason for a low Breg fraction could be due to the dynamic and context-dependent nature of the Breg state. A recent expert view on the matter suggests that Bregs reflect a phenotypic state rather than a hard-wired lineage (47). It is considered that IL-10, the most commonly reported Breg effector molecule, can be immunostimulatory in some contexts. Second, the scarcity of remaining tissue following scRNA-seq, genomic analysis, and imaging hindered our ability to directly measure cytokines, soluble factors, and ligands secreted by TIBs which could better characterize the functional phenotypes of TIBs. Lastly, due to the lack of markers for Tfh and follicular dendritic cells in our mIF or DSP panels, we were not able to further determine whether an LA is a TLS or its maturation status. Nevertheless, this study provides fundamental findings on yet uncharted phenotypic heterogeneity and roles of TIBs in the development and immunopathology of early-stage LUAD, and it constitutes a valuable resource to leverage targets for the development of desperately needed novel immunotherapeutic strategies for early treatment of this morbid malignancy.
Materials and Methods
Multiregional Sampling of Surgically Resected LUADs and Matched Normal Lung Tissues
Sixteen patients with early-stage LUADs (I–IIIA) were evaluated at MD Anderson Cancer Center and underwent standard-of-care surgical resection (Supplementary Table S1). All samples in the study were obtained under waiver of consent from banked or residual tissues approved by MD Anderson institutional review board protocols. Tumor (LUAD) and matched multiregional normal lung samples were collected as we previously described (6).
Single cell library construction and sequencing
Freshly frozen tumor and normal samples (n = 63) were immediately minced and enzymatically digested and red blood cell lysed using optimized protocols as we previously described (6). Briefly, for patient 1, cells were then filtered, counted, and stained with SYTOX Blue viability dye (S34857, Life Technologies), followed by fluorescence-activated single cell sorting (FACS) to eliminate doublets and dead cells and collect viable singlet cells. Cells from patients 2 through 16 (P2-P16) were stained with anti-EPCAM-PE (347198, BD Biosciences;1:50 dilution in ice-cold phosphate-buffered saline, PBS, containing 2% FBS) for 30 minutes with gentle rotation at 4°C. EPCAM-stained cells were then washed, filtered using 40 μm tip filters, stained with SYTOX Blue and processed on a FACS Aria II instrument. Doublets and dead cells were eliminated, and viable (SYTOX-negative) EPCAM-negative singlets were collected in PBS containing 2% FBS. Cells were washed again to eliminate ambient RNA, and a sample was taken for counting by Trypan Blue exclusion (T8154, Sigma Aldrich) before loading on 10X Chromium microfluidic chips. Gene expression libraries were generated according to manufacturer’s instructions using Chromium Next GEM Single Cell 5’ Library & Gel Bead Kits (v1/v3 Chemistry, 10X Genomics). Library quality was assessed using high sensitivity capillary electrophoresis on a Fragment Analyzer (TapeStation: High Sensitivity 5000 from Agilent Technologies). Library pools were denatured and diluted as recommended for sequencing on the Illumina NovaSeq 6000 platforms to achieve a depth of at least 50,000 read pairs per cell for single cell GEX libraries and at least 10,000 reads pairs per cell for single cell V(D)J libraries. The detailed methods on sample processing were described in our recent study (6).
Single cell data processing and analysis
Raw scRNA-seq and scBCR-seq data were pre-processed using CellRanger (v3.0.2, 10x Genomics), followed by quality filtering, doublet removal, data normalization, assessment and correction of batch effects. Briefly, cells expressing less than 500 genes, or more than 6,500 genes were removed. Likely dying or apoptotic cells where >15% of transcripts were derived from the mitochondrial genome were also excluded. Additional possible doublets were further identified and removed. Seurat (version 4.0) (48) was applied for data normalization, unsupervised cell clustering and identification of differentially expressed genes among cell clusters. More details of data processing are described in Supplementary Methods. The cell types and transcriptional states of B cells and PCs were defined based on cluster distribution, cluster specific genes (Supplementary Table S2), and unique expression of canonical B cell/PC-related markers as well as transcriptional factors (Supplementary Methods). Monocle (19) and CytoTRACE (24) were applied to reconstruct the differentiation trajectories and infer cell differentiation states. We constructed cell type-specific gene expression signatures (Supplementary Table S3) for major B cell subtypes (i.e., naïve B cells, memory B cells, PCs) using our scRNA-seq data in the manner as we preciously described (6).
scBCR-seq data was firstly pre-processed by Cell Ranger v3.0.2 for V(D)J sequence assembly and BCR reconstruction using the GRCh38 assembly in Ensembl (refdata-cellranger-vdj-GRCh38-alts-ensembl-2.0.0) as the reference. The Change-O repertoire clonal assignment toolkit (49) was used to define clones, and BCR clonal abundances and diversity were evaluated by estimateAbundance and alphaDiversity functions in Alakazam (v1.1.0), respectively (49). The inverse of Simpson index was used to quantify the clonal diversity. The diversity index (D) for each group is the mean value of overall resampling realizations, and the confidence intervals of clonal diversity are derived from the standard deviation of the resampling realizations. We quantitatively assessed the similarity between BCR repertoires among groups based on the nucleotide sequences of the CDR3 regions. To identify SHM, BCR germline sequences were reconstructed using CreateGermlines.py, and the number of somatic mutations for each sequence was calculated using observedMutations (Shazam v1.1.0) (49). Paired scBCR-seq data were integrated with scRNA-seq based on their matched unique cell barcodes.
Multiplex immunofluorescence, immunohistochemistry staining, and Nanostring GeoMx Digital Spatial Profiling (DSP)
Multiplex immunofluorescence (mIF) staining was performed using four μm-thick formalin fixed, paraffin embedded sample sections from 5 of the 16 patients, with antibodies against a validated panel of 8 markers including pancytokeratin, CD20, CD3, CD4, CD8, CD56, Foxp3, and Ki67. In each sample, five regions of interest (ROIs, 931 × 698 μm size) were selected at a resolution of 20x for image analysis. The primary antibody (Anti-BCA1, clone EPR23400–92, Abcam) was used for CXCL13 immunohistochemistry. For GeoMx DSP, five μm sections were stained with the semi-automated GeoMx DSP standard protein protocol. As visualization markers, we used pancytokeratin (AE1/AE3) for epithelial cells, SYTO 13 for nuclear stain, CD3e for T cells, and CD20 for B cells. For each case, a total of 13 ROIs were selected and selected ROIs were segmented into tumor, T cell-enriched, and B cell-enriched compartments. More details of samples processing, selection of ROIs, segmentation and data analysis are described in Supplementary Methods.
Spatial transcriptomics (ST) data generation
We further performed spatial transcriptomics on 4 samples from two patients (P10 and P14) in our scRNA-seq cohort using the Visium spatial technology from 10x Genomics. First, the formalin-fixed, paraffin-embedded (FFPE) tissue block was placed in a microtome and cut to expose the tissue. After the tissue is exposed, three to four tissue sections of 10 um thickness were then collected for RNA extraction, which was performed using Qiagen RNeasy FFPE Kit. To assess the RNA quality of the tissue, the purified RNA was immediately proceeded to calculate the percentage of total RNA fragments >200 nucleotides (DV200) of RNA extracted from tissue sections using Agilent RNA 6000 Pico Kit. Based on DV200 evaluation, blocks with DV200 >50% were selected for proceeding with sectioning. The tissue blocks were then sectioned by a microtome to generate appropriately sized sections for Visium slides. Sections were collected in a 42°C water bath and were placed within the frames of capture areas on Visium Spatial Gene Expression Slide (PN-1000188, 10x Genomics), with only one section placed within each capture area (6.5 × 6.5 mm). The tissues were then deparaffinized, stained, and decrosslinked, followed by probe hybridization, ligation, release & extension. The Visium spatial gene expression FFPE libraries were constructed using the Visium Human Transcriptome Probe Kit (PN-1000363) and Visium FFPE Reagent Kit (PN-1000361) following the manufacture’s guidance. Constructed libraries were sequenced on the Illumina NovaSeq 6000 platforms to achieve a depth of at least 75,000 mean read pairs per spot and at least 2,000 median genes per spot. More details of ST data analysis are described in Supplementary Method.
Additional statistical analyses
In addition to the bioinformatics approaches and statistical analyses described above, all other statistical analyses were performed using statistical software R v3.6.0. To assess phenotypic relationships and examine co-occurrence patterns (both positive and negative) between TIB cell subsets and various other immune and stromal cell populations in the TME, Spearman’s correlation analysis was applied. To compare the fractions of different B and plasma cells and their subsets across tumor and spatially defined normal tissues, the Mann-Whitney test and paired t test were applied as indicated. All statistical significance testing in this study was two-sided and results were considered statistically significant at P-values or FDR q-values < 0.05. Defaulted “p < 2 × 10−16” reported in R v3.6.0 was used when the p value was too small to illustrate.
Supplementary Material
SIGNIFICANCE.
While TIBs are highly enriched in LUADs, they are poorly characterized. This study provides a much-needed understanding of the transcriptional, clonotypic states and phenotypes of TIBs, unrevealing their potential roles in immunopathology of early-stage LUADs and constituting a roadmap for development of TIB-targeted immunotherapies for treatment of this morbid malignancy.
Financial support:
This study was supported in part by the start-up research fund and the University Cancer Foundation via the Institutional Research Grant Program at the University of Texas MD Anderson Cancer Center (to L. Wang). This study was also supported by National Cancer Institute (NCI) grants U01CA264583 (to H. Kadara and L. Wang), R01CA205608 (to H. Kadara), and 1U2CCA233238 (to A. Spira, S. Dubinett, and H. Kadara), Cancer Prevention and Research Institute of Texas grants RP220101 (to H. Kadara and L. Wang) and RP160668 (to I.I. Wistuba), University of Texas SPORE in Lung Cancer P50CA070907, and NCI P50 core grant CA016672, as well as research funding from Johnson & Johnson (to H. Kadara). L. Wang and H. Kadara are Andrew Sabin Family Foundation fellows. A. Sinjab is supported by a T32CA217789 MD Anderson Cancer Center Translational Genomics and Precision Medicine Training program.
Conflict of interest:
C.S.S. and A.E.S. are employees of Johnson & Johnson. H.K. receives research support from Johnson & Johnson. T.C. has received speakers’ fees from the Society for Immunotherapy of Cancer, Bristol Myers Squibb, Roche and Medscape Oncology; reports consulting/advisory role fees from MedImmune, AstraZeneca, Bristol Myers Squibb, Merck & Co., Genentech, Arrowhead Pharmaceuticals and EMD Serono; reports clinical research funding to MD Anderson Cancer Center from Boehringer Ingelheim, MedImmune, AstraZeneca, EMD Serono, and Bristol Myers Squibb. I.I.W. has provided consulting or advisory roles for AstraZeneca/MedImmune, Asuragen, Bayer, Bristol-Myers Squibb, Genentech/Roche, GlaxoSmithKline, Guardant Health, HTG Molecular Diagnostics, Merck, MSD Oncology, OncoCyte, Novartis, Flame Inc, and Pfizer; has received grants and personal fees from Asuragen, Genentech/Roche, Bristol Myers Squibb, AstraZeneca/MedImmune, HTG Molecular, Merck, and Guardant Health; has received personal fees from GlaxoSmithKline and Oncocyte, Daiichi-Sankyo, Roche, Astra Zeneca, Pfizer and Bayer; has received research funding to his institution from 4D Molecular Therapeutics, Adaptimmune, Adaptive Biotechnologies, Akoya Biosciences, Amgen, Bayer, EMD Serono, Genentech, Guardant Health, HTG Molecular Diagnostics, Iovance Biotherapeutics, Johnson & Johnson, Karus Therapeutics, MedImmune, Merck, Novartis, OncoPlex Diagnostics, Pfizer, Akoya, Takeda, and Novartis.
DATA AVAILABILITY
All scRNA-seq and scBCR-seq data generated by this study are deposited at the European Genome–phenome Archive (EGA) under accession number EGAS00001005021. Access to this shared dataset is controlled by the institutional Data Access Committee, in compliance with the NIH policy for Data Management and Sharing. Further information about EGA can be found on https://ega-archive.org. The data can also be accessed through the online Single Cell Data Portal (https://singlecell.mdanderson.org/BcellLC), an interactive web-based tool we have developed for visualizing our single-cell data. All codes used for analysis and cell annotation are available from https://github.com/dapenghao/B_LUAD. B cell gene signatures derived from this study are provided in Supplementary Tables. Previously published datasets reanalyzed in this study are described in Supplementary Methods. Further information and requests should be directed to and will be fulfilled by the corresponding authors.
REFERENCES
- 1.Goldstraw P, Ball D, Jett JR, Le Chevalier T, Lim E, Nicholson AG, et al. Non-small-cell lung cancer. Lancet 2011;378(9804):1727–40 doi 10.1016/S0140-6736(10)62101-0. [DOI] [PubMed] [Google Scholar]
- 2.Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang N-AA, Andrews MC, et al. Distinct cellular mechanisms underlie anti-CTLA-4 and anti-PD-1 checkpoint blockade. Cell 2017;170(6):1120–33. e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nature Reviews Immunology 2015;15(8):486–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sharonov GV, Serebrovskaya EO, Yuzhakova DV, Britanova OV, Chudakov DM. B cells, plasma cells and antibody repertoires in the tumour microenvironment. Nat Rev Immunol 2020;20(5):294–307 doi 10.1038/s41577-019-0257-x. [DOI] [PubMed] [Google Scholar]
- 5.Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020;577(7791):549–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sinjab A, Han G, Treekitkarnmongkol W, Hara K, Brennan P, Dang M, et al. Resolving the spatial and cellular architecture of lung adenocarcinoma by multiregion single-cell sequencing. Cancer Discov 2021;11(10):2506–23 doi 10.1158/2159-8290.CD-20-1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Travaglini K, Nabhan A, Penland L, Sinha R, Gillich A, Sit R, et al. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 2020;587(7835):619–25 doi 10.1038/s41586-020-2922-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gutzeit C, Chen K, Cerutti A. The enigmatic function of IgD: some answers at last. Eur J Immunol 2018;48(7):1101–13 doi 10.1002/eji.201646547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Han G, Sinjab A, Treekitkarnmongkol W, Lu W, Serrano AG, Hernandez SD, et al. An atlas of epithelial cell states and plasticity in lung adenocarcinoma. bioRxiv 2022:2022.05.13.491635 doi 10.1101/2022.05.13.491635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim N, Kim H, Lee K, Hong Y, Cho J, Choi J, et al. Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat Commun 2020;11(1):2285 doi 10.1038/s41467-020-16164-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leader AM, Grout JA, Maier BB, Nabet BY, Park MD, Tabachnikova A, et al. Single-cell analysis of human non-small cell lung cancer lesions refines tumor classification and patient stratification. Cancer Cell 2021;39(12):1594–609.e12 doi 10.1016/j.ccell.2021.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Campbell JD, Alexandrov A, Kim J, Wala J, Berger AH, Pedamallu CS, et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet 2016;48(6):607–16 doi 10.1038/ng.3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol 2019;37(7):773–82 doi 10.1038/s41587-019-0114-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prat A, Navarro A, Pare L, Reguart N, Galvan P, Pascual T, et al. Immune-Related Gene Expression Profiling After PD-1 Blockade in Non-Small Cell Lung Carcinoma, Head and Neck Squamous Cell Carcinoma, and Melanoma. Cancer Res 2017;77(13):3540–50 doi 10.1158/0008-5472.CAN-16-3556. [DOI] [PubMed] [Google Scholar]
- 15.Hwang S, Kwon AY, Jeong JY, Kim S, Kang H, Park J, et al. Immune gene signatures for predicting durable clinical benefit of anti-PD-1 immunotherapy in patients with non-small cell lung cancer. Sci Rep 2020;10(1):643 doi 10.1038/s41598-019-57218-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patil NS, Nabet BY, Muller S, Koeppen H, Zou W, Giltnane J, et al. Intratumoral plasma cells predict outcomes to PD-L1 blockade in non-small cell lung cancer. Cancer Cell 2022;40(3):289–300 e4 doi 10.1016/j.ccell.2022.02.002. [DOI] [PubMed] [Google Scholar]
- 17.Fehrenbacher L, Spira A, Ballinger M, Kowanetz M, Vansteenkiste J, Mazieres J, et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet 2016;387(10030):1837–46 doi 10.1016/S0140-6736(16)00587-0. [DOI] [PubMed] [Google Scholar]
- 18.Rittmeyer A, Barlesi F, Waterkamp D, Park K, Ciardiello F, von Pawel J, et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet 2017;389(10066):255–65 doi 10.1016/S0140-6736(16)32517-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cao J, Spielmann M, Qiu X, Huang X, Ibrahim DM, Hill AJ, et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 2019;566(7745):496–502 doi 10.1038/s41586-019-0969-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cortes M, Georgopoulos K. Aiolos is required for the generation of high affinity bone marrow plasma cells responsible for long-term immunity. J Exp Med 2004;199(2):209–19 doi 10.1084/jem.20031571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rodriguez-Bayona B, Ramos-Amaya A, Lopez-Blanco R, Campos-Caro A, Brieva J. STAT-3 activation by differential cytokines is critical for human in vivo-generated plasma cell survival and Ig secretion. J Immunol 2013;191(10):4996–5004 doi 10.4049/jimmunol.1301559. [DOI] [PubMed] [Google Scholar]
- 22.van SA, de KS, van dS A, Gartlan K, Sofi M, Light A, et al. The tetraspanin CD37 orchestrates the alpha(4)beta(1) integrin-Akt signaling axis and supports long-lived plasma cell survival. Sci Signal 2012;5(250):ra82 doi 10.1126/scisignal.2003113. [DOI] [PubMed] [Google Scholar]
- 23.Wols HAM. Plasma Cells, Encyclopedia of Life Sciences, 2005. John Wiley & Sons, Ltd., USA. [Google Scholar]
- 24.Gulati G, Sikandar S, Wesche D, Manjunath A, Bharadwaj A, Berger M, et al. Single-cell transcriptional diversity is a hallmark of developmental potential. Science 2020;367(6476):405–11 doi 10.1126/science.aax0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lambrechts D, Wauters E, Boeckx B, Aibar S, Nittner D, Burton O, et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med 2018;24(8):1277–89 doi 10.1038/s41591-018-0096-5. [DOI] [PubMed] [Google Scholar]
- 26.Allie S, Bradley J, Mudunuru U, Schultz M, Graf B, Lund F, et al. The establishment of resident memory B cells in the lung requires local antigen encounter. Nat Immunol 2019;20(1):97–108 doi 10.1038/s41590-018-0260-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kometani K, Nakagawa R, Shinnakasu R, Kaji T, Rybouchkin A, Moriyama S, et al. Repression of the transcription factor Bach2 contributes to predisposition of IgG1 memory B cells toward plasma cell differentiation. Immunity 2013;39(1):136–47 doi 10.1016/j.immuni.2013.06.011. [DOI] [PubMed] [Google Scholar]
- 28.Reimold AM, Iwakoshi NN, Manis J, Vallabhajosyula P, Szomolanyi-Tsuda E, Gravallese EM, et al. Plasma cell differentiation requires the transcription factor XBP-1. Nature 2001;412(6844):300–7 doi 10.1038/35085509. [DOI] [PubMed] [Google Scholar]
- 29.Stein J, Nombela-Arrieta C. Chemokine control of lymphocyte trafficking: a general overview. Immunology 2005;116(1):1–12 doi 10.1111/j.1365-2567.2005.02183.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shalapour S, Karin M. The neglected brothers come of age: B cells and cancer. Seminars in Immunology 2021;52:101479 doi 10.1016/j.smim.2021.101479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shinnakasu R, Inoue T, Kometani K, Moriyama S, Adachi Y, Nakayama M, et al. Regulated selection of germinal-center cells into the memory B cell compartment. Nat Immunol 2016;17(7):861–9 doi 10.1038/ni.3460. [DOI] [PubMed] [Google Scholar]
- 32.Seifert M, Steimle-Grauer S, Goossens T, Hansmann M, Brauninger A, Kuppers R. A model for the development of human IgD-only B cells: Genotypic analyses suggest their generation in superantigen driven immune responses. Mol Immunol 2009;46(4):630–9 doi 10.1016/j.molimm.2008.07.032. [DOI] [PubMed] [Google Scholar]
- 33.Dieu-Nosjean MC, Giraldo NA, Kaplon H, Germain C, Fridman WH, Sautes-Fridman C. Tertiary lymphoid structures, drivers of the anti-tumor responses in human cancers. Immunol Rev 2016;271(1):260–75 doi 10.1111/imr.12405. [DOI] [PubMed] [Google Scholar]
- 34.Workel HH, Lubbers JM, Arnold R, Prins TM, van der Vlies P, de Lange K, et al. A Transcriptionally Distinct CXCL13(+)CD103(+)CD8(+) T-cell Population Is Associated with B-cell Recruitment and Neoantigen Load in Human Cancer. Cancer Immunol Res 2019;7(5):784–96 doi 10.1158/2326-6066.CIR-18-0517. [DOI] [PubMed] [Google Scholar]
- 35.Guo X, Zhang Y, Zheng L, Zheng C, Song J, Zhang Q, et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat Med 2018;24(7):978–85 doi 10.1038/s41591-018-0045-3. [DOI] [PubMed] [Google Scholar]
- 36.Sivakumar S, Lucas FAS, McDowell TL, Lang W, Xu L, Fujimoto J, et al. Genomic Landscape of Atypical Adenomatous Hyperplasia Reveals Divergent Modes to Lung Adenocarcinoma. Cancer Res 2017;77(22):6119–30 doi 10.1158/0008-5472.CAN-17-1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kadara H, Scheet P, Wistuba II, Spira AE. Early Events in the Molecular Pathogenesis of Lung Cancer. Cancer prevention research 2016;9(7):518–27 doi 10.1158/1940-6207.CAPR-15-0400. [DOI] [PubMed] [Google Scholar]
- 38.Shockett P, Stavnezer J. Effect of cytokines on switching to IgA and alpha germline transcripts in the B lymphoma I.29 mu. Transforming growth factor-beta activates transcription of the unrearranged C alpha gene. J Immunol 1991;147(12):4374–83. [PubMed] [Google Scholar]
- 39.Stavnezer J, Kang J. The surprising discovery that TGF beta specifically induces the IgA class switch. J Immunol 2009;182(1):5–7 doi 10.4049/jimmunol.182.1.5. [DOI] [PubMed] [Google Scholar]
- 40.Shalapour S, Lin XJ, Bastian IN, Brain J, Burt AD, Aksenov AA, et al. Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature 2017;551(7680):340–5 doi 10.1038/nature24302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shalapour S, Font-Burgada J, Di Caro G, Zhong Z, Sanchez-Lopez E, Dhar D, et al. Immunosuppressive plasma cells impede T-cell-dependent immunogenic chemotherapy. Nature 2015;521(7550):94–8 doi 10.1038/nature14395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maynard A, McCoach CE, Rotow JK, Harris L, Haderk F, Kerr DL, et al. Therapy-Induced Evolution of Human Lung Cancer Revealed by Single-Cell RNA Sequencing. Cell 2020;182(5):1232–51 e22 doi 10.1016/j.cell.2020.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Laughney AM, Hu J, Campbell NR, Bakhoum SF, Setty M, Lavallee VP, et al. Regenerative lineages and immune-mediated pruning in lung cancer metastasis. Nat Med 2020;26(2):259–69 doi 10.1038/s41591-019-0750-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brandsma CA, Kerstjens HA, van Geffen WH, Geerlings M, Postma DS, Hylkema MN, et al. Differential switching to IgG and IgA in active smoking COPD patients and healthy controls. Eur Respir J 2012;40(2):313–21 doi 10.1183/09031936.00011211. [DOI] [PubMed] [Google Scholar]
- 45.Sinjab A, Han G, Wang L, Kadara H. Field Carcinogenesis in Cancer Evolution: What the Cell Is Going On? Cancer Res 2020;80(22):4888–91 doi 10.1158/0008-5472.CAN-20-1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abbosh C, Venkatesan S, Janes SM, Fitzgerald RC, Swanton C. Evolutionary dynamics in pre-invasive neoplasia. Curr Opin Syst Biol 2017;2:1–8 doi 10.1016/j.coisb.2017.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Engelhard V, Conejo-Garcia JR, Ahmed R, Nelson BH, Willard-Gallo K, Bruno TC, et al. B cells and cancer. Cancer Cell 2021;39(10):1293–6 doi 10.1016/j.ccell.2021.09.007. [DOI] [PubMed] [Google Scholar]
- 48.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 2018;36(5):411–20 doi 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gupta NT, Vander Heiden JA, Uduman M, Gadala-Maria D, Yaari G, Kleinstein SH. Change-O: a toolkit for analyzing large-scale B cell immunoglobulin repertoire sequencing data. Bioinformatics 2015;31(20):3356–8 doi 10.1093/bioinformatics/btv359. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All scRNA-seq and scBCR-seq data generated by this study are deposited at the European Genome–phenome Archive (EGA) under accession number EGAS00001005021. Access to this shared dataset is controlled by the institutional Data Access Committee, in compliance with the NIH policy for Data Management and Sharing. Further information about EGA can be found on https://ega-archive.org. The data can also be accessed through the online Single Cell Data Portal (https://singlecell.mdanderson.org/BcellLC), an interactive web-based tool we have developed for visualizing our single-cell data. All codes used for analysis and cell annotation are available from https://github.com/dapenghao/B_LUAD. B cell gene signatures derived from this study are provided in Supplementary Tables. Previously published datasets reanalyzed in this study are described in Supplementary Methods. Further information and requests should be directed to and will be fulfilled by the corresponding authors.