

Abstract
Development of visceral organs such as the esophagus, lung, liver and stomach are coordinated by reciprocal signaling interactions between the endoderm and adjacent mesoderm cells in the fetal foregut. While the recent successes in recapitulating developmental signaling in vitro has enabled the differentiation of human pluripotent stem cells (hPSCs) into various types of organ-specific endodermal epithelium, the generation of organ-specific mesenchyme has received much less attention. This is a major limitation in ongoing efforts to engineer complex human tissue. Here, we describe protocols to differentiate hPSCs into different types of organ-specific mesoderm leveraging signaling networks and molecular markers elucidated from single-cell transcriptomics of mouse foregut organogenesis. Building on established methods, hPSC-derived lateral plate mesoderm (LPM) treated with either retinoic acid (RA) or RA together with a Hedgehog (HH) agonist generates posterior or anterior foregut splanchnic mesoderm respectively (pFG-SpM or aFG-SpM) after 4 days cultures. These are directed into organ-specific mesenchyme lineages by the combinatorial activation or inhibition of WNT, BMP, RA or HH pathways from days 4 to 7 in cultures. By day 7 the cultures are enriched for different types of mesoderm with distinct molecular signatures: 60–90% pure liver septum transversum/mesothelium-like (STM/mesothelium), 70–80% pure liver-like fibroblasts (LF), and populations of ~35% respiratory-like mesoderm (RM), gastric-like mesoderm (GM), or esophageal-like mesoderm (EM). This protocol can be performed by anyone with moderate experience differentiating hPSCs and provides a novel platform to study human mesoderm development and can be used to engineer more complex foregut tissue for disease modeling and regenerative medicine.
Introduction
The development of foregut organs including lungs, esophagus, stomach, pancreas and liver is orchestrated by a reiterative series of growth factors mediated tissue interaction between the embryonic endoderm and the surrounding splanchnic mesoderm1, 2. Much attention has focused on identifying the molecular pathways that instruct the specification of different epithelial lineages of digestive and respiratory organs3, 4. As a result, there have been tremendous advances in replicating developmental signals in vitro to direct the differentiation of human pluripotent stem cells (hPSC) into different endodermal lineages5–10. In contrast, much less is known about the development of organ-specific splanchnic mesoderm and to date there are few if any approaches to generate these cell types in vitro. Adult visceral organs contain resident fibroblasts, mesothelium, smooth muscle, and in some cases cartilage all derived from the gut tube splanchnic mesoderm. Evidence suggest that in some cases similar cell types in different organs have distinct properties and transcriptional programs11–13, although the developmental origins of this specificity are poorly understood. This is a major limitation towards the goal of engineering human tissue that replicates the cellular complexity of in vivo organs– indeed most hPSC-derived foregut organoids reported to date lack the appropriate mesenchyme6, 9, 10. Here, we describe a series of optimized protocols first published by Han et al.14 to differentiate hPSCs into different types of organ-specific-like mesenchyme progenitors including liver septum transversum/mesothelium-like (STM/mesothelium), liver-like fibroblasts (LF), gastric-like mesoderm (GM), esophageal-like mesoderm (EM) and respiratory-like mesoderm (RM). This novel platform for studying human mesoderm development also has potential applications in disease modeling and regenerative medicine where it can be used to engineer more complex foregut organ tissues.
Development of the protocol:
To develop a protocol for generating organ-specific mesenchyme from hPSCs, we turned to studies of mesoderm development in animal models. In mammals, the mesoderm germ layer is induced by BMP and WNT signals while the endoderm is induced by Nodal/Activin/TGFβ (henceforth referred to as TGFβ) as the tissues progressively emerge through the primitive streak (PS) during gastrulation15, 16. After gastrulation, the bi-layered sheet of endoderm and lateral plate mesoderm (LPM) folds into a primitive gut tube. In the foregut region, the LPM splits into an outer somatic mesoderm layer next to the ectoderm which gives rise to the forelimbs and body wall, and an inner splanchnic mesoderm (SpM) layer that gives rise to the cardiac mesoderm (CM) and the mesoderm surrounding the presumptive visceral organs of the gut tube15, 17–19.
The endoderm and mesoderm of the fetal gut tube are then progressively patterned along the anterior-posterior and dorsal-ventral axes into organ-specific cell types by reciprocal cell signaling between the tissue layers1, 2, 20, 21. While a lot is known about the combinatorial signals that direct different epithelial lineages, until recently the SpM was less well studied, with exception of cardiac tissue which has received a lot of attention3, 22–24. To address this, we recently carried out a temporally resolved single-cell transcriptomics analysis of the developing mouse foregut identifying a diversity of organ-specific mesenchyme subtypes, which can be identified by distinct gene expression profiles (Supplementary Fig. 1)14. Furthermore, by examining the expression patterns of ligand-receptor pairs we computationally inferred the signaling network predicted to control the development of these different SpM progenitors. Leveraging this cell signaling roadmap we developed methods to the directed differentiation of hPSCs into different SpM lineages14.
To develop our protocol, we built on studies by Loh et al.23 reporting a stepwise method of activating developmental signals while also blocking unwanted lineages to efficiently differentiate hPSCs into PS, LPM and then into CM. This was an excellent starting point since in addition to CM, the LPM also gives rise to different populations of foregut SpM. Our mouse single-cell transcriptomic analysis suggested that these alternative LPM fates were controlled by differential RA and HH signaling14. This allowed us to establish a protocol where day 1 PS could be directed into three different SpM lineages by day 4: Cardiogenic (CG-SpM), posterior foregut (pFG-SpM), and anterior foregut (aFG-SpM) (Fig. 1)14. All 3 lineages are induced by a common set of signals; BMP and FGF activation in combination with TGFβ inhibition, and WNT inhibition. With no additional factors the cells adopt a CM-SpM fate, whereas addition of RA induces pFG-SpM, and the addition of both RA and HH induces aFG-SpM until day 4. Leveraging predictions from the mouse single-cell roadmap14, together with empirical testing we then established conditions where different combinatorial activation or inhibition of WNT, BMP, RA or HH pathways from days 4–7 in monolayer culture could further direct, pFG-SpM into liver septum transversum/mesothelium (STM/mesothelium), liver fibroblasts-like (LF) progenitors and gastric-like mesoderm (GM), whereas the aFG-SpM could generate cultures enriched in esophageal-like mesoderm (EM) and respiratory-like mesoderm (RM) lineage, which we were able to identify based on their unique transcriptional signatures14.
Fig. 1. Schematic diagram of protocols for generating organ-specific mesenchyme from hPSCs.

The protocols were carried out by sequentially changing the gut media with growth factors and chemical components according to the stepwise process of organogenesis. On day −1, wells are coated with 1% Geltrex in DMEM/F-12. Then, 70–90% confluent cells are gently dissociated with Accutase, before cells are suspended in mTeSR1 supplemented with Thiazovivin. Cells are then split (1:16–20) on Geltrex coated wells. On day 0, after washing cells with DMEM/F-12, cells are fed with middle primitive streak induction media. On day 1, after washing cells with DMEM/F-12, cells are fed with lateral plate mesoderm induction media. On days 2 and 3, cells are fed with either cardiogenic splanchnic mesoderm induction mesoderm or anterior foregut splanchnic mesoderm induction mesoderm or posterior foregut splanchnic mesoderm induction mesoderm. On day 4, after washing cells with DMEM/F-12, cells are fed with organ-specific mesoderm induction media. On days 5 and 6, cells are fed with organ-specific mesoderm induction media. On day 7, cells are harvested for analyses. CG-LPM; cardiogenic-lateral plate mesoderm, CM; cardiac mesoderm, EM; esophageal-like mesoderm, GM; gastric-like mesoderm, aFG-LPM; anterior foregut SpM, aFG-SpM; anterior foregut SpM, LF; liver fibroblast-like, LPM; lateral plate mesoderm, Mid. PS; middle region of primitive streak, pFG-LPM; posterior foregut LPM, pFG-SpM; posterior foregut SpM, RM, respiratory-like mesoderm, PSC; pluripotent stem cell, SpM; splanchnic mesoderm, STM; septum transversum.
Overview of the Procedure
Figure 1 shows an overview of the protocols in comparison to a published method for generating CM23. Briefly, the main stages of the Procedure are listed below.
Single-cell passage of hPSCs (day −1; Steps 1–16) and differentiation of hPSCs into Mid. PS (day 0–1; Steps 17–22)
ESCs or iPSCs are differentiated in 2D monolayer on Geltrex coated plates (Steps 1–38). The first step is induction of mid. PS mesendoderm by culturing hPSCs in a combination of Activin A, CHIR (Wnt activator), BMP4, bFGF, and PIK90 (PI3K inhibitor) for 24 hours23 (Steps 17–22). This results in a downregulation of pluripotency markers OCT3/4 and SOX2 and a robust induction of the PS mesendoderm marker (TBXT) (Supplementary Fig. 2).
Differentiation of Mid. PS into LPM (days 1–2; Steps 23–27) and LPM into SpM (days 2–4; Steps 28–33)
In the CM protocol described by Loh et al., day 1 PS cells are treated for 24 hours with BMP4, Wnt-C59 (Wnt inhibitor) and A83–01 (TGFβ inhibitor) to generate cardiogenic LPM (CG-LPM)23, followed by culture with BMP4, Wnt-C59, A83–01, and bFGF23 on days 2–4 to make CG-SpM. To divert the LPM away from CM we add RA to this induction cocktail on days 1 to 4 (Steps 23–33). The addition of only RA generated pFG-SpM, whereas adding both RA and the HH agonist PMA patterned the LPM into aFG-SpM (Fig. 1). While all the SpM subtypes express the mesoderm markers VIM and FOXF1, only the CG-SpM robustly expresses cardiac mesoderm markers NKX2–5 and ISL1 (Fig. 2). Furthermore, both the pFG-SpM and aFG-SpM, but not CG-SpM are enriched in RA-responsive gene such as HOXA5 and CYP26A1, while only aFG-SpM expresses HH-responsive genes such as GLI1 and PTCH1 (Table 1 and Fig. 2).
Fig. 2. hPSC differentiation to splanchnic mesoderm subtypes.
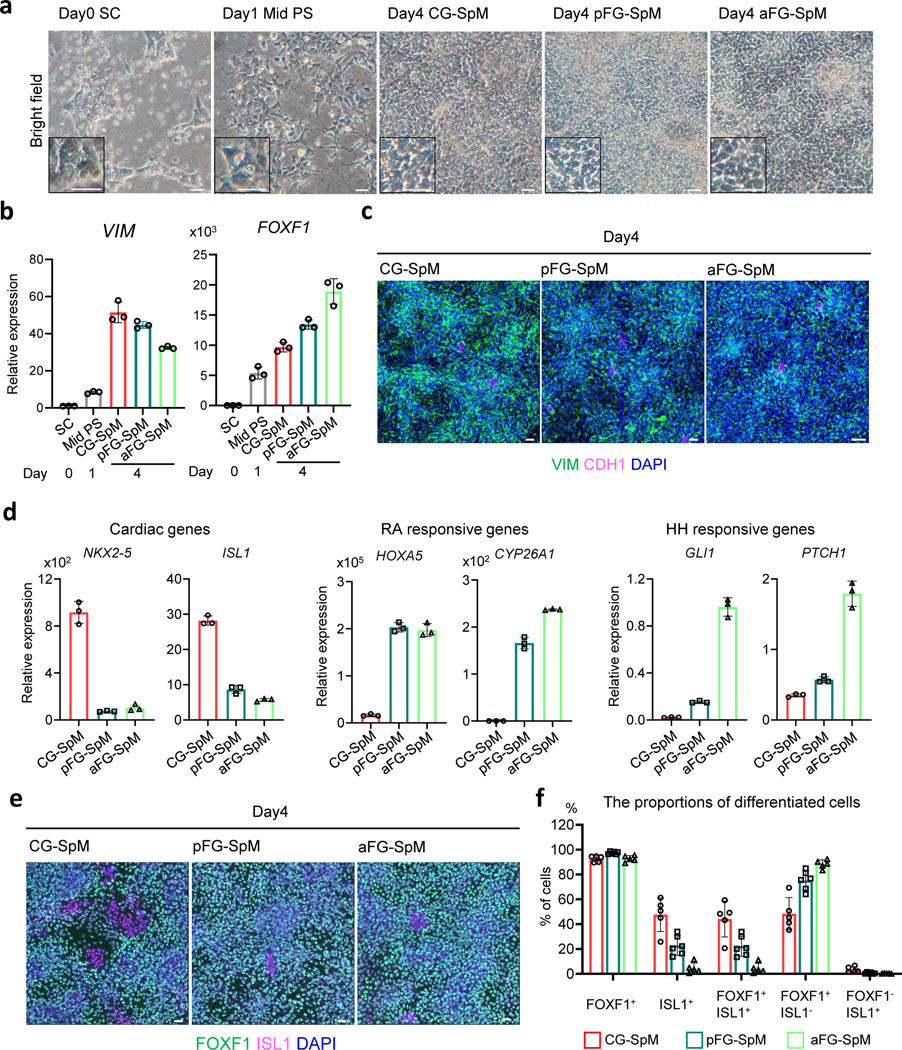
a, Bright field images of differentiating cells from hPSCs from day0 to day4. b, Relative mRNA expression of VIM and FOXF1 by qRT-PCR from day0 to day4. Each bar indicates the average from the 3 independent wells with standard deviation. c, Immunostaining for VIM (green), a pan mesoderm marker, and CDH1 (magenta), an epithelial marker at day 4. The images are maximum intensity projection of confocal stacks. d, Relative mRNA expression of cardiac genes (ISL1 and NKX2–5), RA-responsive genes (HOXA5 and CYP26A1) and HH-responsive genes (GLI1 and PTCH1) by qRT-PCR at day 4. Each bar indicates the average from the 3 independent wells with standard deviation. e, Immunostaining for FOXF1 (green), a splanchnic marker, and ISL1 (magenta), a cardiac mesoderm marker. The images are maximum intensity projection of confocal stacks. f, The average % proportion of differentiated cells positive for the indicated marker by immunocytochemistry. The numbers of marker positive cells were divided by the total number of DAPI positive cells. Mean with S.D. n=3 independent fields at day4. CG-SpM; cardiogenic splanchnic mesoderm, aFG-SpM; anterior foregut SpM, Mid. PS; middle region of primitive streak, pFG-SpM; posterior foregut SpM, SC; stem cell. Each column indicates the average from the 3 independent fields with standard deviation. Scale bar; 50μm
Table 1.
Gene expression profiles of differentiating mesoderm
| Cell type | Gene expression |
|---|---|
|
| |
| d4 CG-SpM | NKX2–5High, ISL1High, HOXA5Low, CYP26A1Low, GLI1Low, PTCH1Low |
| d4 pFG-SpM | NKX2–5Low, ISL1Low, HOXA5High, CYP26A1High, GLI1Low, PTCH1Low |
| d4 aFG-SpM | NKX2–5Low, ISL1Low, HOXA5High, CYP26A1High, GLI1High, PTCH1High |
| d7 Cardiac mesoderm (CM) | NKX2–5+, TBX20+, ACTC1+ |
| d7 Liver STM/mesothelium | WT1+, TBX18+, LHX2+, GATA4+, UPK1B+, UPK3B+ |
| d7 Liver fibroblast (LF) | PITX1+, MSX1+, MSX2+, TBX5+, WNT2+ |
| d7 Gastric mesoderm (GM) | BARX1+, NKX3–2+, FOXF1+ |
| d7 Respiratory mesoderm (RM) | TBX5+, NKX6–1+, WNT2+, FOXF1+ |
| d7 Esophageal mesoderm (EM) | NKX6–1+, MSC+, WNT4+, FOXF1+ |
Differentiation of SpM into organ-specific mesoderm (days 4–7; Steps 34–38)
Treatment of pFG-SpM from day 4 to day 7 with RA and BMP4 generates a cell population with characteristics of liver STM/mesothelium, whereas addition of RA, BMP4 and CHIR induces liver fibroblasts-like cells (LF) (Steps 34–38). On the other hand, treatment of pFG-SpM with RA, PMA from days 4–7 with further addition of NOGGIN (BMP antagonist) on day 6 to day 7 generates gastric-like mesoderm (GM) (Fig. 1) (Steps 34–38). The same conditions that generate GM from pFG-SpM directs aFG-SpM into esophageal-like mesenchyme (EM) (Steps 34–38). Finally, the combination of RA, PMA and BMP4 on days 4–7, with the further addition of CHIR on days 6–7 directs the aFG-SpM into cultures enriched for respiratory-like mesoderm (RM) (Fig. 1) (Steps 34–38). The characteristic and purity of each mesenchyme subtype can be determined by the expression of organ-specific marker genes (Table 1 and Fig. 3)14.
Fig. 3. Characterization of organ-specific-like mesoderm at day7.
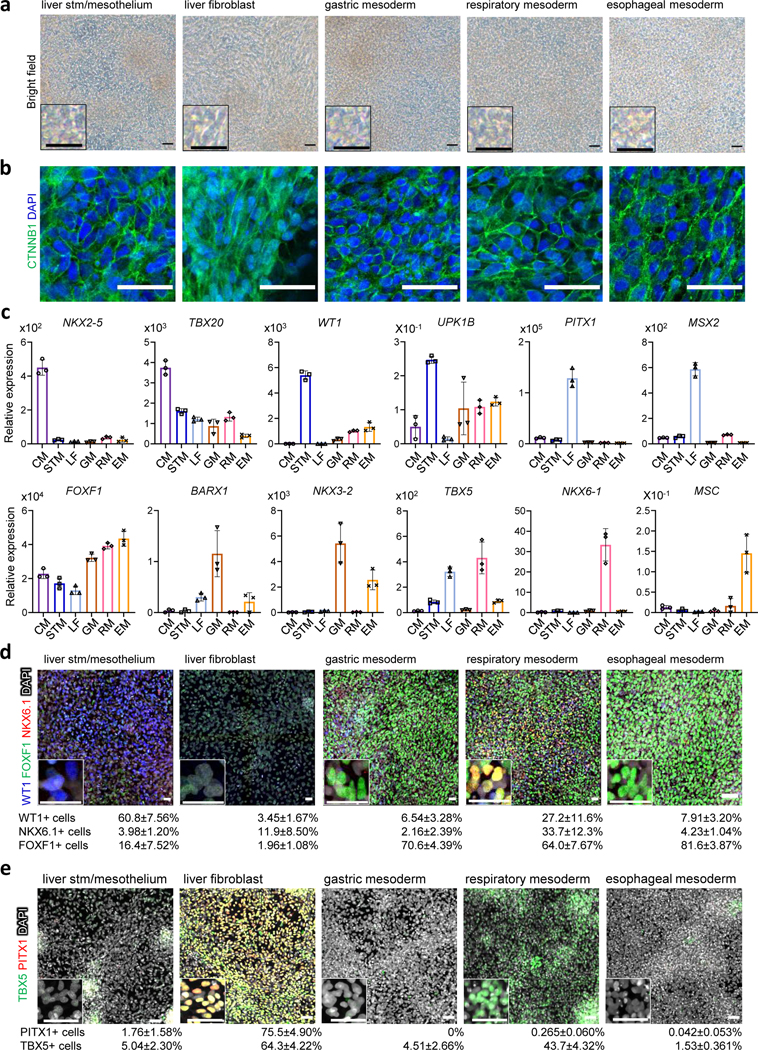
a, Bright field images of differentiated STM/Methothelium-like, liver-like fibroblasts, gastric-like mesoderm, respiratory-like mesoderm, and esophageal-like mesoderm at day7. b, Immunostaining for CTNNB1 (green). The images are maximum intensity projection of confocal stacks. c, Relative mRNA expression of cardiac gene (NKX2–5 and TBX20), liver septum transversum gene (WT1), liver mesothelial gene (UPK1B), liver fibroblast gene (PITX1, MSX2 and TBX5), gastric mesoderm gene (FOXF1, BARX1 and NKX3–2), respiratory mesoderm gene (FOXF1, NKX6–1 and TBX5) and esophageal mesoderm gene (FOXF1 and MSC) by qRT-PCR. Each bar indicates the average from the 3 independent wells with standard deviation. d, Immunostaining for FOXF1 (green), NKX6.1 (red), and WT1 (blue). e, Immunostaining for PITX1 (red) and TBX5 (green). The images in d and e are maximum intensity projection of confocal stacks with the average % proportion of differentiated cells positive for the indicated marker by immunocytochemistry also given. The numbers of marker positive cells were divided by the total number of DAPI positive cells. Mean with S.D. n = 3 independent fields. Scale bar, 50 μm
Applications of the method:
Since our approach to generate organ-specific mesoderm recapitulates key steps of organogenesis, one of the most obvious applications is as platform to study the mechanisms of human splanchnic mesenchyme development, which is virtually unexplored. For example, it is possible to use our system to examine the molecular mechanisms by which these organ-specific mesoderm progenitors differentiate into mature mesenchymal cell types. Indeed, we have begun to explore how the RM can be further differentiated into smooth muscle and cartilage typical of the trachea and airway (Fig. 4). Furthermore, co-culturing hPSC-derived mesoderm with hPSC-derived endoderm would provide a simplified reductionist system to investigate the complex epithelial-mesenchymal crosstalk of human foregut organogenesis in vitro4. Such in vitro culture models are particularly well suited to large scale genomic analysis of the gene regulatory networks governing SpM diversification which would be very difficult to study in vivo.
Fig. 4. Comparison of the protocols for generating trachea/lung-like mesoderm.

a, Overview of the protocols. Two protocols (this paper and Kishimoto and Morimoto27) were compared. b, Relative mRNA expression of trachea/lung mesoderm genes (FOXF1, NKX6–1, TBX5, TBX4, and WNT2) by qRT-PCR. Each column indicates the average from the 3 independent wells with standard deviation. c, Immunostaining for FOXF1 (green) and NKX6.1 (red) at day7. d, Immunostaining for TBX5 (green) at day7. The images in c and d are maximum intensity projection of confocal stacks with the average % proportion of differentiated cells positive for the indicated marker by immunocytochemistry also given. The numbers of marker positive cells were divided by the total number of DAPI positive cells. Mean with S.D. n=3 independent fields. e, Maximum intensity projection of confocal RNAscope for Wnt2 (green), Nkx6–1 (red) in mouse embryo at E9.514, indicating the proposed WNT activity gradient. f, Maximum intensity projection of confocal stacks from SOX9 (green) and SMA (red) immunostaining at day12. g, Relative mRNA expression of chondrocyte progenitor gene, SOX9 and smooth muscle gene ACTA2 by qRT-PCR. Each column indicates the average from the 3 independent wells with standard deviation. Scale bars, 50 μm
Another application would be to use the method to model congenital diseases of the LPM in vitro. For example, we and other have been investigating the developmental role of the LPM in foregut organogenesis and how disruptions to this process can result in life-threatening birth defects such as esophageal atresia and tracheoesophageal fistulas (EA/TEF)26, 27. While ongoing patient genome sequencing is rapidly identifying candidate causative mutations, a major challenge is to determine whether these genes act in the endoderm or mesoderm and how they impact development. By using patient-derived iPSCs in our protocols, investigators can determine whether the patient’s mutation impact the differentiation of mesoderm or endoderm lineages for a wide array of different congenital anomalies affecting foregut organs.
Finally, our method to generate organ-specific mesoderm can be in tissue engineering approaches to increase the cellular complexity of in vitro–generated organoids. To date most protocols to generate hPSC-derived foregut organoids are primarily epithelial with very little mesenchyme6, 9; the addition of mesoderm from our method could solve this limitation. Indeed, we have recently demonstrated that hPSC-derived SpM from our protocol can be incorporated into both gastric and esophageal organoids resulting in in vitro engineered tissue with increase cellular complexity as well as improved growth and maturation of the tissue28.
Comparison with other methods:
There have been a few other reports of protocols that differentiate hPSCs into non-cardiac LPM lineages, such as hepatic or respiratory mesenchyme21, 29–31, but for the most part these are not based on a systematic analysis comparing the development of different mesoderm subtypes as we have done. In Figures 4 and 5, we present a comparison of these other protocols to our methods.
Fig. 5. Comparison of the protocols for generating liver-like mesoderm.
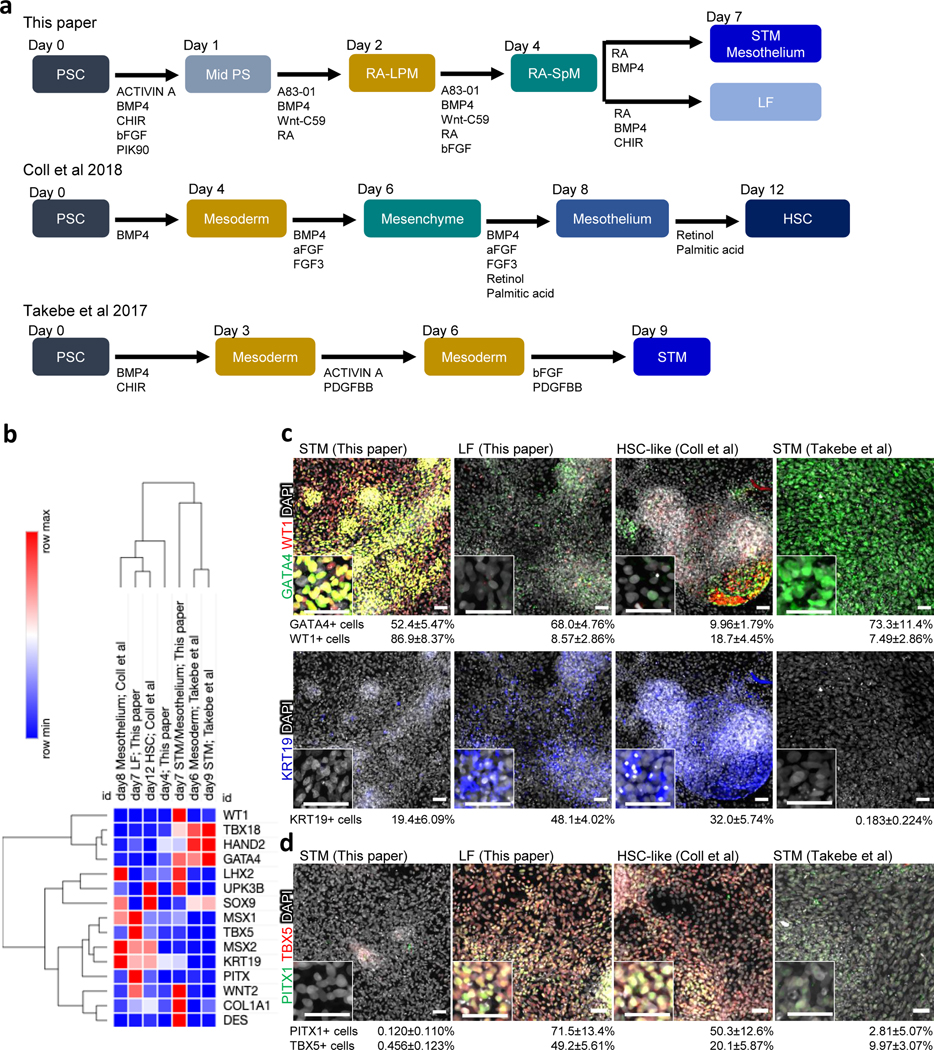
a, Overview comparing three protocols (this paper, Coll et al.30 and Takebe et al.29). b, Heat map based on relative mRNA expression of liver mesenchyme gene expression from qRT-PCR. Each row indicates the average from the three independent wells. c, Immunostaining for GATA4 (green), WT1 (red), and KRT19 (blue). d, Immunostaining for PITX1 (green) and TBX5 (red). The images in c and d are maximum intensity projection of confocal stacks with the average % proportion of differentiated cells positive for the indicated marker by immunocytochemistry also given. The numbers of marker positive cells were divided by the total number of DAPI positive cells. Mean with S.D. n=3 independent fields. Scale bar; 50μm
Previous studies have reported the co-development of a small amount of mesenchyme in lung orgaoinds33–35. However, these protocols were optimized to definitive endoderm (DE) epithelium differentiation and the tissue-specific characteristics of the mesenchyme has not been fully assessed. Part of the problem has been our limited understanding of LPM patterning in the foregut. We and others have recently defined how the foregut mesoderm is patterned along the dorsal-ventral axis into MSC/NKX6–1/WNT4+ dorsal EM that gives rise to esophageal smooth muscle and ventral respiratory mesenchyme (RM) that expresses TBX4/TBX5/NKX6–1/WNT2 and gives rise to smooth muscle of dorsal trachea and SOX9+ chondrocytes on the ventral-lateral trachea and large airways14. In two back-to-back publications, we recently reported similar methods to generate RM: Han et al.14 which report here and Kishimoto and Morimoto27. Figure 4 shows a direct comparison of these methods.
Both protocols generate RM that express typical markers, but their relative expression levels varied between the protocols. Quantitative RT-PCR (qRT-PCR) analysis revealed that NKX6–1 and TBX5 expression was enriched in the cells from our protocol here but that TBX4 and WNT2 were higher in the cells generated by the protocol of Kishimoto and Morimoto27[AU: Here and elsewhere in the paragraph are the changes from “Kishimoto et al. 2020” and “Kishimoto 2020” to Kishimoto and Morimoto27 correct?] (Fig. 4b-d). Immunostaining confirmed that the numbers of NKX6–1+ and TBX5+ cells was greater in our current protocol (NKX6–1+; current protocol 24.8 ± 1.97% and former protocol 13.2±4.28%, TBX5+; current protocol 48.4 ± 6.60% and former protocol 11.2 ± 10.0%). This suggests that the Kishimoto and Morimoto protocol27 generates more ventral RM, whereas the protocol here produced more medial RM, consistent with in vivo expression patterns in the mouse foregut (Fig. 4e) 14, 21. The biggest difference between the two protocols is a higher concentration and longer duration of CHIR treatment (WNT activation) in the Kishimoto and Morimoto protocol27, consistent with recent results in mice that differential WNT activity patterns the tracheal mesenchyme14, 21, 36. Even though the two protocols generated slightly different RM character, prolonged culture until day 12 showed that both cultures were similarly competent to differentiate into SOX9-positive chondrocyte progenitors and smooth muscle actin (SMA)-positive smooth muscle cells (Fig. 4f,g) like differentiated tracheal mesenchyme.
In vivo fetal hepatoblasts derived from the ventral foregut endoderm invade the STM (a transient mesoderm population posterior to the heart) to form the liver bud. The STM then gives rise to mesothelium surrounding the liver bud and interstitial liver fibroblasts (LF), which subsequently become hepatic stellate cells (HSCs)32. Several groups have described methods to differentiate hPSCs into mesenchyme similar to that found in the fetal liver29–31. For example, Takebe et al. reported hPSC-derived liver mesenchyme characteristic of the STM29, while Coll et al. generated PDGFRβ+ HSC-like cells via a mesothelium intermediate30. All three protocols use BMP and FGF, which are known to play a role in hepatic development (Fig. 5a), however only Takebe uses PDGFBB, but not RA, which is used by Coll and our protocol. Moreover, our LF protocol uses Wnt stimulation while Coll uses palmitic acid. Thus, it is possible that these different protocols generate different types of hepatic mesoderm. We therefore performed side-by-side experiments to identify differences (Fig. 5a and Box 1)14, 29, 30.
qRT-PCR analysis showed that the d7 STM/mesothelium from our protocol expressed most of the known in vivo markers of STM (GATA4/WT1/WNT2/TBX18) and mesothelium (LHX2/WT1/UPK1B/UPK3B) suggesting a mixed cell population of cells transitioning from STM to mesothelium, whereas the LF population from our protocol was rather distinct expressing markers characteristic of fetal LFs and HSCs (PITX1/KRT19/MSX1/MSX2/TBX5/COL1A1/DES) (Fig. 5b). In comparison, the STM generated by the Takebe protocol expressed high level of early STM markers (HAND2/GATA4/TBX18) but had low expression of mesothelium and fibroblasts markers, suggesting that it represents an earlier developmental stage than the STM-like population generated by our protocol. In contrast, Coll protocol generated cells that express both mesothelium and liver fibroblasts/HSCs markers but not genes typical of early STM suggesting that these are a mixed cell population of slightly later developmental stage. Hierarchical clustering of the qRT-PCR data indicated that our d7 STM/mesothelium had a transcriptional profile more similar to the Takebe STM, whereas our d7 LF was similar to the Coll d12 HSC-like cells (Fig. 5b). Immunostaining also showed that both our STM/mesothelium and the Takebe d9 STM are enriched in GATA4+ cells (this paper: 52.4±5.47% and Takebe et al.: 73.3±11.4%) (Fig. 5c), but that only our STM/mesothelium was WT1+ (86.9±8.37 %) (Fig. 5c). Furthermore, both our LF and Coll d12 HSC-like cultures contained PITX1+ (this paper: 71.5 ± 13.4% and Coll et al.: 49.2±5.61%) or KRT19+ cells (this paper: 49.2 ± 5.61% and Coll et al.: 20.1 ± 5.87 %) consistent with liver fibroblasts (Fig. 5c,d and Supplementary Fig. 1)14. Thus, these protocols generate similar liver mesenchyme cell types but appear to be at slightly different developmental stages or mixed populations, with our approach being able to generate the full spectrum of mesoderm progenitors found in the developing liver bud.
Experimental design:
To generate organ-specific-like mesenchyme, we differentiated hPSCs in a stepwise process mimicking foregut organogenesis in vivo14. In addition we empirically tested culture conditions including duration, concentration and combinations of growth factors and chemical components to optimize differentiation of each mesenchyme population characterized by the expression of tissue-specific makers. Our differentiation protocols have proven to be effective for both male and female human embryonic stem (ES) cells (H1 and H9)25 as well as induced pluripotent stem cells (iPSC72_3)6. For each differentiation experiments, we prepared at least three replicates to obtain statistically rigorous results.
Limitations:
Two limitations of our current protocols are differentiation efficiency and heterogeneity of the cell populations. While our protocol generates >90% FOXF1-positive population at day 4 for the CG-SpM, pFG-SpM and aFG-SpM (Fig. 2), later the differentiation becomes less efficient for some lineages: 60–90% WT1+ liver septum transversum/mesothelium (STM/mesothelium), 70–80% PITX1+ liver fibroblasts-like (LF), and ~35% NKX6.1+ respiratory-like mesoderm (RM) (Figs. 3d-e, 4c, 5c, d). Moreover, while qRT-PCR shows that the BARX1High/NKX3.2High GM, MSCHigh EM and NKX6–1High/TBX5High RM populations are clearly distinct (Fig. 3c and Table 1), we have not found good antibodies to proteins like BARX1, MSC or NXK3–2 that would allow us to quantify the proportion of cells that express these markers in different populations by immunostaining, despite extensive testing. In the future it will also be important to identify and validate lineage-specific cell-surface proteins that can be used in FACs to easily quantify and purify the different populations. Supplementary Fig. 1c shows the expression of candidate surface protein genes from the published mouse scRNA-seq data, which can also be explored at https://research.cchmc.org/ZornLab-singlecell/.
One possible explanation for incomplete differentiation maybe the fact that we only manipulated a handful of pathways (RA, HH, WNT, and BMP). For example, based on mouse scRNA-seq, in vivo the EM should express Nkx6–1 and Tbx5, but these are missing in the hPSC-derived esophageal-like cultures. We postulate that additional signals, currently absent from our cultures, might participate in organ-specific mesenchyme differentiation, which include PDGF, TGFβ, FGFs and NOTCH. A further analysis of the mouse signaling roadmap might help identify other candidate pathways to optimize the protocol. In addition, prolonged culture should promote more complete differentiation into smooth muscle, mature mesothelium and tissue resident fibroblasts. It is also possible that the human mesenchyme development is just different than mouse, in which case empirical testing of pathways and an examination of single-cell data from early human fetal samples would be helpful.
Materials
!CAUTION When handling cells and reagents, take the necessary precaution. Use proper personal protective equipment (PPE) at biosafety level (BSL)-2 for tissue culture experiment and BSL-1 for non-tissue culture experiment. Refer to specific material safety data sheets for additional information.
Biological materials
Human pluripotent stem cell lines. We used three hPSC lines. WA01-H1 (RRID: CVCL_9771) and WA09-H9 (RRID: CVCL_9773), human embryonic stem cells, were purchased from WiCell (NIH approval number NIHhESC-10–0062). iPSC72_3 (RRID: CVCL_A1BW), human induced pluripotent stem cells, was generated by the CCHMC Pluripotent Stem Cell Facility.
!CAUTION All relevant institutional and governmental regulations for the use of hPSCs must be followed. This work used NIH-approved embryonic stem cell lines and was approved by Embryonic Stem Cell Research Oversight (ESCRO) committee (protocol EIP200117) and CCHMC Biosafety Protocol (protocol IBC2021–0011).
!CAUTION Cell lines should be regularly checked to ensure that they are authentic and not infected with mycoplasma.
Reagents
! CAUTION When handling reagents, take the necessary precautions. Use proper personal protective equipment for BSL-2 for tissue culture work and BSL-1 for non–tissue culture work. Refer to specific reagent material safety data sheets for additional information if unfamiliar with the reagents.
Growth medium and supplements
CRITICAL We have not tested growth medium and supplements from other vendors.
mTeSR1 medium (StemCell Technologies, cat. no. 85850)
DMEM/F12 medium (Thermo Fisher Scientific, cat. no. 11330–032)
Advanced DMEM-F12 (Invitrogen, cat. no. 12634–010)
Matrigel (basement membrane matrix; Corning, cat. no. 354234)
Geltrex (LDEV-Free hESC-qualified Reduced Growth Factor Basement Membrane Matrix, Thermo Fisher Scientific, cat. no. A14133–02)
B27 supplement (50×, without vitamin A; Thermo Fisher Scientific, cat. no. 12587–010)
Penicillin–streptomycin (100×; Thermo Fisher Scientific, cat. no. 15140–122)
N2 supplement (100×; Thermo Fisher Scientific, cat. no. 17502–048)
HEPES buffer (1M; Thermo Fisher Scientific, cat. no. 15630–080)
Glutamax (100×, Thermo Fisher Scientific, cat. no. 35050061)
Enzymes and growth factors
CRITICAL We have not tested enzymes and growth factors from other vendors.
Accutase (Sigma Aldrich, cat. no. A6964)
Dispase (Thermo Fisher Scientific, cat. no. A34181)
Activin A (Cell Guidance Systems, cat. no. GFH6)
Bone morphogenetic protein 4 (BMP4; R&D Systems, cat. no. 314-BP-050)
Basic fibroblast growth factor (bFGF; R&D Systems, cat. no. 233-FB)
CHIR99021 (CHIR; Tocris, cat. no. 4423)
Thiazovivin (Tocris, cat. no. 3845/10)
PIK90 (EMD Millipore, cat. no. 528117–5MG)
CRITICAL As PIK90 is photosensitive, wrap stock tubes and plates in foil and turn the biosafety cabinet light off to prevent light exposure.
2-phospho-L-ascorbic acid trisodium salt (Sigma, cat. no. 49752)
XAV-939 (Selleckchem.com, cat. no. S1180)
A83–01 (Tocris, cat. no. 2939)
CRITICAL As A83–01 is photosensitive, wrap stock tubes and plates in foil and turn the biosafety cabinet light off to prevent light exposure.
Wnt-C59 (Cellagen, cat. no. C7641–10)
Retinoic acid (RA; Sigma, cat. no. R2625)
CRITICAL As RA is photosensitive, wrap stock tubes and plates in foil and turn the biosafety cabinet light off to prevent light exposure.
Purmorphamine (PMA; TOCRIS, cat. no. 4551)
Noggin (R&D systems, cat. no. 6057-NG-100)
Immunostaining reagents
Paraformaldehyde (PFA; Thermo Fisher Scientific, cat. no. T353–500)
Normal donkey serum (Jackson ImmunoResearch Labolatories, Inc, cat. no. 017–000-121)
Tween-20 (Fisher Scientific, cat. no. BP337–100)
Triton-X100 (Sigma-Aldrich, cat. no. X100)
Phosphate buffer solution (PBS; Sigma, cat. no. T8787)
Fluoromount-G fluorescent mounting medium (Southern Biotech, cat. no. 0100–01)
Antibodies (Table 2)
Table 2.
Antibodies
| Antibody | Company | Cat. no. | Species | Dilution | RRID |
|---|---|---|---|---|---|
|
| |||||
| CDH1 | Cell Signaling Technologies | #3195 | Rabbit | 1:400 | AB_2291471 |
| FOXF1 | R&D Systems | AF4798 | Goat | 1:500 | AB_2105588 |
| GATA4 | Santacruz | sc-1237 | Goat | 1:200 | AB_2108747 |
| ISL1 | Abcam | ab109517 | Rabbit | 1:200 | AB_10866454 |
| KRT19 | eBioscience | 14–9898-82 | Mouse | 1:50 | AB_10598673 |
| NKX6.1 | Developmental Studies Hybridoma Bank | F55A12 | Mouse | 1:40 | AB_532379 |
| OCT3/4 | Santacruz | sc-5279 | Mouse | 1:100 | AB_628051 |
| PITX1 | ThermoFisher Scientific | PA5–52600 | Rabbit | 1:100 | AB_2645587 |
| SMA | Sigma | C6198 | Mouse | 1:500 | AB_476856 |
| SOX2 | R&D Systems | AF2018 | Goat | 1:200 | AB_355110 |
| SOX9 | Abcam | AB5535 | Rabbit | 1:500 | AB_2239761 |
| TBX5 | Santacruz | sc-17865 | Goat | 1:200 | AB_2200830 |
| TBXT | Cell Signaling Technologies | #81694 | Rabbit | 1:200 | AB_2799983 |
| VIM | Santacruz | sc-7557 | Goat | 1:200 | AB_793998 |
| WT1 | Abcam | ab89901 | Rabbit | 1:200 | AB_2043201 |
Equipment
Portable Pipet-Aid XP pipette controller (Drummond, cat. no. 4–000-101)
Pipetman single-channel pipettes (2, 20, 200, and 1,000μl; Gilson, cat. no. F144801, cat. no. F123615, cat. no. F123601, and cat. no. F123602)
Horizontal clean bench (Labconco, cat. no. 3600004)
Labgard class II type A2 biological safety cabinet (NuAire, cat. no. NU-425–400)
−80°C Freezer (Thermo Scientific, cat. no. UXF700086D)
Forma Steri-Cycle i160 CO2 incubator (Thermo Scientific, cat. no. 51030533)
Stereomicroscope (Leica, cat. no. S8APO)
Inverted microscope (Nikon, TMS model)
Nunclon delta surface tissue culture dish (6 well; Nunc, cat. no. 140675)
Nunclon delta surface tissue culture dish (12 well; Nunc, cat. no. 150628)
Nunclon delta surface tissue culture dish (24 well; Nunc, cat. no. 142475)
Serological pipettes (5 and 10 ml; Falcon, cat. nos. 357543 and 357551)
Sterilized filter pipette tips (10, 20, 200 and 1250 μl (VWR, cat. nos. 76322–528, 76322–134, 76322–150, and 76322–156)
Millipore 0.22-μm conical sterilization tubes (Millipore, cat. no. SCGP00525)
1.5-ml Low adhesion microcentrifuge tubes (USA scientific, cat. no. 1415–2600)
Cell culture coverslips (sterile plastic; Thermanox; Thermo Scientific, cat. no. 174969)
Microscope slides (Superfrost; Fisher Scientific, cat. no. 12–550-133)
Microscope cover glass (Fisher Scientific, cat. no. 12–545-M)
Hydrophobic pen (ImmeEdge pen; Vector Laboratories, cat. no. H-4000)
Nikon A1 single-photon confocal microscope with NIS-Elements Advanced Research imaging software (Nikon, http://www.microscope.healthcare.nikon.com/en_AOM/products/software/nis-elements)
GraphPad Prism 9.1.2 (GraphPad Software, http://www.graphpad.com/)
Reagent setup
Dispase
Resuspend dispase in DMEM/F12 at a final concentration of 1mg ml-1. After sterilization with filter-sterilization tube, make 10-ml aliquots and store at −20 °C for up to 6 months.
Accutase
Thaw vial on ice or at 4 °C. Make 5-ml aliquots and store at −20 °C.
Aliquots of hESC-qualified Matrigel for hPSCs maintenance.
Thaw a bottle of hESC-qualified Matrigel on ice or at 4 °C. Pre-chill sterile microcentrifuge tubes on ice. Make 270–350 μl of aliquots and store at −80 °C for up to 6 months.
CRITICAL Matrigel has the property to solidify at room temperature (X–Y °C)[AU: Please define room temperature here by replacing ‘X–Y °C’ with the appropriate range]. Matrigel should be aliquoted quickly on ice or at 4 °C.
Aliquots of Geltrex for mesoderm differentiation from hPSCs.
Thaw a bottle of Geltrex on ice or at 4 °C. Pre-chill sterile microcentrifuge tubes on ice. Make 1-ml aliquots and store at −80 °C for up to 6 months.
CRITICAL Geltrex has the property to solidify at room temperature. Geltrex should be aliquoted quickly on ice or at 4 °C.
Matrigel coating plates for hPSC maintenance
Coat six-well plate with hESC-qualified Matrigel according to the manufacturer’s procedure. The dilution is calculated for individual lot based on protein concentration. Thaw Matrigel aliquot on ice and dilute 270–350 μl of the aliquot in 25 ml of ice-cold DMEM/F12 medium to coat four six-well plates. 1 ml per well is required. The plates wrapped in Parafilm can be kept for 2 weeks in 4 °C.
Geltrex coating plates for mesoderm differentiation from hPSCs.
Coat 12-well plates or coverslips placed in 24-well plates with 1% (vol/vol) Geltrex diluted in DMEM/F12 according to the manufacturer’s procedure. Either 1 ml per well in a 12-well plate or 0.5 ml per well in a 24-well plate is required. The plates wrapped in Parafilm can be kept for 2 weeks at 4 °C.
Reconstitution of growth factor and small molecules
Reconstitute growth factors and small molecules as summarized in Table 3. Reconstitution was largely based on manufacturers’ instruction. Divide growth factors into aliquots and store at −80 °C for up to 6 months. Thaw growth factors on ice before use and keep at 4 °C for 2 weeks. We do not re-freeze growth factors once thawed. Divide small molecules into aliquots and store at −20 °C for up to 6 months. Avoid repeating freeze–thaw cycles.
Table 3.
Reconstitution of growth factors and small molecules
| Growth factors and small molecules | Solvent | Stock concentration | Long-term storage |
|---|---|---|---|
|
| |||
| A83–01* | DMSO | 10 mM | −20 °C |
| Activin A | 1XPBS, 0.1% BSA | 100 μg ml−1 | −80 °C |
| bFGF | 1XPBS, 0.1% BSA | 100 μg ml−1 | −80 °C |
| BMP4 | 1XPBS, 0.1% BSA, 4mM HCl | 50 μg ml−1 | −80 °C |
| CHIR99021 | DMSO | 6 mM | −20 °C |
| Noggin | 1XPBS, 0.1% BSA | 200 μg ml−1 | −80 °C |
| PMA | DMSO | 20 mM | −20 °C |
| PIK90* | DMSO | 0.5 mM | −20 °C |
| RA* | DMSO | 20 mM | −80 °C |
| Wnt-C59 | DMSO | 10 mM | −20 °C |
| XAV-939 | DMSO | 2 mM | −20 °C |
A83–01, PIK90 and RA are photosensitive. Wrap aliquots in foil to avoid light.
Gut medium
Supplement advanced DMEM/F12 medium with 1x B27 without vitamin A, 1x N2, 15 mM Hepes, 1xGlutaMAX, 100 unit ml−1 penicillin–streptomycin. Gut medium can be stored at 4 °C for 2 weeks.
Middle primitive streak induction medium (days 0–1)
Add growth factors and small molecules (30 ng ml−1 Activin A, 6 μM CHIR, 40 ng ml−1 BMP4, 20 ng ml−1 bFGF, 100 nM PIK90) to gut medium. The medium, including growth factors and small molecules, should be freshly prepared each day.
Lateral plate mesoderm induction medium (days 1–2)
For CG-LPM induction, add growth factors and small molecules (1μM A83–01, 30 ng ml−1 BMP4, and 1μM Wnt-C59) to gut medium. The medium, including growth factors and small molecules, should be freshly prepared each day. For pFG-LPM induction, add growth factors and small molecules (1μM A83–01, 30 ng ml−1 BMP4, 1μM Wnt-C59, and 2μM RA) to gut medium. The medium, including growth factors and small molecules, should be freshly prepared each day. For aFG-LPM induction, add growth factors and small molecules (1 μM A83–01, 30 ng ml−1 BMP4, 1 μM Wnt-C59, 2 μM RA, and 1 μM PMA) to gut medium. The medium, including growth factors and small molecules, should be freshly prepared each day.
Splanchnic mesoderm induction medium (days 2–4)
For CG-SpM induction, add growth factors and small molecules (1μM A83–01, 30ng ml−1 BMP4, 1μM Wnt-C59, and 20 ng ml−1 bFGF) to gut medium. The medium, including growth factors and small molecules, should be freshly prepared each day. For pFG-SpM induction, add growth factors and small molecules (1 μM A83–01, 30 ng ml−1 BMP4, 1 μM Wnt-C59, 20 ng ml−1 bFGF, and 2 μM RA) to gut medium. The medium, including growth factors and small molecules, should be freshly prepared each day. For aFG-SpM induction, add growth factors and small molecules (1 μM A83–01, 30 ng ml−1 BMP4, 1μM Wnt-C59, 20 ng ml−1 bFGF, 2 μM RA, and 1 μM PMA) to gut medium. The medium, including growth factors and small molecules, should be freshly prepared each day.
Liver septum transversum induction medium (days 4–7)
Add growth factors (2 μM RA and 30 ng ml−1 BMP4) to gut medium. The medium, including growth factors, should be freshly prepared each day.
Liver fibroblast induction medium (days 4–7)
Add growth factors and a small molecule (2μM RA, 30ng ml−1 BMP4, and 6μM CHIR) to gut medium. The medium, including growth factors, should be freshly prepared each day.
Respiratory mesenchyme induction medium (days 4–6)
Add growth factors and a small molecule (2μM RA, 30ng ml−1 BMP4, and 2μM PMA) to gut medium. The medium, including growth factors, should be freshly prepared each day.
Respiratory mesenchyme induction medium (days 6–7)
Add growth factors and a small molecule (2μM RA, 30ng ml−1 BMP4, 2μM PMA, and 1μM CHIR) to gut medium. The medium, including growth factors, should be freshly prepared each day.
Esophageal and gastric mesenchyme induction medium (days 4–6)
Add growth factors and a small molecule (2 μM RA and 2 μM PMA) to gut medium. The medium, including growth factors, should be freshly prepared each day.
Esophageal and gastric mesenchyme induction medium (days 6–7)
Add growth factors and a small molecule (2μM RA, 2μM PMA and 200 ng ml−1 Noggin) to gut medium. The medium, including growth factors, should be freshly prepared each day.
PBST
Add 0.5% (vol/vol) Triton-X100 to PBS. PBST can be stored at room temperature for 6 months.
Normal donkey serum (NDS)
Rehydrate with 10 ml of sterile H2O. Divide into 1-ml aliquots and store at −80 °C for 12 months. Avoid repeated freeze–thaw cycles.
Immunostaining blocking buffer
Add 5% (vol/vol) NDS to PBS. The diluted NDS-PBS can be stored at 4 °C for up to 2 weeks but make fresh blocking buffer before use and keep on ice.
Human pluripotent stem cell lines
Maintain hPSCs in feeder-free condition on hESC-qualified Matrigel coated six-well plates at 37°C in a 5% CO2 tissue incubator. Feed cells with 2 ml/well mTeSR1 medium every day. hPSCs will grow as colonies and reach to 70–90% confluency after 4 days in culture. These cells should be dispase-passaged as cell clumps, commonly at a well/well ratio of 1:6, into hESC-qualified Matrigel coated six-well dishes (see Matrigel coating plates for hPSCs maintenance section above) every 4 days for maintenance. Briefly, wash cells with 2ml/well DMEM/F-12. Incubate the cells with dispase for 6 minutes at 37°C in a 5% CO2 tissue incubator. Aspirate dispase and then, wash the cells with 2 ml/well DMEM/F12 twice. Add 3.2ml/well mTeSR1 medium and scrape cells. Pipette the cell cluster four or five times. Add 1.5 ml/well mTeSR1 to hESC-qualified Matrigel coated 6-well dishes. Add 0.5ml/well cell suspension. Incubate the cells 37°C in a 5% CO2 tissue incubator. Cells should be observed every day and differentiated cells removed by scratching with a pulled-glass Pasteur pipette.
Procedure
CRITICAL All tissue culture steps in the protocol should take place in a sterile environment and be performed with aseptic technique to prevent culture contamination.
Single-cell passage of hPSCs for mesoderm differentiation (day −1) •Timing 24 hours (30–45 minutes for plating cells)
-
1
Pre-coat 12-well plates with Geltrex as described in Reagent setup.
-
2
Pre-warm mTeSR and DMEM/F12 medium at 37 °C. The total volume of medium depends on the numbers of wells: mTeSR1 (2ml/well in 6-well plate) and DMEM/F12 (4.4ml/well in 6-well plate).
-
3
Take a six-well plate of undifferentiated hPSC colonies that are 70–90% confluent (see Reagent setup). Make a note of the confluency as this will determine the subsequent dilution ratio (Step 13). Remove spontaneously differentiated cells by scratching with a pulled -glass Pasteur pipette. Aspirate mTeSR1 from wells.
-
4
Wash wells with 2ml of DMEM/F12 medium. Aspirate DMEM/F12.
-
5
Add 0.6ml Accutase to each well to dissociate hPSC colonies to single cells.
-
6
Incubate at 37°C for 5 min. Observe under a microscope to confirm dissociation to single cells.
? Troubleshooting
-
7
Add 2.4ml pre-warmed DMEM/F12 supplemented with 1μM Thiazovivin to dilute Accutase.
-
8
Triturate hPSC colonies by gentle pipetting to break down any undissociated colonies.
-
9
Centrifuge the cells at 1,300 x g for 3minutes at room temperature.
-
10
Meanwhile, supplement prewarmed mTeSR1 with 1μM Thiazovivin.
-
11
Aspirate media from tube.
CRITICAL STEP Do not aspirate the cells at the bottom of the tube.
-
12
Resuspend cells in prewarmed mTeSR1 supplemented with 1μM Thiazovivin.
-
13
Further diluted the suspended cells in mTeSR1 supplemented with 1μM Thiazovivin. The dilution ratio is determined by the original confluency of the cell in 6-well plate (as noted in Step 3). For wells at 90% confluency, cells are diluted at the ratio of 1:20 (resulting in an approximate plating density of 1.0–2.0×105 cells ml−1). For wells at 70% confluency, the cells are diluted at 1:16 (approximate density of 1.0–2.0×105 cells ml−1).
-
14
Remove the Geltrex from prepared Geltrex coated 12-well or 24-well plate (from Step 1). Plate 1ml or 0.5ml cell suspension in a well of 12-well plate or 24-well plate, respectively.
-
15
Gently rock the plates by hand to ensure that single cells are evenly distributed in the wells and place the 12-well or 24-well plate in a tissue culture incubator.
-
16
Incubate cells overnight in an incubator with 5% CO2 at 37°C.
Differentiation of hPSCs into middle primitive streak (days 0–1) •Timing 24 hours (15–30 minutes for changing medium)
-
17Observe the 12-well or 24-well plate with hPSCs under an inverted microscope. The monolayer of hPSCs should be 30–40% confluent (Fig. 2a). At this time point assay some cells by immunostaining and qRT-PCR:
- Collect cells on coverslips with forceps to check the pluripotency of hPSCs by immunostaining for OCT3/4 and SOX2. The protocol for immunostaining is described in Box 1.
- For rest of the cells proceed to Step 18 to continue differentiation to middle primitive streak.
Table 4.
Primers
| Gene | Forward primer | Reverse primer |
|---|---|---|
|
| ||
| BARX1 | CCAGTGGGAACTTGAACACC | CTGAAGTTCGGCGTGCAG |
| COL1A1 | GACGAAGACATCCCACCAATCA | GGACTCGTCACAGATCACGTC |
| CYP26A1 | GCTGCCTCTCTAACCTGCAC | TGCTTTAGTGCCTGCATGTC |
| DES | AATGACCGCTTCGCCAACTA | GGTTAGTGAGCACCTCCACC |
| FOXA2 | GGGAGCGGTGAAGATGGA | TCATGTTGCTCACGGAGGAGTA |
| FOXF1 | AGCAGCCGTATCTGCACCAGAA | CTCCTTTCGGTCACACATGCTG |
| GATA4 | TAGCCCCACAGTTGACACAC | GTCCTGCACAGCCTGCC |
| GLI1 | GGGATGATCCCACATCCTCAGTC | CTGGAGCAGCCCCCCCAGT |
| HAND2 | CACTTCTGAATTCGCCCAC | TCTCTTTGACCCCCTTTGAG |
| HOXA5 | CGCCCAACCCCAGATCTAC | CGGGCCGCCTATGTTGT |
| ISL1 | AGATTATATCAGGTTGTACGGGATCA | ACACAGCGGAAACACTCGAT |
| KRT19 | CACCAGCCGGACTGAAGAAT | GCAGGTCAGTAACCTCGGAC |
| LHX2 | TCGGGACTTGGTTTATCACCT | GCAAGCGGCAGTAGACCAG |
| MSC | TATGAGAACGGCTACGTGCAC | AGTCCGATTTAAGCGGTGGTT |
| MSX1 | CTCCGCAAACACAAGACGAAC | GGCGGTTCTGGAACCATATCT |
| MSX2 | CGCCAAGACATATGAGCCCT | GTTCTGCCTCCTGCAGTCTT |
| NKX2–5 | TGGAGAAGACAGAGGCGGACAA | ATAGACCTGCGCCTGCGAGAA |
| NKX3–2 | CAACACCGTCGTCCTCG | CCGCTTCCAAAGACCTAGAG |
| NKX6–1 | ATGACAGAGAGTCAGGTCAAGG | CTCCGAGTCCTGCTTCTTCTT |
| OCT3/4 | TGAGAGGCAACCTGGAGAATT | TTTCTTTCCCTAGCTCCTCCC |
| PITX1 | TTCTTGGCTGGGTCGTCT | TCGTCTGACACGGAGCTG |
| PTCH1 | CCACAGAAGCGCTCCTACA | CTGTAATTTCGCCCCTTCC |
| SOX2 | GCTTAGCCTCGTCGATGAAC | AACCCCAAGATGCACAACTC |
| SOX9 | GTAATCCGGGTGGTCCTTCT | GTACCCGCACTTGCACAAC |
| TBX4 | TGATCATCACTAAGGCTGGCAG | ACAGAACTTGTAGCGATGGTCAT |
| TBX5 | ACAAAGTGAAGGTGACGGGCCTTA | ATCTGTGATCGTCGGCAGGTACAA |
| TBX18 | TTAACCTTGTCCGTCTGCCTGAGT | GTAATGGGCTTTGGCCTTTGCACT |
| TBX20 | GGCGACGGAGAACACAATCAA | CTGGGCACAGGACGACTTC |
| TBXT | CAGTGGCAGTCTCAGGTTAAGAAGGA | CGCTACTGCAGGTGTGAGCAA |
| UPK1B | TGGAAGCAACGAACAGTTGA | CTACCGTGTGCGCAGAAA |
| UPK3B | ATACCGTCTGGCTCGTGGTG | CAGGGGCAGCGTCATGTAGT |
| VIM | ATTCCACTTTGCGTTCAAGG | CTTCAGAGAGAGGAAGCCGA |
| WNT2 | CTGTATCAGGGACCGAGAGG | CCCACAGCACATGACTTCAC |
| WT1 | ATAGGCCAGGGCATGTGTATGTGT | AGTTGCCTGGCAGAACTACATCCT |
? Troubleshooting
-
18
Pre-warm middle primitive streak induction media and DMEM/F12 media at 37 °C. The total volume of medium depends on the numbers of wells: 1ml/well in 12-well plate.
-
19
Aspirate media from wells.
-
20
Wash wells with DMEM/F12 media pre-warmed at 37 °C (1 ml/well in a 12-well plate or 0.5 ml/well in a 24-well plate). Aspirate DMEM/F12 media.
-
21
Add middle primitive steak induction media pre-warmed at 37 °C (1 ml/well in a 12-well plate or 0.5 ml/well in a 24-well plate).
-
22
Incubate cells overnight in an incubator with 5% CO2 at 37 °C.
CRITICAL STEP Because PIK90 is photosensitive, wrap stock tubes and plates in foil and turn the biosafety cabinet light off to prevent light exposure.
Differentiation of Mid. PS into LPM (days 1–2) •Timing 24 hours (15–30 minutes for changing medium)
-
23The following day, observe the 12-well or 24-well plate containing middle primitive streak under an inverted microscope. The cells should reach 40–50% confluency (Fig. 2a). Floating dead cells and debris can be observed. At this time point assay some cells by immunostaining and qRT-PCR:
- Collect cells on coverslips with forceps to check the efficiency of mid PS induction by immunostaining for TBXT. The protocol for immunostaining is described in Box 1.
- For rest of the cells proceed to Step 24 to continue differentiation of Mid.PS into LPM.
-
24
Pre-warm lateral plate mesoderm induction media and DMEM/F12 media at 37 °C. The total volume of medium depends on the numbers of wells: lateral plate mesoderm induction media (1ml/well in 12-well plate) and DMEM/F12 media (1ml/well in 12-well plate).
-
25
Aspirate the media. Wash cells with pre-warmed DMEM/F12 media (1 ml/well in a 12-well plate or 0.5 ml/well in a 24-well plate). Aspirate DMEM/F12 media.
-
26
Add pre-warmed lateral plate mesoderm induction media (1 ml/well in a 12-well plate or 0.5 ml/well in a 24-well plate).
-
27
Incubate cells overnight in an incubator with 5% CO2 at 37 °C.
CRITICAL STEP Because RA and A83–01 are photosensitive, wrap stock tubes and plates in foil and turn the biosafety cabinet light off to prevent light exposure.
Differentiation of LPM into SpM (day2 – 4) •Timing 48 hours (15–30 minutes for changing medium)
-
28
After 24 hours, observe the 12-well or 24-well plate containing LPM under an inverted microscope. The monolayer of cells should be 80–100% confluent (Fig. 2a).
-
29
Pre-warm splanchnic mesoderm induction media at 37 °C. The total volume of medium depends on the numbers of wells: 1 ml/well in 12-well plate.
-
30
Aspirate the media. Add pre-warmed splanchnic mesoderm induction media (1 ml/well in a 12-well plate or 0.5 ml/well in a 24-well plate). It is not necessary to wash cells with DMEM/F-12 media.
CRITICAL STEP Because RA and A83–01 are photosensitive, wrap stock tubes and plates in foil and turn off the biosafety cabinet light to prevent light exposure.
-
31
Incubate cells overnight in an incubator with 5% CO2 at 37 °C.
-
32
After 24 hours, aspirate media and add fresh pre-warmed splanchnic mesoderm induction media (1 ml/well in 12-well plate or 0.5ml/well in 24-well plate).
-
33
Incubate cells overnight in an incubator with 5% CO2 at 37 °C.
Differentiation of SpM into organ-specific mesoderm (days 4–7) •Timing 72 hours (15–30 minutes for changing medium)
-
34After 24 hours incubation, observe the cells under inverted microscope. Cells should reach 100% confluency (Fig. 2a). At this time point assay some cells by immunostaining and qRT-PCR:
- Collect cells on coverslips with forceps to check the efficiency of SpM induction by immunostaining for FOXF1 and ISL1. The protocol for immunostaining is described in Box 1.
- For rest of the cells proceed to Step 35 to continue differentiation of SpM into organ-specific mesoderm.
? Troubleshooting
-
35
Pre-warm organ-specific mesoderm induction media and DMEM/F12 media at 37 °C. The total volume of medium depends on the numbers of wells: 1 ml/well in 12-well plate.
-
36
Aspirate the media. Wash cells with pre-warmed DMEM/F12 media (1 ml/well in a 12-well plate or 0.5 ml/well in a 24-well plate). Aspirate DMEM/F12 media.
-
37
Add pre-warmed organ mesoderm (liver STM/LF/GM/RM/EM) induction media (1 ml/well in a 12-well plate or 0.5 ml/well in a 24-well plate).
CRITICAL STEP Because RA is photosensitive, stock tubes and plates can be wrapped in foil and turn the biosafety cabinet light off to prevent light exposure.
-
38Refresh the media every day. On day 7, cells are analyzed by immunostaining and qRT-PCR:
- Collect cells on coverslips with forceps to check the efficiency of organ-specific mesoderm differentiation by immunostaining for WT1, PITX1, FOXF1, NKX6.1, TBX5. The protocol for immunostaining is described in Box 1. Fixed cells can be stored, at least less than 6 months, in PBS at 4 °C.
? Troubleshooting
Timing
Steps 1–16, Single-cell passage of hPSCs for mesoderm differentiation (days −1 to 0): 24 hours (30–45 minutes for plating cells)
Steps 17–22, Differentiation of hPSCs into mid PS (days 0–1): 24 hours (15–30 minutes for changing medium)
Step 17, Immunostaining and qRT-PCR for pluripotency markers (Box 1): 24 hours.
Steps 23–27, Differentiation of mid PS into LPM subtypes (days 1–2): 24 hours (15–30 minutes for changing medium)
Step 23, Immunostaining and qRT-PCR for Mid.PS markers (Box 1): 24 hours.
Steps 28–33, Differentiation of LPM into SpM subtypes (days 2–4): 48 h (15–30 min for changing medium)
Steps 34–38, Differentiation of SpM into organ-specific mesoderm (days 4–7): 72 hours (15–30 minutes for changing medium)
Step 34, Immunostaining and qRT-PCR for SpM markers (Box 1): 24 hours.
Step 38, immunostaining and qRT-PCR for lineage-specific markers (Box 1): 24 hours.
Troubleshooting
Troubleshooting advice can be found in Table 5.
Table 5.
Troubleshooting table
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
|
| |||
| 6 | Not enough dissociation of cells | Cell density is too high | Start experiment with a lower density of hPSCs. |
| Not enough agitation of cells | Tap side of the plate. | ||
| 17 | Low cell density | Low concentration of plated cells | Check plating cell density. |
| Low attachment of the plated cells | Check thiazovivin concentration and expiry date. | ||
| Check protein concentration and expiry data of Geltrex. | |||
| Poor growth of the cells | Check the quality of hPSCs.New line of hPSCs should be thawed. | ||
| 34 | Poor differentiation of hPSCs to SpM | Cell density is too high | Check plated cell density and cell confluency at each process. |
| Check cell distribution in the wells. | |||
| Cells should be evenly distributed. | |||
|
|
|||
| Poor induction of pFG-SpM and aFG-SpM | Cell density is too high | Check plated cell density and cell confluency at each process. | |
| Check cell distribution in the wells. | |||
| Cells should be evenly distributed. | |||
| RA might be degraded by light | Use fresh and photo-protected RA. | ||
| 38 | Cells lift off the plate | Cell density is too high | Check plated cell density and cell confluency at each process. |
| Check cell distribution in the wells. | |||
| Cells should be evenly distributed. | |||
| Poor differentiation of hPSCs to organ-specific mesoderm | Cell density is too high | Check plated cell density and cell confluency at each process. | |
| Check cell distribution in the wells. | |||
| Cells should be evenly distributed. | |||
Anticipated results
The protocol starts from 30–40% confluency of hPSCs at day 0. The cells grow to 100% confluency and differentiate into SpM subtypes by day 4 (Fig. 2a). qRT-PCR and immunostaining analyses confirm that day 0 hPSCs express the pluripotent markers OCT3/4 and SOX2 (Supplementary Fig. 2a and b). At day1, OCT3/4 and SOX2 expression is extinguished and >90% cells express the mid PS mesendoderm marker TBXT (Supplementary Fig. 2c and d). By day 4 of the culture all three SpM subtypes robustly express VIM and ~90% of the cells are FOXF1+, while definitive endoderm (DE) markers FOXA2 and CDH18 are almost undetectable (Fig. 2b-f, and Supplementary Fig. 2e). In the absence of RA or HH activation the protocol generates CG-SpM at day 4 that expresses high levels of cardiac markers NKX2–5 and ISL1 (Fig. 2d, f). RA treatment results in a shift from cardiac to visceral FG-SpM with the expression of RA-responsive genes such as HOXA5, CYP26A1, in both the pFG-SpM and aFG-SpM (Fig. 5d). Activation of HH together with RA induces aFG-SpM, and expression of the HH-responsive genes GLI1 and PTCH1. (Fig. 2d). Optimization experiments not shown here indicate that the pFG-SpM is more predisposed to develop into posterior hepatic and gastric lineages whereas the aFG-SpM is predisposed to more anterior RM and EM. These results are consistent with our previous finding that HH signaling is less activated in liver mesenchyme compared with the RM and EM14.
By day 7 the cultures remain confluent monolayers with the STM/mesothelium, GM, RM and EM cells having a polygonal shape and honeycomb arrangement while LF displays an elongated fibroblastic morphology (Fig. 3a, b). Most day 7 cells of each lineage are positive for VIM, but negative for the DE marker FOXA2, and neuroectoderm marker SOX2 (Supplementary Fig. 3). The different cell population can be identified by a unique set of molecular markers inferred from single-cell transcriptomics of the mouse foregut14, a subset of which are presented in Supplementary Figure S1. Activation of RA/BMP signaling in pFG-SpM promotes robust expression of liver STM/mesothelium specific-genes including WT1 and UPK1B, while RA/BMP/WNT induces a LF fate characterized by co-expression of PITX1, MSX2 and TBX5 (Fig. 3c)14. By contrast, addition of the HH agonist PMA to the d4–7 induction cocktails promotes gastric, respiratory and esophageal fate while suppressing liver fate. pFG-SpM or aFG-SpM treated with RA and HH agonist followed by NOGGIN on days 6–7 displays a BARX1/NKX3–2+ gastric or MSC/NKX3–2+ esophageal identity, respectively (Fig. 3c)14. Finally, aFG-SpM treated with RA, BMP4 and HH agonist followed by d6–7 WNT activation differentiated into RM characterized by the co-expression of NKX6–1, TBX5, TBX4 and WNT2 (Fig. 3c and 4b)14. The lack of marker gene expression from other lineages is also important (e.g., lack of MSC in STM, LF, GM and RM).
When possible, immunostaining should be used to quantify the efficiency of differentiation. We expect 60–90% WT1+ liver STM/mesothelium cells and 70–80% PITX1+ LF cells, while the efficiency of TBX5/NXK6–1+ RM differentiation is 20–40% (Fig. 3d, e)14. Immunostaining also shows the differentiation specificity where each cell population should not express other lineage markers, indicate that for the most part these are not mixed cell populations. As mentioned in the “limitations” section we have not yet identified robust antibody markers to quantify the GM and EM populations. This will be important going forward as will the validation of robust cell-surface markers that can be used in FACs to more easily quantify the differentiation efficiency and further optimize the protocol.
Supplementary Material
Box1 Immunostaining •Timing 24 h.
Transfer coverslip from 24-well plate to new 24-well plate.
Add 4%(vol/vol) paraformaldehyde (PFA) and fix the cells for 30 minutes at room temperature.
Remove PFA and wash the cells on coverslip three times with 0.5 ml PBS.
Add 0.5%(vol/vol) Triton X-100 in PBS and permeabilize the cells for 10 minutes at room temperature.
Aspirate Triton X-100 in PBS and wash the cells on coverslip three times with 0.5 ml PBS.
Add 0.3ml of immunostaining blocking buffer (5% Normal Donkey Serum (NDS) in PBS) and block the cells for 1 hour at room temperature.
Transfer identically treated coverslips from 24-well plate to a 10-cm dish.
Add 50μl primary antibody diluted in immunostaining blocking buffer to the cells on coverslip and incubate the cells overnight at 4°C (Table 2).
Transfer coverslip from 10-cm dish to new 24well plate.
Wash the cells three times with 0.5 ml PBS.
Add 0.3ml secondary antibody and DAPI in immunostaining blocking buffer to the cells on coverslip and incubate the cells for 1 hour at room temperature.
Wash the cells three times with 0.5 ml PBS.
Remove coverslip and mount on a microscopic slide with Fluoromount-G.
Visualize the cells under a confocal microscope.
Acknowledgements
We thank Praneet Chaturvedi for generating plots from the mouse scRNAseq data. We would like to thank all members in Zorn, Takebe, Wells and Morimoto laboratories for reagents and feedback. We also thank Chris Mayhew and Amy Pitstick from the Pluripotent Stem Cell Facility as well as Matt Kofron and Evan Meyer from the Confocal Imaging Core at Cincinnati Children’s Hospital for constant support and guidance. This work was supported by grant NICHD P01HD093363 to A.M.Z. and J.W.M. The NIH Director’s New Innovator Award (DP2 DK128799–01) (T.T.), and the New York Stem Cell Foundation (T.T.) and by a CCHMC CURE award to A.M.Z., J.W.M. and T.T. K.K. is supported by a Uehara Memorial Foundation postdoctoral fellowship and a fund for the Promotion of Joint International Research (A) (18KK0423). K.I is supported by Japan Society for the Promotion of Science Overseas Research Fellowship.
APPENDIX
Appendix
Related links
Key references using this protocol
Han, L. et al. Nat. Commun. 11, 4158 (2020): https://doi.org/10.1038/s41467–020-17968-x
Eicher, A. K. et al. Cell Stem Cell 29, 36–51.e6 (2022): https://doi.org/10.1016/j.stem.2021.10.010
Footnotes
Competing interests
K.K., L.H., M.M. and A.M.Z. have filed patent application on this protocol. K.I., C.F.H., M.M., J.M.W. and T.T, declare no competing interest.
Data availability
The main data discussed in this protocol were generated as part of the studies published in the supporting primary research papers14. The raw datasets of qRT-PCR and immunostaining have been deposited in the online open access figshare repository (10.6084/m9.figshare.19723189). The published mouse foregut scRNA-seq data used to infer the signaling roadmap is available from the Gene Expression Omnibus (GEO) database under accession code: GSE136689. The scRNA-seq data can also be explored at https://research.cchmc.org/ZornLab-singlecell/.
References
- 1.Zorn AM & Wells JM Vertebrate endoderm development and organ formation. Annu Rev Cell Dev Biol 25, 221–251 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kraus MR & Grapin-Botton A. Patterning and shaping the endoderm in vivo and in culture. Curr Opin Genet Dev 22, 347–353 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Kechele DO & Wells JM Recent advances in deriving human endodermal tissues from pluripotent stem cells. Curr Opin Cell Biol 61, 92–100 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Iwasawa K. & Takebe T. Organogenesis in vitro. Curr Opin Cell Biol 73, 84–91 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McCracken KW et al. Wnt/β-catenin promotes gastric fundus specification in mice and humans. Nature 541, 182–187 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McCracken KW et al. Modelling human development and disease in pluripotent stem-cell-derived gastric organoids. Nature 516, 400–404 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Múnera JO et al. Differentiation of Human Pluripotent Stem Cells into Colonic Organoids via Transient Activation of BMP Signaling. Cell Stem Cell 21, 51–64.e56 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spence JR et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature 470, 105–109 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Trisno SL et al. Esophageal organoids from human pluripotent stem cells delineate Sox2 functions during esophageal specification. Cell Stem Cell 23, 501–515.e507 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y. et al. 3D modeling of esophageal development using human PSC-derived basal progenitors reveals a critical role for notch signaling. Cell Stem Cell 23, 516–529.e515 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He S. et al. Single-cell transcriptome profiling of an adult human cell atlas of 15 major organs. Genome Biol 21, 294 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Han X. et al. Construction of a human cell landscape at single-cell level. Nature 581, 303–309 (2020). [DOI] [PubMed] [Google Scholar]
- 13.Yu Q. et al. Charting human development using a multi-endodermal organ atlas and organoid models. Cell 184, 3281–3298.e3222 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Han L. et al. Single cell transcriptomics identifies a signaling network coordinating endoderm and mesoderm diversification during foregut organogenesis. Nat Commun 11, 4158 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferretti E. & Hadjantonakis AK Mesoderm specification and diversification: from single cells to emergent tissues. Curr Opin Cell Biol 61, 110–116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tam PP & Loebel DA Gene function in mouse embryogenesis: get set for gastrulation. Nat Rev Genet 8, 368–381 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Sweetman D, Wagstaff L, Cooper O, Weijer C. & Münsterberg A. The migration of paraxial and lateral plate mesoderm cells emerging from the late primitive streak is controlled by different Wnt signals. BMC Dev Biol 8, 63 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barnes RM, Firulli BA, Conway SJ, Vincentz JW & Firulli AB Analysis of the Hand1 cell lineage reveals novel contributions to cardiovascular, neural crest, extra-embryonic, and lateral mesoderm derivatives. Dev Dyn 239, 3086–3097 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tang X. et al. Transcriptome regulation and chromatin occupancy by E2F3 and MYC in mice. Sci Data 3, 160008 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Y, Bailey D, Yang P, Kim E. & Que J. The development and stem cells of the esophagus. Development 148 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kishimoto K. et al. Bidirectional Wnt signaling between endoderm and mesoderm confers tracheal identity in mouse and human cells. Nat Commun 11, 4159 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loh KM et al. Efficient endoderm induction from human pluripotent stem cells by logically directing signals controlling lineage bifurcations. Cell Stem Cell 14, 237–252 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Loh KM et al. Mapping the pairwise choices leading from pluripotency to human bone, heart, and other mesoderm cell types. Cell 166, 451–467 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Protze SI, Lee JH & Keller GM Human pluripotent stem cell-derived cardiovascular cells: from developmental biology to therapeutic applications. Cell Stem Cell 25, 311–327 (2019). [DOI] [PubMed] [Google Scholar]
- 25.Thomson JA et al. Embryonic stem cell lines derived from human blastocysts. Science 282, 1145–1147 (1998). [DOI] [PubMed] [Google Scholar]
- 26.Edwards NA et al. Developmental basis of trachea-esophageal birth defects. Dev Biol 477, 85–97 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kishimoto K. & Morimoto M. Mammalian tracheal development and reconstruction: insights from in vivo and in vitro studies. Development 148 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eicher AK et al. Functional human gastrointestinal organoids can be engineered from three primary germ layers derived separately from pluripotent stem cells. Cell Stem Cell (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takebe T. et al. Massive and Reproducible Production of Liver Buds Entirely from Human Pluripotent Stem Cells. Cell Rep 21, 2661–2670 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Coll M. et al. Generation of hepatic stellate cells from human pluripotent stem cells enables in vitro modeling of liver fibrosis. Cell Stem Cell 23, 101–113.e107 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Koui Y. et al. An in vitro human liver model by iPSC-derived parenchymal and non-parenchymal cells. Stem Cell Reports 9, 490–498 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Asahina K, Zhou B, Pu WT & Tsukamoto H. Septum transversum-derived mesothelium gives rise to hepatic stellate cells and perivascular mesenchymal cells in developing mouse liver. Hepatology 53, 983–995 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dye BR et al. In vitro generation of human pluripotent stem cell derived lung organoids. Elife 4 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen YW et al. A three-dimensional model of human lung development and disease from pluripotent stem cells. Nat Cell Biol 19, 542–549 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dye BR et al. A bioengineered niche promotes in vivo engraftment and maturation of pluripotent stem cell derived human lung organoids. Elife 5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Snowball J, Ambalavanan M, Whitsett J. & Sinner D. Endodermal Wnt signaling is required for tracheal cartilage formation. Dev Biol 405, 56–70 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The main data discussed in this protocol were generated as part of the studies published in the supporting primary research papers14. The raw datasets of qRT-PCR and immunostaining have been deposited in the online open access figshare repository (10.6084/m9.figshare.19723189). The published mouse foregut scRNA-seq data used to infer the signaling roadmap is available from the Gene Expression Omnibus (GEO) database under accession code: GSE136689. The scRNA-seq data can also be explored at https://research.cchmc.org/ZornLab-singlecell/.